

Abstract
Astrocytes play an essential role in the maintenance and protection of the brain, which we reported was diminished with age. Here, we demonstrate that activation of a purinergic receptor (P2Y-R) signaling pathway, in astrocytes, significantly increases the resistance of astrocytes and neurons to oxidative stress. Interestingly, P2Y-R activation in old astrocytes increased their resistance to oxidative stress to levels that were comparable with stimulated young astrocytes. P2Y-R enhanced neuroprotection was blocked by oligomycin and by Xestospongin C, inhibitors of the ATP synthase and of inositol (1,4,5) triphosphate (IP3) binding to the IP3 receptor, respectively. Treatment of astrocytes with a membrane permeant analog of IP3 also protected astrocytes against oxidative stress. These data indicate that P2Y-R enhanced astrocyte neuroprotection is mediated by a Ca2+-dependent increase in mitochondrial metabolism. These data also reveal a signaling pathway that can rapidly respond to central energy needs throughout the aging process.
Keywords: intracellular Ca2+, mitochondria, IP3, P2Y-R, two-photon, metabolism
Introduction
The classical role of astrocytes is to protect and support neuronal function. They express receptors for a variety of neurotransmitters that are released from either neurons or glial cells (Araque et al., 2001). ATP appears to be the predominant extracellular signaling molecule in astrocytes (Verkhratsky and Kettenmann, 1996; Verkhratsky et al., 1998; James and Butt, 2002). Both metabotropic (P2Y) and ionotropic purinoreceptors (P2X) are expressed in astrocytes, and activation of either receptor subfamily results in an increase in intracellular Ca2+ (Verkhratsky and Kettenmann, 1996; James and Butt, 2002). The metabotropic inositol triphosphate (IP3) signaling pathway provides a mechanism whereby local extracellular signals can be rapidly transduced into increased intracellular ATP. G-protein-linked receptors increase the production of IP3, triggering release of Ca2+ from thapsigargin-sensitive stores in the endoplasmic reticulum. IP3-mediated Ca2+ release can, in turn, increase mitochondrial Ca2+ and, consequently, increase respiration and ATP production (Denton and McCormack, 1985; McCormack et al., 1990; Hajnoczky et al., 1995, 2000). The production of intracellular ATP via Ca2+-induced activation of matrix dehydrogenases is very rapid, occurring at levels 10-fold faster than stimulation by feedback from ATP/ADP pools (Territo et al., 2000). Although mitochondrial Ca2+ uptake can increase ATP production, increased matrix Ca2+ release also sensitizes cells to apoptotic stimuli and induces the opening of the mitochondrial permeability transition pore (MPT) (Bernardi et al., 1992; Petronilli et al., 1993; Zoratti and Szabo, 1995; Byrne et al., 1999; Szalai et al., 1999). Recent reports indicate that Ca2+ sensitivity can be attributed to cyclophilin D, a prolyl isomerase located within the mitochondrial matrix, which plays a critical role in MPT opening (Baines et al., 2005; Basso et al., 2005; Nakagawa et al., 2005). Thus, a purinergic receptor-mediated increase in intracellular Ca2+ release can signal either cell survival or cell death for an astrocyte.
The importance of Ca2+ signaling in aged astrocytes remains mostly unexplored (Landfield and Pitler, 1984; Khachaturian, 1994; Disterhoft et al., 1996; Cotrina and Nedergaard, 2002). Deficiencies in cytosolic Ca2+ buffering of neurons have been implicated in the pathogenesis of seizures (Sloviter, 1989), epilepsy (Miller and Baimbridge, 1983), and neurodegenerative diseases (Heizmann and Braun, 1992; Sutherland et al., 1993). We recently reported that purinergic receptor-mediated increases in intracellular Ca2+ were significantly changed during the aging process (Lin et al., 2007). Several investigators have also reported that mitochondrial Ca2+ levels decrease with age (Leslie et al., 1985; Peterson et al., 1985; Vitorica and Satrustegui, 1986a,b). In this study, we focused our investigation on the potential role of purinergic receptor Ca2+ signaling on astrocyte survival. We show that a brief exposure of astrocytes to extracellular ATP significantly increases the ability of astrocytes, and neurons in coculture, to resist oxidative stress. This protective effect is stimulated by IP3-mediated Ca2+ signaling, is dependent on intracellular mitochondrial ATP production, and is significantly stronger in old astrocytes when compared with nonstimulated young astrocytes. These studies identify astrocyte mitochondria as an important energy source that can be rapidly activated by metabotropic purinergic receptors.
Materials and Methods
Isolation, culture, and growth of astrocytes and neurons.
Primary cultures of astrocytes were prepared as described previously (Lin et al., 2007). C57Bl/6 male mice were purchased from the National Institute of Aging and maintained in standard accredited housing conditions at The University of Texas Health Science Center Animal Facility. Animal protocols were approved by our Institutional Animal Care Use Committee. Astrocytes were cultured in DMEM F-12 media (#11039–021; Invitrogen, Carlsbad, CA) supplemented with 2 mm glutamine, nonessential amino acids, 10% fetal calf serum, 100 U/ml penicillin/streptomycin (#17–602E; Cambrex, Walkersville, MD), and 1% fungizone (#15290–018; Invitrogen) at 37°C in an atmosphere containing 5% CO2. The culture medium was changed every 3–5 d. Confluent cells were gently removed by a trypsin-EDTA solution and replated on 35 mm glass bottom chambers (#04200415C; Bioptechs, Butler, PA) before imaging experiments 3–4 d later. Cells were maintained in plastic culture flasks until the final reseeding of cells onto a glass bottom chamber and were passed up to three times for astrocyte-only experiments. For coculture experiments with neurons, primary astrocytes were plated directly onto corning transwell-clear permeable supports, which physically separates the two cell types by ∼1 mm (#07-200-170; Fisher Scientific, Houston, TX). Astrocytes were cultured in transwells until they were ∼70% confluent. Mouse cortical neurons (#M-CX-300; Cambrex) were then cultured onto six-well mat-tek glass bottom plates and maintained in Neurobasal Medium (#21103-049; Invitrogen), 2 mm l-glutamine (#17-605E; Cambrex), 100 U/ml penicillin/streptomycin (#17-602E; Cambrex), and B-27 with (#17504-044; Invitrogen) and without antioxidants (#10889-038; Invitrogen), which controls glia contamination to <0.5%. The transwell supports with cultured astrocytes were placed in the neuron-seeded glass bottom chambers for 4 d before imaging, and both cell types were then maintained in neuronal media. For neuron-only experiments, the astrocytes on the transwell supports were removed before treatments and imaging.
O2 consumption assay.
O2 consumption was monitored with a Clark electrode (Mitocell S200 micro respirometry system; Strathkelvin Instruments, Motherwell, UK). Astrocytes (500 μl at 105 cells/ml) in buffer (120 mm NaCl, 45 mm KCl, 1 mm CaCl2, 2 mm MgCl2, 10 mm HEPES, pH 7.4) were loaded into a 500 μl MT200A Respirometer Chamber, suspended by a fixed-speed solid-state magnetic stirrer inside the chamber, and maintained at 37°C by a circulating water bath. O2 was monitored for 4 min before injecting 5 μl of ATP (200 μm) into the chamber. O2 consumption was then monitored for another 4 min. Changes in O2 levels were calculated by respirometry software. For control recordings, 5 μl of buffer was injected into chamber. Oligomycin astrocytes were treated with oligomycin (0.01 μm) for 30 min before trypsinization and loading into chamber.
Intracellular ATP determination.
Intracellular ATP was measured using a luciferin-luciferase kit (#22066; Invitrogen). Plated astrocytes were trypsinized and washed three times with PBS buffer (#14190; Invitrogen). An aliquot of cells was set aside to measure the cell density with a hemocytometer. The cells were then suspended in a lysate buffer (4 mm EDTA and 100 mm Tris, pH 7.75) and immediately heated to 100°C for 5 min to minimize enzymatic changes in ATP levels. The supernatant was collected after a 10,000 × g centrifugation for 1 min and used with the luciferin–luciferase kit according to the manufacturer instructions. Luminescence was measured with a Synergy HT Multi-Detection Microplate Reader (BioTek, Burlingame, CA). ATP levels were normalized by cell density.
Imaging acquisition and analysis.
Mitochondrial membrane potentials (ΔΨs) were measured as described previously using the potential-sensitive dye tetra-methyl rhodamine ethyl ester (TMRE, #T-669; Invitrogen, Eugene, OR) (Lin and Lechleiter, 2002). Images were acquired with a 60× oil 1.4 numerical aperture objective on a Nikon (Tokyo, Japan) PCM2000 confocal microscope custom adapted for two-photon imaging. A Ti-sapphire Coherent Mira 900 Laser pumped with a 5W Verdi laser (Coherent, Santa Clara, CA) was used to excite TMRE at 800 nm. A neutral-density filter wheel was used to attenuate the laser intensity so that no detectable photobleaching of TMRE was observed.
Sensitivity of neurons to oxidative stress was assessed by treating cultures with 100 μm tert-butyl hydrogen peroxide (t-BuOOH; #B-2633; Sigma, St. Louis, MO) for 4 h. Neurons were then stained with Hoechst 33342 (10 μg/ml, #H-3570; Invitrogen) to label all cell nuclei and calcein AM (2 μm, #C3100; #C3100; Invitrogen). Neuronal death was quantified by counting the number of nuclei that did not colocalize with calcein-stained cells. Calcein fluorescence is only observed in live cells that have maintained their plasma membrane integrity. Images were acquired on an inverted Nikon TE300 microscope with a Hamamatsu (Bridgewater, NJ) ORCAER camera and Open lab software (Improvision, Lexington, MA).
Ca2+ activity was imaged as described previously (Camacho and Lechleiter, 1995). In brief, cultured astrocytes were incubated with the fura-2 AM (#F-1221; Invitrogen) 30 min before the experiment. Images were acquired with two-photon excitation (800 nm) at the rate of 1.5 images/s. At this wavelength, only the Ca2+-free form of fura-2 is significantly excited. Hence, a Ca2+ increase is observed as a decrease in fluorescence. Images were analyzed with Image J (http://rsb.info.nih.gov/ij/) and ANALYZE software (The Mayo Foundation, Rochester, MN). The D-enantiomer of the IP3-BM ester was synthesized by Stuart Conway in the Cambridge University Chemical Laboratory by using a modification of the protocol of Tsien and coworkers (Li et al., 1997).
One-way ANOVA or a paired t test was used for statistical analysis. Differences with a p value <0.05 were considered statistically significant.
Results
Extracellular ATP protects astrocytes from oxidative stress
The presence of multiple purinergic receptor isoforms on astrocytes suggested to us that an increase in extracellular ATP could affect the sensitivity of astrocytes to oxidative stress during the aging process. To investigate this, primary cultures of astrocytes were prepared from the brains of young (4–6 months of age) and old (26–28 months of age) mice and replated on glass coverslips before each imaging experiment as described previously (Lin et al., 2007). Seeded glass coverslips were then sealed in an open chamber (Bioptechs Delta T) and perfused at 37°C with the potential sensitive dye tetra-methyl rhodamine ethyl ester (TMRE; 200 nm; Invitrogen) to label astrocyte mitochondria. Relative changes in mitochondrial membrane potentials (ΔΨs) were measured every 5 min using two-photon microscopy (800 nm excitation, z-stack of six images in 1 μm steps). Oxidative stress was applied to the cells in the perfusate with t-BuOOH (100 μm). Cell viability was then monitored by the collapse of ΔΨ (TMRE fluorescence) to 10% of its initial value. This value was chosen to ensure the potential indicator remained above the lower limit of fluorescence at de-energization, which has been attributed to TMRE partitioning into the membrane phase (Fink et al., 1998). Images of TMRE-labeled mitochondria from untreated young and old astrocytes are presented in Figures 1a and 2a at the indicated time points during oxidative stress. The time course of ΔΨ collapse is plotted for three astrocytes in each culture (Figs. 1b, 2b). As reported previously (Lin et al., 2007), the time until ΔΨ collapse was significantly faster for old astrocytes (3.62 ± 0.12 h; n = 75 cells; pooled from four experiments) compared with young astrocytes (4.87 ± 0.48 h; n = 15 cells; pooled from two experiments) (Figs. 1e, 2e). To test the impact of purinergic receptor activation, astrocyte cultures were initially exposed to extracellular ATP (10 μm) for 10 min before stressing the cells with t-BuOOH (100 μm). The perfusate again contained TMRE (200 nm) to monitor ΔΨ every 5 min. Under these experimental conditions, we found that the average time for ΔΨ to collapse was significantly increased to 7.35 ± 0.23 h (n = 27 cells pooled from four experiments; p < 0.001) in astrocytes from young mice (Fig. 1c–e). Similarly, a 10 min application of extracellular ATP to astrocytes cultured from old mice significantly increased the average time until ΔΨ collapse to 6.51 ± 0.18 h (n = 24 cells pooled from five experiments; p < 0.001) (Fig. 2c–e). The collapse time for ATP-treated old astrocytes was comparable with treated young astrocytes during oxidative stress (Figs. 1e, 2e). We concluded from these data that a brief application of extracellular ATP to astrocytes established a prolonged period of protection against oxidative stress in astrocytes cultured from both young and old mice.
Figure 1.

P2Y-R activation protects young astrocytes from oxidative stress. a, Images of astrocytes cultured from young mice labeled with the mitochondrial potential-sensitive dye TMRE and exposed to oxidative stress for the indicated times. Each image is a maximum intensity projection of a z-stack of six optical sections (1 μm steps). Plated cells were continuously perfused at 37°C with culture medium containing TMRE (200 nm) and imaged with two-photon microscopy (800 nm). b, Line plots of the mean TMRE fluorescence (ΔΨ) for the cells labeled in a. Fluorescent units are scaled to 100% at 0 h. The times when the TMRE fluorescence collapses to 10% of the initial values are indicated by black arrowheads. c, Images of young astrocytes pre-exposed to extracellular ATP (10 μm) for 10 min before adding t-BuOOH (100 μm) to the perfusate (0 h). d, Line plots of the TMRE fluorescence for the cells indicated in c. e, Histograms of the mean times of ΔΨ collapse for young astrocytes exposed to buffer only (Buff), ATP, or ATP plus Xestospongin C. ***p < 0.001.
Figure 2.

Old astrocytes are also protected from oxidative stress by pretreatment with extracellular ATP. a, Images of astrocytes cultured from old mice with TMRE-labeled mitochondria under oxidative stress for the indicated times. Image acquisition parameters are the same as described in Figure 1. Intensity line plots of the mean TMRE fluorescence (ΔΨ) are shown in b. c, Images of old astrocytes treated with extracellular ATP (10 μm) for 10 min before oxidative stress (t-BuOOH; 100 μm). Intensity line plots of TMRE fluorescence are shown in d. Histograms of the mean times of ΔΨ collapse for old astrocytes exposed to buffer only (Buff), ATP, or ATP plus Xestospongin C are presented in e. ***p < 0.001.
The protective effect of ATP is blocked by pretreatment with Xestospongin C
The primary response of glial cells to extracellular ATP is an increase in intracellular Ca2+ (Verkhratsky and Kettenmann, 1996; James and Butt, 2002). To investigate whether the protective effect of ATP could be initiated by a Ca2+ response, we loaded cultures of astrocytes with the Ca2+ indicator dye fura-2 AM (10 μm for 30 min). Fura-2 fluorescence was imaged with two-photon microscopy using 800 nm excitation (Fig. 3a). At this wavelength, fluorescence is primarily caused by the Ca2+ free form of fura-2. Hence, we expressed increases in Ca2+ as decreases in fura-2 fluorescence (ΔF) normalized by the resting fura-2 fluorescence (Frest). When young astrocyte cultures were exposed to extracellular ATP (10 μm for 10 min), the peak intracellular Ca2+ (−ΔF/F) was increased to 0.44 ± 0.02 (n = 39; pooled from four experiments). A similar increase in the peak Ca2+ amplitude was observed for astrocytes cultured from old mice (−ΔF/F = 0.39 ± 0.03; n = 21; pooled from four experiments). A detailed analysis of the changes that occur in ATP-mediated intracellular Ca2+ signaling in astrocytes during the aging process has been reported (Lin et al., 2007). Here, we tested whether the ATP-induced Ca2+ response could be attributed to the metabotropic pathway, because IP3-mediated Ca2+ release can stimulate mitochondrial energy production as well as sensitize mitochondria to apoptotic stimuli (Hajnoczky et al., 1995, 2000; Szalai et al., 1999). Astrocytes cultured from old mice were pretreated with Xestospongin C (XeC) (25 μm for 30 min), which is a competitive inhibitor of IP3 binding to the IP3 receptor (IP3R) (Gafni et al., 1997). When XeC-treated cultures were exposed to extracellular ATP (10 μm), the peak Ca2+ response was significantly reduced to 0.21 ± 0.03 (n = 30; pooled from four experiments). These data suggested that at least part of the ATP-induced Ca2+ response can be attributed to metabotropic P2Y receptor activation. This is consistent with other reports showing that activation of the ionotropic P2X receptor also increases Ca2+ in cultured astrocytes (Verkhratsky and Kettenmann, 1996; James and Butt, 2002). To determine whether the metabotropic pathway is involved with the protective effect of extracellular ATP, we subsequently exposed the XeC/ATP-treated astrocytes to oxidative stress. As described above, the perfusate contained t-BuOOH (100 μm) and TMRE (200 nm) to monitor ΔΨ. Under these experimental conditions, the average time until ΔΨ collapse was reduced to 5.26 ± 0.25 h (n = 26; pooled from four experiments) in young astrocytes (Fig. 1e) and to 3.97 ± 0.17 h (n = 30; pooled from four experiments) in old astrocytes (Fig. 2e). These values were not significantly different from untreated astrocytes. We concluded that the protective effect of ATP on astrocytes was mediated by P2Y receptors (P2Y-Rs).
Figure 3.

IP3-mediated intracellular Ca2+ release protects young and old astrocytes from oxidative stress. a, Images of cultured young and old astrocytes loaded with fura-2 and imaged with two-photon microscopy at 800 nm excitation. b, IP3-BM (25 μm; black arrowhead) increases intracellular Ca2+ in both young and old astrocytes. Ca2+ increases for the indicated cell numbers are observed as decreases in signal intensity, because the Ca2+ free form of fura-2 is preferentially excited at 800 nm. c, Histogram plots of the peak Ca2+ response for young and old astrocytes. Ca2+ responses were significantly inhibited by pretreatment with XeC (gray bars). d, Intensity line plots of the mean TMRE fluorescence (ΔΨ) for three representative astrocytes pretreated with IP3-BM (25 μm; 20 min) and subsequently exposed to oxidative stress (t-BuOOH; 100 μm). ΔΨ traces from cells that were initially exposed to XeC (25 μm; 1 h) before IP3-BM treatment are presented in the bottom plots. The mean ΔΨ collapse time for each plot is indicated by the vertical dotted lines and black arrowheads. The white arrowheads indicate the mean ΔΨ collapse for buffer alone. e, Histogram plots of the mean times until ΔΨ collapse for astrocytes exposed to IP3-BM or IP3-BM plus XeC. Dotted line histogram bars are presented as control (Buff only) references for the data shown in Figures 1 and 2. ***p < 0.001.
IP3 butyryloxymethyl ester treatment protects astrocytes from oxidative stress
To directly investigate the role of metabotropic IP3/Ca2+ signaling pathway in the protective effect of P2Y-R activation, we examined the Ca2+ response of astrocytes to butyryloxymethyl ester of IP3 (IP3-BM), a membrane permeant analog of IP3 (Li et al., 1997). Cultured astrocytes were loaded with fura-2 and imaged with two-photon microscopy as described above. When cells were treated with IP3-BM (25 μm), an increase in intracellular Ca2+ was observed in astrocytes cultured from both young and old mice (Fig. 3a–c). The peak Ca2+ response was 0.30 ± 0.02 (n = 16; pooled from five experiments) for young astrocytes and 0.24 ± 0.02 (n = 24; pooled from four experiments) for old astrocytes. Overall, the Ca2+ responses of astrocytes treated with IP3-BM were slower than the comparable Ca2+ responses for ATP treatment. This was expected because of the amount of time required to accumulate and activate IP3-BM within cells. After 15 min of IP3-BM treatment, cultured cells were perfused with t-BuOOH, and ΔΨ was monitored with TMRE fluorescence. We found that the time until ΔΨ collapse was significantly increased to 6.48 ± 0.34 h (n = 26; pooled from four experiments) in young astrocytes and to 5.47 ± 0.13 h (n = 33; pooled from four experiments) in old astrocytes (Fig. 3d,e). Again, the time until ΔΨ collapse in IP3-BM-treated old astrocytes was comparable with the time until ΔΨ collapse in treated cultures of young astrocytes. Pretreatment of young and old astrocytes with XeC (25 μm for 30 min) significantly reduced their Ca2+ responses to 0.12 ± 0.01 (n = 28; pooled from four experiments) and 0.10 ± 0.01 (n = 27; pooled from three experiments) (Fig. 3c). Furthermore, XeC completely inhibited the protective effect of IP3-BM in young and old astrocytes. The time until ΔΨ collapse was reduced to 4.13 ± 0.22 h (n = 20; pooled from three experiments) in young astrocytes and to 3.69 ± 0.29 h (n = 24; pooled from three experiments) in old astrocytes (Fig. 3e). Neither of these values were significantly different from those observed in untreated cells. We concluded from these data that IP3-mediated intracellular Ca2+ release protects astrocytes from oxidative stress.
Oligomycin blocks the protective effect of P2Y-R activation against oxidative stress
A potential mechanism by which IP3-mediated intracellular Ca2+ release could provide protection to a cell is by increasing intracellular production of ATP via a Ca2+-mediated increase in mitochondrial respiration (Hansford, 1985; McCormack et al., 1990; Hajnoczky et al., 1995, 2000; Szalai et al., 1999). To initially test this mechanism of action, we pretreated cultured astrocytes with oligomycin (1 μg/ml for 30 min), a specific inhibitor of the mitochondrial ATP synthetase (Merck Index 13, 6902). We found that the time until ΔΨ collapse was 4.64 ± 0.21 h (n = 27; pooled from three experiments) in young astrocytes and 4.05 ± 0.19 h (n = 30; pooled from three experiments) in old astrocytes exposed oxidative stress (t-BuOOH; 100 μm) (Fig. 4). These times until ΔΨ collapse were comparable with control values of untreated astrocytes, suggesting the acute pretreatment of astrocytes with oligomycin did not affect their resistance to oxidative stress. However, when oligomycin-treated astrocytes were exposed to extracellular ATP for 10 min, we found that P2Y-R activation no longer enhanced their resistance to oxidative stress. ATP treatment in old astrocytes made them just as resistant to oxidative injury as young astrocytes. The mean time until ΔΨ collapse was 4.69 ± 0.23 h (n = 34; pooled from three experiments) in young astrocytes and 2.97 ± 0.17 h (n = 8; pooled from three experiments) in old astrocytes (Fig. 4). Together, these data suggest that astrocyte mitochondrial ATP production is required for the protective effect of P2Y-R activation against oxidative stress.
Figure 4.

Oligomycin treatment of young or old astrocytes blocks their P2Y-R-enhanced resistance to oxidative stress. a, Intensity line plots of the mean TMRE fluorescence (ΔΨ) for representative astrocytes pretreated with oligomycin (1 μg/ml; 30 min) and subsequently exposed to oxidative stress (t-BuOOH; 100 μm) with (top traces) and without P2Y-R stimulation (10 μm ATP; 10 min). Vertical dotted lines with black arrowheads indicate the mean ΔΨ collapse times. b, Histograms of the mean ΔΨ collapse times during oxidative stress (t-BuOOH; 100 μm). Dotted line histogram bars are for reference showing control and ATP-enhanced responses from Figures 1 and 2.
P2Y-R activation in astrocytes stimulates O2 consumption and intracellular ATP production
To further investigate the importance of mitochondrial metabolism on the protective effect of P2Y-R activation, we first measured the rate of O2 consumption for astrocytes with and without P2Y-R activation. Primary cultures of astrocytes were grown to ∼70% confluency, gently removed by trypsin EDTA, and resuspended in PBS buffer. Astrocytes (105 cells/ml) were then loaded into a 500 μl Respirometer Chamber (MT200A) and maintained at 37°C by a circulating water bath. We found that the basal rate of O2 consumption in astrocytes was 2.45 ± 0.24 nmol/min (n = 3) (Fig. 5a,b). This rate increased significantly (p < 0.01) to 3.67 ± 0.3 nmol/min (n = 3) when astrocytes were stimulated by a bolus of ATP (2 μm final concentration). The rate of O2 consumption was significantly decreased (p < 0.01) to 0.97 ± 0.21 nmol/min (n = 4) after exposing the astrocytes to the ATP synthetase inhibitor oligomycin (0.01 μm; 30 min). Oligomycin treatment also completely inhibited the effects of extracellular ATP on the rate of O2 consumption (1.19 ± 0.27 nmol/min; n = 3).
Figure 5.
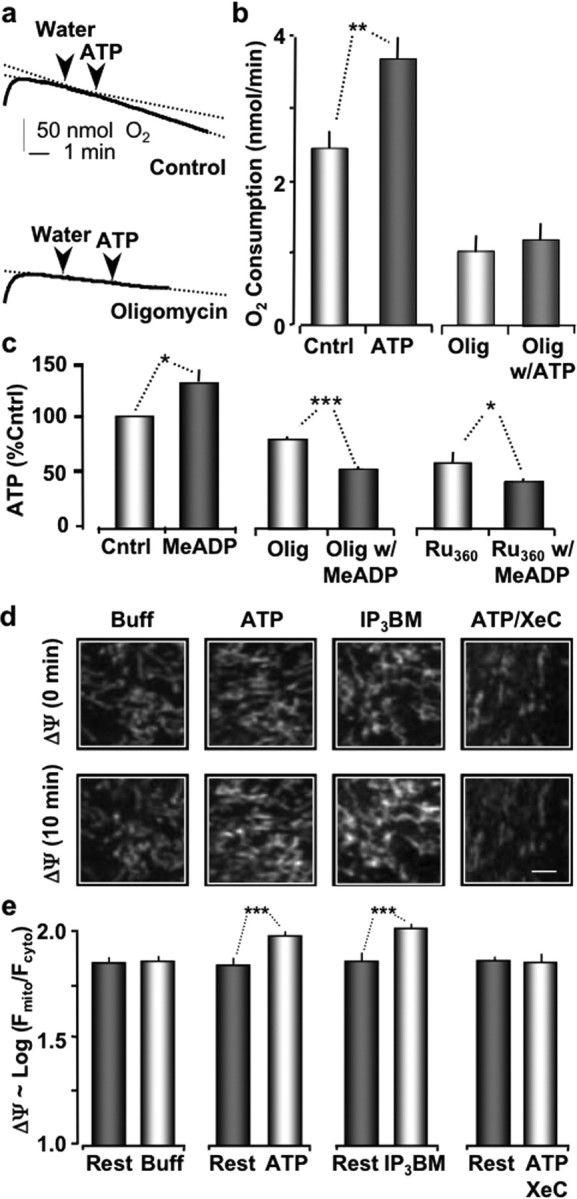
Stimulation of P2Y-Rs increases O2 consumption, ATP production, and increases ΔΨ. a, Plots of O2 levels in suspended astrocytes as labeled. b, Histogram shows the average change in O2 consumption with 2 μm ATP compared with untreated cells (Cntrl). Bars on the right show O2 consumption of astrocytes are preloaded with 0.01 μm oligomycin (30 min). c, Direct measurement of intracellular ATP levels relative to untreated cells (control), P2Y-R stimulated (MeADP), then the same measurements in the presence of oligomycin (oligo) or Ru360. d, Astrocyte mitochondria (TMRE loaded) at 0 min (top panels) and 10 min after treatments as labeled (bottom panels). e, Histogram plot showing average mitochondrial membrane potentials before and after treatment. ***p < 0.001; **p < 0.01; *p < 0.05.
Our next approach to investigate the role of mitochondrial metabolism was to directly measure ATP levels in cultured astrocytes using a luciferin-luciferase assay (Invitrogen). Astrocytes were cultured as described above, trypsinized, and washed with buffer. The cells were then suspended in a lysate buffer and immediately heated to 100°C (5 min) to minimize enzymatic changes in ATP levels. After centrifugation, the ATP-dependent luminescence of the supernatant was measured with a microplate reader (BioTek). We estimated that the resting intracellular ATP concentration in cultured astrocytes was 0.31 ± 0.14 μm ATP/104 cells (n = 3). Treatment of cultured astrocytes with the P2Y1-R specific ligand 2-MeSADP (2 μm) for 10 min increased ATP levels to 0.39 ± 0.16 μm ATP/104 cells. This increase was statistically significant when the data were expressed as a percentage of the control ATP levels for each experiment (p < 0.04; paired Student's t test) (Fig. 5c). Oligomycin (0.01 μm; 30 min) significantly decreased the resting levels of ATP to 82.6 ± 3.2% of control (p < 0.05; n = 3). The same oligomycin pretreatment also completely inhibited P2Y1-R-mediated increases in ATP (Fig. 5c). Finally, we tested whether ruthenium 360 (Ru360; Calbiochem, La Jolla, CA), a polycation that inhibits the electrogenic mitochondrial Ca2+ uniporter (Ying et al., 1991), affected mitochondrial ATP production. We found that Ru360 treatment (1 μm; 30 min) significantly decreased the basal ATP levels to 60.5 ± 10.5% (n = 3) of control values and also completely blocked the ability of P2Y1-R specific ligand, 2-MeSADP, to increase ATP levels (Fig. 5c). Together, these data strongly suggest that P2Y-R stimulation leads to an IP3-mediated intracellular Ca2+ release that increases intracellular production of ATP via a Ca2+-mediated increase in mitochondrial respiration.
Extracellular ATP and IP3-BM treatment increase ΔΨ
Our observation that P2Y-R enhanced protection of astrocytes was mediated by an increase in the intracellular production of ATP suggested that mitochondrial membrane potential (ΔΨ) may also have been increased (Territo et al., 2001; Balaban et al., 2003). We confirmed this by directly measuring the effect of ATP, ATP plus XeC, and IP3-BM treatments on ΔΨ. Cultured cells were labeled with TMRE (200 nm) and imaged with a two-photon microscope as described above. ΔΨ was estimated as the log of (Fmito/Fcyto), where Fmito is the peak fluorescent intensity observed in single mitochondria (Farkas et al., 1989; Lin et al., 2007). The mean values of individual mitochondria from a single cell were used for the mitochondrial potential estimate. Fcyto represents the lowest value of TMRE fluorescence observed within the boundaries of the same cell. Before each treatment, the resting ΔΨ was comparable between experiments. The mean resting ΔΨs for untreated (Buffer), ATP-treated, ATP plus XeC-treated, and IP3-BM-treated were 1.81 + 0.03 (n = 264 mitochondria; pooled from four experiments), 1.79 + 0.03 (n = 269 mitochondria; pooled from four experiments), 1.85 + 0.03 (n = 303 mitochondria; pooled from six experiments), and 1.82 + 0.02 (n = 425 mitochondria; pooled from six experiments), respectively (Fig. 5d,e). Astrocyte cultures were then exposed to their respective treatments, and the same fields of mitochondria were imaged 10 min later. As expected, the control untreated cells exhibited no change. ΔΨ remained at 1.83 ± 0.02 (n = 230 mitochondria; pooled from four experiments). However, treating astrocytes for 10 min with ATP or IP3-BM significantly increased ΔΨ to 1.93 ± 0.02 (n = 346 mitochondria; pooled from four experiments) and 1.98 ± 0.02 (n = 343 mitochondria; pooled from six experiments), respectively. Furthermore, astrocytes pretreated with XeC and then exposed to ATP exhibited no significant change in ΔΨ, which was 1.80 ± 0.04 (n = 460 mitochondria; pooled from six experiments) (Fig. 5d,e). These data are consistent with the model that P2Y-R activation in astrocytes stimulates IP3-mediated intracellular Ca2+ release, which increases mitochondrial matrix Ca2+, stimulating respiration and the subsequent production of intracellular ATP.
P2Y-R activation in astrocytes increases neuroprotection
To determine whether P2Y-R enhanced mitochondrial metabolism in astrocytes affected neuronal resistance to oxidative stress, we prepared cocultures of neurons and astrocytes using corning transwell-clear permeable supports, which physically separates the two cell types by ∼1 mm (#07-200-170; Fisher Scientific). Primary cultures of astrocytes were directly prepared in transwell supports until they were ∼70% confluent. Mouse cortical neurons were then cultured onto glass bottom mat-tek plates and maintained in B-27/Neurobasal medium to control glia contamination to <0.5% (Invitrogen). After 4 d, cocultures were treated with t-BuOOH (100 μm) for 4 h with and without prestimulation of ATP (2 μm; 10 min). Cells were then stained with the DNA intercalating dye Hoechst 33342 (10 μg/ml; #H-3570; Invitrogen) and calcein AM (2 μm; #C3100; Invitrogen). Cell viability was quantified by counting the number of nuclei that did not colocalize with calcein-stained cells, which is only observed in live cells that have maintained their plasma membrane integrity. We found that purinergic receptor activation significantly enhanced neuronal survival from 74 ± 2% (n = 1332 cells; pooled from four experiments) to 87 ± 2% (n = 1914 cells; pooled from five experiments) when cocultured with young astrocytes and from 77 ± 3% (n = 1092 cells; pooled from four experiments) to 87 ± 2% (n = 2510 cells; pooled from six experiments) when cocultured with old astrocytes (Fig. 6a–c). We then tested the neuroprotective effects of isoform-specific purinergic ligands 2-MeSADP (P2Y1-R) and UTP (P2Y2-R). A 10 min pretreatment of astrocytes with either 2-MeSADP (2 μm) or UTP (50 μm) was equally protective against oxidative stress-induced cell death. The percentage of neurons alive after 4.5 h of t-BuOOH treatment was 51 ± 4% (n = 1664 neurons; pooled from four experiments) for untreated controls, 70 ± 2% (n = 2541 neurons; pooled from four experiments) for 2-MeSADP, and 71 ± 3% (n = 1189 neurons; pooled from four experiments) for UTP-treated cocultures (Fig. 6d). P2Y-R-specific ligands also increased the resistance of primary cultures of astrocytes, in the absence of neurons. The percentage of astrocytes alive after 4.5 h of t-BuOOH treatment, in the absence of neurons, was 60 ± 4% (n = 294 cells; pooled from five experiments) for control, 77 ± 4% (n = 294; pooled from five experiments) for 2-MeSADP, and 79 ± 3% (n = 333; pooled from six experiments) for UTP-treated astrocytes.
Figure 6.
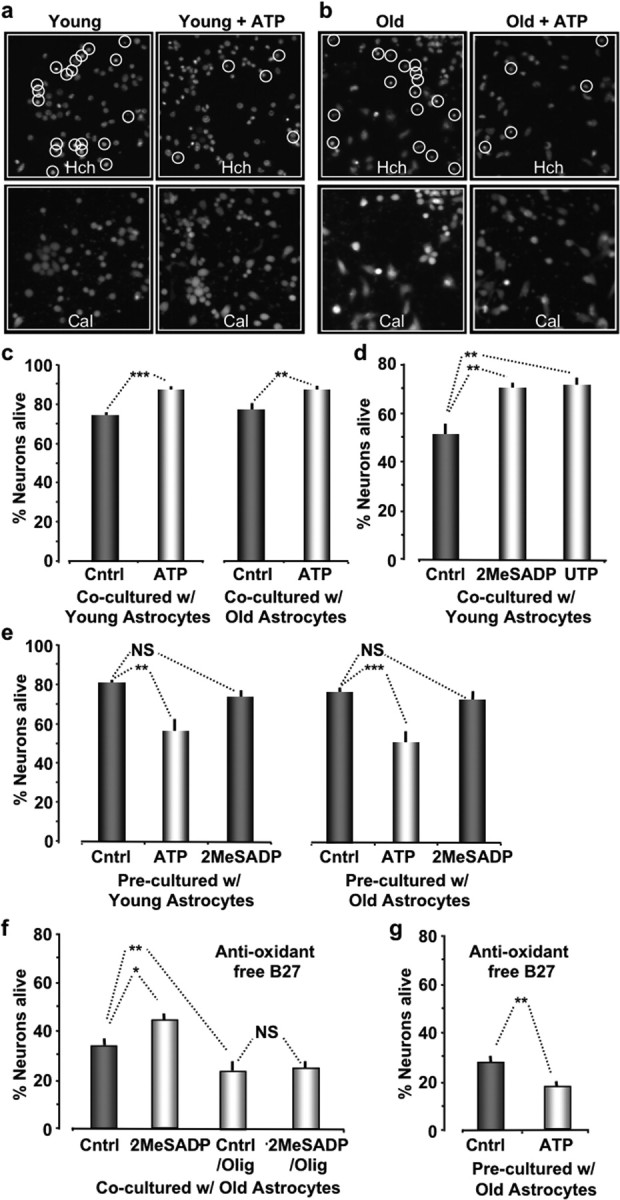
Stimulation of P2Y-Rs in astrocytes enhances their ability to protect neurons from oxidative stress. a, b, Images of cortical neurons cocultured with young (a) and old (b) astrocytes acquired 4 h after incubation with t-BuOOH (100 μm). Cocultures pretreated with ATP (2 μm; 10 min) are presented in the right panels. The top images show Hoechst (Hch) stained nuclei. The bottom images show the same field of neurons stained with calcein (Cal), which only stains the cytoplasm of cells that are alive. Hoechst-labeled nuclei that are calcein negative are circled and counted as cell deaths. c, Histogram plots of the percentage of neurons alive when cocultured with young or old astrocytes. d, Histogram plots of the percentage of neurons alive pretreated with either 2-MeSADP (2 μm) or UTP (50 μm) for 10 min. e, Histogram plots of the percentage of neurons alive that were precultured with astrocytes but were separated before pretreating (10 min) the neurons with either ATP (2 μm ATP; 10 min) or 2-MeSADP (2 μm) and subsequently exposed to oxidative stress (100 μm; t-BuOOH; 4.5 h). f, Histogram plots of percentage of neurons alive cocultured with old astrocytes using anti-oxidant free B-27 supplement for 3 h. g, Same experiment as in e except using anti-oxidant free B-27 supplement. ***p < 0.001; **p < 0.01; *p < 0.05.
Because it is known that B-27 supplement has antioxidant properties, we also repeated the above experiments using an anti-oxidant-free B-27 (also available from Invitrogen) to control glia contamination. Procedures were identical to those described above except that after 4 d of coculturing, dishes were treated with t-BuOOH (100 μm) for only 3 h with and without prestimulation of 2-MeSADP (2 μm; 10 min). Cells were then stained with Hoechst 33342 and calcein AM to ascertain cell viability. We found that the percentage of neurons alive after 3 h of treatment was reduced to 33 ± 2% (n = 1322 cells; pooled from four experiments) (Fig. 6, compare c and f). However, a 10 min treatment of cocultures with 2-MeSADP (2 μm) significantly enhanced neuronal survival to 43 ± 3% (n = 1057 cells; pooled from three experiments; p < 0.03) (Fig. 6f). In addition, oligomycin treatment significantly (0.01 μm; 30 min; p < 0.01) decreased neuronal survival to 25 ± 3% (n = 1409 cells; pooled from three experiments) and completely inhibited the protective effects of 2-MeSADP on neuronal survival (26 ± 3%; n = 1048 cells; pooled from three experiments).
Activation of neuronal P2X receptors has been reported to be neurotoxic (Di Virgilio et al., 1998; Norenberg and Illes, 2000; Koles et al., 2005). Consequently, we further examined the effects of P2Y-R activation on neuronal survival in the absence of astrocytes. Neurons were cocultured with astrocytes in transwell dishes that were removed from the cultures just before treating the neurons with purinergic ligands. Neurons in the absence of astrocytes were then exposed to t-BuOOH (100 μm; 4.5 h with normal B-27, 3 h with antioxidant-free B-27), and cell viability was assessed by the ability of neurons to retain calcein. Under these conditions, we discovered that purinergic receptor activation by ATP treatment decreased cell viability, consistent with published reports (Fig. 6e,g). With normal B-27 supplement, the percentage of neurons alive was significantly decreased to 56 ± 7% (n = 1192 cells; pooled from four experiments; p < 0.005) by ATP treatment, whereas control, untreated neurons were at 80 ± 2% (n = 1049 cells; pooled from four experiments). When the neurons were pretreated with 2-MeSADP, the percentage of neurons alive (74 ± 3%; n = 892 cells; pooled from four experiments) was not significantly different from control (Fig. 6g). The same results were found for neurons initially cocultured with old astrocytes. The percentage of neurons alive was 77 ± 2% (n = 1057 cells; pooled from four experiments) for untreated controls, 49 ± 6% (n = 1197 cells; pooled from four experiments; p < 0.001) for ATP-treated neurons, and 72 ± 3% (n = 1182 cells; pooled from four experiments) for 2-MeSADP-treated neurons (Fig. 6f). We obtained similar results when using the anti-oxidant-free B-27 as a supplement. The percentage of neurons alive was significantly decreased to 18 ± 2% (n = 763 cells; pooled from three experiments; p < 0.01) by ATP treatment, whereas control, untreated neurons were at 27 ± 3% (n = 840 cells; pooled from three experiments) (Fig. 6g). We conclude from these data that the neurotoxic affect of purinergic stimulation is likely mediated by P2X receptors, as reported by others, whereas P2Y-R stimulation in neurons has no significant toxicity.
Discussion
In this study, our studies focused on the capacity of astrocytes to resist oxidative stress and to protect neurons during the aging process. Unexpectedly, we discovered that a brief, transient application of extracellular ATP significantly increased the resistance of astrocytes to oxidative stress. The protective effect initiated by extracellular ATP was active throughout the aging process and made old astrocytes nearly as resistant to oxidative injury as stimulated young astrocytes. Assuming that a significant source of extracellular ATP under physiological conditions is presynaptic release, our data suggest that increased neuronal activity during the aging process can significantly increase the resistance of cells to oxidative stress and, presumably, to the aging process itself.
The protective effect of ATP was inhibited by the pretreatment of astrocytes with XeC, a competitive inhibitor of IP3 binding to the IP3R. These data indicated that the protective effect was mediated by P2Y purinergic receptors and the subsequent production of IP3. This was confirmed by the protective effect of isoform-specific P2Y-R ligands. The ATP stimulated Ca2+ responses were not completely blocked by XeC treatment. This was expected, because P2X purinergic receptors can also increase intracellular Ca2+ by opening plasma membrane Ca2+ permeable channels (Verkhratsky and Kettenmann, 1996; James and Butt, 2002). We directly tested the ability of IP3 to protect astrocytes by pretreatment with a membrane permeant analog of IP3. IP3-BM increased intracellular Ca2+ and significantly increased the time until ΔΨ collapse under conditions of oxidative stress. This protective effect could also be blocked by pretreatment with XeC, strongly supporting the identification of IP3-mediated intracellular Ca2+ release as the specific pathway involved in protection. Recent work by Shinozaki et al. (2005) has also uncovered a protective role for P2Y-R Gq/11 signaling in astrocytes against oxidative stress. However, the underlying mechanism of protection reported by Shinozaki et al. (2005) is very distinct from ours. Protection required prolonged and continuous exposure of astrocytes to extracellular ATP (12–24 h) and could be attributed to the upregulation of oxidoreductase genes, including thioredoxin, and schlafen-1 (Shinozaki et al., 2005). In our case, protection was activated by a transient application of extracellular ATP (10 min) and was dependent on mitochondrial ATP production. ATP treatment in both old and young astrocytes produced similar resistance to oxidative injury. The ability of P2Y-R activation to increase mitochondrial metabolism was further supported by data showing that increases in ΔΨ were only observed in experimental paradigms that resulted in protection against oxidative stress. Ultimately, it will be important to measure mitochondrial pH in both young and old astrocytes, because the proton gradient is responsible for ATP synthase activity. However, our data appear to rule out the possibility that the observed ΔΨ differences between old and young astrocytes can be solely attributed to differences in TMRE lipid partitioning (e.g., because of changing phospholipids content). Rather, the TMRE fluorescent measurements reported here indicate real differences in mitochondrial ΔΨ in old and young astrocytes, although P2Y-R activation enhances neuroprotection to comparable levels.
The observation that mitochondrial Ca2+ uptake via Ca2+ release from the IP3R protected astrocytes was, to some extent, unexpected. It is well established that increased matrix Ca2+ can stimulate Ca2+-sensitive dehydrogenases in the citric acid cycle, which in turn increase the supply of reducing equivalents to the respiratory chain and ultimately increase ATP production (Denton and McCormack, 1985; Hansford, 1985; McCormack et al., 1990; Hajnoczky et al., 1995, 2000; Robb-Gaspers et al., 1998). However, IP3-mediated Ca2+ release has been demonstrated to depolarize mitochondria and to sensitize many cell types to apoptotic stimuli. Specifically, the pro-apoptotic stimuli ceramide and staurosporine sensitize mitochondria to depolarize in response to IP3-mediated Ca2+ release in HEPC2 cells (Szalai et al., 1999). These sensitized cells exhibited significantly increased levels of apoptosis compared with control cells that were not challenged with IP3 stimuli (Szalai et al., 1999). Mitochondrial Ca2+ uptake has also been shown to stimulate reactive oxygen species (ROS) production (Chacon and Acosta, 1991; Paraidathathu et al., 1992; Richter, 1993), and mitochondrial Ca2+ cycling can result in a self-propagating cascade that leads to loss of ATP and ΔΨ (Richter, 1997). Furthermore, oxidative stress induced by t-BuOOH has itself been reported to increase mitochondrial free Ca2+, ROS formation, and stimulate opening of the MPT (Byrne et al., 1999). Consistent with these published reports, we found that IP3-induced intracellular Ca2+ release sensitized human embryonic kidney (HEK293) cells to pro-apoptotic stimuli (our unpublished observations). We induced apoptosis in an HEK293 cell line overexpressing type 1 muscarinic acetylcholine receptors (Lechleiter et al., 1989) by exposing the cells to either t-BuOOH (100 μm; 3 h) or ceramide (40 μm; 12 h). For both stimuli, apoptotic cell death was significantly higher in the presence of ACh (1 μm). It is not clear why astrocytes are not sensitized to apoptotic stimuli by IP3-mediated Ca2+ release. The appropriate Ca2+ binding targets in the mitochondria of astrocytes may be absent or they may be actively inhibited in astrocytes. ADP is known to be a more effective inhibitor of the MPT than ATP (Halestrap and Davidson, 1990; Halestrap et al., 1997; Kantrow et al., 2000). Consequently, it is possible that the adenine nucleotide translocator of astrocytes is less sensitive to Ca2+ and ADP levels. Higher mitochondrial potentials are also known to inhibit the mitochondrial permeability transition pore opening (Bernardi et al., 1992; Petronilli et al., 1993; Zoratti and Szabo, 1995). However, our measurements show that ΔΨ is significantly lower in old astrocytes compared with young astrocytes (Lin et al., 2007), whereas IP3-BM-enhanced neuroprotection is comparable. Thus, it appears unlikely that inherently higher ΔΨs in astrocytes could account for their lack of sensitivity to apoptotic stimuli during IP3-mediated Ca2+ release. Another possible explanation is that the ability of astrocytes to rapidly stimulate oxidative metabolism is itself protective. This could minimize the initial depolarization, which generally leaves the mitochondrial permeability transition pore more susceptible to apoptotic stress (Bernardi, 1996). Enhanced respiration is known to increase the ability of mitochondria to accommodate large Ca2+ influxes without diminution of ΔΨ (Carafoli, 1987; Gunter and Pfeiffer, 1990). In addition, work from Bruce-Keller et al. (1999) indicated that superoxide scavenging by the mitochondrial-specific superoxide dismutase (MnSOD) enhances the ability of mitochondria to sequester Ca2+ by preventing the loss of ΔΨ. The preservation of ΔΨ by MnSOD is apparently sufficient to modulate programmed cell death. Regardless of the underlying reason, astrocyte mitochondria do not appear to depolarize in response to IP3-stimulated Ca2+ release. In fact, our data quite clearly demonstrate that ΔΨ is increased by mitochondrial Ca2+ uptake.
It is generally presumed that metabolic activity generates oxidative stress via an increase in the production of ROS (Paraidathathu et al., 1992; Dawson et al., 1993; Richter, 1997). This only occurs when there is an imbalance between the generation and scavenging of ROS. In addition, ROS increases with pH and often falls under conditions of increased ATP production, presumably because the proton gradient is being partially consumed. The mitochondrial theory of aging proposes that degeneration of physiological processes over the course of a lifetime is fundamentally a result of the accumulation of oxidative damage (Sohal and Brunk, 1992; Shigenaga et al., 1994; Sohal and Dubey, 1994; Warner, 1994; Cortopassi and Wang, 1995; Gadaleta et al., 1998; Cortopassi and Wong, 1999). Little is known about the cumulative effects of damage on astrocytes, the primary function of which is to protect and support neuronal activity. However, it seems likely that a degradation of their supportive and neuroprotective functions would by itself contribute to the aging process (Amin and Pearce, 1997; Robb and Connor, 1998). Mitochondrial dysfunction also plays a key role in a variety of forms of cell death, including ischemia (Pastorino et al., 1993; Zahrebelski et al., 1995), excitotoxic neurodegeneration (Beal et al., 1993), oxidant-induced stress (Dawson et al., 1993), and apoptosis (Newmeyer et al., 1994; Petit et al., 1995; Zamzami et al., 1996; Kroemer, 1997). Our observation that mitochondrial Ca2+ uptake in astrocytes stimulates cell survival places the IP3-stimulated Ca2+ signaling system of these cells in a unique regulatory position. It is particularly well adapted for the role of transducing information about local synaptic activity and metabolic status into appropriate trophic and/or protective responses. It is also well positioned to permit a rapid response to neuronal injury. Astrocytes are known to increase their metabolic activity in response to neuronal injury in a process referred to as reactive gliosis (Cotrina and Nedergaard, 2002).
The specific underlying process (s) that become energy-limited during oxidative stress is unknown. One clue comes from the use of transwell dishes for our astrocyte-neuronal protection assays. This configuration unequivocally shows that oxidant protection is mediated by a diffusible messenger. Given the sensitivity of astrocyte neuroprotection to the inclusion of anti-oxidants in the glia-suppressant, B-27, it is reasonable to speculate that the diffusible substance is also an anti-oxidant. Consequently, a prominent candidate has to be the antioxidant, glutathione (GSH). Neuronal de novo synthesis of GSH is critically dependent on GSH efflux from astrocytes as the source of cysteine (Sagara et al., 1993, 1996; Wang and Cynader, 2000). In addition, de novo synthesis of GSH in astrocytes is controlled by two ATP-dependent enzymes, glutamate cysteine ligase and glutathione synthetase (Suzuki and Kurata, 1992; Papadopoulos et al., 1997). Oxidative stress would rapidly deplete GSH levels and stimulate energy-dependent de novo synthesis in astrocytes. We suggest that the increased energy load overwhelms glycolysis. Astrocyte mitochondrial ATP production is then well positioned to rapidly and abundantly respond to the increased energy demand during stress.
The identification of G-protein-coupled, IP3-Ca2+ signaling as a key pathway in the metabolic regulation of astrocytes suggests that it is an attractive therapeutic target to address many neuropathological processes. Regardless of their potential restorative roles, our data show that astrocytes increase their resistance to oxidative stress in response to a transient exposure to extracellular ATP. Although the source of extracellular ATP could be either glial or neuronal, this metabotropic-mediated protective mechanism can also be viewed as the molecular basis for why increased glia-neuronal activity is physiologically beneficial. Quite literally, brief episodes of neuronal activity are likely to increase the ability of the neurons to resist degeneration and premature cell death and, as a consequence, significantly slow degeneration of the brain during the aging process.
Footnotes
This work was supported by National Institute on Aging Grant AG19316. We thank Patricia Camacho for her critical comments and careful critique of this manuscript.
References
- Amin N, Pearce B. Peroxynitrite-induced toxicity in cultured astrocytes. Brain Res. 1997;773:227–230. doi: 10.1016/s0006-8993(97)00955-4. [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Bose S, French SA, Territo PR. Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. Am J Physiol Cell Physiol. 2003;284:C285–C293. doi: 10.1152/ajpcell.00129.2002. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Beal F, Hyman T, Horoshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- Bernardi P. The permeability transition pore. Control points of a cyclosporinA-sensitive mitochondrial channel involved in cell death. Biochim Biophys Acta. 1996;1275:5–9. doi: 10.1016/0005-2728(96)00041-2. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zoratti M. Modulation of mitochondrial permeability transition pore. Effects of protons and divalent cations. J Biol Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- Bruce-Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holtsberg FW, Parthasarathy S, Steiner SM, Mattson MP. Anti-death properties of TNF against metabolic poisoning: mitochondrial stabilization by MnSOD. J Neuroimmunol. 1999;93:53–71. doi: 10.1016/s0165-5728(98)00190-8. [DOI] [PubMed] [Google Scholar]
- Byrne AM, Lemasters JJ, Nieminen AL. Contribution of increased mitochondrial free Ca2+ to the mitochondrial permeability transition induced by tert-butylhydroperoxide in rat hepatocytes. Hepatology. 1999;29:1523–1531. doi: 10.1002/hep.510290521. [DOI] [PubMed] [Google Scholar]
- Camacho P, Lechleiter JD. Calreticulin inhibits repetitive intracellular Ca2+ waves. Cell. 1995;82:765–771. doi: 10.1016/0092-8674(95)90473-5. [DOI] [PubMed] [Google Scholar]
- Carafoli E. Intracellular calcium homeostasis. Annu Rev Biochem. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- Chacon E, Acosta D. Mitochondrial regulation of superoxide by Ca2+: an alternate mechanism for the cardiotoxicity of doxorubicin. Toxicol Appl Pharmacol. 1991;107:117–128. doi: 10.1016/0041-008x(91)90336-d. [DOI] [PubMed] [Google Scholar]
- Cortopassi G, Wang E. Modelling the effects of age-related mtDNA mutation accumulation; complex I deficiency, superoxide and cell death. Biochim Biophys Acta. 1995;1271:171–176. doi: 10.1016/0925-4439(95)00025-y. [DOI] [PubMed] [Google Scholar]
- Cortopassi GA, Wong A. Mitochondria in organismal aging and degeneration. Biochim Biophys Acta. 1999;1410:183–193. doi: 10.1016/s0005-2728(98)00166-2. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Nedergaard M. Astrocytes in the aging brain. J Neurosci Res. 2002;67:1–10. doi: 10.1002/jnr.10121. [DOI] [PubMed] [Google Scholar]
- Dawson L, Gores J, Nieminen A, Herman B, Lemasters J. Mithochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:C961–C967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- Denton RM, McCormack JG. Ca2+ transport by mammalian mitochondria and its role in hormone action. Am J Physiol. 1985;259:E543–E554. doi: 10.1152/ajpendo.1985.249.6.E543. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR, Jr, Mogul DJ. Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 1996;59:413–420. doi: 10.1016/0024-3205(96)00320-7. [DOI] [PubMed] [Google Scholar]
- Di Virgilio F, Chiozzi P, Falzoni S, Ferrari D, Sanz JM, Venketaraman V, Baricordi OR. Cytolytic P2X purinoceptors. Cell Death Differ. 1998;5:191–199. doi: 10.1038/sj.cdd.4400341. [DOI] [PubMed] [Google Scholar]
- Farkas DL, Wei MD, Febbroriello P, Carson JH, Loew LM. Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys J. 1989;56:1053–1069. doi: 10.1016/S0006-3495(89)82754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink C, Morgan F, Loew LM. Intracellular fluorescent probe concentrations by confocal microscopy. Biophys J. 1998;75:1648–1658. doi: 10.1016/S0006-3495(98)77607-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta MN, Cormio A, Pesce V, Lezza AM, Cantatore P. Aging and mitochondria. Biochimie. 1998;80:863–870. doi: 10.1016/s0300-9084(00)88881-1. [DOI] [PubMed] [Google Scholar]
- Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanism by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Krishnamurthy R, Szalai G. Mitochondrial calcium signaling driven by the IP3 receptor. J Bioenerg Biomembr. 2000;32:15–25. doi: 10.1023/a:1005504210587. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Hansford RG. Relation between mitochondrial calcium transport and control of energy metabolism. Rev Physiol Biochem Pharmacol. 1985;102:1–72. doi: 10.1007/BFb0034084. [DOI] [PubMed] [Google Scholar]
- Heizmann CW, Braun K. Changes in Ca2+-binding proteins in human neurodegenerative disorders. Trends Neurosci. 1992;15:259–264. doi: 10.1016/0166-2236(92)90067-i. [DOI] [PubMed] [Google Scholar]
- James G, Butt AM. P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur J Pharmacol. 2002;447:247–260. doi: 10.1016/s0014-2999(02)01756-9. [DOI] [PubMed] [Google Scholar]
- Kantrow SP, Tatro LG, Piantadosi CA. Oxidative stress and adenine nucleotide control of mitochondrial permeability transition. Free Radic Biol Med. 2000;28:251–260. doi: 10.1016/s0891-5849(99)00238-5. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium hypothesis of Alzheimer's disease and brain aging. Ann NY Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- Koles L, Furst S, Illes P. P2X and P2Y receptors as possible targets of therapeutic manipulations in CNS illnesses. Drug News Perspect. 2005;18:85–101. doi: 10.1358/dnp.2005.18.2.886479. [DOI] [PubMed] [Google Scholar]
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226:1089–1092. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Lechleiter J, Peralta E, Clapham D. Diverse functions of muscarinic acetylcholine receptor subtypes. Trends Pharmacol Sci. 1989;([Suppl]):34–38. [PubMed] [Google Scholar]
- Leslie SW, Chandler LJ, Barr EM, Farrar RP. Reduced calcium uptake by rat brain mitochondria and synaptosomes in response to aging. Brain Res. 1985;329:177–183. doi: 10.1016/0006-8993(85)90523-2. [DOI] [PubMed] [Google Scholar]
- Li W, Schultz C, Llopis J, Tsien RY. Membrane-permeant esters of inositol polyphosphates, chemical syntheses and biological applications. Tetrahedron. 1997;53:12017–12040. [Google Scholar]
- Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J Biol Chem. 2002;277:31134–31141. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- Lin DT, Wu J, Holstein D, Upadhyay G, Rourk W, Muller E, Lechleiter JD. Ca(2+) signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol Aging. 2007;28:99–111. doi: 10.1016/j.neurobiolaging.2005.11.004. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton PM. Role of calcium ions regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Miller JJ, Baimbridge KG. Biochemical and immunohistochemical correlates of kindling-induced epilepsy: role of calcium binding protein. Brain Res. 1983;278:322–332. doi: 10.1016/0006-8993(83)90264-0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Farschon DM, Reed JC. Cell-free apoptosis in Xenopus egg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Norenberg W, Illes P. Neuronal P2X receptors: localization and functional properties. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:324–339. doi: 10.1007/s002100000311. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Koumenis IL, Dugan LL, Giffard RG. Vulnerability to glucose deprivation injury correlates with glutathione levels in astrocytes. Brain Res. 1997;748:151–156. doi: 10.1016/s0006-8993(96)01293-0. [DOI] [PubMed] [Google Scholar]
- Paraidathathu T, de Groot H, Kehrer JP. Production of reactive oxygen by mitochondria from normoxic and hypoxic rat heart tissue. Free Radic Biol Med. 1992;13:289–297. doi: 10.1016/0891-5849(92)90176-h. [DOI] [PubMed] [Google Scholar]
- Pastorino G, Snyder W, Serroni A, Hoek B, Farber L. Cyclosporin and carnitine prevent the anoxic death of cultured hepatocytes by inhibiting the mitochondrial permeability transition. J Biol Chem. 1993;268:13791–13798. [PubMed] [Google Scholar]
- Peterson C, Nicholls DG, Gibson GE. Subsynaptosomal distribution of calcium during aging and 3,4-diaminopyridine treatment. Neurobiol Aging. 1985;6:297–304. doi: 10.1016/0197-4580(85)90007-7. [DOI] [PubMed] [Google Scholar]
- Petit PX, Lecoeur H, Zorn E, Cauguet C, Mignotte B, Gougeon M. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol. 1995;130:157–167. doi: 10.1083/jcb.130.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Cola C, Bernardi P. Modulation of the mitochondrial cyclosporinA-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+ J Biol Chem. 1993;268:1011–1016. [PubMed] [Google Scholar]
- Richter C. Pro-oxidants and mitochondrial Ca2+: their relationship to apoptosis and oncogenesis. FEBS Lett. 1993;325:104–107. doi: 10.1016/0014-5793(93)81423-w. [DOI] [PubMed] [Google Scholar]
- Richter C. Reactive oxygen and nitrogen species regulate mitochondrial Ca2+ homeostasis and respiration. Biosci Rep. 1997;17:53–66. doi: 10.1023/a:1027387301845. [DOI] [PubMed] [Google Scholar]
- Robb SJ, Connor JR. An in vitro model for analysis of oxidative death in primary mouse astrocytes. Brain Res. 1998;788:125–132. doi: 10.1016/s0006-8993(97)01543-6. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara J, Makino N, Bannai S. Glutathione efflux from cultured astrocytes. J Neurochem. 1996;66:1876–1881. doi: 10.1046/j.1471-4159.1996.66051876.x. [DOI] [PubMed] [Google Scholar]
- Sagara JI, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61:1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki Y, Koizumi S, Ishida S, Sawada J, Ohno Y, Inoue K. Cytoprotection against oxidative stress-induced damage of astrocytes by extracellular ATP via P2Y1 receptors. Glia. 2005;49:288–300. doi: 10.1002/glia.20118. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Calcium-binding protein (calbindin D28k) and parvalbumin immuno-histochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol. 1989;280:183–196. doi: 10.1002/cne.902800203. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Brunk UT. Mitochondrial production of pro-oxidants and cellular senescence. Mutat Res. 1992;275:295–304. doi: 10.1016/0921-8734(92)90033-l. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Dubey A. Mitochondrial oxidative damage, hydrogen peroxide release, and aging. Free Radic Biol Med. 1994;16:621–626. doi: 10.1016/0891-5849(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Sutherland MK, Wong L, Somerville MJ, Yoong LKK, Bergeron C, Parmentier M, McLachlan DR. Reduction of calbindin-28k mRNA levels in Alzheimer as compared to Huntington hippocampus. Mol Brain Res. 1993;18:32–42. doi: 10.1016/0169-328x(93)90171-k. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kurata M. Effects of ATP level on glutathione regeneration in rabbit and guinea-pig erythrocytes. Comp Biochem Physiol B. 1992;103:859–862. doi: 10.1016/0305-0491(92)90205-6. [DOI] [PubMed] [Google Scholar]
- Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am J Physiol Cell Physiol. 2000;278:C423–C435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, and light scattering. J Biol Chem. 2001;276:2586–2599. doi: 10.1074/jbc.M002923200. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends Neurosci. 1996;19:346–352. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Vitorica J, Satrustegui J. The influence of age on the calcium-efflux pathway and matrix calcium buffering power in brain mitochondria. Biochim Biophys Acta. 1986a;851:209–216. doi: 10.1016/0005-2728(86)90127-1. [DOI] [PubMed] [Google Scholar]
- Vitorica J, Satrustegui J. Involvement of mitochondria in the age-dependent decrease in calcium uptake of rat brain synaptosomes. Brain Res. 1986b;378:36–48. doi: 10.1016/0006-8993(86)90284-2. [DOI] [PubMed] [Google Scholar]
- Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- Warner HR. Superoxide dismutase, aging, and degenerative disease. Free Radic Biol Med. 1994;17:249–258. doi: 10.1016/0891-5849(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Ying WL, Emerson J, Clarke MJ, Sandai DR. Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry. 1991;30:4949–4952. doi: 10.1021/bi00234a016. [DOI] [PubMed] [Google Scholar]
- Zahrebelski G, Nieminen A, Al-Ghoul K, Qian T, Herman B, Lemaster JJ. Progression of subcellular changes during chemical hypoxia to cultured rat hepatocytes: a laser scanning confocal microscopic study. Hepatology. 1995;21:1361–1372. [PubMed] [Google Scholar]
- Zamzami N, Susin SA, Marchetti P, Hirsch T, Monterrey-Gomez I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]