

Abstract
Multiple molecular mechanisms influence nerve regeneration. Because serine proteases were shown to affect peripheral nerve regeneration, we performed nerve crush experiments to study synapse reinnervation in adult mice lacking the serpin protease nexin-1 (PN-1). PN-1 is a potent endogenous inhibitor of thrombin, trypsin, tissue plasminogen activators (tPAs), and urokinase plasminogen activators. Compared with the wild type, a significant delay in synapse reinnervation was detected in PN-1 knock-out (KO) animals, which was associated with both reduced proliferation and increased apoptosis of Schwann cells. Various factors known to affect Schwann cells were also altered. Fibrin deposits, tPA activity, mature BDNF, and the low-affinity p75 neurotrophin receptor were increased in injured sciatic nerves of mutant mice. To test whether the absence of PN-1 in Schwann cells or in the axon caused delay in reinnervation, PN-1 was overexpressed exclusively in the nerves of PN-1 KO mice. Neuronal PN-1 expression did not rescue the delayed reinnervation. The results suggest that Schwann cell-derived PN-1 is crucial for proper reinnervation through its contribution to the autocrine control of proliferation and survival. Thus, the precise balance between distinct proteases and serpins such as PN-1 can modulate the overall impact on the kinetics of recovery.
Keywords: protease nexin-1, Schwann cell, BDNF, fibrin, nerve crush, serine proteases
Introduction
After nerve crush, axonal outgrowth depends on Schwann cell guidance of the growth cone through an altered environment to reinnervate the synaptic target. Growth cone migration is also promoted by neurotrophic factors, cell adhesion molecules, and extracellular matrix (ECM) proteins (Fawcett and Keynes, 1990). Degradation of the ECM and tissue remodeling are influenced by the balance between serine proteases and their inhibitors. Various studies have demonstrated the presence of plasminogen activators (PAs) in neurons and the involvement of the PA system in axonal outgrowth (Krystosek and Seeds, 1981, 1984). The PA system is rapidly induced after nerve injury in sensory neurons, in which it plays an important role in sciatic nerve transection and regeneration (Niclou et al., 1998). Moreover, tissue PA (tPA), urokinase (uPA), and plasminogen knock-out (KO) mice show a delay in functional recovery after sciatic nerve crush, implying that PAs are necessary for timely functional recovery of regenerating peripheral nerves (Siconolfi and Seeds, 2001a,b).
Protease nexin-1 (PN-1) is a 43 kDa protein of the serpin superfamily (Sommer et al., 1987; Festoff et al., 1996) secreted in the extracellular space, where it binds and inhibits several serine proteases, including thrombin, trypsin, plasmin, tPA, and uPA (Baker et al., 1980; Monard, 1993; Knauer et al., 2000). Its expression is finely regulated both spatially and temporally during development and in adulthood (Mansuy et al., 1993). In vivo, PN-1 regulates proteolytic activity in both the CNS and PNS (Monard et al., 1992; Scotti et al., 1994). PN-1 expression has also been detected in rat aortic smooth muscle cells (Richard et al., 2004) and mouse vessels (M. M. Lino, unpublished data). Other PN-1 sources in the peripheral nervous system are Schwann cells and fibroblasts, whereas microglia and macrophages do not express it but take it up (D. Monard, unpublished data). Changes in PN-1 expression have been detected in various pathological states (e.g., Alzheimer's disease, amyotrophic lateral sclerosis, and ictus), suggesting that an imbalance in proteolytic activity contributes to the pathological changes associated with these diseases (Wagner et al., 1989; Sisodia et al., 1990; Golde et al., 1992; Vaughan et al., 1994; Chou et al., 1998). Mechanistically, PN-1 has been shown to promote neurite outgrowth and regulate survival or death of injured neurons (Monard, 1988; Zurn et al., 1988; Houenou et al., 1995; Smith-Swintosky et al., 1995; Vaughan et al., 1995; Donovan et al., 1997). Injury of the rat sciatic nerve triggers PN-1 production in Schwann cells localized at the distal site of the lesion (Meier et al., 1989), possibly to protect structures from prolonged and extensive proteolytic attacks.
The present study examined the role of PN-1 in nerve regeneration after sciatic nerve crush. We found a delay in reinnervation and functional recovery in PN-1 KO mice after such a lesion. The absence of PN-1 led to formation of fibrin deposits, known to reduce Schwann cells proliferation and migration (Akassoglou et al., 2002, 2003). After sciatic nerve crush, tPA activity was higher in PN-1 KO mice than in the wild type (WT), paralleling higher levels of mature BDNF and an increase in Schwann cell death. Moreover, the delay in reinnervation and functional recovery persisted in transgenic mice overexpressing PN-1 exclusively in the nerve. These results suggest that PN-1 expression in Schwann cells is crucial for the proper kinetics of reinnervation. They further point to an important autocrine function of Schwann cell PN-1 in proper reinnervation by contributing to their guidance function.
Materials and Methods
Experimental animals.
The PN-1 knock-in (KI) reporter mice, PN-1 KO, and mice overexpressing PN-1 exclusively in neurons (PN-1 KO/Tg) were generated as described previously (Luthi et al., 1997; Meins et al., 2001; Kvajo et al., 2004). Mice were backcrossed for 10 generations in the C57BL/6 line. Heterozygous mating generated PN-1 KO and wild-type littermates. Genotyping was performed by PCR of DNA from tail biopsies. Wild-type C57BL/6 were purchased from Charles River (Arbresle, France). All experimental animals were 12–16 weeks of age. All animal experiments were approved by the Swiss veterinary authorities.
Surgery.
Mice were anesthetized with a mixture of ketamine and xylazine injected intraperitoneally. The right lower leg was shaved and sterilized with 70% ethanol. The right sciatic nerve was exposed at the sciatic notch. The crush was produced by tightly compressing the sciatic nerve at the sciatic notch with flattened forceps (Atanasoski et al., 2001, 2006a). As opposed to a complete cut, a crush produces degeneration of distal axons, which subsequently regenerate from the proximal segment. A total lack of hindlimb movement together with the absence of the toe-spreading reflex and lateral leg extension confirmed the completeness of the nerve crush. The distal nerve stumps were recovered at 4, 7, 12, and 20 d and 2 months after the crush. Contralateral nerves were used as unlesioned control samples.
Measurement of motor nerve conduction velocity and fibrillation potential.
Conduction velocities and fibrillation potentials of the gastrocnemius nerve were recorded from anesthetized mice. A group of WT and PN-1 KO mice of 3 and 16 weeks of age (n = 3 per genotype per age) were analyzed. A monopolar needle recording electrode was inserted into the gastrocnemius muscle, and a ground electrode was placed as reference in the opposite leg. The sciatic nerve was sectioned at the level of the hip, and the distal stump was stimulated via a suction electrode. The compound muscle action potential in response to sciatic supramaximal stimulation (0.02 ms) was recorded, and the latency between nerve stimulation and the foot of the compound action potential was measured. The nerve conduction velocity (MCV) was determined as follows: MCV = distance between stimulating and recording electrodes/stimulus-response latency.
Immunohistochemistry.
PN-1 immunostaining was performed using the Discovery XT automated stainer (Ventana Medical Systems, Tucson, AZ), with DAB Map detection kit (Ventana Medical Systems). Paraffin sections 6 μm thick were used after fixation in Bouin's fixative.
PN-1 antigen retrieval was achieved by a 96 min pretreatment in CC2 buffer (Ventana Medical Systems reagent). Slides were incubated for 1 h at 37°C with the primary antibody: mouse anti-rat PN-1 (dilution, 1:100). They were then treated for 20 min at 37°C with Ventana Medical Systems detection (fast red substrate) and then incubated for 20 min with the Umap anti-Rb Multimer Alkaline Phosphatase. The signal was amplified using UltraMap AP XT kit (Ventana Medical Systems).
PN-1 KO, PN-1 KO/Tg, and wild-type littermate pairs were examined histologically using sections processed under identical conditions.
Silver esterase staining was performed on fresh frozen triceps surae as described by Frey et al. (2000). Immunohistochemistry for p75 (anti-p75 was a gift from Dr. L. Reichhardt, San Francisco, CA) and fibrinogen (sheep α-human fibrinogen; United States Biological, Swampscott, MA) was performed on fixed tissue prepared from 4% paraformaldehyde-perfused mice using standard procedures. Fixed tissue was immersed in 30% sucrose/PBS overnight at 4°C before embedding in Tissue-Tek optimal cutting temperature (Sakura Finetek Europe, Zoeterwoude, Netherlands) and freezing in dry ice. β-Galactosidase histochemistry was performed on sections from PN-1 KI mice as described previously (Kvajo et al., 2004).
Proliferation and apoptosis.
Mice were injected intraperitoneally with bromodeoxyuridine (BrdU; 100 μg/g of body weight) 2 h and 1 h before being killed. For BrdU/S100 staining, sections were processed as described previously (Atanasoski et al., 2004, 2006b). For S100/terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated UTP nick end labeling (TUNEL), longitudinal sections were fixed in 2% paraformaldehyde for 10 min at room temperature, blocked in PBS containing 10% goat serum, 0.1% BSA, and 1% Triton X-100, and incubated with a primary rabbit antiserum against S100 (1:300; Dako, Glostrup, Denmark) at 4°C overnight. The probes were then exposed to donkey anti-rabbit FITC secondary antibody (1:200; Jackson ImmunoResearch, West Grove, PA) for 60 min at room temperature. The sections were equilibrated in TdT buffer (30 mm Trizma base, pH 7.2, 140 mm sodium cacodylate, and 1 mm cobalt chloride) before addition of TdT and biotinylated dUTP. The sections were incubated at 37°C for 90 min, and then the reaction was stopped by incubating in 2× SSC (300 mm sodium chloride and 30 mm sodium citrate) for 10 min at room temperature. TUNEL staining was detected by incubating the probes with streptavidin–Cy3 (cyanine 3) (1:200; Jackson ImmunoResearch) for 60 min at room temperature. Sections were mounted in AF1 (Citifluor, Canterbury, UK) supplemented with 4,6-diamidino-2-phenylindole (DAPI; 1:1000; Roche Diagnostics, Mannheim, Germany). Immunoreactivity was visualized by epifluorescence microscopy and documented using a Hamamatsu (Bridgewater, NJ) Color Chilled 3CCD Camera. Images were processed with Adobe Photoshop 7.0 for Macintosh (Adobe Systems, San Jose, CA).
Immunoblot.
Sciatic nerves were homogenized in lysis buffer (radioimmunoprecipitation assay plus complete mixture of protease inhibitors; Roche Diagnostics). Eighty micrograms of proteins were subjected to 15% SDS-PAGE (30:1 acrylamide/bisacrylamide) and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes were blocked (PBS–0.2% Tween 20 and 5% skimmed milk) and incubated overnight at 4°C with a mouse 4B3 anti-rat PN-1 antibody (1:1000) (Mansuy et al., 1993) and anti-actin (1:3000; NeoMarkers, Fremont, CA). Blots were developed using SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology, Lausanne, Switzerland).
Histological analysis.
For semithin sections and electron microscopy, 2 month post-sciatic nerve crush mice were anesthetized as described above and perfused with 0.1 m phosphate buffer, pH 7.2, containing 4% paraformaldehyde and 3% glutaraldehyde. Samples were postfixed overnight in the same fixative at 4°C, washed in phosphate buffer, treated with 2% OsO4 overnight at 4°C, dehydrated in an ascending acetone series, infiltrated with Spurr, and incubated at 60°C for polymerization. Semithin sections (0.5 μm) were stained with 0.1% toluidine blue and examined by electron microscopy.
Statistical analysis.
For reinnervation analysis, the reinnervation rate (total number of reinnervated synapses relative to the total number of synapses) was counted on 10 sections through each triceps surae muscle (on average, one of every four sections). Three to four mice per genotype were used for each time point. Statistical analysis was performed using the two-tailed unpaired Student's t test.
BrdU-positive cells were counted in two adjacent 40× objective fields, each covering an area of 0.25 mm2, 2.5–3.0 mm distal to the crush lesion. Three animals per genotype and six sections per mouse were analyzed for a total of ∼1000 S100-positive cells per mouse. The percentages of S100/BrdU-positive cells were determined (mean ± SD) and compared using the two-tailed unpaired Student's t test. To quantify Schwann cell apoptosis, two or more longitudinal sections of sciatic nerve were mounted on a slide. All nuclei on the sections were counted at 40× as well as all TUNEL-positive cells, 2.5–3.0 mm distal to the crush lesion. Three animals per genotype were analyzed, and the percentages of S100/TUNEL-positive cells were determined (mean ± SD) and compared using the two-tailed unpaired Student's t test.
Gel zymography.
Gel zymography was performed as described previously (Lantz and Ciborowski, 1994). Mice were deeply anesthetized and transcardially perfused with ice-cold PBS at different time points after crush surgery. One centimeter of sciatic nerve including the crush site and the comparable site of the contralateral uncrushed nerve were removed, homogenized in 10 mm Tris-Cl, pH 6.8, and centrifuged. Dilutions of the cleared homogenate (6 μg of protein) and 30 μg of recombinant tPA (Genentech, San Francisco, CA) were separated by electrophoresis at 4°C on 10% polyacrylamide-SDS gels copolymerized with casein (1 mg/ml; Sigma-Aldrich, Steinheim, Germany) and plasminogen (2.5 U/ml; Chromogenix, Mölndal, Sweden). After electrophoresis, the SDS was extracted from the gel using 2.5% Triton X-100, and the gel was incubated in 0.1 m Tris, pH 8.1, at 37°C, followed by staining with 0.125% Coomassie blue in 50% methanol/10% acetic acid. Destaining with the same solvent revealed transparent zones of lysis against the dark casein background at molecular weights of 65 and 43 kDa, corresponding to tPA and uPA.
Pro-BDNF and BDNF ELISA assays.
Because available assays did not distinguish between BDNF and its precursor form, a test to evaluate pro-BDNF was first developed. The amount of pro-BDNF thus determined in a parallel assay was subtracted from the total amount of BDNF (precursor and mature forms) measured by BDNF Emax Immunosystem (Promega, Madison, WI) according to the manufacturer's instructions to quantify the mature BDNF in samples containing 15 μg of protein from crushed or intact nerves.
The pro-BDNF ELISA assay developed detected only the precursor form of the neurotrophin. For this purpose, pro-BDNF protein produced using conditioned media from COS-7 cells transfected with furin-resistant pro-BDNF (Fayard et al., 2005) was quantified with the BDNF Emax Immunosystem and used as standard. After coating a 96-well plate with mouse anti-BDNF (provided in BDNF Emax Immunosystem), nerve lysates and 25, 50, 100, 200, and 250 pg/ml pro-BDNF standard were added. Detection and quantification of pro-BDNF in standards and samples were performed exactly as described for the BDNF Emax Immunosystem, except that the secondary antibody was a rabbit polyclonal anti-pro-BDNF specific for the precursor form (Millipore, Lucerne, Switzerland).
Functional testing.
Functional testing to study recovery of toe pressure sensitivity was performed on conscious mice as described previously (Siconolfi and Seeds, 2001b). Mice were tested every other day (n = 8).
Results
PN-1 expression in Schwann cells
Using PN-1 KI reporter mice, we analyzed PN-1 expression by monitoring 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) staining in nerve and muscle. PN-1 expression started 3 d after birth (P3), increased at P6, and is almost vanished at P28 (Fig. 1A). PN-1 was not expressed in the sciatic nerve of adult mice but was strongly upregulated 4 d postcrush (dpc) (Fig. 1B–D). The expression persisted up to 12 d after the crush (Fig. 1B,C) and began to decrease after 20 d (data not shown). Four days after lesion, some proliferating Schwann cells reexpressed PN-1 (Fig. 1D). This PN-1 upregulation was also detected at the protein level (Fig. 1E). In the muscle, both in the control and after nerve crush, PN-1 expression was detected exclusively in blood vessels (data not shown).
Figure 1.
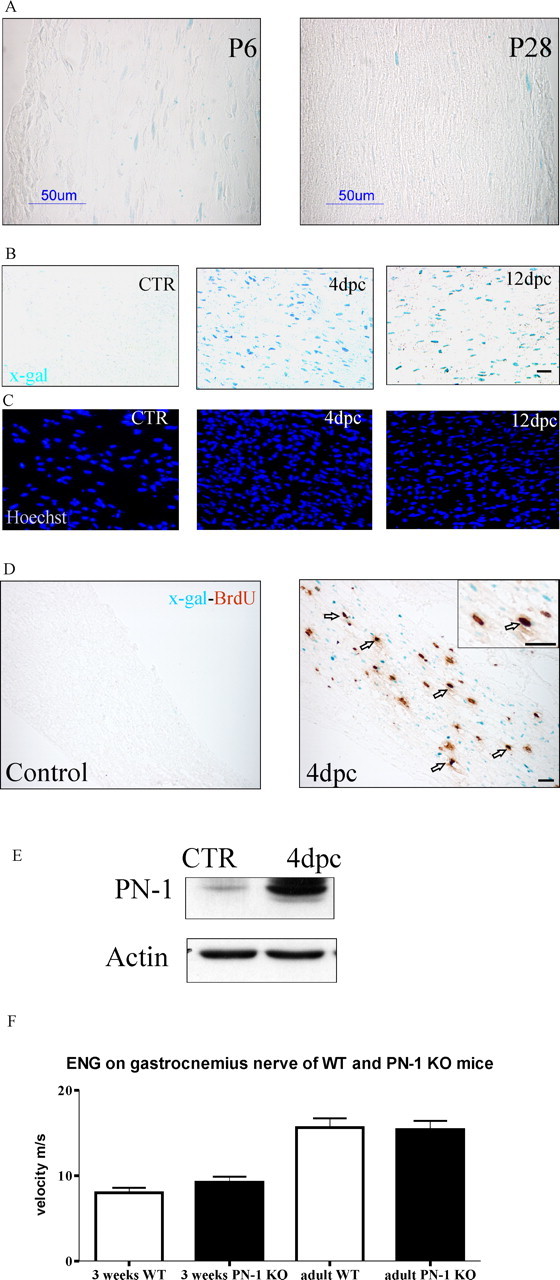
PN-1, nerve function and upregulation after sciatic nerve crush. A, PN-1 is expressed during myelination. X-gal staining of longitudinal sections of sciatic nerves at 6 and 28 d after birth is shown. PN-1 is expressed in many Schwann cells during myelination, whereas its expression is strongly reduced when myelination is almost completed. B, C, PN-1 is reexpressed 4 and 12 dpc. X-gal staining of longitudinal sections of uninjured and the distal stump of injured sciatic nerves (B) and Hoechst staining illustrating also Schwann cell proliferation after crush (C). D, Colabeling of BrdU and X-gal on longitudinal sections of the intact and distal stump of an injured sciatic nerve. Arrows point to double-stained Schwann cell nuclei. Scale bars: A, B, D, 50 μm. E, Immunoblot showing the presence of PN-1 protein in uninjured and 4 dpc sciatic nerves. F, Electroneurogram (ENG) analysis: PN-1 deficiency does not lead to changes in gastrocnemius nerve conduction velocity in 3-week-old and adult mice. CTR, Control. Error bars represent SD.
Nerve conduction velocity does not differ between WT and PN-1 KO mice
Myelin alteration and/or internodal distance affect the velocity of the nerve impulse conduction (Stoffel and Bosio, 1997). Fibrillation potentials and conduction velocities in the sciatic nerves were measured by myography and electroneurography as functional tests for alterations in myelin and/or motor neurons in PN-1 KO and WT mice. Functional innervation was ascertained in PN-1 KO mice from the absence of fibrillation potentials in gastrocnemius muscle. WT muscles denervated for 7 d served as positive controls. Conduction velocity was not affected in PN-1 mutant mice, neither in 3-week-old nor in adult (3–5 months) mice (Fig. 1F). Given that myelination is concluded at ∼3–4 weeks, this means that myelination developed normally in PN-1 KO mice. The absolute conduction velocities measured by our electromyograph both in WT and mutant animals were lower than published values (16 vs 42 m/s); this is because our stimulus-response latencies (∼1.5 ms) included the synaptic delay at the neuromuscular junction (NMJ) (∼0.5–0.7 ms). Taking synaptic delay into account, our velocity estimates are comparable with those published by others (Higashimori et al., 2006).
Axonal regeneration of sciatic nerve in triceps surae is delayed in PN-1 KO mice
The p75 low-affinity neurotrophin receptor is known to be strongly upregulated in Schwann cells after denervation. Consequently, p75 immunoreactivity was analyzed at the NMJ to investigate denervation in PN-1 KO mice. As expected, prominent p75 upregulation was detected both in wild-type and KO animals 4 d after crush (Fig. 2A,B). Twelve days after crush, p75 immunoreactivity had vanished in wild-type NMJs but was still very high in PN-1 KO mice (Fig. 2C–F). Twenty days after crush, both KO and wild-type muscle were negative (data not shown). Axonal degeneration and regeneration was also confirmed by GAP-43 (growth-associated protein 43) immunohistochemistry (data not shown).
Figure 2.
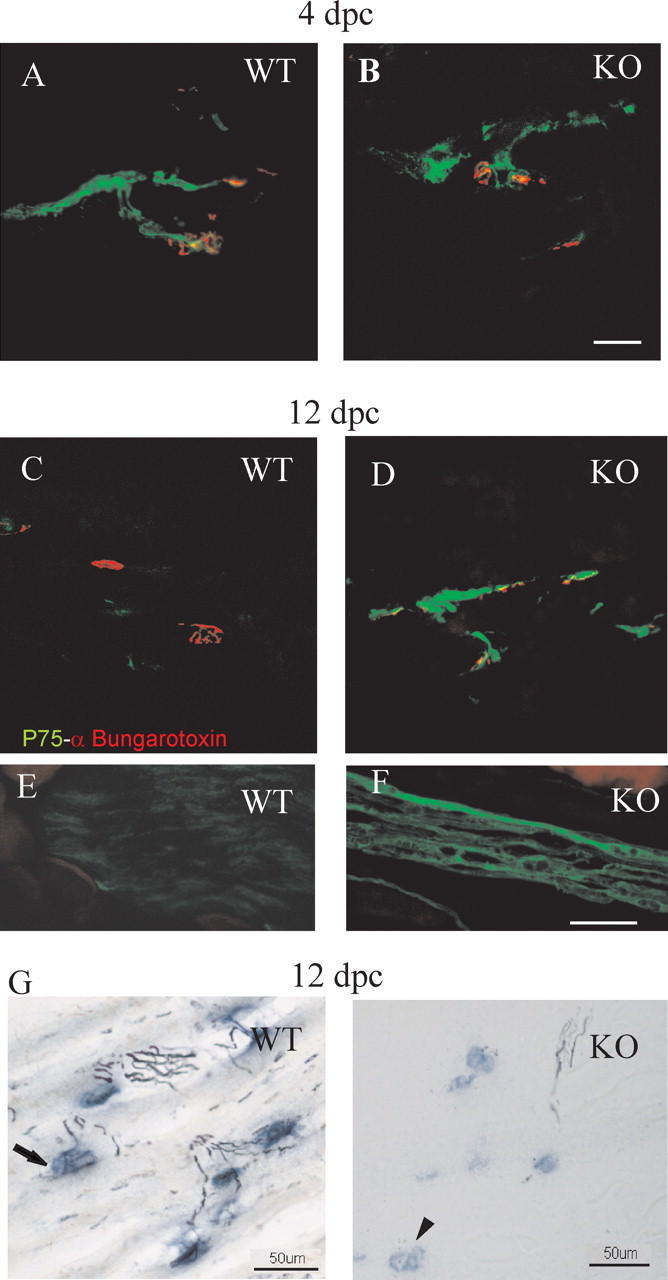
Delayed axonal regeneration in the absence of PN-1. A, B, Longitudinal sections of gastrocnemius at 4 dpc. p75 (green) and α-bungarotoxin (red) double labeling indicate similar denervation responses at NMJs. C–F, At 12 dpc, the same double labeling suggests delayed recovery at NMJs (C, D) and at the nerves of PN-1 KO mice (E, F). G, Silver esterase staining at 12 dpc shows delayed reinnervation in PN-1 KO NMJs (synapses, blue; nerves, black). The arrow indicates an innervated synapse in WT, and the arrowhead indicates a denervated synapse in KO. Scale bars: A–D (in B), G, 50 μm; E, F (in F), 5 μm.
The triceps surae muscle was analyzed by silver esterase staining to study the reinnervation rate. Four days after crush, no signs of reinnervation were observed in either wild-type or PN-1 KO mice (data not shown). Twelve days after crush, wild-type mice had a significantly higher reinnervation rate than PN-1 KO mice (809.8 ± 110 vs 148.7 ± 61.1 synapses; *p < 0.04) (Fig. 2G). Twenty days after crush, most of the NMJs were reinnervated in both wild-type and PN-1 KO mice (942.5 ± 41.7 vs 970.7 ± 87.2 synapses; p > 0.05).
PN-1 KO mice show a delayed functional recovery after sciatic nerve crush
To further substantiate the effect of PN-1 absence on nerve regeneration, we studied sensory and motor function recovery following the procedures as described previously (Devor and Govrin-Lippmann, 1979; Siconolfi and Seeds, 2001b). Before surgery, all wild-type and mutant mice showed equal sensitivity and response to the test. After the crush, the responses were totally abolished in the injured hindlimb. Recovery in responsiveness of the first four digits was significantly delayed in the PN-1 KO mice (Fig. 3A). Wild-type mice could reuse the injured leg 2–4 d earlier than the PN-1 KO mice. On average, wild-type mice showed a complete recovery of reflexes after 15 d compared with 18 d for the mutant mice. These results suggest that the absence of PN-1 delays functional recovery after peripheral nerve injury.
Figure 3.
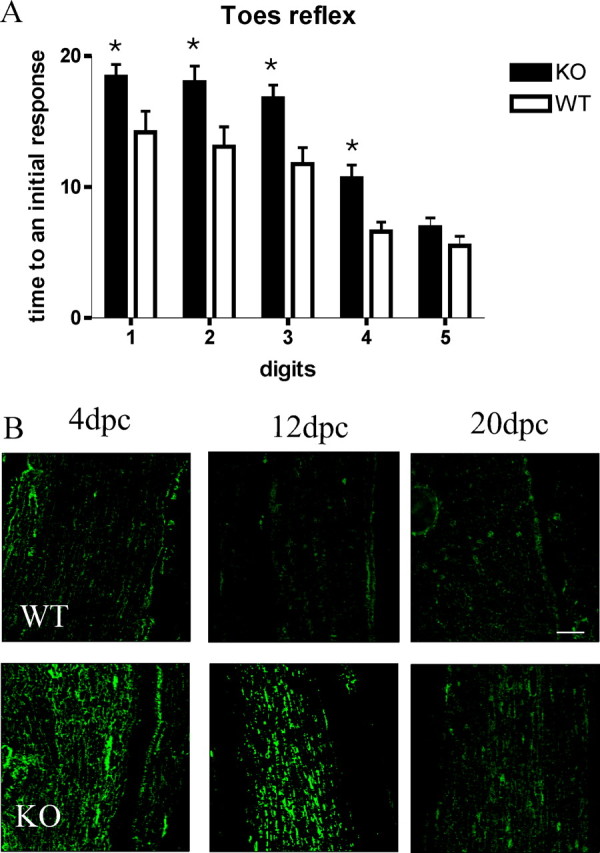
A, Recovery of response to pinch in hindfoot digits after sciatic nerve crush. WT and PN-1 KO mice received a pinch by forceps at the distal portion of each hindfoot digit. Vocalization and foot withdrawal were scored as a positive response. Time (in days) to elicit an initial response was recorded for each digit. PN-1 KO mice show a delayed recovery to pinch. Data are mean ± SD values obtained from eight animals in three different experiments. *p < 0.05; two-way ANOVA. B, Immunostaining reveals increased and prolonged fibrin(ogen) deposition in PN-1 KO mice after sciatic nerve injury. Scale bar, 100 μm.
Fibrin accumulates in PN-1 KO mice after nerve injury
Accumulation of fibrin deposits in the sciatic nerve has been reported to affect Schwann cell migration, known to be a key issue for proper reinnervation (Akassoglou et al., 2002, 2003). Fibrin formation and accumulation is mainly caused by the cleavage of fibrinogen by thrombin (Walsh and Ahmad, 2002), and PN-1 is a potent thrombin inhibitor (Monard, 1993). Nerve crush was shown to induce a time-dependent increase in thrombin activity (Smirnova et al., 1996) and to cause fibrin deposition (Boyd and Gordon, 2001; Akassoglou et al., 2002). We, therefore, examined the presence of fibrin in the regenerating nerves of wild-type and PN-1 KO mice. Immunostaining on longitudinal sections revealed obviously more fibrin deposits in PN-1 KO animals than in the wild type at all postcrush time points (Fig. 3B).
Absence of PN-1 alters Schwann cell proliferation and apoptosis after sciatic nerve crush
We next evaluated whether the delay in reinnervation in mice lacking PN-1 is associated with changes in Schwann cells. Their proliferation rate was compared in wild-type and PN-1 KO mice 4, 12, and 20 d after lesion. Longitudinal sections of the sciatic nerve were stained for BrdU incorporation, the Schwann cell marker S100, and DAPI. The proliferation rate was determined as the number of BrdU-positive cells relative to the total number of cells within a defined area (0.25 mm2) (see Materials and Methods). Compared with wild-type mice, a significant decrease in Schwann cell proliferation rate was measured at 4 and 12 d after injury in the PN-1 KO mice (p < 0.05 and p < 0.01, respectively) (Fig. 4A,B). The apoptosis rate, determined using triple TUNEL, S100, and DAPI staining, showed a significant increase in PN-1 KO mice 12 d after crush (p < 0.05) (Fig. 4C,D). No differences were detected 4 and 20 d after injury.
Figure 4.

Reduced proliferation and increased apoptosis in PN-1 KO Schwann cells. A, Longitudinal sections of uninjured and the distal stump of crushed (12 dpc) sciatic nerves. Nerves were taken from control and PN-1 KO animals. Sciatic nerves were double labeled with an anti-BrdU monoclonal antibody (green) and a rabbit antiserum against the Schwann cell marker S100β (red). B, Quantitative analysis of cell proliferation in WT (gray bar) and PN-1 KO (black bar) sciatic nerves 4, 12, and 20 dpc. Data are mean ± SD values obtained from three animals per time point and genotype. Significant differences are present at both 4 and 12 dpc. C, Longitudinal sections of uninjured and the distal stump of crushed (12 dpc) sciatic nerves. Sciatic nerves were triple labeled with TUNEL (red), a rabbit antiserum against the Schwann cell marker S100β (green), and Hoechst (blue). D, Quantitative analysis of Schwann cell apoptosis in WT (gray bar) and PN-1 KO (black bar) sciatic nerves 4, 12, and 20 dpc. Data are mean ± SD values obtained from three animals per time point and genotype. Significant differences occurred at 12 dpc. Scale bars, A, C, 20 μm. *p < 0.05; **p < 0.01; Student's t test.
Normal remyelination
To check whether the late-stage postinjury remyelination process was affected by the absence of PN-1, histological analysis was performed 2 months after the crush. The overall morphology and the ultrastructure of the sciatic nerve cross sections appeared normal in both genotypes (Fig. 5A,C,E). Both wild-type and PN-1 KO crushed nerves showed thinner myelin sheaths than unlesioned nerves and a similar myelination state, indicating that the remyelination process was ongoing (Fig. 5B,D,F).
Figure 5.
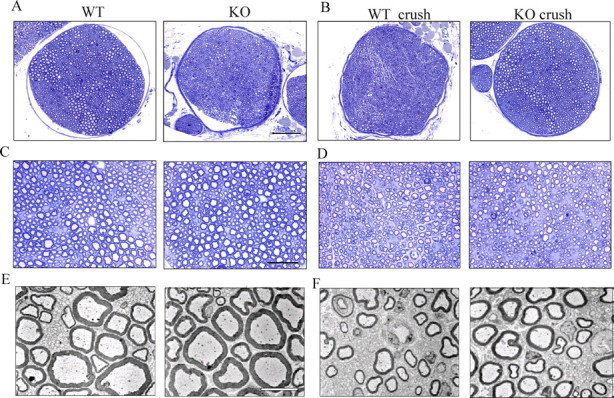
Myelinated peripheral nerves are maintained in the absence of PN-1. Transverse semithin sections of adult mouse nerves stained with toluidine blue are shown. A, C, Nerve bundle of uninjured sciatic nerve of WT and PN-1 KO mice. B, D, Nerve bundle of the distal stump of crushed nerves of WT and PN-1 KO mice 2 months after sciatic nerve crush. Magnification: A, B, 20×; C, D, 40×. E, F, Ultrastructure of PN-1 KO and WT mice control axons (E) and 2 months after nerve crush (F), both at magnifications of 1400×. Scale bars: A, B (in A), 20 μm; C, D (in C), 10 μm; E, F (in E), 5 μm.
tPA activity increases after sciatic nerve crush
Nerve injury induces a strong activation of the PA/plasminogen system at the level of both mRNA and enzymatic activities (Siconolfi and Seeds, 2001a). Because PN-1 is also a potent tPA inhibitor, tPA activity was measured in sciatic nerves before and after injury using gel zymography. A greater increase in tPA proteolytic activity was detected after nerve crush in PN-1 KO mice at 4, 12, and 20 d after crush (Fig. 6A)
Figure 6.
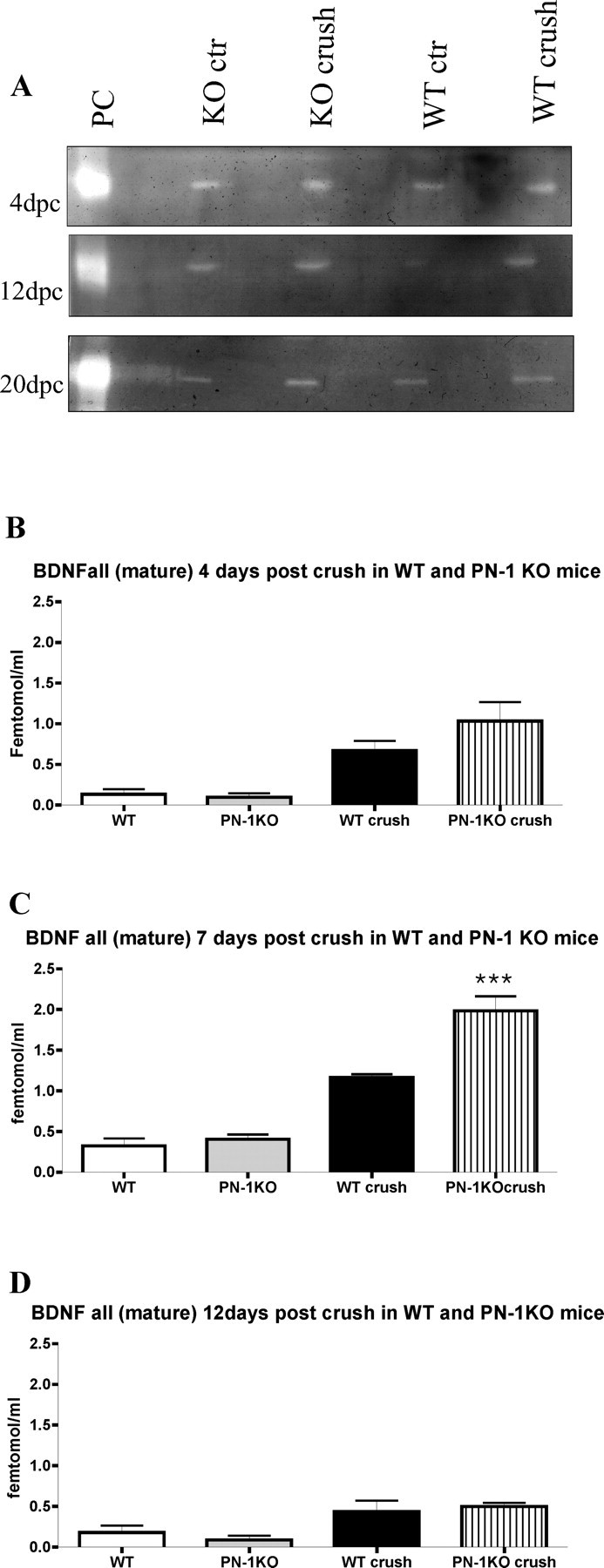
PN-1 KO mice show increased tPA activity and mature BDNF after sciatic nerve crush. A, Zymography of uninjured and distal stump crush sciatic nerves 4, 12, and 20 dpc. B–D, Quantitative analysis of mature BDNF of uninjured and distal stump crush sciatic nerves 4, 7, and 12 dpc. Data are mean ± SD values obtained from four animals per time point and genotype (BDNF determinations were performed in triplicate). ***p < 0.0001; one-way ANOVA. ctr, Control.
Absence of PN-1 increases the level of mature BDNF caused by sciatic nerve crush
tPA has been implicated in the processing of pro-BDNF into mature BDNF (Pang et al., 2004), which was reported to increase 4 d after nerve injury. The BDNF level is highest after 7 d but then slowly decreases to normal (Marcinkiewicz et al., 1998). Because we recorded a greater increase in tPA activity after sciatic nerve crush in PN-1 KO than in WT mice (Fig. 6A), we evaluated the levels of mature BDNF. Using a method distinguishing mature BDNFs from pro-BDNF forms, mature BDNFs were measured in control and crushed sciatic nerves at 4, 7, 12, and 20 d after lesion. An increase in BDNFs was detected at 4 d after crush, with a peak at 7 d (p < 0.0001) (Fig. 6B,C). At this stage, the levels in mice lacking PN-1 were significantly higher than those in wild-type animals. The levels decreased by 12 d. Interestingly, all BDNF found in crushed sciatic nerves was in the mature form. No differences in either total or processed BDNF were detected in intact nerves from wild-type or PN-1 KO animals (Fig. 6B–D).
Schwann cell-derived PN-1 regulates the kinetics of reinnervation
To assess whether neuron-derived PN-1 is sufficient to rescue the reinnervation delay observed in our model, we crossed PN-1 KO mice with mice overexpressing rat PN-1 exclusively in neurons (Meins et al., 2001). The difference in molecular weight easily distinguished the wild type from the Thy-1 promoter-driven transgenic protein, which is expressed in the muscle (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and not upregulated after lesion (data not shown).
Compared with wild-type animals, a slight, although not statistically significant, reduced reinnervation was observed at 12 d after crush in parental transgenic mice overexpressing PN-1 in the wild-type background (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material). At 12 dpc, p75 immunoreactivity was also slightly increased, but 20 d after crush, no difference in reinnervation could be detected (data not shown).
High expression of rat PN-1 protein level was detected in the sciatic nerve of PN-1 KO/Tg (Fig. 7A). PN-protein expression is more prominent in the nerve of the PN-1 KO/Tg than in WT. This level, because of the constitutive activity of the Thy-1 promoter, is certainly not lower than the one induced by crush in the WT animals (Fig. 1D). Neuronal PN-1 expressed in the PN-1 KO/Tg nerve is secreted and taken up by the muscle, as detected by immunohistochemistry (Fig. 7B). The delayed reinnervation phenotype was still observed in progeny PN-1 KO/Tg mice 12 d after crush (942.5 ± 41.7 vs 278 ± 51.4 synapses; **p < 0.001), as was the high p75 immunoreactivity (results not shown), similarly to the changes observed in the PN-1 KO mice. Twenty days after crush, p75 immunoreactivity had returned to normal levels, although reinnervation was still reduced (data not shown). As in PN-1 KO animals, decreased proliferation and an increased apoptotic rate were detected in Schwann cells (Fig. 7C,D). The sensory and motor function recovery after peripheral nerve lesion was still delayed in these PN-1 KO/Tg mice (Fig. 7E). These data suggest that high PN-1 expression restricted to neurons is not sufficient to rescue the delay of reinnervation observed in PN-1 KO mice.
Figure 7.

Overexpression of PN-1 in the nerve did not rescue reinnervation delay in PN-1 KO mice. A, PN-1 sciatic nerve protein expression in WT (1), PN-1 KO (2), and PN-1 KO/Tg (3) mice. PN-1 KO/Tg mice express a high level of rat PN-1 in the nerve. B, PN-1 immunodetection in WT, PN-1 KO, and PN-1 KO/Tg gastrocnemius muscle. Neuronal overexpressed PN-1 in PN-1 KO/Tg mice is secreted and taken up by the muscle fibers. Scale bar, 50 μm. C, Quantitative analysis of cell proliferation in WT (gray bar) and PN-1 KO/Tg (black bar) sciatic nerves at 4 and 12 dpc. Data are mean ± SD values obtained from three animals per time point and genotype. Significant differences occurred at 4 and 12 dpc. D, Quantitative analysis of Schwann cell apoptosis in WT (gray bar) and PN-1 KO/Tg (black bar) sciatic nerves at 4 and 12 dpc. Data are mean ± SD values obtained from three animals per time point and genotype. *p < 0.05; **p < 0.01; Student's t test. E, Recovery of response to pinch in hindfoot digits after sciatic nerve crush. WT, PN-1 KO, and PN-1 KO/Tg mice received a pinch to the distal portion of each hindfoot digit by forceps. Vocalization and foot withdrawal were noted as positive responses. Time (in days) to elicit an initial response was recorded for each digit. PN-1 KO mice show a delayed response to pinch compared with WT. *p < 0.05; **p < 0.01; two-way ANOVA.
Discussion
The results of this study show that PN-1 deficiency slows down the kinetics of reinnervation and the recovery of sensory and motor functions in the hindlimb after peripheral nerve injury. The data indicate that various factors are involved in this phenomenon: reduced proliferation and increased apoptosis of Schwann cells, increased and longer-lasting fibrin deposition, and higher levels of mature BDNF in the nerves. Altogether, we provide evidence that tight control of extracellular proteolytic activity is needed for efficient reinnervation. Furthermore, because neuronal PN-1 overexpression did not rescue the observed delay, Schwann cell-derived PN-1 is most likely one of the key regulatory factors for proper timing of recovery.
Functional nerve regeneration requires sprouting, axonal elongation, and new myelin synthesis. The rates of dedifferentiation, proliferation, and redifferentiation into myelinating Schwann cells (Kioussi and Gruss, 1996) are known to influence the rate of nerve regeneration (Son and Thompson, 1995a,b; Son et al., 1996). Various factors regulate the reaction to injury.
For example, the ECM molecules of the basal lamina are crucial for both axonal elongation and remyelination (Bunge et al., 1986; Reichardt and Tomaselli, 1991). Schwann cells react to changes in the composition of the ECM after nerve lesion. Among those, injury leads to accumulation of fibrin associated with rupture of the blood–brain barrier. Fibrin deposition and clearing are of crucial importance for Schwann cell migration and differentiation. In mice deficient in fibrinogen, injury induces slow proliferating Schwann cells with a high myelinating potential (Akassoglou et al., 2002, 2003). In mice lacking tPA, excess of fibrin exacerbates axonal degeneration, indicating a beneficial tPA fibrinolytic function (Akassoglou et al., 2000, 2002, 2003). In vitro, fibrin excess blocks Schwann cell migration, supporting observations in vivo that it prevents their redifferentiation and myelinating function (Akassoglou et al., 2002). In the present study, significant and sustained postinjury fibrin deposition was also observed in mice lacking PN-1 (Fig. 3), in which fibrin deposits persist despite the general increase in tPA activity (Fig. 6A) (Kvajo et al., 2004). The high amount of fibrin observed despite a higher level of tPA activity may be explained by the strong inhibition of thrombin by PN-1 (Baker et al., 1980; Monard, 1993; Knauer et al., 2000), which has been shown to increase after sciatic nerve injury (Friedmann et al., 1999; Hirakawa et al., 2003, 2004). This suggests that generation of fibrin overcomes the tPA fibrinolytic effect in mice lacking PN-1. The reduced Schwann cell proliferation detected (Fig. 4A,B) appears to contradict the earlier observation that absence of fibrin leads to a reduction in BrdU incorporation (Akassoglou et al., 2002). Fibrin induces Schwann cells to produce p75 receptor, which influences their apoptosis rate (Chao et al., 1998; Ferri and Bisby, 1999; Syroid et al., 2000). Furthermore, contradictory reports indicate that this receptor modulates their myelinating state (Boyd and Gordon, 2001; Cosgaya et al., 2002; Tolwani et al., 2004). In the present study, high upregulation of p75 receptor was already detected at 4 d after crush before apoptosis was evident. It cannot, therefore, explain the decrease in proliferation observed in the mutant mice at this stage. The influence of p75 upregulation could, however, become significant at 12 d after crush, when a higher decrease in proliferation correlated with a significant increase in apoptosis (Fig. 4). A comparative quantitative evaluation of fibrin deposition in the different studies is not possible. However, it cannot be excluded that different levels of fibrin accumulation could differentially affect the proliferative response of the Schwann. Additional studies will be needed to clarify these issues (Akassoglou et al., 2002, 2003).
Alternative mechanisms could explain an effect of PN-1 on fine tuning of nerve regeneration. Activation of protease-activated receptor 1 (PAR1) by thrombin perturbs neurite outgrowth and induces apoptosis and degeneration in motoneuron cultures (Smirnova et al., 1998; Turgeon et al., 1998). PAR1 increases in the proximal and distal parts of the sectioned sciatic nerve and in denervated muscle (Niclou et al., 1998), and overactivation of this receptor in the absence of PN-1 could consequently contribute to the delayed axonal regrowth. Interestingly, tPA, uPA, and plasminogen KO mice show delayed functional recovery after sciatic nerve crush (Siconolfi and Seeds, 2001a,b). Such data are in line with observations in vitro that higher levels of tPA localize at the growth cone and contribute to its motility (Krystosek and Seeds, 1981, 1984; Garcia-Rocha et al., 1994). Surprisingly, our experiments gave similar results in the absence of PN-1. Thus, removing either a protease or one of its inhibitors leads to the same result. This seemingly contradictory situation indicates, at least in this postlesion case, that the function of PN-1 is probably mediated by its much more potent inhibitory impact on thrombin fibrinogenic activity. In PN-1 KO mice, the increased tPA activity could lead to an increase in plasmin activity insufficient to switch the equilibrium toward fibrin degradation but nevertheless sufficient to enhance conversion of the BDNF precursor to mature BDNF (Pang et al., 2004). Mature BDNF, which increases after sciatic nerve crush (Marcinkiewicz et al., 1998), is an important regulatory factor for correct repair after injury (Zhang et al., 2000; Tolwani et al., 2004). It is, therefore, interesting that higher levels of mature BDNF were detected in mice lacking PN-1 after injury (Fig. 6). This could contribute to the observed delayed reinnervation through various mechanisms. First, mature BDNF can bind to p75, the level of which remains high in the mutant mice, thus contributing to the apoptosis (Boyd and Gordon, 2001, 2002; Yeiser et al., 2004) detected at 12 d after crush. Second, a high level of the neurotrophin can cause direct inhibition of axonal regeneration. In fact, BDNF is required for peripheral nerve regeneration and remyelination after injury (Sendtner et al., 1992; Zhang et al., 2000; Tolwani et al., 2004), but its beneficial effect seems to be dose dependent, because high doses of BDNF, acting through the p75 receptor, can significantly inhibit motor axonal regeneration (Boyd and Gordon, 2001, 2002). Finally, a much more speculative hypothesis cannot be excluded. The similarity of phenotype between the tPA, uPA, or plasminogen KO mice and the PN-1-deficient mice could indicate that the amount of serine protease/serpin complex formation is also involved. Because the complex between PN-1 and its target proteases has a much higher affinity to the low-density lipoprotein receptor proteins than the individual partners, one cannot exclude that this interaction could influence distinct cellular responses (Knauer et al., 1997).
In any case, neuronal overexpressed PN-1 can be secreted by the nerve (Fig. 7B) and is certainly more prominent in PN-1 KO/Tg than the level detected after crush in WT animals (Figs. 1D, 7A). Nevertheless, it does not rescue the observed regeneration delay. This further supports the hypotheses that Schwann cell-derived PN-1, possibly in an autocrine mechanism, is a key regulatory factor for correct and timely recovery after injury.
In conclusion, Schwann cell-derived PN-1 is required for a normal reinnervation after sciatic nerve crush. Thus, the precise tuning of distinct proteases via serpins such as PN-1 can modulate different pathways affecting the kinetics of recovery (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Footnotes
This work was supported by the Swiss National Science Foundation (S.A., U.S.) and the National Center of Competence in Research Neural Plasticity and Repair (U.S.). We are grateful to Dr. A. Matus and P. King for comments on the manuscripts. We thank Katja Wichmann for technical assistance.
References
- Akassoglou K, Kombrinck KW, Degen JL, Strickland S. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol. 2000;149:1157–1166. doi: 10.1083/jcb.149.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akassoglou K, Yu WM, Akpinar P, Strickland S. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron. 2002;33:861–875. doi: 10.1016/s0896-6273(02)00617-7. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Akpinar P, Murray S, Strickland S. Fibrin is a regulator of Schwann cell migration after sciatic nerve injury in mice. Neurosci Lett. 2003;338:185–188. doi: 10.1016/s0304-3940(02)01387-3. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Shumas S, Dickson C, Scherer SS, Suter U. Differential cyclin D1 requirements of proliferating Schwann cells during development and after injury. Mol Cell Neurosci. 2001;18:581–592. doi: 10.1006/mcne.2001.1055. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Notterpek L, Lee HY, Castagner F, Young P, Ehrengruber MU, Meijer D, Sommer L, Stavnezer E, Colmenares C, Suter U. The protooncogene Ski controls Schwann cell proliferation and myelination. Neuron. 2004;43:499–511. doi: 10.1016/j.neuron.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Boller D, De Ventura L, Koegel H, Boentert M, Young P, Werner S, Suter U. Cell cycle inhibitors p21 and p16 are required for the regulation of Schwann cell proliferation. Glia. 2006a;53:147–157. doi: 10.1002/glia.20263. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Scherer SS, Sirkowski E, Leone D, Garratt AN, Birchmeier C, Suter U. ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. J Neurosci. 2006b;26:2124–2131. doi: 10.1523/JNEUROSCI.4594-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JB, Low DA, Simmer RL, Cunningham DD. Protease-nexin: a cellular component that links thrombin and plasminogen activator and mediates their binding to cells. Cell. 1980;21:37–45. doi: 10.1016/0092-8674(80)90112-9. [DOI] [PubMed] [Google Scholar]
- Boyd JG, Gordon T. The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J Neurobiol. 2001;49:314–325. doi: 10.1002/neu.10013. [DOI] [PubMed] [Google Scholar]
- Boyd JG, Gordon T. A dose-dependent facilitation and inhibition of peripheral nerve regeneration by brain-derived neurotrophic factor. Eur J Neurosci. 2002;15:613–626. doi: 10.1046/j.1460-9568.2002.01891.x. [DOI] [PubMed] [Google Scholar]
- Bunge RP, Bunge MB, Eldridge CF. Linkage between axonal ensheathment and basal lamina production by Schwann cells. Annu Rev Neurosci. 1986;9:305–328. doi: 10.1146/annurev.ne.09.030186.001513. [DOI] [PubMed] [Google Scholar]
- Chao M, Casaccia-Bonnefil P, Carter B, Chittka A, Kong H, Yoon SO. Neurotrophin receptors: mediators of life and death. Brain Res Brain Res Rev. 1998;26:295–301. doi: 10.1016/s0165-0173(97)00036-2. [DOI] [PubMed] [Google Scholar]
- Chou SM, Taniguchi A, Wang HS, Festoff BW. Serpin=serine protease-like complexes within neurofilament conglomerates of motoneurons in amyotrophic lateral sclerosis. J Neurol Sci. 1998;160(Suppl 1):S73–S79. doi: 10.1016/s0022-510x(98)00202-0. [DOI] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science. 2002;298:1245–1248. doi: 10.1126/science.1076595. [DOI] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R. Maturation of axonal sprouts after nerve crush. Exp Neurol. 1979;64:260–270. doi: 10.1016/0014-4886(79)90267-x. [DOI] [PubMed] [Google Scholar]
- Donovan FM, Pike CJ, Cotman CW, Cunningham DD. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci. 1997;17:5316–5326. doi: 10.1523/JNEUROSCI.17-14-05316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Keynes RJ. Peripheral nerve regeneration. Annu Rev Neurosci. 1990;13:43–60. doi: 10.1146/annurev.ne.13.030190.000355. [DOI] [PubMed] [Google Scholar]
- Fayard B, Loeffler S, Weis J, Vogelin E, Kruttgen A. The secreted brain-derived neurotrophic factor precursor pro-BDNF binds to TrkB and p75NTR but not to TrkA or TrkC. J Neurosci Res. 2005;80:18–28. doi: 10.1002/jnr.20432. [DOI] [PubMed] [Google Scholar]
- Ferri CC, Bisby MA. Improved survival of injured sciatic nerve Schwann cells in mice lacking the p75 receptor. Neurosci Lett. 1999;272:191–194. doi: 10.1016/s0304-3940(99)00618-7. [DOI] [PubMed] [Google Scholar]
- Festoff BW, Smirnova IV, Ma J, Citron BA. Thrombin, its receptor and protease nexin I, its potent serpin, in the nervous system. Semin Thromb Hemost. 1996;22:267–271. doi: 10.1055/s-2007-999018. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann I, Faber-Elman A, Yoles E, Schwartz M. Injury-induced gelatinase and thrombin-like activities in regenerating and nonregenerating nervous systems. FASEB J. 1999;13:533–543. doi: 10.1096/fasebj.13.3.533. [DOI] [PubMed] [Google Scholar]
- Garcia-Rocha M, Avila J, Armas-Portela R. Tissue-type plasminogen activator (tPA) is the main plasminogen activator associated with isolated rat nerve growth cones. Neurosci Lett. 1994;180:123–126. doi: 10.1016/0304-3940(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- Higashimori H, Carlsen RC, Whetzel TP. Early excision of a full-thickness burn prevents peripheral nerve conduction deficits in mice. Plast Reconstr Surg. 2006;117:152–164. doi: 10.1097/01.prs.0000186537.62939.07. [DOI] [PubMed] [Google Scholar]
- Hirakawa H, Okajima S, Nagaoka T, Takamatsu T, Oyamada M. Loss and recovery of the blood-nerve barrier in the rat sciatic nerve after crush injury are associated with expression of intercellular junctional proteins. Exp Cell Res. 2003;284:196–210. doi: 10.1016/s0014-4827(02)00035-6. [DOI] [PubMed] [Google Scholar]
- Hirakawa H, Okajima S, Nagaoka T, Kubo T, Takamatsu T, Oyamada M. Regional differences in blood-nerve barrier function and tight-junction protein expression within the rat dorsal root ganglion. NeuroReport. 2004;15:405–408. doi: 10.1097/00001756-200403010-00004. [DOI] [PubMed] [Google Scholar]
- Houenou LJ, Turner PL, Li L, Oppenheim RW, Festoff BW. A serine protease inhibitor, protease nexin I, rescues motoneurons from naturally occurring and axotomy-induced cell death. Proc Natl Acad Sci USA. 1995;92:895–899. doi: 10.1073/pnas.92.3.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kioussi C, Gruss P. Making of a Schwann. Trends Genet. 1996;12:84–86. doi: 10.1016/0168-9525(96)81411-9. [DOI] [PubMed] [Google Scholar]
- Knauer DJ, Majumdar D, Fong PC, Knauer MF. SERPIN regulation of factor XIa. The novel observation that protease nexin 1 in the presence of heparin is a more potent inhibitor of factor XIa than C1 inhibitor. J Biol Chem. 2000;275:37340–37346. doi: 10.1074/jbc.M003909200. [DOI] [PubMed] [Google Scholar]
- Knauer MF, Hawley SB, Knauer DJ. Identification of a binding site in protease nexin I (PN1) required for the receptor mediated internalization of PN1-thrombin complexes. J Biol Chem. 1997;272:12261–12264. doi: 10.1074/jbc.272.19.12261. [DOI] [PubMed] [Google Scholar]
- Krystosek A, Seeds NW. Plasminogen activator release at the neuronal growth cone. Science. 1981;213:1532–1534. doi: 10.1126/science.7197054. [DOI] [PubMed] [Google Scholar]
- Krystosek A, Seeds NW. Peripheral neurons and Schwann cells secrete plasminogen activator. J Cell Biol. 1984;98:773–776. doi: 10.1083/jcb.98.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, Albrecht H, Meins M, Hengst U, Troncoso E, Lefort S, Kiss JZ, Petersen CC, Monard D. Regulation of brain proteolytic activity is necessary for the in vivo function of NMDA receptors. J Neurosci. 2004;24:9734–9743. doi: 10.1523/JNEUROSCI.3306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantz MS, Ciborowski P. Zymographic techniques for detection and characterization of microbial proteases. Methods Enzymol. 1994;235:563–594. doi: 10.1016/0076-6879(94)35171-6. [DOI] [PubMed] [Google Scholar]
- Luthi A, Van der PH, Botteri FM, Mansuy IM, Meins M, Frey U, Sansig G, Portet C, Schmutz M, Schroder M, Nitsch C, Laurent JP, Monard D. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J Neurosci. 1997;17:4688–4699. doi: 10.1523/JNEUROSCI.17-12-04688.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy IM, Van der PH, Schmid P, Meins M, Botteri FM, Monard D. Variable and multiple expression of protease Nexin-1 during mouse organogenesis and nervous system development. Development. 1993;119:1119–1134. doi: 10.1242/dev.119.4.1119. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz M, Savaria D, Marcinkiewicz J. The pro-protein convertase PC1 is induced in the transected sciatic nerve and is present in cultured Schwann cells: comparison with PC5, furin and PC7, implication in pro-BDNF processing. Brain Res Mol Brain Res. 1998;59:229–246. doi: 10.1016/s0169-328x(98)00141-7. [DOI] [PubMed] [Google Scholar]
- Meier R, Spreyer P, Ortmann R, Harel A, Monard D. Induction of glia-derived nexin after lesion of a peripheral nerve. Nature. 1989;342:548–550. doi: 10.1038/342548a0. [DOI] [PubMed] [Google Scholar]
- Meins M, Piosik P, Schaeren-Wiemers N, Franzoni S, Troncoso E, Kiss JZ, Brosamle C, Schwab ME, Molnar Z, Monard D. Progressive neuronal and motor dysfunction in mice overexpressing the serine protease inhibitor protease nexin-1 in postmitotic neurons. J Neurosci. 2001;21:8830–8841. doi: 10.1523/JNEUROSCI.21-22-08830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monard D. Cell-derived proteases and protease inhibitors as regulators of neurite outgrowth. Trends Neurosci. 1988;11:541–544. doi: 10.1016/0166-2236(88)90182-8. [DOI] [PubMed] [Google Scholar]
- Monard D. Tinkering with certain blood components can engender distinct functions in the nervous system. Perspect Dev Neurobiol. 1993;1:165–168. [PubMed] [Google Scholar]
- Monard D, Suidan HS, Nitsch C. Relevance of the balance between glia-derived nexin and thrombin following lesion in the nervous system. Ann NY Acad Sci. 1992;674:237–242. doi: 10.1111/j.1749-6632.1992.tb27492.x. [DOI] [PubMed] [Google Scholar]
- Niclou SP, Suidan HS, Pavlik A, Vejsada R, Monard D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur J Neurosci. 1998;10:1590–1607. doi: 10.1046/j.1460-9568.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–491. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci. 1991;14:531–570. doi: 10.1146/annurev.ne.14.030191.002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard B, Arocas V, Guillin MC, Michel JB, Jandrot-Perrus M, Bouton MC. Protease nexin-1: a cellular serpin down-regulated by thrombin in rat aortic smooth muscle cells. J Cell Physiol. 2004;201:138–145. doi: 10.1002/jcp.20103. [DOI] [PubMed] [Google Scholar]
- Scotti AL, Hoffmann MC, Nitsch C. The neurite growth promoting protease nexin 1 in glial cells of the olfactory bulb of the gerbil: an ultrastructural study. Cell Tissue Res. 1994;278:409–413. doi: 10.1007/BF00414183. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992;360:757–759. doi: 10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- Siconolfi LB, Seeds NW. Induction of the plasminogen activator system accompanies peripheral nerve regeneration after sciatic nerve crush. J Neurosci. 2001a;21:4336–4347. doi: 10.1523/JNEUROSCI.21-12-04336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siconolfi LB, Seeds NW. Mice lacking tPA, uPA, or plasminogen genes showed delayed functional recovery after sciatic nerve crush. J Neurosci. 2001b;21:4348–4355. doi: 10.1523/JNEUROSCI.21-12-04348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL. Evidence that beta-amyloid protein in Alzheimer's disease is not derived by normal processing. Science. 1990;248:492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- Smirnova IV, Ma JY, Citron BA, Ratzlaff KT, Gregory EJ, Akaaboune M, Festoff BW. Neural thrombin and protease nexin I kinetics after murine peripheral nerve injury. J Neurochem. 1996;67:2188–2199. doi: 10.1046/j.1471-4159.1996.67052188.x. [DOI] [PubMed] [Google Scholar]
- Smirnova IV, Zhang SX, Citron BA, Arnold PM, Festoff BW. Thrombin is an extracellular signal that activates intracellular death protease pathways inducing apoptosis in model motor neurons. J Neurobiol. 1998;36:64–80. [PubMed] [Google Scholar]
- Smith-Swintosky VL, Zimmer S, Fenton JW, Mattson MP. Protease nexin-1 and thrombin modulate neuronal Ca2+ homeostasis and sensitivity to glucose deprivation-induced injury. J Neurosci. 1995;15:5840–5850. doi: 10.1523/JNEUROSCI.15-08-05840.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer J, Gloor SM, Rovelli GF, Hofsteenge J, Nick H, Meier R, Monard D. cDNA sequence coding for a rat glia-derived nexin and its homology to members of the serpin superfamily. Biochemistry. 1987;26:6407–6410. doi: 10.1021/bi00394a016. [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Schwann cell processes guide regeneration of peripheral axons. Neuron. 1995a;14:125–132. doi: 10.1016/0896-6273(95)90246-5. [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron. 1995b;14:133–141. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- Son YJ, Trachtenberg JT, Thompson WJ. Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 1996;19:280–285. doi: 10.1016/S0166-2236(96)10032-1. [DOI] [PubMed] [Google Scholar]
- Stoffel W, Bosio A. Myelin glycolipids and their functions. Curr Opin Neurobiol. 1997;7:654–661. doi: 10.1016/s0959-4388(97)80085-2. [DOI] [PubMed] [Google Scholar]
- Syroid DE, Maycox PJ, Soilu-Hanninen M, Petratos S, Bucci T, Burrola P, Murray S, Cheema S, Lee KF, Lemke G, Kilpatrick TJ. Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J Neurosci. 2000;20:5741–5747. doi: 10.1523/JNEUROSCI.20-15-05741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolwani RJ, Cosgaya JM, Varma S, Jacob R, Kuo LE, Shooter EM. BDNF overexpression produces a long-term increase in myelin formation in the peripheral nervous system. J Neurosci Res. 2004;77:662–669. doi: 10.1002/jnr.20181. [DOI] [PubMed] [Google Scholar]
- Turgeon VL, Lloyd ED, Wang S, Festoff BW, Houenou LJ. Thrombin perturbs neurite outgrowth and induces apoptotic cell death in enriched chick spinal motoneuron cultures through caspase activation. J Neurosci. 1998;18:6882–6891. doi: 10.1523/JNEUROSCI.18-17-06882.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan PJ, Su J, Cotman CW, Cunningham DD. Protease nexin-1, a potent thrombin inhibitor, is reduced around cerebral blood vessels in Alzheimer's disease. Brain Res. 1994;668:160–170. doi: 10.1016/0006-8993(94)90521-5. [DOI] [PubMed] [Google Scholar]
- Vaughan PJ, Pike CJ, Cotman CW, Cunningham DD. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J Neurosci. 1995;15:5389–5401. doi: 10.1523/JNEUROSCI.15-07-05389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SL, Geddes JW, Cotman CW, Lau AL, Gurwitz D, Isackson PJ, Cunningham DD. Protease nexin-1, an antithrombin with neurite outgrowth activity, is reduced in Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:8284–8288. doi: 10.1073/pnas.86.21.8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh PN, Ahmad SS. Proteases in blood clotting. Essays Biochem. 2002;38:95–111. doi: 10.1042/bse0380095. [DOI] [PubMed] [Google Scholar]
- Yeiser EC, Rutkoski NJ, Naito A, Inoue J, Carter BD. Neurotrophin signaling through the p75 receptor is deficient in traf6−/− mice. J Neurosci. 2004;24:10521–10529. doi: 10.1523/JNEUROSCI.1390-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Luo XG, Xian CJ, Liu ZH, Zhou XF. Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents. Eur J Neurosci. 2000;12:4171–4180. [PubMed] [Google Scholar]
- Zurn AD, Nick H, Monard D. A glia-derived nexin promotes neurite outgrowth in cultured chick sympathetic neurons. Dev Neurosci. 1988;10:17–24. doi: 10.1159/000111951. [DOI] [PubMed] [Google Scholar]