

Abstract
Single-channel analysis revealed the existence of neuronal L-type Ca2+ channels (LTCCs) with fundamentally different gating properties; in addition to LTCCs resembling cardiac channels, LTCCs with anomalous gating were identified in a variety of neurons, including cerebellar granule cells. Anomalous LTCC gating is mainly characterized by long reopenings after repolarization following strong depolarizations or trains of action potentials. To elucidate the unknown molecular nature of anomalous LTCCs, we performed single-channel patch-clamp recordings from cerebellar granule cells of wild-type, Cav1.3−/− and Cav1.2DHP−/− [containing a mutation in the Cav1.2 α1 subunit that eliminates dihydropyridine (DHP) sensitivity] mice. Quantitative reverse transcription-PCR revealed that Cav1.2 accounts for 89% and Cav1.3 for 11% of the LTCC transcripts in wild-type cerebellar granule cells, whereas Cav1.1 and Cav1.4 are expressed at insignificant levels. Anomalous LTCCs were observed in neurons of Cav1.3−/− mice with a frequency not different from wild type. In the presence of the DHP agonist (+)-(S)-202-791, the typical prepulse-induced reopenings of anomalous LTCCs after repolarization were shorter in Cav1.2DHP−/− neurons than in Cav1.3−/− neurons. Reopenings in Cav1.2DHP−/− neurons in the presence of the DHP agonist were similar to those in wild-type neurons in the absence of the agonist. These data show that Cav1.2α1 subunits are the pore-forming subunits of anomalous LTCCs in mouse cerebellar granule cells. Given the evidence that Cav1.2 channels are specifically involved in sustained Ras-MAPK (mitogen-activated protein kinase)-dependent cAMP response element-binding protein phosphorylation and LTCC-dependent hippocampal long-term potentiation (LTP) (Moosmang et al., 2005), we discuss the hypothesis that anomalous rather than cardiac-type Cav1.2 channels are specifically involved in LTCC-dependent and gene transcription-dependent LTP.
Keywords: calcium channels, L-type, LTP, transgenic mice, single channel, cerebellar granule cells
Introduction
Voltage-gated L-type Ca2+ channels (LTCCs) play a key role in neuronal Ca2+ signaling. In contrast to P/Q-, N-, and R-type channels, which mediate neurotransmitter release in nerve terminals, LTCCs are located at somatodendritic compartments (Hell et al., 1993; Davare et al., 2001; Obermair et al., 2004) and control activity-dependent neuronal gene expression, which underlies processes like circadian rhythms, neuronal survival, and long-term memory (Deisseroth et al., 2003).
Brain LTCCs are mainly formed by Cav1.2 and Cav1.3α1 subunits (Sinnegger-Brauns et al., 2004). Both isoforms show a broad expression pattern and coexist in many neurons, where they typically form clusters on cell bodies and along dendrites (Hell et al., 1993; Obermair et al., 2004). They possess different biophysical and biochemical properties and thus can serve different neuronal functions. Cav1.3 channels activate more rapidly and at more negative voltages than Cav1.2 (Koschak et al., 2001), which allows them to participate in the stabilization of upstate potentials and the control of neuronal firing (Hernandez-Lopez et al., 2000; Olson et al., 2005). In contrast, Cav1.2 is the major LTCC isoform responsible for activity-dependent gene transcription, hippocampal late-long-term potentiation (L-LTP) (Clark et al., 2003; Moosmang et al., 2005), and memory (Moosmang et al., 2005). Interaction with different intracellular proteins can result in different regulatory properties of Cav1.2 and Cav1.3 LTCCs (Olson et al., 2005; Zhang et al., 2005, 2006).
Single-channel analysis revealed the existence of two fundamentally different gating patterns of brain LTCCs, which should give rise to distinct intracellular calcium signals in response to neuronal activity. In addition to LTCCs closely resembling cardiac channels, LTCCs with anomalous gating properties were identified in cerebellar granule cells (Forti and Pietrobon, 1993), hippocampal neurons (Kavalali and Plummer, 1994), sensory neurons (Ferroni et al., 1996), and motoneurons (Hivert et al., 1999). The “anomalous” gating properties were mainly characterized by low open probability during depolarizations, which further decreased with increasing test potentials, and typical long reopenings after repolarization following strong depolarizations (Forti and Pietrobon, 1993; Hivert et al., 1999), including stimuli mimicking trains of action potentials (APs) (Ferroni et al., 1996; Schjott and Plummer, 2000). There are no reports of anomalous gating of recombinant Cav1.2 and Cav1.3 channels. At present, the nature of the pore-forming subunit of anomalous LTCCs is unknown. This lack of structural information represents a major drawback for additional studies of the molecular basis and physiological relevance of anomalous LTCCs.
The contribution of Cav1.2 and Cav1.3 isoforms to LTCC currents in different neurons is difficult to assess using pharmacological tools, as a consequence of incomplete selectivity of non-dihydropyridine (DHP) LTCC blockers (Ishibashi et al., 1995; Dobrev et al., 1999) and state-dependent block by the highly selective DHPs (Helton et al., 2005). No pharmacological approach is available to determine which of the LTCC isoforms underlies anomalous LTCC gating.
Here, we exploited two previously developed mouse models to unequivocally demonstrate that Cav1.2 channels form the anomalous LTCCs in cerebellar granule cells.
Materials and Methods
Animals.
Cav1.2DHP−/− and Cav1.3−/− mice were generated as described previously (Platzer et al., 2000; Sinnegger-Brauns et al., 2004) and backcrossed for at least five generations into C57Bl/6N mice. To minimize use and breeding of genetically modified animals, the number of experiments was kept to a minimum. All animal experiments were approved by the Austrian Bundesministerium für Bildung, Wissenschaft, und Kultur.
Cell culture.
Cerebellar granule cells were grown in primary culture from postnatal day 6 (P6) mice as described previously (Fletcher et al., 2001). Cells were plated on poly-l-lysine-coated coverslips and maintained in basal Eagle's medium supplemented with 10% FCS, 25 mm KCl, 2 mm glutamine, and 50 g/ml gentamycin. Cytosine βD-arabino furanoside was added 24 h after plating the cells to a final concentration of 10 μm to prevent proliferation of nonneuronal cells. The preparation was highly enriched with granule cells. Electrophysiological recordings were performed from cells grown 2–8 d in vitro, and granule cells were morphologically identified by their small size, oval or round soma, and bipolar neurites. For quantitative reverse transcription-PCR (qRT-PCR), experiment cells were grown 2 d in culture before total RNA was isolated.
RNA isolation and cDNA preparation.
Total RNA from wild-type mouse cerebellar granule cells was prepared using the RNAqueousR-4PCR kit (Ambion, Foster City, CA), for skeletal muscle and whole-eye preparations using the E.Z.N.A. Total RNA kit (Omega Bio-Tek, Winooski, VT) according to the manufacturer instructions. Total cochlea RNA (P4 mice) was isolated as described by Michna et al. (2003). RNA integrity was checked by the presence of clear 28S and 18S rRNA bands after loading 1–2 μg of RNA on a denaturating gel (1%). Total RNA was DNaseI (Ambion) treated to remove contaminating genomic DNA before reverse transcription. One microgram of total RNA was reverse transcribed using RevertAid H Minus M-MuLV first-strand cDNA Synthesis kit with both oligo (dT) primers and random hexamer primers (MBI; Fermentas, Hanover, MD).
qRT-PCR.
The relative abundance of different LTCC mRNAs in wild-type mouse cerebellar granule cells was assessed by TaqMan quantitative PCR (50 cycles) using a standard curve method based on PCR products of known concentration. TaqMan Gene Expression Assays, designed to span exon–exon boundaries, were purchased from Applied Biosystems (Foster City, CA). Assay identification numbers are as follows: Cav1.1, Mm00489257_m1; Cav1.2, Mm00437917_m1; Cav1.3, Mm01209919_m1; Cav1.4, Mm00490443_m1; β-actin, Mm00607939s1.
For each LTCC gene expression assay, flanking primer pairs were designed to amplify the templates for the standard curves using cDNA from cerebellar granule cells. The following primer pairs were used: Cav1.1-F, GTTACATGAGCTGGATCACACAG; Cav1.1-R, ATGAGCATTTCGATGGTGAAG; Cav1.2-F, CATCACCAACTTCGACAACTTC; Cav1.2-R, CAGGTAGCCTTTGAGATCTTCTTC; Cav1.3-F, ACATTCTGAACATGGTCTTCACAG; Cav1.3-R, AGGACTTGATGAAGGTCCACAG; Cav1.4-F, CTCTTCATCTGTGGCAACTACATC; Cav1.4-R, GTACCACCTTCTCCTTGGGTACTA. The PCR products were run on a 1% TBE (90 mm Tris-borate and 2 mm EDTA, pH 8.3) Seakem gel, excised, and eluted using Nucleospin Extract II columns (Macherey-Nagel, Düren, Germany). PCR product specificity was tested by restriction-enzyme analysis. The DNA concentration of the PCR products was determined photometrically, and the molecular weight was calculated. The standard curve was generated using a 10-fold serial dilution starting from 107 to 10 copies of PCR product.
Twenty nanograms of total RNA equivalents of cDNA and the specific TaqMan Gene Expression Assay were used for each 20 μl reaction in TaqMan Universal PCR Master Mix (Applied Biosystems). RNA samples without RT and samples without template were routine controls. cDNA concentrations of all individual experiments were comparable as revealed by the reference β-actin transcript. Analyses were performed using the Mx4000 Multiplex Quantitative PCR System (Stratagene, La Jolla, CA). Data are presented as mean ± SD.
Electrophysiological recordings.
Single-channel patch-clamp recordings followed standard techniques. All recordings were obtained in the cell-attached configuration at room temperature (∼25°C). Single-channel currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Foster City, CA) at sampling rates of 5 kHz and low-pass filtered at 1 kHz. Borosilicate glass pipettes were pulled using a Sutter P-97 microelectrode puller, coated with Sylgard (Dow Corning, Kaiserslautern, Germany), fire polished, and showed typical resistances of 2.5–10 MΩ when filled with internal solution. The pipette solution contained the following (in mm): 90 BaCl2, 10 tetraethylammonium (TEA)-Cl, 15 CsCl, 10 HEPES, pH 7.4, with TEA-OH. The bath solution contained the following (in mm): 140 K-gluconate, 5 EGTA, 35 d-glucose, 10 HEPES, pH 7.4, with KOH. High-potassium bath solution was used to zero the membrane potential outside the patch. The dihydropyridine agonist (+)-(S)-202-791 (a gift from Dr. Hof, Sandoz, Basel, Switzerland) was added (3 μm) to the bath solution in all recordings. The holding potential was always −40 mV, and depolarizing test pulses were delivered every 4 s. Test pulses of 500 ms to different voltages were applied followed by a 296 ms voltage step to −40 mV. Linear leak and capacitative currents were subtracted manually for every single trace.
LTCCs with cardiac-like and anomalous gating were identified as described previously (Forti and Pietrobon, 1993). Briefly, the anomalous channel was identified by the presence of reopenings during repolarization to −40 mV after a depolarizing voltage step to +40 mV. In the presence of the dihydropyridine calcium channel agonist (+)-(S)-202-791, anomalous channels could further be distinguished from cardiac-like channels on the basis of the unitary current amplitude and shorter channel openings. The open probability was calculated from experiments containing only one single channel in the patch after exclusion of null sweeps. The decay of the averaged single-channel currents during repolarization at −40 mV was best fitted with a single exponential function. To verify the best exponential fit, data sets were also fitted by a biexponential function. However, in none of the experiments, the χ2 value was further reduced by using a biexponential instead of a monoexponential function.
Statistics.
Origin 6.1 (Microcal, Northampton, MA) and Prism 4.03 (Graphpad Software, San Diego, CA) were used for linear and nonlinear curve fitting and statistical data analysis. All data are presented as mean ± SE for the indicated number of experiments. Unless stated otherwise, statistical significance was determined by unpaired student's t test.
Results
Cerebellar granule cells express Cav1.2 and Cav1.3 but not Cav1.1 and Cav1.4 mRNA
Forti and Pietrobon (1993) have shown previously in a single-channel study that functionally different LTCCs exist in rat cerebellar granule cells in primary culture. In these cells, neuronal LTCCs with a classical cardiac-like gating pattern coexist with anomalous LTCCs characterized by multiple peculiar voltage-dependent gating properties, including long reopenings at negative voltages after a depolarization. LTCCs with anomalous gating and LTCCs with cardiac-like gating were observed with similar frequency in cell-attached patches (Forti and Pietrobon, 1993). Long reopenings of DHP-sensitive Ca2+ channels at negative voltages were also reported in mouse cerebellar granule cells (Slesinger and Lansman, 1991).
To identify the potential pore-forming subunits of anomalous LTCCs, we first analyzed the relative fraction of Cav1 channels expressed in mouse cerebellar granule cells using qRT-PCR. Quantitation of mRNA levels from four independent primary cultures of cerebellar granule cells showed that Cav1.2 channels are the major LTCCs expressed in these neurons accounting for 89 ± 2.4% of total Cav1 mRNA; the remaining 11 ± 2.4% were Cav1.3 channels, whereas Cav1.1 and Cav1.4 reached only negligible expression levels of 0.03 ± 0.03 and 0.002 ± 0.001% (Fig. 1). However Cav1.1. and Cav1.4 expression was observed in control tissues (Fig. 1 C,D). The expression profile in cerebellar granule cells excludes Cav1.1 and Cav1.4 as the main potential pore-forming subunits of anomalous LTCCs. Despite the significantly lower relative abundance of Cav1.3 mRNA, this isoform cannot be ruled out as the molecular substrate of anomalous channels, because differences in protein stability could result in a higher relative abundance of Cav1.3 α1 subunit protein than predicted from its mRNA, and there might be a higher number of functional CaV1.3 channels in the membrane than CaV1.2. Moreover, although unlikely (Schramm et al., 1999), the possibility that the pore-forming subunits of anomalous LTCCs are formed by yet unidentified DHP-sensitive Ca2+ channel subunits cannot be ruled out.
Figure 1.

Expression of different LTCC α1-subunit mRNAs in cerebellar granule cells and control tissues quantified by qRT-PCR. The data for each LTCC α1-subunit isoform are presented as percentage of the total amount of LTCCs. The relative amount of each LTCC transcript in any RNA preparation was assessed by standard curves (see Materials and Methods for details). A, Cultured cerebellar granule cells. B–D, Cochlea (B) (expressing predominantly Cav1.3α1; n = 2), skeletal muscle (C) (expressing almost exclusively Cav1.1α1; n = 2), and a crude eye preparation (D) (expressing also retinal Cav1.4α1; n = 2) served as positive controls for α1 subunits expressed at lower densities than Cav1.2 in cerebellar granule cells.
To unequivocally identify the molecular nature of anomalous LTCCs, we performed single-channel recordings in the presence of DHP agonist on cerebellar granule cells in primary culture from Cav1.3−/− mice lacking Cav1.3 channels and Cav1.2DHP−/− mice containing a mutation in the Cav1.2 α1 subunit that eliminates DHP agonist sensitivity without causing obvious changes in Cav1.2 functional properties and expression levels (Sinnegger-Brauns et al., 2004).
Single LTCC recordings in cerebellar granule cells of Cav1.3−/− mice
We first performed cell-attached single-channel recordings from cerebellar granule cells of Cav1.3−/− mice to test the hypothesis that Cav1.3α1 subunits are the pore-forming subunits of the anomalous LTCCs. Experiments were performed in the presence of the Ca2+ channel activator (+)-(S)-202-791 using 90 mm Ba2+ as charge carrier. Cells were held at a holding potential of −40 mV, and single-channel activity was elicited by applying 500 ms depolarizing pulses to various test potentials. As shown by the traces in Figure 2 A from two representative patches containing only one channel, LTCCs with cardiac-like (left panel) and anomalous (right panel) gating pattern were both present in neurons from Cav1.3−/− mice. In accordance with previous reports from rat cerebellar granule cells (Forti and Pietrobon, 1993), the anomalous LTCCs recorded in neurons of Cav1.3−/− mice were distinguishable from cardiac-like LTCCs by their smaller unitary current and conductance (Fig. 2 A,B) and by their unusual voltage-dependent properties (Figs. 2 A,C, 3). The average single-channel conductance of anomalous LTCCs (20.3 ± 0.5 pS; n = 4; i = −1.24 ± 0.04 pA at 0 mV) was slightly but significantly (p = 0.001) smaller than the average single-channel conductance measured for cardiac-like LTCCs (22.4 ± 0.2 pS; n = 7; i = −1.47 ± 0.02 pA at 0 mV). The open probability (po) of anomalous LTCCs showed a characteristic anomalous bell-shaped voltage dependence (as shown in Fig. 2 C for the single anomalous LTCC in Fig. 2 A, right panel) that reached a maximum and then decreased with increasing voltage, in contrast with the increase in po typically observed in the same voltage range for cardiac-like LTCCs (Fig. 2 A, left panel). Note in the representative traces in Figure 2 A (right panel) the short openings and long closings and the very low open probability at +30 mV of the anomalous channel despite the presence of the DHP agonist in contrast with the typical long openings, short closings, and high po of the cardiac-like channel.
Figure 2.

Gating properties of single LTCCs in cerebellar granule cells of Cav1.3−/− mice. Cell-attached single-channel recordings with 90 mm Ba2+ as charge carrier in the presence of the Ca2+ channel activator (+)-(S)-202-791 from two patches of cerebellar granule cells of Cav1.3−/− mice: one containing a single LTCC with cardiac-like gating and the other a single LTCC with anomalous gating. Depolarizations of 500 ms were applied every 4 s from a holding potential of −40 mV to various test potentials (TP). A, Representative unitary current traces of the LTCC with cardiac-like (left) and the LTCC with anomalous gating (right) at 0, +10, +20, and +30 mV. Note the typical short openings, long closings, and low open probability of the anomalous LTCC at high voltages, in contrast with the typical long openings, short closings, and high open probability increasing with increasing voltage of the classical LTCC with cardiac-like gating. Cells: 1310_02, cardiac-like; 1510_07, anomalous. B, Unitary I–V relationships of the two representative LTCCs in A with cardiac-like (open symbols; g = 23 pS) and anomalous (closed symbols; g = 19 pS) gating. C, Voltage dependence of po of the single anomalous channel shown in A. For each voltage, the values indicated are averages of the open probability measured in each sweep with activity (n = 5–26).
Figure 3.
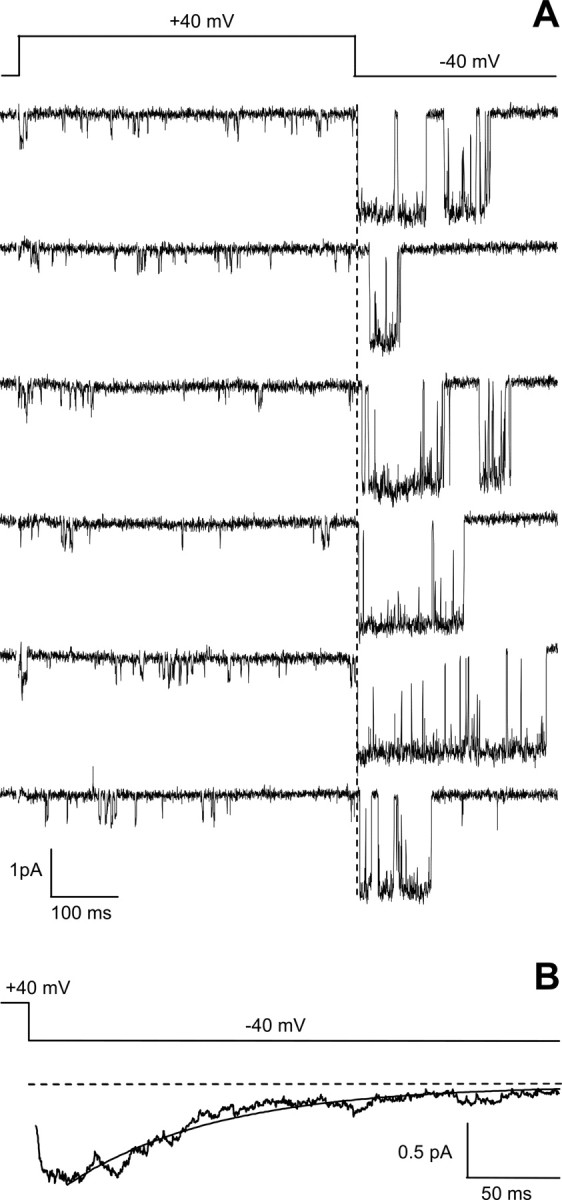
Characteristic reopenings of anomalous LTCCs in cerebellar granule cells of Cav1.3−/− mice in the presence of the Ca2+ channel activator (+)-(S)-202-791. Cell-attached single-channel recordings in the presence of the Ca2+ channel activator (+)-(S)-202-791 from a patch containing two anomalous LTCCs on a Cav1.3−/− cerebellar granule cell are shown. Single-channel activity was elicited by the voltage protocol shown in the top part of the figure. A, The representative traces show low activity of two anomalous LTCCs during a depolarization to +40 mV. Reopenings of the anomalous channels after repolarization at −40 mV after the predepolarization are also clearly revealed. B, Normalized average single-channel current during the repolarization was obtained from 53 averaged traces with reopenings. The gray line is the best fit to a single exponential. Cell, 1410_04.
A third property that unequivocally identified anomalous LTCCs in Cav1.3−/− cerebellar granule cells were the long prepulse-induced reopenings at negative potentials. To reveal prepulse-induced reopenings, cells were pulsed to a negative potential of −40 mV after a depolarizing prepulse to +40 mV lasting 500 ms. Reopenings of anomalous channels as observed in cerebellar granule cells of Cav1.3−/− mice in the presence of the Ca2+ channel activator (+)-(S)-202-791 are shown in Figure 3 A. Note the remarkably long openings separated by brief closings recorded at −40 mV after the depolarizing test pulse, during which channel activity was characterized by short openings separated by long closings. In agreement with previous reports (Forti and Pietrobon, 1993; Hivert et al., 1999), the average current at −40 mV (Fig. 3 B) shows the typical rising phase immediately after the prepulse (reflecting the kinetics of reopening from a closed state outside the activation pathway) and the typical slow decay toward zero (reflecting mainly the duration of the long-opening gating mode induced by the prepulse). For the representative experiment in Figure 3, the slow decay was best fit by a single exponential with a time constant of 83.9 ms. On average, the time constant of the decay was 98.6 ± 17.4 ms (n = 5).
To test the possibility of spontaneous channel openings at −40 mV, 820 ms control pulses to −40 mV were alternated with the prepulse protocol. No spontaneous channel activity was observed in a large majority of sweeps in the single experiments (∼97% of sweeps); if seen (in only 5 of 17 experiments), active sweeps were mainly present at the end of the experiment. These experiments were not used for additional analysis.
To compare the frequency of anomalous channels in wild-type and Cav1.3−/− cerebellar granule cells, we analyzed patches with up to three LTCCs (80% of all patches) as well as patches with a maximum of three cardiac-like and two anomalous channels. Anomalous LTCCs were observed with similar frequency in wild-type (in 8 of 21 patches; 38%) and Cav1.3−/− (in 12 of 25 patches; 48%) mouse cerebellar granule cells.
Our data clearly show that anomalous LTCCs are present in cerebellar granule cells of Cav1.3−/− mice and indicate that the anomalous LTCCs in mouse cerebellar granule cells must mainly be formed by either Cav1.2α1 subunits or a yet uncharacterized α1 subunit. Given the similar frequency of observation of anomalous LTCCs in wild-type and Cav1.3−/− mice, the possibility that Cav1.3α1 subunits also contribute to anomalous channel activity appears unlikely. To exclude this possibility and test the hypothesis that Cav1.2α1 subunits are the pore-forming subunits of anomalous LTCCs, we performed cell-attached patch-clamp recordings in the presence of DHP agonist on cerebellar granule cells from Cav1.2DHP−/− mice.
Single LTCC recordings in cerebellar granule cells of Cav1.2DHP−/− mice
In Cav1.2DHP−/− mice, a targeted α1-subunit mutation abolishes the modulation of Cav1.2 LTCCs by DHP activators (Sinnegger-Brauns et al., 2004) and therefore prevents the typical agonist-induced prolonged openings of Cav1.2 channels. If Cav1.2α1 subunits are the pore-forming subunits of anomalous LTCCs, the typical reopenings of anomalous LTCCS measured in neurons from Cav1.2DHP−/− mice in the presence of the DHP Ca2+ channel activator (+)-(S)-202-791 should be similar to those measured in neurons of wild-type mice in the absence of DHP agonist and shorter than those in neurons from Cav1.3−/− mice in the presence of (+)-(S)-202-791.
Typical reopenings of single anomalous LTCCs recorded at −40 mV after a depolarizing pulse to +40 mV in cerebellar granule cells from Cav1.2DHP−/− mice in the presence of DHP agonist are shown in Figure 4 A. The reopenings, although remarkably long compared with the very short unresolved openings recorded at +40 mV, were clearly shorter than those recorded in the presence of agonist in CaV1.3−/− neurons (compare Fig. 3). The decay of the average current at −40 mV for the representative experiment in Figure 4 was best fit by a single exponential with a time constant of 21.1 ms (Fig. 4 B). In Cav1.2DHP−/− mice, very long reopenings as observed in Cav1.3−/− mice (Fig. 3) were completely absent (n = 12). Accordingly, the time constant of decay of the average current at −40 mV in Cav1.2DHP−/− neurons in the presence of DHP agonist was on average 33.4 ± 6.1 ms (n = 5), and the corresponding pooled average current (Fig. 5) was best fit by a single exponential with a time constant of 28.4 ms. The pooled average current of reopenings of anomalous LTCCs in Cav1.3−/− neurons in the presence of DHP agonist decayed more slowly and was best fit by a single exponential with time constant of 104.3 ms (Fig. 5).
Figure 4.
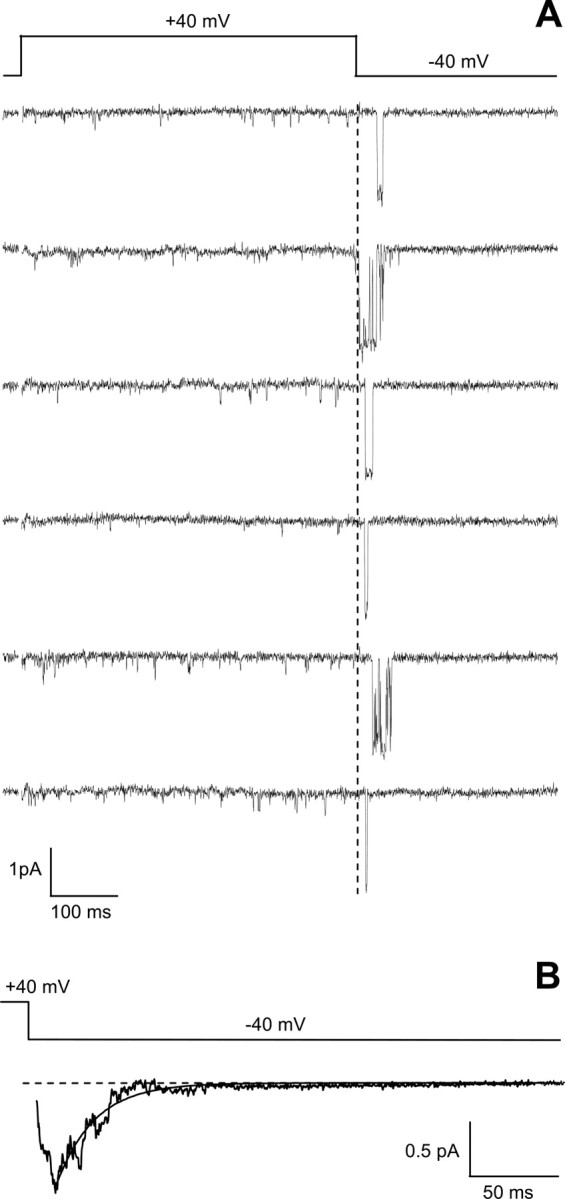
Characteristic reopenings of anomalous LTCCs in cerebellar granule cells of Cav1.2DHP−/− mice in the presence of the Ca2+ channel activator (+)-(S)-202-791. Cell-attached single-channel recordings were obtained in the presence of the Ca2+ channel activator (+)-(S)-202-791 from a patch containing two anomalous LTCCs on a cerebellar granule cell from Cav1.2DHP−/− mice. Single-channel activity was elicited by the same voltage protocol as in Figure 3 A. Both of the representative traces (A) and the normalized average single-channel current during the repolarization at −40 mV (B; n = 74) show clearly shorter reopenings of the anomalous channel in Cav1.2DHP−/− mice compared with Cav1.3−/− mice (see Fig. 3). Cell, 270804_04.
Figure 5.

Averaged single-channel currents at negative potential after predepolarization from Cav1.3−/− and Cav1.2DHP−/− cerebellar granule cells in the presence of the Ca2+ channel activator (+)-(S)-202-791. A normalized ensemble average current at −40 mV (after a 500 ms depolarization to +40 mV) pooled from several patches containing (≥2 in most patches) anomalous LTCCs, six patches (n = 236 traces) from Cav1.2DHP−/− mice and five patches (n = 205 traces) from Cav1.3−/− mice, is shown. In both cases, the decaying average was best fitted by a single exponential as indicated by the gray line. The time constants of the best-fitting exponentials were 104.3 and 28.4 ms for Cav1.3−/− and Cav1.2DHP−/−, respectively. The difference was significant (p < 0.0001; F test).
Figure 6 A shows typical reopenings of single anomalous LTCCs recorded at −40 mV after a depolarizing pulse to +40 mV in cerebellar granule cells from wild-type mice in the absence of DHP agonist. The reopenings in the absence of agonist were very similar to those recorded in CaV1.2−/− neurons in the presence of agonist (compare Fig. 4). The slow decay of the average current at −40 mV for the representative experiment in Figure 6 was best fit by a single exponential with a time constant of 35.7 ms (Fig. 6 B). The time constant of the decay of the average current of reopenings of anomalous LTCCs at −40 mV in wild-type neurons in the absence of DHP agonist was on average 44.1 ± 4.7 ms (n = 7), a value not significantly different from that in Cav1.2DHP−/− neurons in the presence of DHP agonist.
Figure 6.
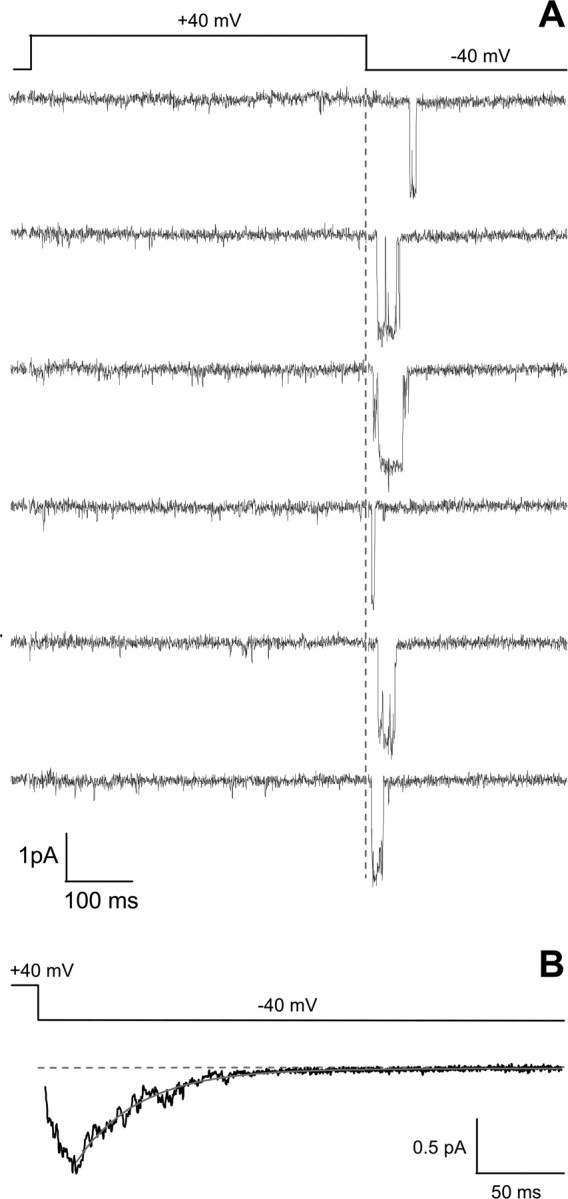
Characteristic reopenings of anomalous LTCCs in cerebellar granule cells of wild-type mice in the absence of agonist. Cell-attached single-channel recordings were obtained from a patch containing two anomalous LTCCs on a cerebellar granule cell from wild-type mice in the absence of DHP agonist. Single-channel activity was elicited by the same voltage protocol as in Figure 3 A. Both of the representative traces (A) and the normalized average single-channel current during the repolarization at −40 mV (B; n = 38) are similar to those of the anomalous channel in Cav1.2DHP−/− mice in the presence of DHP agonist (see Fig. 4). Cell, 150107_07.
Together, our data support the conclusion that Cav1.2α1 subunits are the pore-forming subunits of anomalous LTCCs.
Discussion
Molecular nature of anomalous LTCC channels
In our study, two mouse models were investigated as tools to elucidate the molecular nature of the anomalous LTCC in cerebellar granule cells: Cav1.3−/− mice lacking Cav1.3 channels (Platzer et al., 2000) and Cav1.2DHP−/− mice containing a mutation in the Cav1.2α1 subunit that eliminates sensitivity to DHP agonists and antagonists (Sinnegger-Brauns et al., 2004). We concluded that Cav1.2α1 subunits are the pore-forming subunits of anomalous LTCCs on the basis of the following main findings: (1) LTCCs with anomalous gating were observed in cerebellar granule cells of Cav1.3−/− mice with a frequency not different from wild-type mice; (2) the typical prepulse-induced reopenings of anomalous LTCCs measured after repolarization in Cav1.2DHP−/− neurons in the presence of DHP agonist were shorter than in Cav1.3−/− neurons and were similar to those measured in wild-type neurons in the absence of DHP agonist; and (3) because of the very low expression of Cav1.1 and Cav1.4α1 mRNA, these isoforms are unlikely to contribute to LTCC activity in neonatal cerebellar granule cells.
Our conclusion is important because it represents an essential first step to reveal the yet unknown molecular mechanism giving rise to anomalous Cav1.2 gating. From a biophysical point of view, the essential feature of the kinetic scheme accounting for the anomalous gating is the presence of a nonadsorbing closed state outside the activation pathway connected to the open state through a voltage-dependent transition (Forti and Pietrobon, 1993; Hivert et al., 1999). The biophysical data are equally consistent with this anomalous closed state being an open-pore blocked state of Cav1.2 (Slesinger and Lansman, 1991; Hivert et al., 1999) or an intrinsic conformation of a peculiar Cav1.2 variant. In the hypothesis of an open-pore blocked state, the finding that the anomalous gating remains intact in excised patches (Kavalali et al., 1997; Hivert et al., 1999) excludes block by a diffusible intracellular blocker and suggests that the charged blocking particle is either part of the channel or of a protein in close association with the channel. Interestingly, a similar kinetic scheme with a voltage-dependent transition between the open state and a closed state outside the activation pathway could simulate the so-called resurgent Na+ current, characterized by an anomalous surge of transient current after repolarization (Raman and Bean, 1997, 2001). Grieco et al. (2002) have shown that the cytoplasmic tail of the Na+ channel β4 subunit acts as an open-pore blocker and suggested that the anomalous closed state giving rise to the resurgent current corresponds to the open channel blocked by the β4 subunit. If open-pore block is also the basis of the anomalous gating of Cav1.2 channels, the lack of inactivation of anomalous LTCCs during prolonged strong depolarizations (Forti and Pietrobon, 1993; Kavalali and Plummer, 1994; Hivert et al., 1999), in contrast with the fast inactivation of resurgent Na+ channels, implies reversible open-pore block of anomalous Cav1.2 channels even at high positive voltages in contrast with the irreversible block of Na+ channels at these voltages (in addition to the very slow classical inactivation of LTCCs in contrast with the fast inactivation of Na+ channels).
LTCCs with anomalous gating have been found, thus far, only in neuronal cells [cerebellar, hippocampal, sensory, and motor neurons (Slesinger and Lansman, 1991; Forti and Pietrobon, 1993; Thibault et al., 1993; Kavalali and Plummer, 1994; Ferroni et al., 1996; Hivert et al., 1999)]. Extensive single-channel recordings (our unpublished observations) have not revealed the presence of anomalous LTCCs in either cardiac ventricular myocytes [that express only Cav1.2 LTCCs (Mangoni et al., 2003)] or pituitary GH3 cells [that express both Cav1.2 and Cav1.3 (Fomina et al., 1996)]. Whereas anomalous Cav1.2 channels appear to be neuron specific, it remains an open question whether the anomalous gating is an intrinsic property of an unknown/not yet characterized neuron-specific Cav1.2α1 splice variant or of a Cav1.2α1 subunit with a neuron-specific posttranslational modification. Alternatively, the anomalous gating may derive from assembly of Cav1.2α1 subunits with a not yet characterized neuron-specific protein or accessory subunit. Somewhat favoring the latter hypotheses is the observation by Forti and Pietrobon (1993) of a single anomalous LTCC that abruptly switched from the anomalous (with prevailing conductance of 24 pS) to the cardiac-like (with prevailing conductance of 27 pS) gating pattern. Also somewhat favoring the interaction with a neuron-specific protein is the consistency of the anomalous gating with open-pore block by a nondiffusible particle and the recent evidence that open-pore block by the cytoplasmic tail of an accessory subunit is at the basis of the resurgent Na+ current (Grieco et al., 2002). Moreover, although no major attempts have been made thus far to systematically characterize single-channel properties of different Cav1.2 splice variants, the available functional studies have not revealed recombinant Cav1.2 channels with anomalous gating.
Physiological potential of anomalous Cav1.2 channels in neurons
Activity-dependent gene expression and long-term forms of synaptic plasticity are among the most interesting neuronal functions specifically regulated by LTCCs. In particular, sustained Ras-mitogen-activated protein kinase (MAPK)-dependent cAMP response element (CRE)-binding protein (CREB) phosphorylation and associated CRE-mediated gene transcription depend selectively on Ca2+ influx through LTCCs (Hardingham et al., 1999; Dolmetsch et al., 2001; Wu et al., 2001; West et al., 2002) and specifically on Ca2+ influx through Cav1.2 channels (Moosmang et al., 2005). CREB-dependent transcription is generally regarded as an important step in the generation of the long-lasting forms of synaptic plasticity that are associated with learning and long-term memory (West et al., 2002; Thomas and Huganir, 2004). Interestingly, the induction of L-LTP, the long-lasting form of LTP that requires gene transcription and protein synthesis, depends mostly on Ca2+ influx through LTCCs in hippocampal CA1 neurons (Moosmang et al., 2005; Raymond and Redman, 2006), again with a specific involvement of Cav1.2 channels (Clark et al., 2003; Moosmang et al., 2005).
In different brain regions, depending on the LTP-induction protocol, the crucial Ca2+ influx leading to LTP induction can occur through LTCCs (Grover and Teyler, 1990; Kapur et al., 1998; Weisskopf et al., 1999), NMDA receptors (Cavus and Teyler, 1996; Bauer et al., 2002), or both channels (Cavus and Teyler, 1996; Magee and Johnston, 1997). Invariably, a sustained postsynaptic depolarization (of 20–30 mV) with superimposed bursts of APs appears as the postsynaptic voltage stimulus necessary for induction of LTCC-dependent LTP. Schjott and Plummer (2000) have shown that such a voltage stimulus applied to hippocampal pyramidal cells leads to an overall activation of LTCCs with anomalous gating (Lp channels) that is much larger and long-lasting than that of LTCCs with cardiac-type gating. Moreover, they have shown that the burst of APs induces a shift from short- to long-duration reopenings of anomalous LTCCs and consequently increases their open probability compared with that during the same plateau depolarization without APs. This finding is consistent with the voltage-dependent equilibrium between gating modes proposed by Forti and Pietrobon (1993) as the basis of the long reopenings of anomalous Cav1.2 channels. Although a voltage-dependent equilibrium between mode 1 and mode 2 has been described also for cardiac Cav1.2 channels (Pietrobon and Hess, 1990), the voltage range controlling the transition from the short-opening to the long-opening mode is shifted to lower voltages for anomalous LTCCs, and the potentiation lasts much longer (Forti and Pietrobon, 1993; Kavalali and Plummer, 1996; Hivert et al., 1999), resulting in a larger and more prolonged activation after trains of APs, increasing with frequency and duration of the train (Ferroni et al., 1996; Schjott and Plummer, 2000). The biophysical properties of anomalous Cav1.2 channels include a very low open probability during the APs and the opposite dependence on repolarization voltage of the duration of the long-opening mode (increasing with increasing voltage) and of the unitary current and mean open time of reopenings (decreasing with increasing voltage). Because of these properties, the temporal coincidence of a burst of APs with a small postsynaptic depolarization resulting from summation of EPSPs appears as a particularly efficient stimulus to maximally activate anomalous LTCCs after the AP burst (Forti and Pietrobon, 1993; Hivert et al., 1999). Thus, the anomalous gating may allow LTCCs to function as Hebbian coincidence detectors and might also confer specificity of AP signaling to the nucleus (Adams and Dudek, 2005).
We therefore propose that anomalous Cav1.2 channels are the channels specifically involved in the long-lasting LTCC-dependent and gene transcription-dependent forms of LTP, likely as a consequence of their specific coupling to sustained Ras-MAPK-dependent CREB phosphorylation and associated CRE-mediated gene transcription. This working hypothesis is based on the following: (1) the evidence that Cav1.2 and not Cav1.3 are the LTCCs specifically involved in the sustained Ras-MAPK-dependent CREB phosphorylation that is associated to CRE-mediated gene transcription (Moosmang et al., 2005) and in the gene transcription-dependent hippocampal L-LTP (Clark et al., 2003; Moosmang et al., 2005); (2) our present evidence that Cav1.2α1 and not Cav1.3α1 subunits are the pore-forming subunits of anomalous LTCCs; and (3) the peculiar biophysical properties of anomalous Cav1.2 channels that make them more suitable than Cav1.2 channels with cardiac-type gating to generate a large and sustained Ca2+ influx in response to the postsynaptic voltage stimulus necessary for induction of LTCC-dependent LTP.
Footnotes
This work was supported by Italian Ministry of University and Research Grants Prin2005 and FIRB2002 (D.P.), European Union Grant “EUROHEAD” LSHM-CT-2004-504837 (D.P.), Fonds zur Förderung der Wissenschaftlichen Forschung Grants P-17159 (J.S.) and P17807-B05 (G.J.O.), Tyrolean Science Fund Grant UNI-0404/353 (A.K.), and the University of Innsbruck.
References
- Adams JP, Dudek SM. Late-phase long-term potentiation: getting to the nucleus. Nat Rev Neurosci. 2005;6:737–743. doi: 10.1038/nrn1749. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- Clark NC, Nagano N, Kuenzi FM, Jarolimek W, Huber I, Walter D, Wietzorrek G, Boyce S, Kullmann DM, Striessnig J, Seabrook GR. Neurological phenotype and synaptic function in mice lacking the Cav1.3 alpha subunit of neuronal L-type voltage-dependent Ca2+ channels. Neuroscience. 2003;120:435–442. doi: 10.1016/s0306-4522(03)00329-4. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Milde AS, Andreas K, Ravens U. The effects of verapamil and diltiazem on N-, P- and Q-type calcium channels mediating dopamine release in rat striatum. Br J Pharmacol. 1999;127:576–582. doi: 10.1038/sj.bjp.0702574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Ferroni A, Galli A, Mazzanti M. Functional role of low-voltage-activated dihydropyridine-sensitive Ca channels during the action potential in adult rat sensory neurones. Pflügers Arch. 1996;431:954–963. doi: 10.1007/s004240050091. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Tottene A, Lennon VA, Wilson SM, Dubel SJ, Paylor R, Hosford DA, Tessarollo L, McEnery MW, Pietrobon D, Copeland NG, Jenkins NA. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001;15:1288–1290. doi: 10.1096/fj.00-0562fje. [DOI] [PubMed] [Google Scholar]
- Fomina AF, Levitan ES, Takimoto K. Dexamethasone rapidly increases calcium channel subunit messenger RNA expression and high voltage-activated calcium current in clonal pituitary cells. Neuroscience. 1996;72:857–862. doi: 10.1016/0306-4522(95)00580-3. [DOI] [PubMed] [Google Scholar]
- Forti L, Pietrobon D. Functional diversity of L-type calcium channels in rat cerebellar neurons. Neuron. 1993;10:437–450. doi: 10.1016/0896-6273(93)90332-l. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Afshari FS, Raman IM. A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar Purkinje neurons. J Neurosci. 2002;22:3100–3107. doi: 10.1523/JNEUROSCI.22-08-03100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22:789–798. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RW, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and Class D L-type calcium channel α1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[β]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hivert B, Luvisetto S, Navangione A, Tottene A, Pietrobon D. Anomalous L-type calcium channels of rat spinal motoneurons. J Gen Physiol. 1999;113:679–694. doi: 10.1085/jgp.113.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi H, Yatani A, Akaike N. Block of P-type Ca2+ channels in freshly dissociated rat cerebellar Purkinje neurons by diltiazem and verapamil. Brain Res. 1995;695:88–91. doi: 10.1016/0006-8993(95)00815-8. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel MF, Gray R, Johnston D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR. Selective potentiation of a novel calcium channel in rat hippocampal neurones. J Physiol (Lond) 1994;480:475–484. doi: 10.1113/jphysiol.1994.sp020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR. Multiple voltage-dependent mechanisms potentiate calcium channel activity in hippocampal neurons. J Neurosci. 1996;16:1072–1082. doi: 10.1523/JNEUROSCI.16-03-01072.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. J Neurosci. 1997;17:5334–5348. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 calcium channels in cardiac pacemaker activity. Proc Natl Acad Sci USA. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michna M, Knirsch M, Hoda JC, Muenkner S, Langer P, Platzer J, Striessnig J, Engel J. Cav1.3 (alpha1D) Ca2+ currents in neonatal outer hair cells of mice. J Physiol (Lond) 2003;553:747–758. doi: 10.1113/jphysiol.2003.053256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha 1C (Cav1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, Hess P. Novel mechanism of voltage-dependent gating in L-type calcium channels. Nature. 1990;346:651–655. doi: 10.1038/346651a0. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol (Lond) 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjott JM, Plummer MR. Sustained activation of hippocampal Lp-type voltage-gated calcium channels by tetanic stimulation. J Neurosci. 2000;20:4786–4797. doi: 10.1523/JNEUROSCI.20-13-04786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm M, Vajna R, Pereverzev A, Tottene A, Klockner U, Pietrobon D, Hescheler J, Schneider T. Isoforms of alpha1E voltage-gated calcium channels in rat cerebellar granule cells–detection of major calcium channel alpha1-transcripts by reverse transcription-polymerase chain reaction. Neuroscience. 1999;92:565–575. doi: 10.1016/s0306-4522(99)00013-5. [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig J. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest. 2004;113:1430–1439. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA, Lansman JB. Reopening of Ca2+ channels in mouse cerebellar neurons at resting membrane potentials during recovery from inactivation. Neuron. 1991;7:755–762. doi: 10.1016/0896-6273(91)90278-8. [DOI] [PubMed] [Google Scholar]
- Thibault O, Porter NM, Landfield PW. Low barium and calcium induce a sustained high probability of repolarisation openings of L-type calcium channels in hippocampal neurons: physiological implications. Proc Natl Acad Sci USA. 1993;90:11792–11796. doi: 10.1073/pnas.90.24.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci. 1999;19:10512–10519. doi: 10.1523/JNEUROSCI.19-23-10512.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 2001;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. Cav1.2 and Cav1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci. 2006;23:2297–2310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of Cav1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]