

Abstract
Type I lissencephaly, a genetic disease characterized by disorganized cortical layers and gyral abnormalities, is associated with severe cognitive impairment and epilepsy. Two genes, LIS1 and doublecortin (DCX), have been shown to be responsible for a large proportion of cases of type I lissencephaly. Both genes encode microtubule-associated proteins that have been shown to be important for radial migration of cortical pyramidal neurons. To investigate whether DCX also plays a role in cortical interneuron migration, we inactivated DCX in the ganglionic eminence of rat embryonic day 17 brain slices using short hairpin RNA. We found that, when DCX expression was blocked, the migration of interneurons from the ganglionic eminence to the cerebral cortex was slowed but not absent, similar to what had previously been reported for radial neuronal migration. In addition, the processes of DCX-deficient migrating interneurons were more branched than their counterparts in control experiments. These effects were rescued by DCX overexpression, confirming the specificity to DCX inactivation. A similar delay in interneuron migration was observed when Doublecortin-like kinase (DCLK), a microtubule-associated protein related to DCX, was inactivated, although the morphology of the cells was not affected. The importance of these genes in interneuron migration was confirmed by our finding that the cortices of Dcx, Dclk, and Dcx/Dclk mutant mice contained a reduced number of such cells in the cortex and their distribution was different compared with wild-type controls. However, the defect was different for each group of mutant animals, suggesting that DCX and DCLK have distinct roles in cortical interneuron migration.
Keywords: interneurons, DCX, DCLK, lissencephaly, tangential migration, RNAi
Introduction
The development of the cerebral cortex, the largest and most complex part of the mammalian brain, involves the highly coordinated generation, migration, and differentiation of its constituent neurons: the excitatory pyramidal cells that project to distant targets, and the inhibitory nonpyramidal cells, the cortical interneurons. Pyramidal neurons are generated in the germinal ventricular zones lining the lateral ventricles and migrate radially to take their positions in the cortex in an orderly manner (for review, see Rakic, 1990; Nadarajah and Parnavelas, 2002; Kriegstein and Noctor, 2004). The GABA-containing cortical interneurons originate in the ganglionic eminences (GE) of the ventral telencephalon and follow tangential migratory routes to populate the cortex (for review, see Marin and Rubenstein, 2003; Metin et al., 2006).
In humans, defects in radial migration result in lissencephaly (smooth brain). Type I lissencephaly is typically associated with mental retardation and intractable epilepsy and is characterized by massive disorganization of neurons throughout the cortex (Dobyns et al., 1996). Four genes have so far been shown to be responsible for most cases of type I lissencephaly. They are the following: LIS1, doublecortin (DCX), reelin (RELN), and Aristaless-related homeobox gene (ARX) (Reiner et al., 1993; des Portes et al., 1998; Gleeson et al., 1998; Hong et al., 2000; Kitamura et al., 2002).
Although humans with DCX mutations show alterations in cortical lamination, a genetic deletion in mice has shown a milder defect involving mainly the hippocampus (Corbo et al., 2002). In contrast, RNA interference (RNAi)-mediated knock-down of DCX in rats and mice showed impairment in radial migration of cortical neurons (Bai et al., 2003; Ramos et al., 2006). These studies suggested that other related genes might functionally compensate for DCX during brain development. One such homolog is Doublecortin-like kinase (DCLK). Two groups have recently shown that, although mice with a Dclk mutation do not have obvious cortical migration defects, mutant mice for both Dcx and Dclk demonstrate perinatal lethality, disorganized neocortical layering, and profound cytoarchitectural defects of the hippocampus caused by the disruption of radial neuronal migration. These results suggest that Dcx and Dclk have genetically compensatory roles in neuronal migration (Deuel et al., 2006; Koizumi et al., 2006a).
Here, we sought to determine whether DCX and DCLK also play a role in the migration of the cortical interneurons. Using short hairpin RNA (shRNA) to inactivate DCX in the GE of rat brain slices, we observed that the migration of interneurons into the cortex was impaired. This defect was rescued by coelectroporation with a DCX overexpression plasmid. In addition, DCX-deficient interneurons showed increased branching compared with their counterparts in control experiments. Similar results were obtained after inactivation of DCLK with a specific RNAi construct. The importance of these genes in interneuron migration was confirmed by examining the cortices of mutant mice for Dcx, Dclk, and both Dcx/Dclk during prenatal corticogenesis and at birth. This analysis showed a significant decrease in the number of interneurons and a change in their distribution compared with controls, but the defect was different for each group of mutant animals, suggesting that DCX and DCLK have different roles in cortical interneuron migration.
Materials and Methods
Dissociated cell cultures and transfections.
All animal procedures were performed in accordance with institutional guidelines. Brains of embryos, removed from pregnant Sprague Dawley albino rats at different stages during the last week of gestation [embryonic day 1 (E1); the day vaginal plug was found], were used for the preparation of brain slice cultures and for dissociated cortical cell cultures. The brains of E16 rat embryos were removed and their cortices were microdissected and placed in a solution containing 0.05% trypsin and 100 μg/ml DNase1 in Neurobasal medium. After incubation in 5% CO2 at 37°C for 15 min, they were washed in Neurobasal medium with 10% fetal bovine serum and dissociated with a fire-polished Pasteur pipette. The resulting suspension was centrifuged at 3000 rpm for 3 min. The cell pellet was resuspended in Neurobasal medium containing 2 mm l-glutamine, penicillin/streptomycin, and B27 (diluted 1:50). A total of 2 × 105 cells was plated onto each 13-mm-diameter glass coverslip coated with laminin and poly-l-lysine (100 μg/ml each). Dissociated cells were transfected at 2 d postplating, using Lipofectamine 2000 according to the guidelines of the manufacturer (Invitrogen, Paisley, UK). The cultures were processed for immunochemistry 48 h after transfection. All materials were purchased from Invitrogen, unless otherwise stated.
Electroporation of brain slices and RNAi.
We used the same shRNAs as those used by Bai et al. (2003) for DCX and Shu et al. (2006) for DCLK, which target the 3′-untranslated region (UTR) sequence of rat Dcx and Dclk genes, respectively. As controls, we used shRNAs that targeted the same region but contained two or three point mutations and, thus, did not affect DCX or DCLK mRNA stability. Annealed oligonucleotides were cloned in the psiStrike U6 promoter-driven vector according to manufacturer's instructions (Promega, Southampton, UK). Because the green fluorescent protein (GFP) gene in this vector is under the control of the cytomegalovirus (CMV) promoter, the expression in brain slices was too faint to be detected. To visualize transfected cells, the shRNA was cotransfected with the CAG-IRES-EGFP vector (a kind gift from Dr. M. Hoshino, Kyoto University, Kyoto, Japan). To ensure that all green cells expressed the shRNA, the plasmids were cotransfected in a ratio of 3:1.
Brains slices were cultured as described previously (Alifragis et al., 2004). Briefly, medial coronal brain slices, obtained from E16 or E17 rats, were placed onto nitrocellulose filters (0.45 μm; Millipore, London, UK). Fifty to 60 nl of plasmids (2 μg/μl) were then microinjected by pressure (General Valve Picospritzer, Fairfield, NJ) using pulled glass micropipettes (Drummond Scientific, Broomall, PA). To focally electroporate the GE, a vertically orientated platinum wire electrode (negative pole) was positioned over it, whereas a horizontal one was positioned underneath the nitrocellulose filter (positive pole). The electrodes were connected to a dual-pulse isolated stimulator (Intracel, Herts, UK). The parameters set on the electroporator (Electro Square Porator, ECM 830; BTX, San Diego, CA) were as follows: 100 Hz, 70 V, 5 ms pulse length, three to five pulses every 100 ms. Slices were then transferred in culture medium containing DMEM:F12 (Sigma, St. Louis, MO), 5% fetal bovine serum, 1× N-2, 100 μm l-glutamine, 2.4 g/L d-glucose (Sigma), 5 U/ml penicillin, and 5 mg/ml streptomycin in a humidified 5% CO2 incubator at 37°C. Two or 3 d later, they were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, overnight at 4°C, mounted onto glass slides and examined with a Leica (Nussloch, Germany) TCS SP2 confocal microscope. Images were processed using MetaMorph imaging software (Universal Imaging, West Chester, PA) and Adobe Photoshop.
Antibodies and immunohistochemistry.
The primary antibodies used were anti-DCX (goat; C-18; Santa Cruz Biotechnology, Santa Cruz, CA; 1:100), anti-calbindin (rabbit; Swant, Bellinzona, Switzerland; 1:1000), anti-GABA (rabbit; Sigma; 1:1000), anti-DCLK (rabbit; 1:100) (Lin et al., 2000), and anti-ARX (rabbit; 1:1000) (Poirier et al., 2004). To visualize DCX, immunolabeling was performed on free-floating sections. E16 rat brains were fixed in 4% paraformaldehyde overnight, embedded in 4% agarose, and sectioned at 60–80 μm with a Vibroslice (Campden Instruments, Leicester, UK). Sections were blocked with 10% normal goat serum in PBS and 0.2% Triton X-100 for 1 h, and incubated in primary antibody (see above) overnight at 4°C. After three washes in PBS, sections were incubated with FITC anti-goat or anti-rabbit rhodamine-conjugated secondary antibody (Invitrogen) in a 1:400 dilution at room temperature for 2 h, washed, and mounted with Citifluor antifading solution (Agar, Essex, UK). Images were acquired on a Leica TCS SP2 confocal system and reconstructed using MetaMorph imaging software (Universal Imaging).
Dclk−/−, Dcx−/y, and Dcx−/y;Dclk−/− mutant mice have been described previously (Corbo et al., 2002; Deuel et al., 2006). For interneuron analysis, E16 and postnatal day 0 (P0) brains were fixed with 4% paraformaldehyde, cryoprotected with 24% sucrose and sectioned at 15 μm using a cryostat (Bright Instruments, Huntingdon, UK). Immunoperoxidase labeling was performed as described previously (Andrews et al., 2006).
Results
DCX inactivation causes migration delay of interneurons in rat brain slices
Previous studies have reported the presence of DCX in migrating neurons during cortical development (Francis et al., 1999; Gleeson et al., 1999). To assess whether DCX is expressed in cortical interneurons, we used immunohistochemistry with a specific antibody against DCX in the developing rat cerebral cortex. Examination of sections taken during the period of early corticogenesis showed that DCX is abundantly expressed throughout the cortical mantle including a small number of cells in the ventricular zone (VZ) (Fig. 1A). Conspicuously present were a stream of horizontally oriented cells at the level of the lower intermediate zone (IZ) (Fig. 1A). Double-labeling experiments showed that many of these cells, as well as a number present in the VZ, also contained GABA (Fig. 1A1–A3), suggesting that DCX is expressed in migrating cortical interneurons. The DCX/GABA+ cells in the VZ represent most likely migrating interneurons that seek the cortical proliferative zone before moving to their positions in the cortical plate (Nadarajah et al., 2002).
Figure 1.
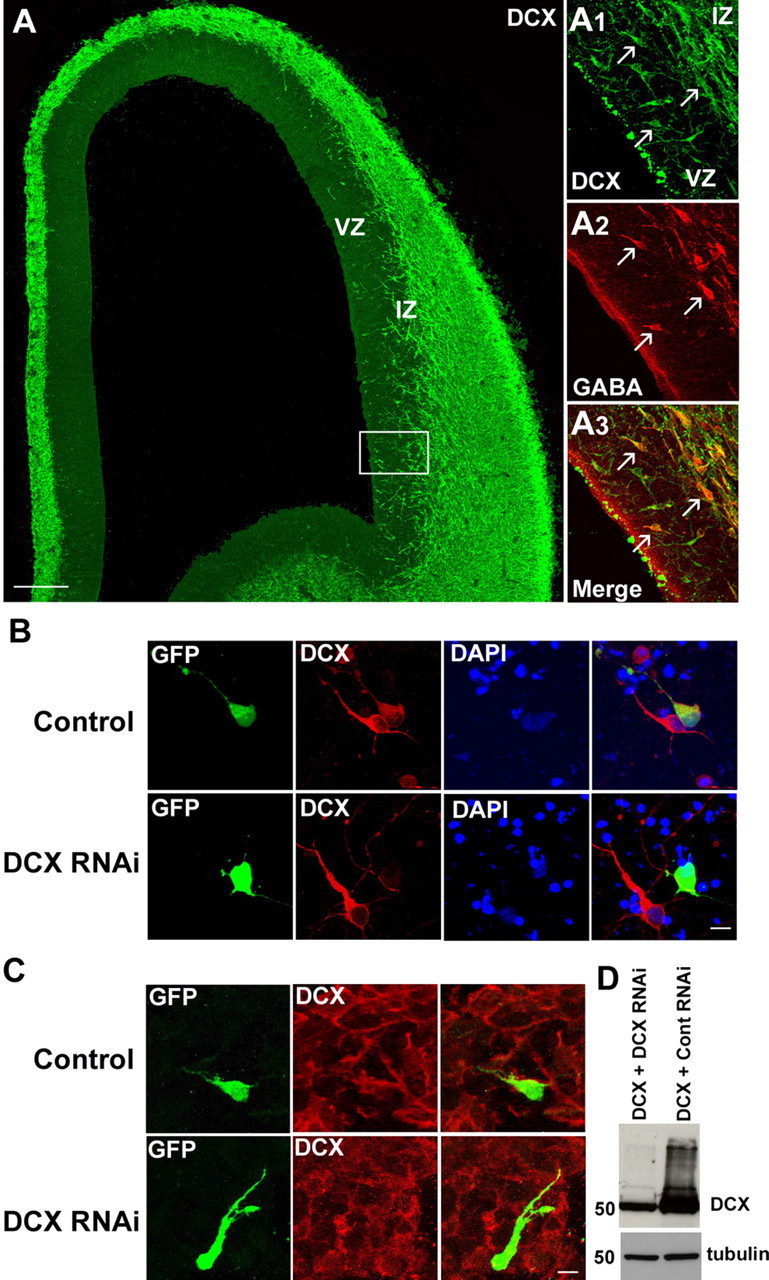
DCX protein expression in embryonic rat forebrain and DCX silencing using RNAi. A, DCX expression in a coronal section through an E16 rat brain. Labeling was abundant throughout the cortical mantle, including a stream of horizontally oriented cells at the level of the lower IZ and a small number of cells in the VZ. A1–A3 (boxed area in A), Double-labeling showed that many of the DCX+ cells in these zones were also positive for GABA. B, Representative confocal images of DCX protein expression in dissociated cortical cells transfected with either DCX or a control RNAi (3UTR3mhp). Each shRNA plasmid also encodes GFP, enabling identification of transfected cells. Forty-eight hours after transfection, cells expressing the control shRNA were still immunopositive for DCX, whereas most of the cells transfected with DCX RNAi were DCX deficient. DAPI, 4′,6′-Diamidino-2-phenylindole. C, After injection and focal electroporation with either DCX RNAi or a control RNAi together with CAG-IRES-EGFP, brain slices were immunolabeled using DCX antibody. A large majority of the cells transfected with DCX RNAi were negative for DCX labeling, further demonstrating the efficacy of the shRNA. D, COS-7 cells were transfected with a DCX-expressing plasmid and with either DCX RNAi or a control RNAi. Two days after transfection, cell lysates were subjected to Western blot analysis using DCX antibody. α-Tubulin detection shows that similar amounts of proteins were loaded on the gel. Scale bars: A, 100 μm; B, C, 20 μm.
To investigate the potential role of DCX in cortical interneuron migration, we examined the effect of DCX inactivation in the GE using RNAi. A recent study, which used in utero electroporation of shRNA to silence DCX in the developing rat cerebral cortex, showed severe radial migration defects (Bai et al., 2003). We focally electroporated the GE of rat brain slices with the same shRNAs as those used by these authors. To confirm that our construct was able to inactivate DCX, cells dissociated from the cortex of E16 rat embryos were transfected with the DCX or control (3UTRmhp) RNAi construct. Cells were fixed 48 h after transfection and immunolabeled with anti-DCX antibody. We found that all cells transfected with the mutated construct expressed DCX, but the vast majority of cells expressing the shRNA were negative for DCX immunolabeling (Fig. 1B). Similar results were obtained when sections electroporated with the RNAi constructs were processed for DCX immunohistochemistry (Fig. 1C). This was further verified by transfecting COS-7 cells with both a DCX construct, expressed under a CMV promoter, and either DCX RNAi or a control RNAi and detecting protein extracts with DCX antibodies (Fig. 1D).
After injection and focal electroporation of shRNA in the GE of E17 rats, we kept brain slices in culture for 2–3 d, and then analyzed the positions of GFP-labeled cells using confocal microscopy. We observed that control slices contained an abundance of cells scattered in the ventral forebrain and along previously described paths of migrating interneurons in the cortex. However, slices in which DCX was silenced contained numerous labeled cells in the ventral forebrain and an apparently reduced number of cells in the cortex compared with control slices (Fig. 2A). To quantify differences in the disposition of labeled neurons in the cortex of slices electroporated with DCX or control RNAi constructs, the cortical mantle was divided into three sectors as shown in Figure 2B. Estimates of the percentage of labeled cells contained in each sector showed that the most lateral one (sector I) contained nearly all DCX− cells compared with control (95.1 ± 2.8% for DCX RNAi and 85.1 ± 4.1% for the control), with a very small proportion present in sector II (4.9 ± 2.8 vs 14.5 ± 4.1%) and none in sector III (Fig. 2B). Similar results were obtained in electroporation experiments in slices taken from E16 rat brains (data not shown). The observation that, after 2–3 d in culture, nearly all labeled cells in the RNAi-treated slices were found in the lateral cortex, and very few were detected medial to it, suggests a delay in the migration of DCX-silenced interneurons into the cortex.
Figure 2.
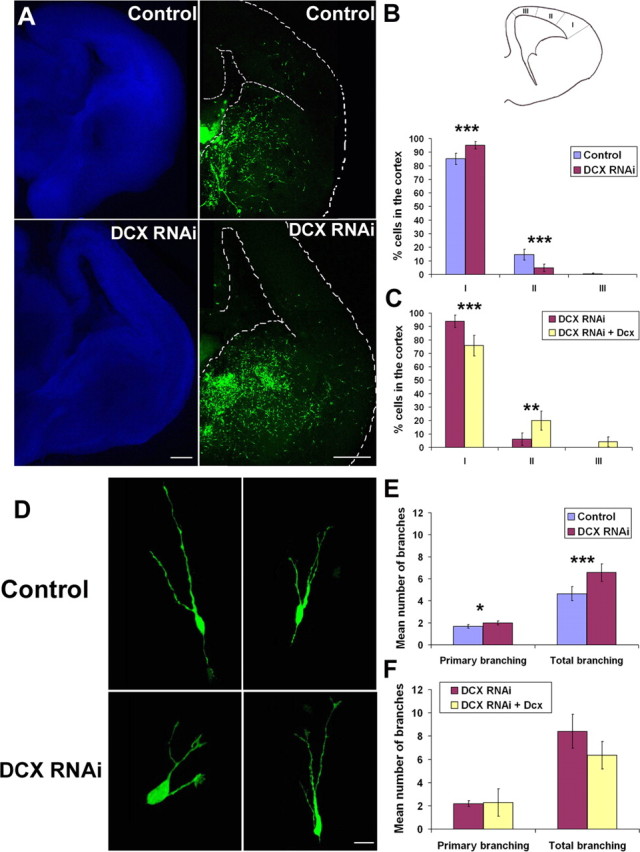
Effect of DCX RNAi on interneuron migration and morphology. A, The ganglionic eminence in an E17 rat brain slice was injected and focally electroporated with either DCX RNAi or a control RNAi (3UTR3mhp) together with CAG-IRES-EGFP. Cells transfected with DCX RNAi still migrated to the cortex, but were reduced in number. B, The cortex was divided into three sectors (I–III), and the number of GFP+ cells was counted in each sector. The result was expressed as a percentage of cells detected in each sector divided by the total number of cells that migrated into the cortex. The vast majority of DCX-silenced cells were located in sector I, whereas control cells were more spread in the three sectors (28 and 25 sections analyzed for DCX RNAi and the control, respectively). ***p < 0.001, χ2 test. C, The ganglionic eminence of an E17 rat brain slice was injected with DCX RNAi and either CAG-IRES-EGFP vector alone or CAG-DCX (n = 15 sections analyzed for both). In the presence of the rescue construct, there was a significant decrease of the number of cells in sector I of the cortex combined with an increase in cell numbers in sectors II and III. ***p < 0.001; **p < 0.01, χ2 test. D, Morphology of the DCX-inactivated cells. Representative confocal images of GFP-positive cells migrating into the cortex of embryonic rat brain slices show a higher complexity of branching in DCX-silenced cells. E, The number of primary processes arising from the cell bodies and total branches were counted for GFP-positive cells migrating tangentially in the cortex (n = 48 cells from 14 sections for DCX RNAi and n = 77 cells from 16 sections for the control RNAi). The histogram illustrates that, although the numbers of primary processes were similar in the two groups of cells, those inactivated for DCX tended to have more branches than control cells. ***p < 0.001; *p < 0.05, Student's t test. F, Rescue of the branching defect after coelectroporation of either DCX RNAi and the CAG vector alone (n = 44 cells from 10 sections) or DCX RNAi and CAG-DCX plasmids (n = 11 cells from 4 sections). Scale bars: A, 100 μm; D, 15 μm. Error bars indicate SEM.
To confirm the specificity of this defect, we attempted a rescue experiment by cotransfecting DCX shRNA together with either the CAG-IRES-EGFP vector alone, or a CAG-DCX construct without the 3′-UTR and, thus, insensitive to DCX RNAi. Figure 2C shows that there was significantly more migration from the GE into the cortex in the presence of the rescue construct. The proportion of cells in sector I decreased after the introduction of DCX (93.9 ± 4.7% for DCX RNAi and 75.8 ± 7.6% for the rescue), but it showed an increase in sector II (6.1 ± 4.7% for DCX RNAi vs 20 ± 7.1%) and in sector III (4.2 ± 3.5% for the rescue).
DCX-deficient interneurons display increased branching
We then examined the morphology of the GFP-labeled migrating interneurons in the cortex. Figure 2D illustrates typical examples of DCX-silenced cells showing increased branching in comparison with neurons seen in control slices. In our analysis, we counted the number of primary processes emanating from the somata of migrating cells into the cortex of electroporated brain slices as well as the total number of branches of each cell. As shown in Figure 2E, although the mean number of primary processes was comparable in the two groups (2 ± 0.2% for the DCX RNAi-expressing cells vs 1.7 ± 0.2% for the controls), there was a significant increase in the mean number of branches of cells inactivated for DCX (6.6 ± 0.8%) compared with those taken from control slices (4.6 ± 0.6%). To further assess the specificity of this change in neuron branching, we performed similar analysis on cells taken from slices coelectroporated with DCX shRNA together with the CAG-IRES-EGFP vector alone or the rescue construct (Fig. 2F). Although the mean number of primary processes did not change (2.2 ± 0.2% for DCX RNAi vs 2.3 ± 1.2% for the rescue), the mean number of branches decreased after the introduction of the DCX rescue construct (8.4 ± 1.4% for DCX RNAi vs 6.4 ± 1.2% for the rescue). This finding indicates that the observed change in cell morphology is specifically related to DCX inactivation.
DCLK inactivation causes a delay in interneuron migration, but no branching defect
Two studies have recently shown that, although Dcx-deficient mice do not display any major migration disorders, mice mutated for both Dcx and Dclk have disorganized neocortical layering, suggesting that these genes have compensatory roles in neuronal migration (Deuel et al., 2006; Koizumi et al., 2006a). This observation prompted us to investigate whether Dclk is also required for cortical interneuron migration. Thus, we performed similar experiments as for Dcx and used a shRNA located in the 3′-UTR region of the rat Dclk gene, as described by Shu et al. (2006). Because both genes share very high homology, we first confirmed that DCLK RNAi specifically inactivates DCLK, and that DCX RNAi does not have any effect on DCLK protein expression on dissociated cultures prepared from embryonic rat cortex (Fig. 3A). As shown in Figure 3B, electroporation of DCLK shRNA produced results very similar to DCX RNAi with the great majority of cells located in cortical sector I (91.3 ± 5.7% for DCLK RNAi and 82.5 ± 3.3% for the control) and only few cells in sector II (5.4 ± 4.6% for DCLK RNAi and 16.1 ± 3.2% for the control) and sector III (3.3 ± 3.6% for DCLK RNAi and 1.4 ± 1% for the control). Electroporation of both DCX and DCLK shRNAs together produced a more severe phenotype. In most sections, cells were located primarily in the ventral forebrain and very few had reached the cortex (Fig. 3C). As previously, we counted the number of primary processes and total branches of GFP+ cells in the cortex of the slices electroporated with either DCLK shRNA or both DCX/DCLK shRNAs (Fig. 3D). Interestingly, the morphology of DCLK− cells did not differ significantly from those in control slices (mean number of primary branches, 1.8 ± 0.3%; mean number of total branches, 4.7 ± 0.9) (Fig. 3D). However, DCX/DCLK RNAi gave similar results as DCX RNAi with no difference of primary branching (1.8 ± 0.2% for DCX/DCLK RNAi vs 1.8 ± 0.1% for the control), but a higher number in total branching (5.5 ± 0.9% for DCX/DCLK RNAi vs 4.6 ± 0.6% for the control). These results suggest that, although both DCX and DCLK genes are important for interneuron migration, their involvement in this process may be different.
Figure 3.

DCLK and DCX are both necessary for proper interneuron migration. A, Rat E16 cortical cell cultures were transfected with DCLK shRNA, a mutated DCLK shRNA, or DCX shRNA and labeled using antibodies directed against DCLK or DCX. DCLK RNAi led to specific knock-down of DCLK, whereas DCX RNAi did not have any effect on DCLK expression. Similarly, DCX RNAi had a specific effect on DCX expression. B, Electroporation of DCLK RNAi in the GE of E17 rat brain slices. As previously, the cortex was divided into three sectors and the number of cells were counted in each (22 and 38 sections analyzed for DCLK RNAi and control, respectively). Similar to DCX RNAi, there was a significant increase in the proportion of cells in sector I of the cortex combined with a decrease in sectors II and III. **p < 0.01; *p < 0.05, χ2 test. C, Electroporation of both DCX and DCLK shRNAs together led to even less migration in the cortex compared with controls (3UTR3mhp and DCLKm). D, No branching defect was found to be associated with DCLK RNAi. As previously, the number of primary processes and total branching were counted for GFP+ cells migrating tangentially into the cortex (n = 42 cells from 8 sections for DCLK RNAi alone; n = 55 cells from 11 sections for DCX/DCLK RNAi; n = 56 cells from 18 sections for the control). The histogram shows that, although the number of primary processes or total branching for DCLK RNAi was not different from the control, the combined DCX/DCLK shRNAs showed increased branching, similar to DCX RNAi alone. Scale bars: A, 10 μm; C, 40 μm. Error bars indicate SEM.
To assess whether DCX and DCLK exert their effects through different pathways, we tried to rescue DCLK RNAi migration defects by overexpressing DCX in E17 rat brain slices. This resulted in significantly more migration (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). We also found, similar to results of in vitro migration assays (Tanaka et al., 2004), that DCX overexpression alone enhances migration (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), suggesting that the level of DCX protein is important for the process of interneuron migration. These results indicate that DCX, if expressed at a sufficient level, rescues, at least in part, DCLK RNAi-induced migration defects. However, these experiments do not rule out the possibility that other aspects of interneuron migration (e.g., final positioning in the developing cortex or differentiation) cannot be rescued by DCX overexpression.
Genetic interaction between Dcx and Dclk in interneuron migration
To determine whether DCX and DCLK also play a role in interneuron migration in vivo, we examined the cortices of Dcx, Dclk, and Dcx/Dclk mutant mice at the time of birth (P0) (Fig. 4A–J). At this stage, most interneurons have migrated into the cortex from the ventral telencephalon (Lavdas et al., 1999). Using calbindin as a marker of interneurons (Anderson et al., 1997; Lopez-Bendito et al., 2004), we assessed the number and distribution of these cells in mutant cortices and in wild-type littermates. We counted the number of calbindin-positive cells in 200 μm coronal strips of dorsal and lateral neocortex in sections taken halfway along the rostrocaudal axis. No difference was observed between dorsal and lateral cortex (data not shown). This analysis revealed a decrease of ∼25% (p < 0.0001; N = 3 for each genotype) in the number of calbindin-positive cells in mutant mice for Dcx or Dclk and of 34% (p < 0.0001; N = 3) in the Dcx−/y;Dclk−/− mutant mice (Fig. 4I). We then examined the distribution of interneurons in these groups of mice by dividing the cortical plate (CP) into 10 equally spaced divisions, bin 1 corresponding to the deepest part of the cortex and bin 10 at the marginal zone as indicated in Figure 4H, and estimated the percentage of labeled cells in each bin. Figure 4, J1 and J2, shows noticeable differences in interneuron distribution in both Dcx−/y and Dclk−/− single knock-out mice. Dcx−/y mice had more cells in the superficial layers of the CP (bins 7, 8, and 10) (Fig. 4J1), whereas Dclk mutants had fewer cells in these layers (bins 7, 9, and 10) (Fig. 4J2); the latter showed an excess of cells in the deeper layers, especially in bins 3, 4, and 5, whereas Dcx−/y had fewer cells in the same depth of the CP (bins 5 and 6). Although there was a significant decrease in the total number of calbindin-positive cells in the cortex of Dcx−/y;Dclk−/− animals, their distribution was very similar to that observed in wild-type controls (Fig. 4J3). The only differences were a decrease in the proportion of cells in bins 7 and 10 and an increase in bin 4. Surprisingly, these differences are more similar to what was observed for Dclk−/− mutants rather than Dcx−/y mutants. Together, these results suggest that both DCLK and DCX play a role in interneuron migration in vivo, but these roles are likely to be different for the two genes. The fact that Dcx−/y;Dclk−/− mutant mice have a similar distribution to the wild-type suggests that these roles may be partly complementary.
Figure 4.

Interneuron defects in Dcx, Dclk, and Dcx/Dclk mutant mice. A–H, Calbindin labeling in coronal sections from P0 mutant animals. I, Quantification of the number of calbindin-positive cells in the cortex of mutant mice. The histogram depicts the average number of calbindin-positive cells in a 200-μm-wide radial strip of dorsal and lateral cortex in wild-type and mutant brain sections at P0. All three genotypes show a significantly decreased number of calbindin-positive cells in the cortex. ***p < 0.001, Student's t test. J1–J3, The cortical plate (a 400-μm-wide radial strip) at dorsomedial level was divided into 10 equally spaced bins (as shown in H). The number of cells in each bin was expressed as a percentage of the total number of cells. For each genotype, the distribution was different from the wild-type. ***p < 0.001; **p < 0.01; *p < 0.05, χ2 test. Scale bars: A–D, 400 μm; B–H, 200 μm. Error bars indicate SEM.
We confirmed the number and distribution of interneurons in the cortices of Dcx, Dclk, and Dcx/Dclk mutant mice at P0 by using ARX, a different marker of these cells (Kitamura et al., 2002; Poirier et al., 2004). Similar to the analysis of calbindin-labeled cells, the cortices of all three groups of mutants showed reduced numbers of ARX-positive cells (data not shown). Moreover, analysis of their distribution confirmed that there was an increase in the percentage of ARX+ cells in lower layers (bins 3–6) and a decrease in upper layers (bins 8 and 10), especially in the cortices of Dclk−/− and Dcx−/y;Dclk−/− mutants (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). ARX labeling showed slightly different cell distribution compared with calbindin labeling in the Dcx knock-outs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This may be attributable to slightly different cell populations labeled with the two markers.
We also examined calbindin-stained sections taken from brains of two E16 Dclk−/− and two Dcx−/y;Dclk−/− embryos. Similar to newborn animals, there was a decrease in the number of calbindin-positive cells in the cortices of these mice, and this deficiency was more pronounced in the Dcx−/y;Dclk−/− embryos than in Dclk-deficient mice (data not shown). We also observed that, although the streams of migrating interneurons were present in Dclk−/− and Dcx−/y;Dclk−/− at this stage of development, they appeared to be more spread out and not as well confined as in wild-type embryos (data not shown).
Discussion
The main findings to emerge from this study are as follows: (1) Inactivation of DCX and DCLK in the GE of rat brain slices using focal electroporation of shRNAs resulted in impaired migration of cortical interneurons. (2) Dcx-deficient interneurons showed increased branching compared with their counterparts in control slices. (3) Analysis of cortices of Dcx, Dclk, and Dcx/Dclk mutant mice during prenatal corticogenesis and at birth showed a significant decrease in the number of interneurons and a change in their distribution compared with controls, but the defect was different for each group of mutant animals.
Interneuron migration is delayed after DCX RNAi
We inactivated DCX in the GE of rat embryonic brain slices using RNA interference. This approach has a number of advantages to conventional mouse knock-out studies in that it inactivates DCX in a desired subset of cells in otherwise normal tissue and in a discrete window of time. We found that the migration of DCX-inactivated cells from the GE to the cortex was delayed in comparison with control RNAi-transfected cells. In addition, we noted that these cells displayed increased complexity of branching. Because RNAi can have off-target effects (Scherer and Rossi, 2003), we assessed the specificity of this result by introducing a DCX-overexpression construct lacking the 3′-UTR. This successfully rescued both the delay in migration and the branching defect. Recent reports have suggested that DCX overexpression increases the rate of cerebellar neuronal migration in vitro (Tanaka et al., 2004). Our overexpression experiments (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), as well as the fact that, in the presence of the rescue construct, more cells reached medial positions in cortical slices compared with vector alone (Fig. 2, compare C, B), also support these findings.
Lis1, another gene associated with most cases of type 1 lissencephaly, appears to have a similar role to Dcx in cortical interneuron migration. Specifically, McManus et al. (2004) have reported that LIS1-deficient interneurons migrate more slowly than wild-type cells in brain slices prepared from E14.5 Lis1 knock-out mice. The defects in cortical interneuron migration that result from inactivation of DCX or LIS1 resemble the reported impairment in radial migration of pyramidal neurons after DCX RNAi in combination with in utero electroporation in rat embryonic brain (Bai et al., 2003). Specifically, these authors have shown that inactivation of DCX leads to the presence of cells in the IZ, halfway in their migration to the CP, mimicking the human phenotype of subcortical laminar heterotopia. They also described cell morphology defects, although this may be attributable to an excess number of multipolar cells at the level of the IZ before their acquiring bipolar form at the start of their radial movement to the CP (Bai et al., 2003; Tabata and Nakajima, 2003). Together, these results indicate that, although the mechanisms involved in the radial movement of pyramidal neurons are different from those that guide interneurons in their tangential journeys, microtubule-associated proteins such as DCX and LIS1 are necessary for both modes of migration.
It is interesting that DCX RNAi-mediated knock-down induced branching defects in migrating interneurons. Branching defects have also been reported for migrating interneurons taken from Dcx knock-out mice, leading to disorganized migration (Kappeler et al., 2006). Interestingly, these authors did not observe any differences in speed or distance of migration, but they used explant cocultures and fluorescent dyes on brain slices to assess migratory dynamics of interneurons. We cannot comment as to whether the branching abnormality observed in our experiments is responsible for the observed delay in the migration of these cells. However, we can suggest that if DCX-deficient cells have more branches, their ability to respond to environmental cues, and thus follow a definite path, might be compromised. A recent imaging study of migrating cortical interneurons has clearly documented complex morphological transformations at their leading edge that precede the forward translocation of their nucleus and perinuclear cytoplasm (Bellion et al., 2005). Thus, it is conceivable that alterations in cell branching, especially of the leading processes, can lead to migration impairment.
DCLK RNAi results in a decrease in the number of interneurons in the cortex, but no defect in branching
Recent studies have shown that DCLK is involved in radial neuronal migration (Deuel et al., 2006; Koizumi et al., 2006a). This gene was first identified because of its high homology to DCX, but has not yet been associated with any human neuronal migration disorders. It has been shown that both DCLK and DCX have microtubule stabilization properties, but DCLK also encodes a kinase domain in the C-terminal part of the protein (Burgess and Reiner, 2000; Lin et al., 2000). The two genes show overlapping expression patterns, although DCLK is more widely expressed in the brain (Sossey-Alaoui and Srivastava, 1999; Burgess and Reiner, 2000). In addition, DCLK encodes multiple transcripts, which are all differently regulated and may have different functions (Hevroni et al., 1998; Burgess et al., 1999; Silverman et al., 1999; Vreugdenhil et al., 2001; Burgess and Reiner, 2002). Two groups have recently reported that, although Dcx or Dclk mutant mice do not show defects in radial neuronal migration, mutants for both Dcx and Dclk genes display cortical lamination defects, suggesting that they can functionally compensate each other (Deuel et al., 2006; Koizumi et al., 2006a). In the present study, DCLK inactivation in embryonic rat brain slices showed results very similar to those described for DCX. Specifically, interneurons were observed to be delayed in their migration to the cortex, but, interestingly, DCLK-inactivated cells did not display any significant changes in their branching compared with cells analyzed from control slices. These results suggest that either the branching defect observed with DCX RNAi is independent of the delay in migration or that DCX and DCLK act through different mechanisms.
Cortical interneurons in Dclk and Dcx mutant mice
Recent studies have reported that Dcx or Dclk single knock-outs do not show any apparent malformations of the cortex (Corbo et al., 2002; Deuel et al., 2006; Kappeler et al., 2006; Koizumi et al., 2006a). Furthermore, one of these studies (Kappeler et al., 2006) reported no abnormalities in the number and distribution of cortical interneurons in Dcx mutants at different prenatal and postnatal ages. Here, we examined the Dcx−/y, Dclk−/−, as well as the Dcx−/y;Dclk−/− mutants for differences in cortical interneuron number and distribution. For each genotype, fewer interneurons were present in the cortex compared with wild-type controls. The distribution of these interneurons was also affected, with more cells present in the upper layers of the cortex for Dcx−/y and fewer neurons in these layers for Dclk−/−. We have not examined later developmental stages because Dcx−/y;Dclk−/− mice die soon after birth. We performed a similar analysis at one embryonic stage (E16), a time of extensive interneuron migration within the cortex. Similar to the results at birth, we noted a decrease in the total number of interneurons in the cortex and disorganization in their distribution in comparison with wild-type controls (data not shown).
It is interesting to note that defects in migration observed in Dcx−/y and Dclk mutant mice apply to tangentially migrating interneurons, but not to radially moving pyramidal cells. This difference may be accounted by different roles that these genes play in the two modes of migration. Thus, although DCX and DCLK seem to compensate each other in radial migration, our present results suggest that this is not the case in tangential migration. It is also possible that differences in distance of migration for the two forms of movement account for the differential response of the two neuronal cell types to the lack of Dcx and Dclk. Thus, interneurons that arise in the ventral telencephalon travel much longer distances compared with their pyramidal counterparts, and any subtle defect in form or movement is more likely to become apparent at the end of their long and tortuous journey. These results are consistent with what has been described for the long rostral migratory stream (RMS) (Koizumi et al., 2006b). This stream in the postnatal mammalian brain contains cells that migrate from the anterior region of the lateral ventricle to the olfactory bulb. In their study of Dcx knock-out mice, the authors found morphological alterations of the RMS and delayed cell migration that is independent of direction or responsiveness to Slit chemorepulsion. Thus, defects in migration have been demonstrated in neurons that follow especially long migratory routes.
Previous studies had suggested that the major phenotypes observed in mutants for proteins involved in neuronal migration, such as Reelin, p35, Cdk5, LIS1, or DCX, were primarily attributable to defective radial rather than tangential migration. However, current evidence suggests that many of these mutants display defects in both modes of migration (McManus et al., 2004; Kappeler et al., 2006; this study). Impairment in the migration, disposition, and proper integration of interneurons in the cortical circuitry may explain, at least in part, the epilepsy and mental retardation observed in lissencephaly.
Footnotes
This work was supported by Wellcome Trust Programme Grant 074549 (J.G.P.). G.F. was supported in part by a Prix Jeune Chercheur from the Fondation Bettencourt–Schueller and J.S.L. by National Institute of Child Health and Human Development Grant K12 HD051959-01. We are grateful to Anastasia Liapi, Mary Rahman, Daniel Cianter, and Luis Hernandez for technical help and to Clare Faux and William Andrews for critical reading of this manuscript.
References
- Alifragis P, Liapi A, Parnavelas JG. Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their GABA phenotype. J Neurosci. 2004;24:5643–5648. doi: 10.1523/JNEUROSCI.1245-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Andrews W, Liapi A, Plachez C, Camurri L, Zhang J, Mori S, Murakami F, Parnavelas JG, Sundaresan V, Richards LJ. Robo1 regulates the development of major axon tracts and interneuron migration in the forebrain. Development. 2006;133:2243–2252. doi: 10.1242/dev.02379. [DOI] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bellion A, Baudoin JP, Alvarez C, Bornens M, Metin C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J Neurosci. 2005;25:5691–5699. doi: 10.1523/JNEUROSCI.1030-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Martinez S, Reiner O. KIAA0369, doublecortin-like kinase, is expressed during brain development. J Neurosci Res. 1999;58:567–575. [PubMed] [Google Scholar]
- Burgess HA, Reiner O. Doublecortin-like kinase is associated with microtubules in neuronal growth cones. Mol Cell Neurosci. 2000;16:529–541. doi: 10.1006/mcne.2000.0891. [DOI] [PubMed] [Google Scholar]
- Burgess HA, Reiner O. Alternative splice variants of Doublecortin-like kinase are differentially expressed and have different kinase activities. J Biol Chem. 2002;277:17696–17705. doi: 10.1074/jbc.M111981200. [DOI] [PubMed] [Google Scholar]
- Corbo JC, Deuel TA, Long JM, LaPorte P, Tsai E, Wynshaw-Boris A, Walsh CA. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci. 2002;22:7548–7557. doi: 10.1523/JNEUROSCI.22-17-07548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Deuel TA, Liu JS, Corbo JC, Yoo SY, Rorke-Adams LB, Walsh CA. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Andermann E, Andermann F, Czapansky-Beilman D, Dubeau F, Dulac O, Guerrini R, Hirsch B, Ledbetter DH, Lee NS, Motte J, Pinard JM, Radtke RA, Ross ME, Tampieri D, Walsh CA, Truwit CL. X-linked malformations of neuronal migration. Neurology. 1996;47:331–339. doi: 10.1212/wnl.47.2.331. [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Hevroni D, Rattner A, Bundman M, Lederfein D, Gabarah A, Mangelus M, Silverman MA, Kedar H, Naor C, Kornuc M, Hanoch T, Seger R, Theill LE, Nedivi E, Richter-Levin G, Citri Y. Hippocampal plasticity involves extensive gene induction and multiple cellular mechanisms. J Mol Neurosci. 1998;10:75–98. doi: 10.1007/BF02737120. [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- Kappeler C, Saillour Y, Baudoin JP, Tuy FP, Alvarez C, Houbron C, Gaspar P, Hamard G, Chelly J, Metin C, Francis F. Branching and nucleokinesis defects in migrating interneurons derived from doublecortin knockout mice. Hum Mol Genet. 2006;15:1387–1400. doi: 10.1093/hmg/ddl062. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo SI, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006a;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Higginbotham H, Poon T, Tanaka T, Brinkman BC, Gleeson JG. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat Neurosci. 2006b;9:779–786. doi: 10.1038/nn1704. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PT, Gleeson JG, Corbo JC, Flanagan L, Walsh CA. DCAMKL1 encodes a protein kinase with homology to Doublecortin that regulates microtubule polymerization. J Neurosci. 2000;20:9152–9161. doi: 10.1523/JNEUROSCI.20-24-09152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bendito G, Sturgess K, Erdelyi F, Szabo G, Molnar Z, Paulsen O. Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb Cortex. 2004;14:1122–1133. doi: 10.1093/cercor/bhh072. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- McManus MF, Nasrallah IM, Pancoast MM, Wynshaw-Boris A, Golden JA. Lis1 is necessary for normal non-radial migration of inhibitory interneurons. Am J Pathol. 2004;165:775–784. doi: 10.1016/S0002-9440(10)63340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metin C, Baudoin JP, Rakic S, Parnavelas JG. Cell and molecular mechanisms involved in the migration of cortical interneurons. Eur J Neurosci. 2006;23:894–900. doi: 10.1111/j.1460-9568.2006.04630.x. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Parnavelas J. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002;3:423–432. doi: 10.1038/nrn845. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Ventricle-directed migration in the developing cerebral cortex. Nat Neurosci. 2002;5:218–224. doi: 10.1038/nn813. [DOI] [PubMed] [Google Scholar]
- Poirier K, Van Esch H, Friocourt G, Saillour Y, Bahi N, Backer S, Souil E, Castelnau-Ptakhine L, Beldjord C, Francis F, Bienvenu T, Chelly J. Neuroanatomical distribution of ARX in brain and its localisation in GABAergic neurons. Mol Brain Res. 2004;122:35–46. doi: 10.1016/j.molbrainres.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Rakic P. Principles of neural cell migration. Experientia. 1990;46:882–891. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cereb Cortex. 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- Scherer LJ, Rossi JJ. Approaches for the sequence-specific knockdown of mRNA. Nat Biotechnol. 2003;21:1457–1465. doi: 10.1038/nbt915. [DOI] [PubMed] [Google Scholar]
- Shu T, Tseng HC, Sapir T, Stern P, Zhou Y, Sanada K, Fischer A, Coquelle FM, Reiner O, Tsai LH. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. 2006;49:25–39. doi: 10.1016/j.neuron.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Silverman MA, Benard O, Jaaro H, Rattner A, Citri Y, Seger R. CPG16, a novel protein serine/threonine kinase downstream of cAMP-dependent protein kinase. J Biol Chem. 1999;274:2631–2636. doi: 10.1074/jbc.274.5.2631. [DOI] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Srivastava AK. DCAMKL1, a brain-specific transmembrane protein on 13q12.3 that is similar to doublecortin (DCX) Genomics. 1999;56:121–126. doi: 10.1006/geno.1998.5718. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreugdenhil E, Engels B, Middelburg R, van Koningsbruggen S, Knol J, Veldhuisen B, de Kloet ER. Multiple transcripts generated by the DCAMKL gene are expressed in the rat hippocampus. Mol Brain Res. 2001;94:67–74. doi: 10.1016/s0169-328x(01)00213-3. [DOI] [PubMed] [Google Scholar]