

Abstract
Plasma membrane Ca2+ ATPase 2 (PMCA2) is a fast, highly effective mechanism to control resting cytosolic Ca2+ and Ca2+ excursions in neurons and other excitable cells. The strong expression of PMCA2 in the cerebellum and the cerebellar behavioral deficits presented by PMCA2−/− knock-out mice all point to its importance for cerebellar circuit dynamics. Here, we provide direct functional evidence for the influence of presynaptic PMCA2-mediated Ca2+ extrusion for short-term plasticity at cerebellar parallel fiber to Purkinje neuron synapses. Dramatic structural alterations to the Purkinje neurons in the absence of PMCA2 also suggest a strong influence of this fast PMCA2 isoform for development and maintenance of cerebellar function.
Keywords: plasma membrane Ca2+ ATPase, synapse, cerebellum, parallel fibers, plasticity, presynaptic
Introduction
Plasma membrane Ca2+ ATPases (PMCAs), a family of P-type Ca2+ ATPases, extrude Ca2+ from the cytosol against its inward gradient using energy derived from ATP hydrolysis (Carafoli, 1992). Their high affinity for Ca2+ makes them a highly efficient route for Ca2+ efflux even at intracellular Ca2+ levels ([Ca2+]i) close to rest, but also during [Ca2+]i transients (Thayer et al., 2002).
There are four PMCA isoforms, PMCAs 1–4. Each is the product of a different gene and all are distributed in a cell-specific manner. PMCAs 2 and 3 are enriched within excitable cells where their faster activation and extrusion rates, compared with the more ubiquitously expressed PMCA isoforms 1 and 4 (Brini et al., 2003), are ideally suited to control fast [Ca2+]i dynamics.
This is particularly true in the cerebellum where the predominant PMCA isoforms 2 and 3 are expressed throughout the molecular (ML) and granule cell (GC) layers (Burette et al., 2003). Furthermore, the importance of PMCA2 for cerebellar function is indicated from studies on the PMCA2 knock-out mouse. In addition to hearing loss, these mice exhibit vestibular and gait abnormalities and a reduced thickness of the cerebellar ML (Kozel et al., 1998).
As a first step toward a mechanistic understanding of the specific role for PMCA2 in cerebellar function, we investigated the well characterized parallel fiber (PF) to Purkinje neuron (PF–PN) synapse. This synapse has been used to develop the “residual Ca2+ hypothesis” (Katz and Miledi,1968; Zucker and Regehr, 2002) to explain its pronounced paired-pulse facilitation (PPF) and complex [Ca2+]i dynamics (Atluri and Regehr, 1996). Previous studies indicate the importance of presynaptic Ca2+ extrusion by the high capacity, plasmalemmal Na+/Ca2+ exchangers (NCX) for the control of residual [Ca2+]i and synaptic transmission at this and other synapses (Regehr, 1997; Jeon et al., 2003; Kim et al., 2005). In contrast, the contribution of higher affinity, lower capacity Ca2+ extrusion by PMCAs is less well characterized.
Here, using the PMCA2 knock-out mouse, we have directly determined the role of PMCA2-mediated Ca2+ extrusion for short-term plasticity at the PF–PN synapse. PMCA2 was richly expressed at these synapses in wild-type (WT) cerebellum, both at the PF presynaptic terminals and within PN spines. Absence of PMCA2 in the PMCA2 knock-out mouse led to enhanced and prolonged PPF that coincided with an enhanced time for recovery of the PF presynaptic Ca2+ transient. Together, these results strongly supported a role for PMCA2-mediated Ca2+ extrusion from the PF terminal to control presynaptic residual [Ca2+]i and thereby influence PF–PN synapse behavior. Unexpectedly, lack of PMCA2 also dramatically altered the morphology of the PNs, a feature that will undoubtedly alter their postsynaptic integration properties.
Our results begin to explain how loss of a specific isoform of a Ca2+ extrusion protein at both presynaptic and postsynaptic sites can influence cerebellar circuit dynamics and function.
Materials and Methods
Mice.
PMCA2 knock-out mice were a generous gift from Dr. Gary Shull (University of Cincinnati, Cincinnati, OH) (Kozel et al., 1998). Two colonies based on FVBN and Swiss B6 background strains were used interchangeably throughout the study without any detectable differences. The genotype of each mouse was determined, preweaning as described previously (Kozel et al., 1998). All animal husbandry and procedures followed RIKEN Brain Science Institute (Saitama, Japan)-approved guidelines.
Biocytin immunohistochemistry.
PNs were filled with biocytin (Sigma, Poole, UK) and processed (Uusisaari et al., 2007) before visualization with a C1si spectral confocal (Nikon, Tokyo, Japan). Images were collected at 1 μm depth intervals, reconstructed, and analyzed with ImagePro 6 (Media Cybernetics, Bethesda, MD).
Electrophysiology.
Two-hundred seventy-micrometer-thick sagittal slices or 300-μm-thick coronal slices were prepared from age-matched littermate male and female WT and homozygous (PMCA2−/−) and heterozygous (PMCA2+/−) mice (on the same day), 21–26 d old, after rapid anesthesia with halothane as described previously (Matsukawa et al., 2003). Slices were maintained at 24°C, or temporarily at 35°C (TC324B; Harvard Apparatus, Hamden, CT) in a flow (3 ml/min) of artificial CSF (aCSF), equilibrated with 95% O2 and 5% CO2, containing (in mm) 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.3 MgCl2, 2.4 CaCl2, 26 NaHCO3, and 10 glucose. Whole-cell recordings from PN soma under visual control used glass electrodes (3–4 MΩ) containing (in mm) 5 KCl, 20 KOH, 3.48 MgCl2, 4 NaCl, 120 K gluconate, 10 HEPES, 8 sucrose, 10 EGTA, 4 Na2ATP, and 0.4 Na3GTP or, where biocytin was included, 10 NaCl, 10 NaCl, 136 K gluconate, 10 HEPES, 0.2 EGTA, 4 Na2ATP, 4 MgATP, 5 glutathione, and 8 biocytin, pH 7.3, with osmolarity 305 mOsm/L. Voltage-clamp (Axopatch 200B; Molecular Devices, Menlo Park, CA) maintained cells at −65 mV, less than −0.5 nA holding current. Pairs of EPSCs in response to PF stimulation (∼10 μA) at 0.03 Hz with varying intervals between the two stimuli, used a glass aCSF-filled electrode (200–600 kΩ) placed in the outer ML. All off-line analysis used pClamp 9 (Molecular Devices) and Prism version 3.03 (GraphPad Software, San Diego, CA) for statistical comparisons and curve fitting.
Ca2+ Imaging.
Ca2+ green dextran (Kd, 591 nm) (Invitrogen, Eugene, OR) was introduced into PFs using 2–3 MΩ glass electrodes [filled with 5% Ca2+ green dextran (10,000 molecular weight) in 126 mm NaCl] with 900 current pulses (50–70 μA), 5 Hz, to the outer ML before incubation in aCSF (as above) at 32°C for <2 h (Beierlein et al., 2004).
Using a 10× water-immersion objective and excitation light (450–490 nm) (halogen lamp; Moritex, Tokyo, Japan) we selected recording sites in rapidly perfused slices at 35°C (see above) where the calcium indicator dye was loaded into a bundle of PFs. A stimulating electrode (as above, 130–300 kΩ) remote from the depot of dye delivered a single stimulus to the PFs. Emitted light (>525 nm) averaged across 20 stimulations was collected with a cooled CCD camera (Sensicam; PCO, Kelheim, Germany) at 100 Hz (8 × 8 binning), with custom-written macros (ImagePro; Media Cybernetics) and a linear correction for dye bleaching (<0.2%/s) applied. Exponential decay fits of the fluorescence transients used Origin software (OriginLab, Northampton, MA) and statistical comparisons were made with Prism.
Solutions.
EPSCs were evoked in the presence of 50 μm picrotoxin (Sigma) and prevented by 20 μm 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX; Tocris Cookson, Bristol, UK) whereas presynaptic Ca2+ transients were insensitive to NBQX (n = 3). Carboxyeosin (CE; Invitrogen) and picrotoxin were stored as a 1000× stock solution in DMSO and ethanol, respectively.
Results
PPF at the PF–PN synapse is enhanced in PMCA2−/− mouse cerebellum
Extensive studies within the PF–PN presynapse have indicated that transmitter release (Mintz et al., 1995) and the time course of PPF (Atluri and Regehr, 1996) are directly related to the recovery kinetics of the presynaptic [Ca2+]i transient. The presence of PMCA2 within PF presynaptic terminals ought to influence their [Ca2+]i dynamics and, in doing so, should influence PPF. In the absence of isoform specific inhibitors of PMCA2, we took advantage of a molecular deletion of the PMCA2 gene (Atp2b) in the PMCA2 knock-out mouse (Kozel et al., 1998). We compared the amplitude and time course of PPF of EPSCs in PNs from WT and PMCA2−/− mice as an indirect indicator of the rate of the recovery of the PF presynaptic residual [Ca2+]i.
Robust PPF (Fig. 1A) [seen as an increase in the paired-pulse ratio (PPR)] of evoked EPSCs occurred in WT PNs (19 cells, six animals) after paired stimulation over interstimulus intervals (ISIs) of between 25 and 250 ms. PPF was significantly enhanced in PNs from PMCA2−/− slices (19 cells, six animals) (Fig. 1B, compare filled (WT) versus open (PMCA2−/− cells)] (p < 0.005, one-way ANOVA). The enhanced PPF occurred in the absence of any detectable differences in the amplitude of the first EPSC or the stimulus intensity required to evoke them (p = 0.58 and 0.57, respectively, t tests). The enhanced PPF was predicted by the residual Ca2+ hypothesis (Atluri and Regehr, 1996; Zucker and Regehr, 2002) if PMCA2 contributed to Ca2+ extrusion from the terminal. Moreover, absence of PMCA2-mediated Ca2+ extrusion should prolong [Ca2+]i within the PF terminal and therefore not only enhance the amount of PPF, but also slow the time for its recovery (Atluri and Regehr, 1996). The time course of PPF recovery was fitted best with a two-phase exponential decay (Zucker and Regehr, 2002) and both phases of PPF decay were slower in the PMCA2−/− synapses compared with WT. From the mean data (Fig. 1B), the faster, first decay half-time, T1, was 19 ms in WT cells compared with 49 ms in the PMCA2−/− cells. Likewise, the slower, second decay half-time, T2, was 122 ms in the WT cells and increased to 366 ms in the PMCA2−/− cells. Amplitude normalized paired-pulse ratios clearly showed the longer time required for the recovery of PPF at PMCA2−/− synapses compared with WT (Fig. 1C). Figure 1D shows the significant differences between the mean PPF T1 and T2 values from individual WT and PMCA2−/− synapses (p < 0.005 and p < 0.05, both t tests, n = 17). A separate set of experiments conducted at near-physiological temperature, 35°C (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), showed a similar result.
Figure 1.

PF-evoked EPSCs in PNs from PMCA2−/− mice exhibited enhanced PPF that recovered more slowly than WT PPF. A, PPF of the PF input to PNs from WT (left) and PMCA2−/− (right) mice, at varying ISIs. Traces represent responses averaged over 17 cells. B shows the average PPR as a function of ISI for all cells, WT (filled symbols and lines) and PMCA2−/− (open symbols), where dotted lines show extrapolated values determined from the exponential fits. C shows recovery of amplitude normalized PPR for WT and PMCA2−/− (filled and open symbols and lines, respectively). Recovery of PPR fitted with a double exponential in all cases. The first half-time for PPR decay, T1, increased from 21 to 51 ms and the second, slower half-time for PPR decay, T2, increased from 286 to 650 ms. D shows means from individual recovery phases of PPR (T1 and T2) for WT (filled bars) and PMCA2−/− (open bars). *p < 0.05; **p < 0.01. All values are mean ± SEM.
The enhanced amplitude and slowed recovery of PPF at PF–PN synapses in PMCA2−/− mice indicated a functional contribution from PMCA2-mediated efflux for short-term plasticity at this synapse. However, to be certain that our findings were specific and not simply an adaptive feature to the loss of PMCA2, we used carboxyeosin, a moderately specific inhibitor of P-type ATPases (Gatto et al., 1995). As shown in Figure 2A, 10 μm CE enhanced the amplitude of PPF at the PF–PN synapse and slowed its recovery in WT PNs (two-way ANOVA, p < 0.005; n = 3), but had no effect on PF–PN PPF in PMCA2−/− PNs (two-way ANOVA, p = 0.58; n = 3) (Fig. 2B). CE did not significantly alter the amplitude of the first EPSC in WT or PMCA2−/− cells (t test, p = 0.15; n = 6). Because CE replicated, at least qualitatively, the observed effects of PMCA2 (Atp2b) knock-out, PMCA2 likely contributes to PF presynaptic Ca2+ dynamics. We therefore sought to test this directly by recording presynaptic stimulus-evoked Ca2+ transients in WT and PMCA2−/− PF populations.
Figure 2.

Carboxyeosin, an inhibitor of PMCAs, enhanced PPF of PF-evoked EPSCs in PNs from WT but not PMCA2−/− mice. A, PPF of the PF input to a representative PN from a WT mouse, at varying ISIs, before (left) and after (middle) treatment with 10 μm CE. Note the increase in amplitude of the second and subsequent EPSCs compared with control. The far right panel shows the increase in the average PPR as a function of ISI for all WT cells, before (filled squares) and after treatment with CE (filled triangles) (n = 3). The lines represent double exponential fits to the mean data, and the dotted lines show extrapolated values determined from the exponential fits. CE also slowed the recovery of the PPF in WT cells. The first half-time for PPR decay, T1, in the WT cells increased from 25 to 41 ms, and the second, slower half-time for PPR decay, T2, increased from 102 to 204 ms in the presence of CE. CE application lasted 20 min. B, Results from the same experiment as in A, but from cells from PMCA2−/− mice, where the mean PPF (n = 3; far right) was not further enhanced by the presence of CE (open squares vs open triangles), nor its recovery slowed. The left and middle panels show traces from a representative PMCA2−/− cell. All values are means ± SEM.
The recovery of stimulus-evoked Ca2+ transients was slowed in PF presynaptic terminals from PMCA2−/− mice
We loaded bundles of PFs with the calcium indicator Ca2+ green dextran to image the dynamics of presynaptic Ca2+ transients (Fig. 3A). After a single stimulation to a PF bundle (Fig. 3A, asterisk, top left), presynaptic Ca2+ rapidly increased to similar peak levels in WT and PMCA2−/− bundles (p = 0.97, t test; n = 7) and then recovered to baseline. The kinetics of the recovery of normalized, single-stimulation Ca2+ transients at 35°C in six slices from three WT animals and from seven slices from three PMCA2−/− animals were fitted best with a double exponential (Fig. 3B). The slower time constant of the Ca2+ transient recovery, T2, was much larger in the PMCA2−/− terminals compared with WT (Fig. 3C) (p < 0.01, t test; n = 6). These direct Ca2+ measurements identified a clear contribution from PMCA2-mediated Ca2+ efflux to the rate of recovery of residual presynaptic PF [Ca2+]i and were similar to previous findings in another presynaptic compartment, the calyx of Held, where CE most effectively slowed the slower phase of the Ca2+ transient recovery (Kim et al., 2005). In turn, the demonstration of a PMCA2-dependent component to the recovery of presynaptic Ca2+ transients provided an explanation for the enhanced amplitude and slowed recovery of PPF at the PMCA2−/− PF–PN synapses.
Figure 3.
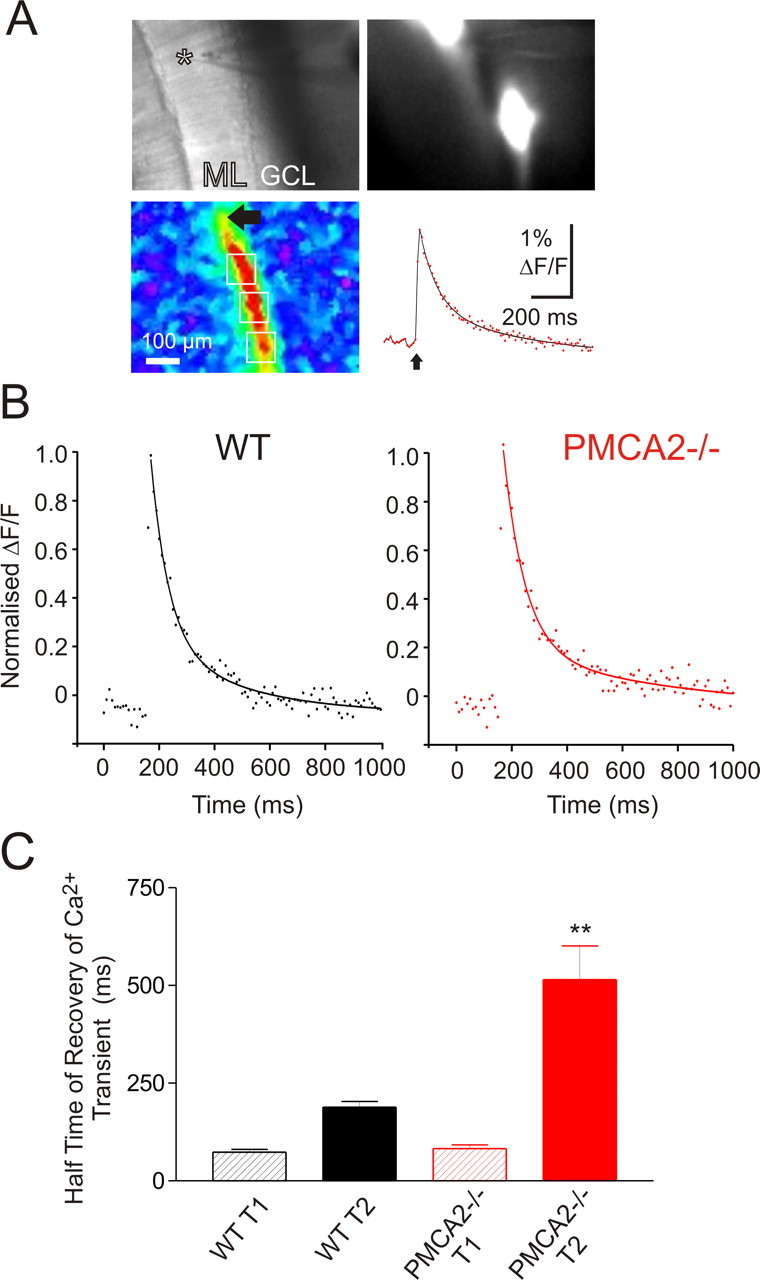
Recovery of presynaptic PF [Ca2+]i transients was slowed in slices from PMCA2−/− mice. A, PF stimulation-induced presynaptic Ca2+ transients. Top left, Transmission image. Asterisk denotes tip of stimulation electrode. GCL, Granule cell layer. Top right, Resting fluorescence image. Note two dye deposits and resting fluorescence of associated PF bundles. Bottom left, Pseudocolored map of peak ΔF/F signal, the baseline normalized change in fluorescence, induced by a single electrical stimulation of PFs. Bottom right, The time course of the Ca2+ transients extracted from regions of interest (white boxes, bottom left) along the PF bundle “beam”-shaped responsive area. Ca2+ signals are averages >20 stimulations. B, Time course of the average, normalized PF Ca2+ transient for WT (left, in black) and PMCA2−/− (right, in red). Each point represents the average response obtained from n = 6 slices normalized to their peak amplitude determined by double exponential fits (lines). C, Recovery time constants from double exponential fits. Values are mean ± SEM of the individual fast (T1) and slow (T2) time constants of recovery fitted individually to all PF presynaptic Ca2+ transients. **p < 0.01.
PNs from PMCA2−/− mouse cerebellum show structural differences compared with WT
Although the amplitude of PF–PN EPSCs and Ca2+ transients were similar between WT and PMCA2−/− slices, PNs from PMCA2−/− slices exhibited an increased input resistance (mean values increased from 47.1 ± 6.1 MΩ in WT PNs to 135.3 ± 18.2 MΩ in PMCA2−/− PNs; p < 0.00005, t test; n = 23) and strikingly faster EPSC decays (mean values of the EPSC decay time constant determined from a standard exponential fit decreased from 14.1 ± 1.0 in WT PNs to 5.6 ± 0.4 ms in PMCA2−/− PNs; p < 0.0001, t test; n = 18) (Fig. 1A). Because changes in EPSC waveform can be caused by alterations of dendritic filtering or by morphological constrains that determine diffusion of synaptically released glutamate (Barbour et al., 1994; Takahashi et al., 1996), we reconstructed individual PNs filled with biocytin (Fig. 4) and detected a clear difference between the overall shape of the WT (nine cells, three animals) and PMCA2−/− (11 cells, three animals) PNs. The PNs from the PMCA2−/− mice presented a smaller cell body and a more disordered dendritic tree with excessive fine branching at their lateral extremities that, in contrast to the WT PNs, rarely reached the pial surface (Fig. 4B).
Figure 4.
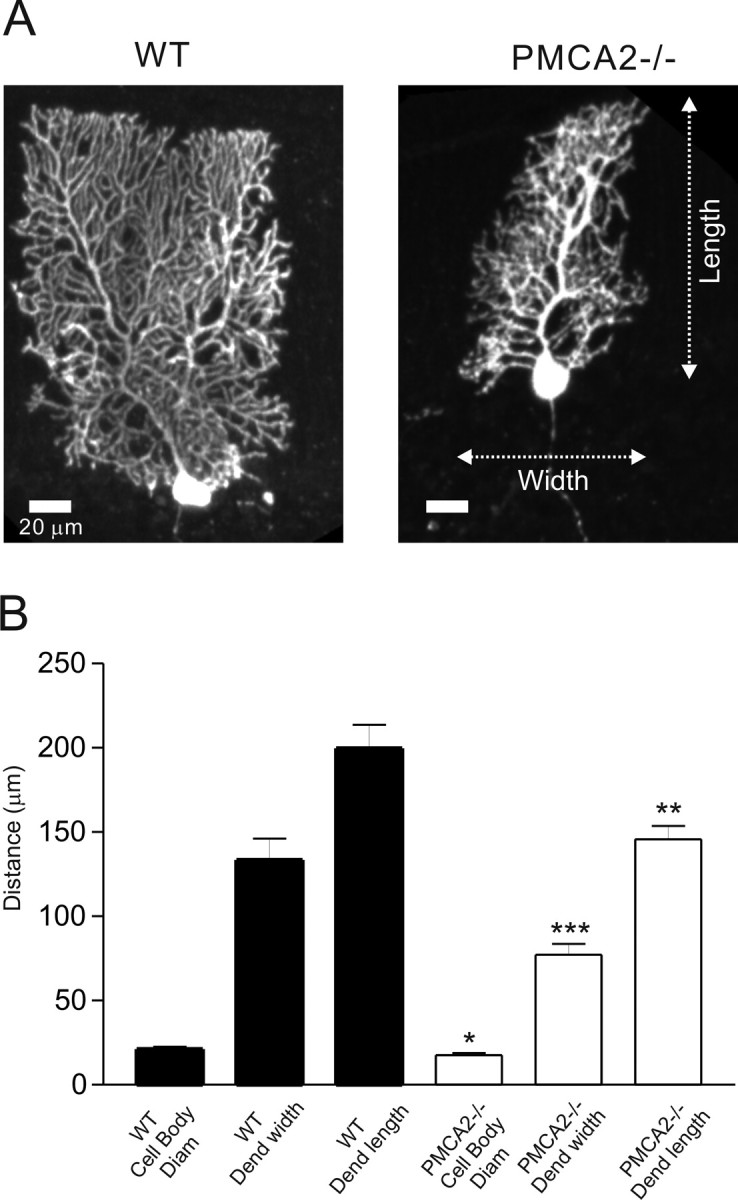
Purkinje neuron morphology was altered in PMCA2−/− mice. A, representative WT and PMCA2−/− PNs. B, Basic morphological measurements from n = 20 reconstructed cells; mean values ± SEM are *p < 0.05, **p <0.01, and ***p < 0.001.
Discussion
PMCA2 is highly expressed throughout the cerebellum and particularly within the PN cell body membrane where it contributes to ∼20% of Ca2+ extrusion during large excursions in [Ca2+]i (Fierro et al., 1998). The high turnover, high affinity, and rapidly activating properties of PMCA2 (Brini et al., 2003) led us to propose a role for this PMCA for efficient [Ca2+]i homeostasis in presynaptic terminals.
We investigated the functional significance of presynaptic PMCA2 by taking advantage of the PMCA2 knock-out mouse and the well studied phenomenon of PPF at the PF–PN synapse. At this synapse, the recovery time course of PPF coincides with the recovery of the residual component of the presynaptic [Ca2+]i transient (Atluri and Regehr, 1996) within the PF terminal. Therefore, if PMCA2 normally accelerated the recovery of terminal [Ca2+]i, we predicted that its absence should prolong the presence of residual [Ca2+]i and so increase the magnitude and duration of PPF. Consistent with this hypothesis, absence of PMCA2 enhanced PPF and also slowed the rate of its recovery and the rate of recovery of the PF presynaptic Ca2+ transient. Interestingly, the time scale over which these changes occurred was similar to those predicted from modeled dynamics of [Ca2+]i and [Na+]i within the PF terminal, where PMCA-mediated Ca2+ extrusion contributed rather early after a peak in cytosolic [Ca2+]i when the high capacity NCX was in its reverse mode (Regehr, 1997). Previously, NCX in forward mode, Ca2+ extrusion, was shown to contribute to the recovery of the presynaptic Ca2+ transient in the calyx of Held (Kim et al., 2005) and in the NCX2 knock-out mouse, where its absence enhances PPF at hippocampal synapses (Jeon et al., 2003). Interestingly, both studies revealed contributions from NCX to the function of these very different synapses on time scales similar to those observed here. However, knock-out of the K+ dependent Na+/Ca2+ exchanger (NCKX2) also impaired cerebellar motor learning, but not hippocampal synapse PPF (Li et al., 2006), and indicates that interplay between Ca2+ extrusion mechanisms is synapse dependent.
Importantly, lack of PMCA2 at the PF–PN synapse clearly influenced the magnitude and time course of PPF and, although the most plausible explanation for this was the slowed recovery of [Ca2+]i within the PF terminals, we recognized that our results could also reflect a reduction in release probability at this synapse (Mintz et al., 1995), perhaps via adaptive changes within the PMCA2−/− mouse. This might explain why we paradoxically observed an enhanced extrapolated PPF value at time 0 (Fig. 1B, right) when slow removal of [Ca2+]i from the terminal by PMCA2 would not be expected to contribute. However, because pharmacological inhibition of PMCA similarly enhanced PPF and slowed its recovery in WT, but not PMCA2−/− synapses, adaptive changes seem less likely. Instead, we may speculate that removal of PMCA2 activity at these synapses causes a small elevation in basal [Ca2+]i that alters the capacity and kinetics of [Ca2+]i buffering within the terminal, but is insufficient to significantly alter evoked release. Further understanding of the complex interplay between Ca2+ extrusion and buffering mechanisms that determine [Ca2+]i dynamics within the PF terminal would benefit greatly from a detailed modeling approach. Notwithstanding the lack of a complete set of parameters required for a detailed realistic model of PF [Ca2+]i dynamics, our results have indicated a clear contribution of PMCA2 to presynaptic Ca2+ handling and short-term plasticity at the PF–PN synapse.
The rich expression of PMCA2 within PNs and their dendritic spines (Burette et al., 2003) raises the possibility that postsynaptic Ca2+ homeostasis and downstream signaling may be impaired in the absence of PMCA2. Indeed, the slower PMCA2 “b” splice variant interacts with the postsynaptic density protein 95 in vitro (DeMarco and Strehler, 2001) and, previously, PMCA activity has been shown to influence Ca2+ dynamics in hippocampal spines (Scheuss et al., 2006). Somewhat to our surprise, reconstruction of individual PNs from the PMCA2−/− mice did reveal a gross and dramatic alteration in their dendritic tree, a feature that must surely influence their postsynaptic integration properties. Indeed, EPSCs decayed faster in PMCA2−/− PNs, as predicted by alterations in dendritic filtering or morphological constraints that determine diffusion of synaptically released glutamate (Barbour et al., 1994; Takahashi et al., 1996), but there were no other detectable changes in the postsynaptic responses of PMCA2−/− PNs.
However, any changes that would depend on postsynaptic calcium dynamics were likely masked by the high EGTA concentration in our recording electrode. A number of possibilities exist to explain how lack of PMCA2 could have such a dramatic effect on PN structure. The disruption of the plasticity and timing of the presynaptic PF input to the PNs, as we have shown here, could of itself directly influence PN development (Landis and Mullen, 1978). The high levels of PMCA2 in the PN also suggest an important contribution from Ca2+ extrusion during the developmental shaping of the PN dendritic tree, especially at a time of intense synaptogenesis, when PMCA2 levels rise (Jensen et al., 2004). Alternatively, recent reports that the PMCA2−/− cerebellum expresses fewer homer/metabotropic glutamate receptor 1 (mGluR1)/IP3-receptor (IP3R) complexes (Kurnellas et al., 2006) and that loss of IP3–R-mediated signaling through mGluR1 activation in GCs prevents BDNF-mediated outgrowth of PNs (Hisatsune et al., 2006) provides a possible explanation for the stunted appearance of the PN dendritic tree in PMCA2−/− mice.
In summary, we demonstrated a specific, functional contribution to short-term cerebellar synapse plasticity from a fast Ca2+-extrusion mechanism, PMCA2, through its influence on presynaptic [Ca2+]i dynamics. Additional studies are required to clarify the functions of PMCA2 at postsynaptic compartments, whether as part of a signaling complex to influence dendritic expansion or purely as a Ca2+ extrusion protein.
Footnotes
This work was supported by Biotechnology and Biological Sciences Research Council Grant BBS/0338, the Physiological Society, the British Council, and RIKEN intramural funding. We thank all members of the Laboratory for Neuronal Circuit Dynamics, RIKEN Brain Science Institute for their valuable contributions to this work.
References
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gee KR, Martin VV, Regehr WG. Presynaptic calcium measurements at physiological temperatures using a new class of dextran-conjugated indicators. J Neurophysiol. 2004;92:591–599. doi: 10.1152/jn.00057.2004. [DOI] [PubMed] [Google Scholar]
- Brini M, Coletto L, Pierobon N, Kraev N, Guerini D, Carafoli E. A comparative functional analysis of plasma membrane Ca2+ pump isoforms in intact cells. J Biol Chem. 2003;278:24500–24508. doi: 10.1074/jbc.M300784200. [DOI] [PubMed] [Google Scholar]
- Burette A, Rockwood JM, Strehler EE, Weinberg RJ. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J Comp Neurol. 2003;467:464–476. doi: 10.1002/cne.10933. [DOI] [PubMed] [Google Scholar]
- Carafoli E. The Ca2+ pump of the plasma membrane. J Biol Chem. 1992;267:2115–2118. [PubMed] [Google Scholar]
- DeMarco SJ, Strehler EE. Plasma membrane Ca2+-atpase isoforms 2b and 4b interact promiscuously and selectively with members of the membrane-associated guanylate kinase family of PDZ (PSD95/Dlg/ZO-1) domain-containing proteins. J Biol Chem. 2001;276:21594–21600. doi: 10.1074/jbc.M101448200. [DOI] [PubMed] [Google Scholar]
- Fierro L, DiPolo R, Llano I. Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J Physiol (Lond) 1998;510:499–512. doi: 10.1111/j.1469-7793.1998.499bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto C, Hale CC, Xu W, Milanick MA. Eosin, a potent inhibitor of the plasma membrane Ca pump, does not inhibit the cardiac Na-Ca exchanger. Biochemistry. 1995;34:965–972. doi: 10.1021/bi00003a031. [DOI] [PubMed] [Google Scholar]
- Hisatsune C, Kuroda Y, Akagi T, Torashima T, Hirai H, Hashikawa T, Inoue T, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor type 1 in granule cells, not in Purkinje cells, regulates the dendritic morphology of Purkinje cells through brain-derived neurotrophic factor production. J Neurosci. 2006;26:10916–10924. doi: 10.1523/JNEUROSCI.3269-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TP, Buckby LE, Empson RM. Expression of plasma membrane Ca2+ ATPase family members and associated synaptic proteins in acute and cultured organotypic hippocampal slices from rat. Brain Res Dev Brain Res. 2004;152:129–136. doi: 10.1016/j.devbrainres.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H, Shin HS. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron. 2003;38:965–976. doi: 10.1016/s0896-6273(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK, Lee SH. Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci. 2005;25:6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T, Duffy JJ, Doetschman T, Miller ML, Cardell EL, Shull GE. Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+-ATPase isoform 2. J Biol Chem. 1998;273:18693–18696. doi: 10.1074/jbc.273.30.18693. [DOI] [PubMed] [Google Scholar]
- Kurnellas MP, Lee AK, Li H, Deng L, Ehrlich DJ, Elkabes S. Molecular alterations in the cerebellum of the plasma membrane calcium ATPase 2 (PMCA2)-null mouse indicate abnormalities in Purkinje neurons. Mol Cell Neurosci. 2006;34:178–188. doi: 10.1016/j.mcn.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis SC, Mullen RJ. The development and degeneration of Purkinje cells in pcd mutant mice. J Comp Neurol. 1978;177:125–143. doi: 10.1002/cne.901770109. [DOI] [PubMed] [Google Scholar]
- Li XF, Kiedrowski L, Tremblay F, Fernandez FR, Perizzolo M, Winkfein RJ, Turner RW, Bains JS, Rancourt DE, Lytton J. Importance of K+-dependent Na+/Ca2+-exchanger 2, NCKX2, in motor learning and memory. J Biol Chem. 2006;281:6273–6282. doi: 10.1074/jbc.M512137200. [DOI] [PubMed] [Google Scholar]
- Matsukawa H, Wolf AM, Matsushita S, Joho RH, Knöpfel T. Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking potassium channels Kv3.1 and Kv3.3. J Neurosci. 2003;23:7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Regehr WG. Interplay between sodium and calcium dynamics in granule cell presynaptic terminals. Biophys J. 1997;73:2476–2488. doi: 10.1016/S0006-3495(97)78276-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Yasuda R, Sobczyk A, Svoboda K. Nonlinear [Ca2+] signaling in dendrites and spines caused by activity-dependent depression of Ca2+ extrusion. J Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Sarantis M, Attwell D. Postsynaptic glutamate uptake in rat cerebellar Purkinje cells. J Physiol (Lond) 1996;497:523–530. doi: 10.1113/jphysiol.1996.sp021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SA, Usachev YM, Pottorf WJ. Modulating Ca2+ clearance from neurons. Front Biosci. 2002;7:d1255–d1279. doi: 10.2741/A838. [DOI] [PubMed] [Google Scholar]
- Uusisaari M, Obata K, Knöpfel T. Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol. 2007;97:901–911. doi: 10.1152/jn.00974.2006. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]