

Abstract
Hyperphosphorylation of the microtubule-associated protein tau is a key event in the development of Alzheimer's disease (AD) neuropathology. Acute stress can induce hippocampal tau phosphorylation (tau-P) in rodents, but the mechanisms and pathogenic relevance of this response are unclear. Here, we find that hippocampal tau-P elicited by an acute emotional stressor, restraint, was not affected by preventing the stress-induced rise in glucocorticoids but was blocked by genetic or pharmacologic disruption of signaling through the type 1 corticotropin-releasing factor receptor (CRFR1). Conversely, these responses were exaggerated in CRFR2-deficient mice. Parallel CRFR dependence was seen in the stress-induced activation of specific tau kinases. Repeated stress exposure elicited cumulative effects on tau-P and its sequestration in an insoluble, and potentially pathogenic, form. These findings support differential regulatory roles for CRFRs in an AD-relevant form of neuronal plasticity and may link datasets documenting alterations in the CRF signaling system in AD and implicating chronic stress as a risk factor in age-related neurological disorders.
Keywords: Alzheimer's disease, antalarmin corticotropin-releasing factor, corticotropin-releasing hormone, hippocampus, restraint stress, tau phosphorylation
Introduction
Alzheimer's disease (AD) is defined neuropathologically by the accumulation of β-amyloid plaques and neurofibrillary tangles (NFTs), the latter consisting of hyperphosphorylated forms of the microtubule-associated protein tau. Hyperphosphorylated tau exhibits reduced ability to bind and stabilize microtubules and can self-aggregate to form insoluble paired helical filaments (PHFs), which comprise NFTs (Gustke et al., 1992; Bramblett et al., 1993; Alonso et al., 1996). The incidence of NFTs is positively correlated with cognitive deficit and neuronal loss in AD (Arriagada et al., 1992; Gomez-Isla et al., 1997), and the discovery that mutations in the tau gene underlie autosomal dominant forms of frontotemporal dementia suggests that pathological changes in tau can serve as a principal cause of neurodegeneration and cognitive impairment (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998).
Exposure to a range of environmental insults, or stresses, can activate tau kinases and induce tau phosphorylation (tau-P) in the rodent CNS (Korneyev et al., 1995; Papasozomenos, 1996; Korneyev, 1998; Yanagisawa et al., 1999; Planel et al., 2001, 2004; Arendt et al., 2003; Feng et al., 2005). This effect has been reported consistently in the hippocampal formation, a key structure in learning and memory, and the initial site of tau pathology in AD (Braak and Braak, 1991). Although acute stress-induced tau-P is reversible, the mechanisms that govern this phenomenon are unknown, and it is unclear whether and how it may be manifest under chronic stress conditions. Addressing these questions may better define the elusive links between the stress axis and AD-related pathogenic processes, as increased exposure and/or sensitivity to stress in humans and rodent models confers increased risk of dementia and AD neuropathology (Wilson et al., 2003; Jeong et al., 2006).
Warranting consideration in this respect are glucocorticoids, dominant stress hormones whose elevated levels in aging have been linked to increased neuronal vulnerability in hippocampus (Sapolsky et al., 1985, 1986). However, acute stress-induced tau-P is reportedly unaffected in adrenalectomized mice (Korneyev et al., 1995), suggesting that glucocorticoid secretion may not be pivotally involved. Alternatively, the corticotropin-releasing factor (CRF) signaling system plays an essential role in initiating pituitary–adrenal responses to stress and has been implicated as a transmitter/modulator in CNS systems that mediate complementary autonomic and behavioral adjustments, earning consideration as a general mediator/integrator of stress adaptations (Chadwick et al., 1993). CRF and related ligands (urocortins 1–3) exert their biological effects via two G-protein-coupled receptors [CRF receptor 1 (CRFR1) and CRFR2] that are differentially distributed in brain (Van Pett et al., 2000), and exert convergent effects on a range of stress-related endpoints (Bale and Vale, 2004). CRFR ligands can confer neuroprotection, in vitro, by altering amyloid precursor protein processing and suppressing tau kinases, and reduced central CRF expression has been documented early in AD progression (Rehman, 2002; Bayatti and Behl, 2005). Nevertheless, the nature of any association between the CRF system and AD-related pathogenesis remains to be elucidated. Here, we provide evidence to support a push–pull involvement of CRFRs in regulating stress-induced tau-P and tau kinase activity in murine hippocampus.
Materials and Methods
CRFR knock-out mice.
Mutant mice and littermate wild-type (wt) controls were bred from heterozygote breeder pairs of established lines backcrossed to founder mice to achieve a pure C57BL/6 background (Smith et al., 1998; Bale et al., 2000). Genotype was determined by PCR, and males were used for experimentation at 15–22 weeks of age. Pregnant females used for generating CRFR1-deficient mice received drinking water supplemented with corticosterone (10 μg/ml; Sigma-Aldrich, St. Louis, MO) from embryonic day 12 to postnatal day 14 to prevent early mortality as a result of pulmonary dysplasia (Smith et al., 1998). Because CRFR1−/− mice exhibit adrenal cortical agenesis, experimental animals were reinstated on corticosterone for 21 d before testing to allow the normal nocturnal bias in appetitive behavior to approximate the circadian fluctuation in circulating hormone levels. To assess effectiveness of the replacement regimen, plasma corticosterone levels were determined by RIA from blood samples collected when the mice were killed. The Salk Institute Institutional Animal Care and Use Committee approved all experimental protocols.
Adrenalectomy and corticosterone replacement.
Twelve-week-old male wt C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) underwent adrenalectomy (ADX) via bilateral incisions on the dorsolateral flanks under isoflurane anesthesia. ADX mice received replacement corticosterone (10 μg/ml) in drinking water containing 0.9% saline immediately after surgery. Animals were used in stress experiments 21 d after surgery.
Restraint stress.
Acute restraint stress involved placing mice in ventilated 50 ml conical tubes for 30 min; repeated stress involved 14 consecutive daily exposures. Animals were killed at various intervals ranging from 20 min to 24 h after stress. Control mice were handled comparably but were not otherwise manipulated.
Intracerebroventricular injections.
CRFR1−/− and CRFR2−/− mice, along with age-matched wt controls (n = 3/group), were anesthetized with isoflurane and implanted stereotaxically with 26 ga guide cannulas (Plastics One, Wallingford, CT) aimed to terminate above the lateral ventricle. Cannulas were affixed to the skull with dental acrylic adhering to jeweler's screws partially driven into the skull and sealed with stylets. After 7 d of recovery, stylets were replaced with 33 ga injection cannulas, and 2 h later the animals were remotely injected with 0.5 μg of synthetic mouse/human CRF in 2 μl of saline, or vehicle alone, over ∼1 min. To approximate the time frame used in acute stress experiments, animals were killed 40 min after intracerebroventricular injection and perfused for immunohistochemistry, as described below. CRF was provided by Dr. J. Rivier (Salk Institute).
In vivo pharmacology.
The small-molecule CRFR1-selective antagonist antalarmin (Webster et al., 1996) was administered (20 mg/kg, i.p. injection) 20 min before stress exposure. All animals were handled twice daily for 28 d before experimentation and received daily mock intraperitoneal injections to minimize stress of injection at testing. Antalarmin was solubilized in equal volumes of absolute ethanol and Cremaphor EL (Sigma-Aldrich), as described previously (Webster et al., 1996; Pernar et al., 2004). This stock solution was diluted in prewarmed (50°C) distilled water and adjusted to a final concentration of 4 mg/ml immediately before injection.
Western blot analysis.
Mice were anesthetized with sodium pentobarbital (40 mg/kg), which has been demonstrated to not influence tau-P over the time frame used here (Papasozomenos, 1996). After sedation, animals were decapitated, and the hippocampus was rapidly dissected and frozen on dry ice. Hippocampal tissues were homogenized in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris-HCl, pH 7.4, 0.1% SDS, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4, and 1 μm okadaic acid). Before homogenization, protease inhibitors PMSF, NaF (1 mm), aprotinin, leupeptin, and pepstatin (1 μg/ml each) were added. RIPA fractions were obtained by centrifuging twice at 40,000 × g for 20 min, and the supernatant was collected. For analysis of tau solubility (repeated stress), sequential fractionation of RAB and RIPA extracts were performed as described previously (Higuchi et al., 2002; Kraemer et al., 2003). In this case, tissues were first homogenized in high-salt RAB (0.1 m MES, 0.75 NaCl, 1 mm EGTA, and 0.5 mm MgSO4) and centrifuged at 40,000 × g for 40 min. The supernatant was collected (soluble RAB fraction), and pellets were resuspended in RIPA buffer to obtain detergent-soluble fractions. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL). Proteins were then boiled in sample buffer containing SDS, BME, and glycerol at 95°C for 5 min. Six micrograms of protein were then separated by 12% SDS-PAGE. Proteins were transferred to nitrocellulose membrane (0.2 μm; Bio-Rad, Hercules, CA) and incubated in primary antibodies diluted in 5% milk-PBS-T overnight at 4°C. Primary antibodies were detected with either anti-mouse or -rabbit horseradish peroxidase-linked secondary antibodies (1:1000; EMD Biosciences, La Jolla, CA) and developed with an enhanced chemiluminescence Western blot detection kit (Supersignal West Pico; Pierce Biotechnology). Background subtraction was performed, and quantitative band intensity readings were obtained using NIH Image software.
Antibodies.
Well characterized phospho-specific antibodies were used for detection of phosphorylated residues on mouse tau. For Western blots, T181, S199, S212, T231, S422 (1:1000; Biosource, Camarillo, CA), S202/T205 (1:500; AT8; Pierce Biotechnology), and S396/404 (1:1000; PHF-1; gift from Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY). These antibodies were chosen based on their ability to resolve target bands at the appropriate molecular weight for phosphorylated tau (i.e., ∼50–75 kDa). Phospho-specific antibodies against S217, S262, S356, and S409 phosphorylated tau (Biosource) were also tested but were found to resolve only very high-molecular-weight target bands (i.e., >100 kDa) and were therefore excluded from the analysis. Antibody PHF-1 (1:500) was used for detection of phosphorylated tau in immunohistochemical analyses. Specificity of PHF-1 in mouse tissue was confirmed by pretreating sections from stressed mice with alkaline phosphatase (40 mg/ml; Sigma-Aldrich), which eliminated detectable PHF-1 labeling in all experimental groups (data not shown). For assessment of tau kinases, specific antibodies to phosphorylation sites or activator proteins were used. This included total glycogen synthase kinase-3β (GSK-3β; 1:2500; BD Biosciences, San Diego, CA), activated GSK-3β (pY216; 1:1000; BD Biosciences), inactive GSK-3β (pS9; 1:1000; Cell Signaling Technology, Danvers, MA), cyclin-dependent kinase 5 (cdk5; 1:1000; EMD Biosciences), cdk5 activator proteins, p25 and p35 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), phosphorylated c-Jun-N-terminal kinase (JNK; 1:1000; Cell Signaling Technology), mitogen-activated protein (MAP) kinases [extracellular signal-regulated kinases 1 and 2 (ERK1/2); 1:500; Cell Signaling Technology], and the catalytic subunit of protein phosphatase 2A (PP2A-c; 1:5000; BD Biosciences). β-Actin (1:2000; Sigma-Aldrich) was used as a control for protein loading.
Immunohistochemistry.
Mice were perfused with 4% paraformaldehyde as described previously (Bittencourt and Sawchenko, 2000). Thirty-micrometer-thick frozen sections were cut on a sliding microtome and stored at −20°C in cryoprotectant solution (20% glycerol and 30% ethylene glycol in 0.1 m phosphate buffer). PHF-1 was used to detect tau-P in free-floating sections containing hippocampus using Mouse-on-Mouse Immunodetection Kit reagents (Vector Laboratories, Burlingame, CA) to avoid detection of endogenous mouse Ig. Endogenous peroxidase was quenched with 0.3% hydrogen peroxide, followed by 1% sodium borohydride to reduce free aldehydes. Reaction product was developed using a nickel-enhanced glucose oxidase method (Shu et al., 1988).
Statistical analyses.
Integrated intensity readings from Western blots were analyzed using either a one- or two-way ANOVA using Prism4 software (GraphPad, San Diego, CA). Resultant data were plotted on bar graphs, with data expressed as mean ± SEM percentage of control values.
Results
Time course of tau-P after acute restraint stress
We initially determined whether increased tau-P was observable in response to acute restraint, an acknowledged “emotional” stressor (Sawchenko et al., 2000). Western analysis was used to examine tau-P at several AD-relevant N- and C-terminal sites [S181, S199, S202/T205 (AT8), T212, T231, S396/404 (PHF-1), and S422] in hippocampal extracts from C57BL/6 mice killed at various intervals after a single 30 min episode of restraint stress. Relative to basal (nonstressed) values, all sites exhibited significant increases in phosphorylation that were apparent at the termination of stress (0 min), with peak elevations (2- to 10-fold) achieved 20–40 min later and sustained through 60 min (Fig. 1). By 90 min, levels were reduced to or near those of unstressed controls. These results demonstrate that a representative emotional stressor induces rapid and reversible increases in tau-P at multiple AD-relevant sites. Increments in tau-P were quite stable over 20–60 min after stress, and the 20 min time point was selected for subsequent analyses.
Figure 1.

Time course of stress-induced tau phosphorylation. Western blots of hippocampal extracts probed for tau-P at select AD-relevant sites in unstressed controls (C) and mice killed 0, 20, 40, 60, or 90 min after a single 30 min exposure to restraint stress. For all epitopes examined, signal under basal conditions was low but rose immediately after stress to levels that were markedly increased (2- to 10-fold) over control levels through 60 min and diminished thereafter. The 20 min poststress time point was adopted for use in subsequent experiments. β-Actin was used as a loading control.
Glucocorticoid involvement in stress-induced tau phosphorylation?
Because of the dominant role of glucocorticoids in mediating stress effects and their implication in neuronal loss (Sapolsky et al., 1985, 1986) and pathology in mouse models of AD (Green et al., 2006), we tested whether stress-induced tau-P was dependent on stress-induced glucocorticoid secretion. Although a previous study used immunoassay and found no effect of ADX on PHF-1 reactivity after acute cold water stress (Korneyev et al., 1995), phosphatase inhibitors were not used, and only soluble fractions of tau protein were examined. We examined stress-induced tau-P responses at the AT8 and PHF-1 sites in hippocampal extracts from ADX and control mice exposed to acute restraint stress (Fig. 2). Robust stress-induced tau-P responses were observed in ADX mice that did not significantly differ from those of intact controls at either the AT8 or PHF-1 phosphorylation sites (each p > 0.10). These results confirm that acute stress-induced tau-P is not dependent on glucocorticoid secretion.
Figure 2.

Role of glucocorticoids in stress-induced tau-P. Western blot analysis of hippocampal tau-P at the AT8 and PHF-1 sites under control (C) and acute stress (S) conditions in intact and ADX mice. Quantitative analysis, expressed as mean ± SEM percentage of integrated intensity values of intact unstressed controls, reveals that intact and ADX animals manifest comparably robust stress-induced increments in tau-P at each site, whose magnitude did not differ significantly. **Differs significantly from intact, unstressed controls (p < 0.001); ns, nonsignificant (p > 0.05). n = 3 mice per condition. β-Actin was used as a loading control.
CRFRs differentially regulate stress-induced tau phosphorylation
The CRF family of signaling molecules is broadly involved in physiological and behavioral responses to stress (Chadwick et al., 1993) and undergoes alterations early in AD progression (Davis et al., 1999). However, the nature of its involvement in AD neuropathology is unclear. We next investigated the role of CRFRs in acute stress-induced tau phosphorylation using mice deficient in CRFR1 (Smith et al., 1998) or CRFR2 (Bale et al., 2000). Western analyses indicated a tendency for CRFR1−/− mice to exhibit higher basal levels of tau-P than unstressed wt animals at several sites, although this difference was statistically reliable only at the AT8 epitope (p < 0.01) (Fig. 3). More importantly, CRFR1-deficient mice failed to exhibit significant increases in tau-P at any site at 20 min after stress, compared with age-matched wt controls. In contrast, CRFR2 knock-outs displayed normal basal levels of tau-P (p > 0.05 vs wt) but showed robust responses to acute stress that commonly exceeded those seen in wt animals. Specifically, phosphorylation responses of CRFR2−/− mice were significantly greater than those of wt animals at the S181 (p < 0.01), S199 (p < 0.001), T212 (p < 0.05), T231 (p < 0.05), and PHF-1 (p < 0.01) sites. Phosphorylation responses of CRFR2 null mice at the AT8 and S422 sites did not differ significantly from those of wt stressed animals (p > 0.05).
Figure 3.

Differential regulation of stress-induced tau-P by CRFR status. Phosphorylation responses of seven AD-relevant tau sites in hippocampal extracts from wt, CRFR1−/−, and CRFR2−/− mice killed at 20 min after acute restraint stress (S) or no treatment (C). Quantitative analysis revealed that wt animals displayed the expected increases in tau-P after stress at all epitopes. CRFR1−/− animals displayed elevated basal levels of phosphorylation at the AT8 and PHF-1 sites but did not manifest significantly increased responses after stress at any site. Conversely, CRFR2−/− mice showed normal basal levels of tau-P at all sites but exaggerated responses to stress at five of the seven epitopes assayed. Data are presented as mean ± SEM percentage of wt control values. *Differs significantly from unstressed wt condition (p < 0.01); **p < 0.001; †differs from wt stressed group (p < 0.05). n = 3 mice per condition. β-Actin was used as a loading control.
To probe the localization of tau-P responses, immunohistochemical methods were used to examine the distribution of PHF-1 reactivity and its stress and CRFR dependence. Immunolabeling results were highly compatible with biochemical data in showing prominent upregulation of PHF-1 staining in wt mice in response to stress, which was attenuated and exaggerated in CRFR1- and CRFR2-deleted animals, respectively (Fig. 4A). In the dentate gyrus of stressed wt animals, PHF-1 positive cell bodies were seen primarily in the hilus (polymorph and subgranular regions), but also in deep aspects of the granule cell layer. We also observed PHF-1 positive mossy fibers and a band of punctate (presumably axonal) elements in the inner third of the molecular layer. In Ammon's Horn, dominant features included labeled perikarya scattered mainly throughout the pyramidal layer and proximal dendritic zones and bands of axon terminal-like puncta engulfing the pyramidal cell layer and in stratum lacunosum-moleculare. Radially oriented processes, some traceable to labeled cell bodies and presumably representing dendritic labeling, were seen in stressed wt and CRFR2−/− animals. Alterations in immunostaining as a function of stress and genotype were manifest as differences in the number and/or intensity of labeled elements, with no discernible differences in distribution.
Figure 4.
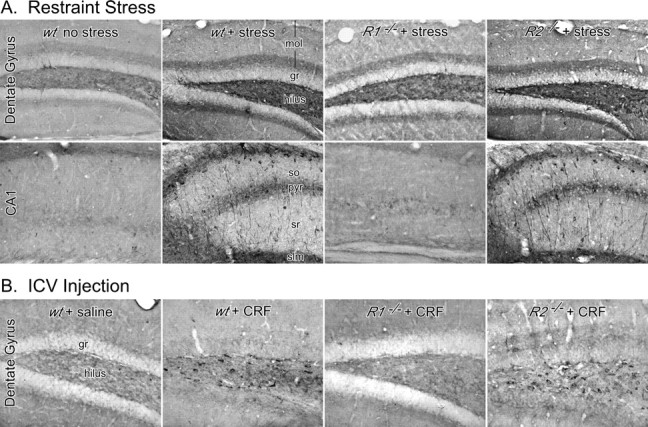
Cellular localization of stress- and peptide-induced tau-P. A, Immunoperoxidase staining for PHF-1 immunoreactivity in the dentate gyrus (top) and CA1 field (bottom) of mouse hippocampus as a function of stress status and genotype. Phosphorylation is localized to distinct perikaryal/dendritic and axonal elements (see Results) and, importantly, varies with stress exposure and CRFR status in a manner identical to that seen by Western analysis (Fig. 3). B, PHF-1 in the dentate gyrus of mice injected intracerebroventricularly with synthetic CRF. Tau-P varied as a function of genotype in a manner similar to that seen in stressed animals (A). gr, Granule cell layer; mol, molecular layer; pyr, pyramidal layer; slm, stratum lacunosum-moleculare; sr, stratum radiatum.
To determine whether CRF is capable of independently eliciting hippocampal tau-P, wt and CRFR-deficient mice were implanted with lateral ventricular cannulas for intracerebroventricular injection. Resultant PHF-1 immunoreactivity was examined 40 min after administration of CRF or saline vehicle (Fig. 4B). The general pattern of results was similar to that observed in response to stress, in that wt and CRFR2−/− mice treated with peptide displayed robust increases in hippocampal tau-P, whereas labeling in CRFR1−/− animals was comparable with the low level seen in saline-injected controls. In the dentate gyrus, PHF-1 positive cell bodies were seen in the hilus and deep aspects of the granule cell layer (Fig. 4B). In Ammon's Horn, we observed PHF-1 labeling in mossy fibers and in the form of punctate pericellular labeling throughout the pyramidal cell layer and, more sporadically, in the dendritic zone, with particular concentration at the septal pole of the hippocampus (data not shown). In contrast to stress effects, however, we did not observe labeling of pyramidal neurons or their processes after intracerebroventricular CRF injections. These findings indicate that central CRF administration at least partially recapitulates the effects of stress and demonstrates a similar dependence on CRFR integrity.
Effects of pharmacologic blockade of CRFR1
Interpretation of data derived from conventional knock-out animals may be complicated by developmental or indirect effects of lifelong lack of expression of the targeted gene. This is particularly true of CRFR1-deficient mice, which exhibit chronically impaired pituitary–adrenal function (Smith et al., 1998). Despite efforts to mitigate such effects by steroid replacement perinatally and immediately before experimentation (see Materials and Methods), confidence in the assertion of a regulatory role for CRFR1 in this context would be bolstered if the effects were maintained in the face of acute disruption of receptor function. We therefore assessed the effect of antalarmin, a small-molecule, selective CRFR1 antagonist (Webster et al., 1996), on basal and stress-induced tau-P. Neither antalarmin nor the administration vehicle significantly altered basal levels of tau-P at the AT8 or PHF-1 sites, relative to untreated controls (Fig. 5). However, antalarmin treatment prevented stress-induced increments in phosphorylation at both sites (lanes 5–6; each p > 0.10 vs untreated controls). Phosphorylation responses in stressed, vehicle-treated animals were comparable with those of stressed, untreated controls and significantly elevated over vehicle control levels (p < 0.01). These findings support a specific involvement of CRFR1 signaling in stress-induced tau-P.
Figure 5.

CRFR1 antagonist blocks stress-induced tau-P. Quantitative Western blot analysis of tau-P at the AT8 and PHF-1 sites in hippocampal extracts from mice pretreated with vehicle or the small-molecule CRFR1 antagonist antalarmin and subjected to acute stress or no additional treatment, expressed as a percentage of levels in untreated controls (C). At each site, antagonist treatment did not affect basal levels of phosphorylation but blocked the stress-induced increment. The vehicle used for drug administration had no significant effect under basal or stress conditions. **Differs significantly from unstressed controls (p < 0.001); †Differs from untreated stressed group (p < 0.05). n = 3 per condition. β-Actin was used as a loading control.
Modulation of tau kinase activity by stress and CRFRs
To identify potential mediators of acute stress-induced tau-P, we used antibodies specific to active and inactive states of kinases implicated in tau-P in the same tissue extracts used above to interrogate changes in stress-induced kinase activation and their CRFR dependence (Fig. 6). Several kinases examined, including the active (phosphorylated at Y216 or pY216), but not the inactive (pS9), or total (unphosphorylated) form of GSK-3β, the pT183/Y185 form of the 46 and 54 kDa c-Jun N-terminal protein kinases (JNK46/54), and the pT202/Y204 form of the mitogen-activated protein kinases, ERK2, but not ERK1, displayed upregulation in response to acute restraint. The time courses of stress effects seen on these kinases were similar to those shown in Figure 1 for stress-induced tau-P (data not shown). Relative levels of cdk5 were unchanged from steady state over the poststress intervals examined, but one of its regulatory proteins, p35, was robustly upregulated. We were unable to reproducibly detect the p35-truncated product, p25.
Figure 6.

Modulation of tau kinase activity by stress and CRFR status. Western blot analyses using phosphoepitope-specific antibodies to interrogate stress- and/or genotype-dependent variations in the activation state of GSK-3, JNK46/54, and ERK1/2; relative levels of cdk5 and its activator protein, p35, were also evaluated. The behavior of each kinase group recapitulates aspects of the general pattern of tau-P responses in the same design. Thus, the activated (pY216) form of GSK-3β is upregulated by stress in wt mice, and this response is blocked CRFR1-deficient animals and exaggerated in CRFR2 mutants. The inhibitory (pS9) form and total (unphosphorylated) GSK3 are unresponsive over these conditions. Both phospho-JNK isoforms were also stress responsive, and more so in CRFR2 knock-outs; these kinases were distinguished by very high basal phosphorylation levels in CRFR1−/− mice, which may relate to elevated tau-P observed in this condition at the AT8 site (see Fig. 3). Levels of phospho-ERKs, particularly ERK1, were relatively unresponsive. Levels of cdk5 were stable across the conditions in force here, but its p35 activator protein was strongly upregulated by stress in wt and CRFR2−/− mice. Data are presented as mean ± SEM percentage of unstressed wt values. *Differs significantly from wt control (p < 0.01); **p < 0.001. †Differs significantly from wt stress condition (p < 0.05). β-Actin was used as a loading control.
When tested in hippocampal extracts from wt and knock-out mice, each of the stress-responsive kinase forms or regulators also exhibited modulation as a function of CRFR status that mirrored some or all of the effects of genotype on restraint-induced tau-P. The activated (pY216) form of GSK-3β, implicated in phosphorylating tau at S199, S212, T231, and PHF-1 sites, was most similar in that the stress-induced increment seen in wt mice was not evident in CRFR1−/− animals and was exaggerated in CRFR2−/− mice. Phosphorylation responses of both JNK isoforms were also significantly greater in CRFR2 knock-outs than in wt controls (p < 0.05). These kinases also exhibited pronounced elevations in basal phosphorylation in CRFR1-deficient mice, whose magnitude rivaled or exceeded stress-induced levels in wt mice. This may relate to the elevated tau-P levels seen under this condition at the AT8 and PHF-1 sites, although less-marked elevations of phosphorylated GSK-3β and ERK2, and of p35 levels, in unstressed CRFR1−/− mice may also contribute in this regard. Overall, these results identify several tau kinases as potential effectors of CRFR-dependent effects of acute emotional stress on tau-P.
Tau phosphorylation in response to repeated stress
Because past and present data characterize acute stress-induced tau-P as a transient phenomenon, its relevance to neuropathology may be questioned. Data from animal models and AD brain have demonstrated that NFTs and other manifestations of tau pathology are dependent on aberrantly phosphorylated tau being sequestered into insoluble cellular fractions (Iqbal et al., 1994). We therefore used sequential fractionation in the absence (RAB buffer; see Materials and Methods) and presence (RIPA buffer) of detergents to compare the persistence and solubility of phosphorylated tau in animals subjected to acute versus repeated (14 consecutive daily exposures) restraint stress (Fig. 7). Groups of mice in each condition were killed 20 min or 24 h after their final or only stress episode. Results from the acute stress condition replicated findings detailed above in showing increased phosphorylation at the AT8 and PHF-1 sites 20 min after stress. At 24 h after stress, relative levels of tau-P were indistinguishable from unstressed animals (lane 3). In terms of solubility, phosphorylated tau induced by acute stress was detected only in the soluble fraction; that is, no additional signal was evident after further extraction with detergent (lanes 6–8). In contrast, under repeated stress conditions, comparably elevated AT8 and PHF-1 signal were present in soluble fractions at both 20 min and 24 h after the final restraint episode. In addition, extraction of detergent-soluble proteins (RIPA) revealed significant occurrence of phosphorylated tau at both time points (lanes 9–10). These results suggest that repeated stress leads to chronic elevations in phosphorylated tau and a shift in its disposition toward more insoluble, and potentially pathogenic, forms.
Figure 7.

Repeated stress results in chronic elevations and reduced solubility of phosphorylated tau. Western analysis of hippocampal tau-P at the PHF-1 and AT8 sites of mice killed 20 min or 24 h after 30 min acute restraint or 14 consecutive daily exposures. Analysis was performed on both soluble and detergent-soluble fractions. Under acute stress (AS) conditions, tau-P is transient and contained wholly within the soluble fraction, whereas repeated stress (RS) results in elevated levels of tau-P at both time points and the appearance of a significant portion of phospho-tau in the detergent-soluble fraction. These data suggest that the effects of repeated emotional stress on tau-P are cumulative and associated with increased sequestration in the cellular fraction known to contain the bulk of PHFs in the AD brain. Note that for this analysis, data are expressed as mean ± SEM optical density, rather than as a percentage of control values, to reflect the lack of detectable signal in detergent-soluble extracts under control conditions. *Differs significantly from control (p < 0.01); **p < 0.001. β-Actin was used as a loading control. C, Control.
Phosphatase involvement in restraint-induced tau phosphorylation?
Alterations in tau phosphatase activity have been implicated as contributing to stress-induced tau-P (Planel et al., 2001, 2004, 2007). To determine whether similar changes are associated with acute or repeated restraint stress, we analyzed the same extracts used in the preceding analysis (Fig. 7), for alterations in the catalytic subunit of the dominant tau phosphatase, PP2A (PP2A-c). We found no evidence of PP2A-c in detergent-soluble RIPA fractions under any experimental condition (data not shown). In soluble RAB fractions, relative levels of PP2A-c from hippocampi of acute stressed mice did not differ reliably from controls (lanes 1–3) but were significantly elevated at both 20 min (p < 0.001) and 24 h (p < 0.01; lanes 4–5; see supplemental Fig. 1, available at www.jneurosci.org as supplemental material) after repeated stress. Although additional characterization is needed, these findings identify PP2A as a potential contributor to alterations in tau-P under repeated stress conditions.
Discussion
Our findings extend the range of insults that provoke tau-P to include a representative emotional stressor and provide evidence on its mechanism (Fig. 8). We fail to implicate stress-induced glucocorticoid secretion in this phenomenon but provide data from pharmacologic and genetic manipulation to support a differential involvement of CRFRs. The abrogation of stress-induced tau-P in CRFR1-deficient animals and the enhancement observed in CRFR2 mutants were paralleled by altered activities of specific tau kinases. Furthermore, the demonstration that repeated stress exerts cumulative increases in tau-P and results in its sequestration in insoluble forms defines a possible underpinning for evidence implicating chronic stress as a risk factor in age-related neurological disorders, including AD.
Figure 8.

CRFR involvement in stress-induced tau phosphorylation. Schematic summary of the progression of events leading to neurofibrillary tangle formation in AD, adapted with permission from Drewes (2004). Indicated on this are the present findings supporting a differential involvement of CRFRs, acting via specific tau kinases, in mediating acute stress-induced tau-P (red circles). Although acute stress effects are transient and readily reversible, repeated stress exposure produces cumulative increases in phosphorylated tau, a portion of which is sequestered in detergent-soluble fractions. It remains to be determined whether long-term increases in stress exposure and/or sensitivity may result in development of the paired helical filaments and tangles that represent a defining feature of AD neuropathology.
Relationship to previous studies
Tau normally binds and stabilizes neuronal microtubules, facilitating their roles in cellular structure, polarity, and transport (Stamer et al., 2002). Phosphorylation can disrupt these activities and promote cytoskeletal destabilization (Sengupta et al., 1998). Aberrantly phosphorylated forms of tau aggregate into PHFs, and these aggregate into insoluble NFTs, a defining feature of AD (Kopke et al., 1993). In this light, the observation that acute stress results in tau-P at AD-relevant sites defines one potential means by which stress exposure may translate into neuropathology. This effect has been elicited by a range of strenuous challenges, including heat shock (Papasozomenos, 1996), starvation (Yanagisawa et al., 1999; Planel et al., 2001), forced swimming in cold water (Korneyev et al., 1995; Okawa et al., 2003; Feng et al., 2005; Yoshida et al., 2006), glucoprivation (Planel et al., 2004), ether inhalation (Ikeda et al., 2007), and hibernation (Arendt et al., 2003; Hartig et al., 2007). Two broad categories of stress models are now commonly recognized. These may be termed “physiological” and “emotional” and are distinguished by the sensory modalities that register the challenges, the patterns of neuronal activation they induce in brain, the extent to which they invoke affective responses, and the circuitry that mediates adaptive responses to them (Sawchenko et al., 1996; Watts, 1996; Herman and Cullinan, 1997; Dayas et al., 2001). Whereas the stressors shown previously to elicit tau-P fall mainly or exclusively in the physiological category, restraint is a prototypic emotional stressor. Because established models of anxiety, fear, and social stress share key features with restraint such as a capacity to engage a stereotyped set of interconnected limbic forebrain cell groups (Duncan et al., 1996; Campeau et al., 1997; Martinez et al., 2002), the present findings suggest that the generality of stress-induced tau-P may extend into the realm of stresses encountered in everyday experience.
Several challenges that elicit tau-P are associated with marked reductions in body temperature (7–10°C), which can differentially modulate tau kinase and phosphatase activities, leading Planel et al. (2001, 2004) to hypothesize that hypothermia may be a common underlying mechanism. Restraint also results in reduced core temperature, although of lesser magnitude (0.5–2°C) (Clement et al., 1989; Turek and Ryabinin, 2005; Meijer et al., 2006). It remains to be determined whether the correlation between body temperature and tau-P noted in other paradigms (Planel et al., 2007) extends to emotional stressors.
Whereas human tau is normally phosphorylated at 2–3 mol/mol of protein, PHF-tau from AD brain is hyperphosphorylated at a 7–10 molar ratio (Kopke et al., 1993). Murine tau can be phosphorylated and form PHFs in vitro (Kampers et al., 1999). We find that restraint provokes tau-P at each of seven epitopes examined, all but one of whose responses is differentially modulated by CRFR status. Coupled with the finding that central CRF administration stimulates PHF-1 phosphorylation in a CRFR1-dependent manner, we suggest a mechanism involving mediation by CRFR1, which is normally restrained by CRFR2-based signaling.
Signaling intermediates
Complementary monitoring of the activation status of tau kinases identified several candidate effectors. GSK-3β has been implicated in catalyzing tau-P at S199, S212, T231, S396, and, to a lesser extent, S202/T205, and confers PHF-like changes (Liu et al., 2003). Its activity is stimulated or inhibited by phosphorylation at Y216 and S9, respectively (Cohen and Frame, 2001). We found stress-induced increments in activated GSK-3β whose time course and genotype dependence paralleled tau-P responses, without significant variation in the inhibitory form (cf. Okawa et al., 2003) or total (unphosphorylated) GSK3 levels. The signaling intermediates that link CRFR ligand binding to alterations in GSK3 activity are unclear. Though commonly associated with a Gs–cAMP–protein kinase A mechanism, other signaling pathways can be activated downstream of CRFRs by different G-proteins in a cell type- and ligand-dependent manner (Grammatopoulos and Chrousos, 2002; Arzt and Holsboer, 2006; Hillhouse and Grammatopoulos, 2006). Contributing to the lack of clarity is uncertainty as to the proximate mechanism of tyrosine phosphorylation of GSK, with some reports identifying this as an autophosphorylation event (Cole et al., 2004) and others implicating distinct tyrosine kinases (Pyk2 and Fyn) in this regard (Lee et al., 1998; Hartigan et al., 2001).
Similar stress- and genotype-dependent changes were observed in levels of activated JNK, implicated in tau-P at the AT8 and PHF-1 epitopes (Atzori et al., 2001). High basal levels of phospho-JNKs in CRFR1−/− mice paralleled, and may explain, elevated AT8 and PHF-1 phosphorylation in this genotype. The MAP kinases ERK1 and ERK2, which can target all tau sites examined here except T231 (Drewes et al., 1992), were relatively unresponsive. Relative levels of cdk5, a major tau kinase active at T231, AT8, and PHF-1 sites (Patrick et al., 1999), were also stable across conditions, but one of its activator proteins, p35, exhibited strong CRFR-dependent stress responsiveness. Consistent with previous findings (Okawa et al., 2003), we did not detect the truncated p35 product, p25, which is a more potent cdk5 activator (Patrick et al., 1999).
Alterations in the activity of tau phosphatases, notably PP2A, have also been implicated in stress-induced tau-P (Planel et al., 2001, 2004). Here, we find that repeated stress increases relative levels of the catalytic subunit of PP2A, a finding that has been associated with diminished enzymatic activity and potent autoregulation (Baharians and Schonthal, 1998; Planel et al., 2001). Our failure to discern an acute stress effect on PP2A-c levels is not necessarily indicative of a lack of phosphatase involvement under these conditions.
Issues of interpretation
Two aspects of our findings warrant further consideration. First, CRFR2−/− mice displayed exaggerated responses to stress at five of seven phosphorylation sites and in three activated kinases. Although these data support an interaction with CRFR1 in regulating tau-P, whether they represent convergent or parallel effects is uncertain. In preliminary studies, we find that mice deficient in both CRFRs fail to manifest acute restraint-induced tau-P, supporting some degree of interdependence (R. A. Rissman and P. E. Sawchenko, unpublished observations). This interpretation is consistent with evidence that CRF, but not CRFR1 expression or binding, is upregulated in brain regions of CRFR2-deficient mice, and that CRFR1 antagonists can normalize aspects of the behavioral phenotype of these mutants (Kishimoto et al., 2000; Bale and Vale, 2003). These results suggest that exaggerated stress-induced tau-P of CRFR2 mutants does not result from alterations in CRFR1 expression or distribution. Second, although the consistent lack of stress responsiveness of CRFR1−/− mice suggests a target for intervention in tau pathologies, this is offset by a tendency toward increased basal tau-P (AT8 and PHF-1 sites) and activated kinase (notably JNK) expression in CRFR1 mutants. In addition, the need to supplement CRFR1-deficient mice with corticosterone during the perinatal period complicates interpretation by exposing animals to higher glucocorticoid levels during the stress-hyporesponsive period (Baram et al., 1997) and the switch in the dominant isoform of tau that occurs at this time (Kosik et al., 1989). It is noteworthy in both respects that acute pharmacological interference with CRFR1 blocked restraint-induced increments in tau-P without affecting basal levels, suggesting that the elevated baselines seen in knock-outs may result from chronic receptor deficiency.
CRF system involvement in AD
Exposure to stress can impact learning, memory, and hippocampal morphology and function, and may confer increased risk of AD (Wilson et al., 2003). Attention has focused on glucocorticoids as likely mediators of such effects, because increased circulating levels of these hormones in aging have been linked to a range of adverse effects in hippocampus (Sapolsky et al., 1985, 1986). Nevertheless, the present findings are consistent with those of Korneyev et al. (1995) in demonstrating that stress-induced tau-P proceeds unabated in animals lacking the capacity to mount a glucocorticoid response, implicating instead the CRF signaling system as integrally involved.
CRF is best known as a hypothalamic neuropeptide that governs the endocrine arm of the stress response (Vale et al., 1981). Aspects of its central distribution and actions suggest a general involvement in integrating hormonal, autonomic, and behavioral adaptations (Chadwick et al., 1993), motivating consideration of a role for this system in stress-induced tau-P. Alteration in CRF immunoreactivity is a prominent neurochemical change occurring early in AD progression (Davis et al., 1999), in brain regions vulnerable to AD neuropathology (Powers et al., 1987; Raadsheer et al., 1995; Pedersen et al., 2002; Rehman, 2002; Swaab et al., 2005). It has been suggested that these changes result from sequestration of peptide by a specific binding protein (CRF-BP) (Behan et al., 1995), which can reversibly neutralize CRF bioactivity. Accordingly, competitive antagonists of CRF-BP action enhance performance on a range of learning and memory tasks (Behan et al., 1995). In addition, marked increases in CRF binding have been described in cortical areas of the AD brain (De Souza et al., 1986). However, this work predated the cloning and characterization of CRFRs, CRF-BP, and additional ligands that interact differentially with them.
CRFR1-deficient mice exhibit impaired hormonal and behavioral responses to stress, whereas CRFR2 knock-outs are hyperresponsive on many of the same measures (Smith et al., 1998; Timpl et al., 1998; Bale et al., 2000; Coste et al., 2000; Kishimoto et al., 2000). Subsequent work indicates that CRFR interactions are not necessarily starkly differential but has generally supported convergent influences of the two receptor mechanisms on many stress-related endpoints (Bale and Vale, 2004). The circuitry that provides for their interaction the present context is unclear. CRFR1 mRNA is expressed prominently in the pyramidal layer of Ammon's Horn and the hilus of the dentate gyrus, whereas CRFR2 transcripts are weakly expressed throughout the principal cell layers of both structures (Van Pett et al., 2000). Unfortunately, the lack of validated antisera has precluded histochemical characterization of receptor protein disposition. Regarding ligands, extrinsic CRF-containing inputs to hippocampus have not been described, leaving local interneurons (Chen et al., 2004) and the CSF (Arborelius et al., 1999) as possible sources for delivering CRF to hippocampal CRFRs. Among the urocortins, only a sparse urocortin 1-immunoreactive input to hippocampus has been documented (Bittencourt and Sawchenko, 2000). The apparent paucity of CRFR2 ligands in hippocampal afferents suggests that their hypothesized interaction with CRFR1-dependent mechanisms may occur outside the hippocampus.
Effects of repeated stress
Acute stress-induced tau-P has been consistently characterized as a transient and reversible phenomenon. As such, it would seem well positioned to contribute to the rapid dendritic/synaptic remodeling seen in the hippocampus in response to stress (Fuchs et al., 2006), but its pathogenic relevance has remained uncertain (see Fig. 8). The present finding that repeated stress exposure results in sustained elevations in tau-P, a portion of which is sequestered in a detergent-soluble form, supports such a potential, because the bulk of dispersed PHFs from the AD brain reside in this same fraction (Iqbal et al., 1984; Rubenstein et al., 1986). It remains to be determined whether protracted stress exposure can lead to PHF/NFT pathology in animal models. We think it more likely that stress-induced alterations in tau-P and solubility may enhance the vulnerability of affected neurons to other experiential or genetic factors that play on tau dynamics. This view is coarsely analogous to the “endangerment” hypothesis to explain the role of glucocorticoids in the age-related impairment of hippocampal function (Sapolsky et al., 1985, 1986). Studies performed over a lifespan, or in aging, will be required to fully appreciate the contribution of stress exposure and/or sensitivity to tau-related neuropathologies and the capacity of CRFR signaling mechanisms to modulate them.
Footnotes
This work was supported by National Institutes of Health Grant DK026741, the Clayton Medical Research Foundation, and the Ellison Medical Foundation and American Federation for Aging Research (R.A.R.). K.-F.L., W.V., and P.E.S. are senior investigators of the Clayton Medical Research Foundation. We thank Drs. J. Serrats, D. Brown, Z. F. Yuan, and J. Radley for helpful discussions and K. Trulock and C. Arias for technical assistance. We also thank Dr. G. Chrousos for providing antalarmin, Dr. R. Valentino for antalarmin protocols, Dr. P. Davies for the PHF-1 antibody, and Dr. G. Drewes for permission to reproduce portions of his graphic incorporated in Figure 8.
References
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Arzt E, Holsboer F. CRF signaling: molecular specificity for drug targeting in the CNS. Trends Pharmacol Sci. 2006;27:531–538. doi: 10.1016/j.tips.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol. 2001;60:1190–1197. doi: 10.1093/jnen/60.12.1190. [DOI] [PubMed] [Google Scholar]
- Baharians Z, Schonthal AH. Autoregulation of protein phosphatase type 2A expression. J Biol Chem. 1998;273:19019–19024. doi: 10.1074/jbc.273.30.19019. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. Increased depression-like behaviors in corticotropin-releasing factor receptor-2-deficient mice: sexually dichotomous responses. J Neurosci. 2003;23:5295–5301. doi: 10.1523/JNEUROSCI.23-12-05295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- Baram TZ, Yi S, Avishai-Eliner S, Schultz L. Development neurobiology of the stress response: multilevel regulation of corticotropin-releasing hormone function. Ann NY Acad Sci. 1997;814:252–265. doi: 10.1111/j.1749-6632.1997.tb46161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayatti N, Behl C. The neuroprotective actions of corticotropin releasing hormone. Ageing Res Rev. 2005;4:258–270. doi: 10.1016/j.arr.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Behan DP, Heinrichs SC, Troncoso JC, Liu XJ, Kawas CH, Ling N, De Souza EB. Displacement of corticotropin releasing factor from its binding protein as a possible treatment for Alzheimer's disease. Nature. 1995;378:284–287. doi: 10.1038/378284a0. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Sawchenko PE. Do centrally administered neuropeptides access cognate receptors?: an analysis in the central corticotropin-releasing factor system. J Neurosci. 2000;20:1142–1156. doi: 10.1523/JNEUROSCI.20-03-01142.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- Campeau S, Falls WA, Cullinan WE, Helmreich DL, Davis M, Watson SJ. Elicitation and reduction of fear: behavioural and neuroendocrine indices and brain induction of the immediate-early gene c-fos. Neuroscience. 1997;78:1087–1104. doi: 10.1016/s0306-4522(96)00632-x. [DOI] [PubMed] [Google Scholar]
- Chadwick D, Marsh J, Ackrill K. Chichester, NY: Wiley; 1993. Corticotropin-releasing factor. [Google Scholar]
- Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JG, Mills P, Brockway B. Use of telemetry to record body temperature and activity in mice. J Pharmacol Methods. 1989;21:129–140. doi: 10.1016/0160-5402(89)90031-4. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste SC, Kesterson RA, Heldwein KA, Stevens SL, Heard AD, Hollis JH, Murray SE, Hill JK, Pantely GA, Hohimer AR, Hatton DC, Phillips TJ, Finn DA, Low MJ, Rittenberg MB, Stenzel P, Stenzel-Poore MP. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:403–409. doi: 10.1038/74255. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Marin DB, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Neuropeptide abnormalities in patients with early Alzheimer disease. Arch Gen Psychiatry. 1999;56:981–987. doi: 10.1001/archpsyc.56.11.981. [DOI] [PubMed] [Google Scholar]
- Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 2001;14:1143–1152. doi: 10.1046/j.0953-816x.2001.01733.x. [DOI] [PubMed] [Google Scholar]
- De Souza EB, Whitehouse PJ, Kuhar MJ, Price DL, Vale WW. Reciprocal changes in corticotropin-releasing factor (CRF)-like immunoreactivity and CRF receptors in cerebral cortex of Alzheimer's disease. Nature. 1986;319:593–595. doi: 10.1038/319593a0. [DOI] [PubMed] [Google Scholar]
- Drewes G. MARKing tau for tangles and toxicity. Trends Biochem Sci. 2004;29:548–555. doi: 10.1016/j.tibs.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Drewes G, Lichtenberg-Kraag B, Doring F, Mandelkow EM, Biernat J, Goris J, Doree M, Mandelkow E. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992;11:2131–2138. doi: 10.1002/j.1460-2075.1992.tb05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Knapp DJ, Breese GR. Neuroanatomical characterization of Fos induction in rat behavioral models of anxiety. Brain Res. 1996;713:79–91. doi: 10.1016/0006-8993(95)01486-1. [DOI] [PubMed] [Google Scholar]
- Feng Q, Cheng B, Yang R, Sun FY, Zhu CQ. Dynamic changes of phosphorylated tau in mouse hippocampus after cold water stress. Neurosci Lett. 2005;388:13–16. doi: 10.1016/j.neulet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Flugge G, Czeh B. Remodeling of neuronal networks by stress. Front Biosci. 2006;11:2746–2758. doi: 10.2741/2004. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Chrousos GP. Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrinol Metab. 2002;13:436–444. doi: 10.1016/s1043-2760(02)00670-7. [DOI] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer's disease. J Neurosci. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M, Mandelkow E. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992;307:199–205. doi: 10.1016/0014-5793(92)80767-b. [DOI] [PubMed] [Google Scholar]
- Hartig W, Stieler J, Boerema AS, Wolf J, Schmidt U, Weissfuss J, Bullmann T, Strijkstra AM, Arendt T. Hibernation model of tau phosphorylation in hamsters: selective vulnerability of cholinergic basal forebrain neurons—implications for Alzheimer's disease. Eur J Neurosci. 2007;25:69–80. doi: 10.1111/j.1460-9568.2006.05250.x. [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Xiong WC, Johnson GV. Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun. 2001;284:485–489. doi: 10.1006/bbrc.2001.4986. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VM. Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron. 2002;35:433–446. doi: 10.1016/s0896-6273(02)00789-4. [DOI] [PubMed] [Google Scholar]
- Hillhouse EW, Grammatopoulos DK. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev. 2006;27:260–286. doi: 10.1210/er.2005-0034. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Ishiguro K, Fujita SC. Ether stress-induced Alzheimer-like tau phosphorylation in the normal mouse brain. FEBS Lett. 2007;581:891–897. doi: 10.1016/j.febslet.2007.01.064. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Zaidi T, Thompson CH, Merz PA, Wisniewski HM. Alzheimer paired helical filaments: bulk isolation, solubility, and protein composition. Acta Neuropathol (Berl) 1984;62:167–177. doi: 10.1007/BF00691849. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso AC, Gong CX, Khatoon S, Singh TJ, Grundke-Iqbal I. Mechanism of neurofibrillary degeneration in Alzheimer's disease. Mol Neurobiol. 1994;9:119–123. doi: 10.1007/BF02816111. [DOI] [PubMed] [Google Scholar]
- Jeong YH, Park CH, Yoo J, Shin KY, Ahn SM, Kim HS, Lee SH, Emson PC, Suh YH. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer's disease model. FASEB J. 2006;20:729–731. doi: 10.1096/fj.05-4265fje. [DOI] [PubMed] [Google Scholar]
- Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer's disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Radulovic J, Radulovic M, Lin CR, Schrick C, Hooshmand F, Hermanson O, Rosenfeld MG, Spiess J. Deletion of crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:415–419. doi: 10.1038/74271. [DOI] [PubMed] [Google Scholar]
- Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- Korneyev A, Binder L, Bernardis J. Rapid reversible phosphorylation of rat brain tau proteins in response to cold water stress. Neurosci Lett. 1995;191:19–22. doi: 10.1016/0304-3940(95)11546-3. [DOI] [PubMed] [Google Scholar]
- Korneyev AY. Stress-induced tau phosphorylation in mouse strains with different brain Erk 1 + 2 immunoreactivity. Neurochem Res. 1998;23:1539–1543. doi: 10.1023/a:1020980004539. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Kuan CY, Wen ZD, Yang SD. The naturally occurring PKC inhibitor sphingosine and tumor promoter phorbol ester potentially induce tyrosine phosphorylation/activation of oncogenic proline-directed protein kinase FA/GSK-3alpha in a common signalling pathway. J Protein Chem. 1998;17:15–27. doi: 10.1023/a:1022582312954. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, Xu H, Wang JZ. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem. 2003;87:1333–1344. doi: 10.1046/j.1471-4159.2003.02070.x. [DOI] [PubMed] [Google Scholar]
- Martinez M, Calvo-Torrent A, Herbert J. Mapping brain response to social stress in rodents with c-fos expression: a review. Stress. 2002;5:3–13. doi: 10.1080/102538902900012369. [DOI] [PubMed] [Google Scholar]
- Meijer MK, Spruijt BM, van Zutphen LF, Baumans V. Effect of restraint and injection methods on heart rate and body temperature in mice. Lab Anim. 2006;40:382–391. doi: 10.1258/002367706778476370. [DOI] [PubMed] [Google Scholar]
- Okawa Y, Ishiguro K, Fujita SC. Stress-induced hyperphosphorylation of tau in the mouse brain. FEBS Lett. 2003;535:183–189. doi: 10.1016/s0014-5793(02)03883-8. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC. Heat shock induces rapid dephosphorylation of tau in both female and male rats followed by hyperphosphorylation only in female rats: implications for Alzheimer's disease. J Neurochem. 1996;66:1140–1149. doi: 10.1046/j.1471-4159.1996.66031140.x. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Wan R, Zhang P, Mattson MP. Urocortin, but not urocortin II, protects cultured hippocampal neurons from oxidative and excitotoxic cell death via corticotropin-releasing hormone receptor type I. J Neurosci. 2002;22:404–412. doi: 10.1523/JNEUROSCI.22-02-00404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernar L, Curtis AL, Vale WW, Rivier JE, Valentino RJ. Selective activation of corticotropin-releasing factor-2 receptors on neurochemically identified neurons in the rat dorsal raphe nucleus reveals dual actions. J Neurosci. 2004;24:1305–1311. doi: 10.1523/JNEUROSCI.2885-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer's disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Powers RE, Walker LC, DeSouza EB, Vale WW, Struble RG, Whitehouse PJ, Price DL. Immunohistochemical study of neurons containing corticotropin-releasing factor in Alzheimer's disease. Synapse. 1987;1:405–410. doi: 10.1002/syn.890010504. [DOI] [PubMed] [Google Scholar]
- Raadsheer FC, van Heerikhuize JJ, Lucassen PJ, Hoogendijk WJ, Tilders FJ, Swaab DF. Corticotropin-releasing hormone mRNA levels in the paraventricular nucleus of patients with Alzheimer's disease and depression. Am J Psychiatry. 1995;152:1372–1376. doi: 10.1176/ajp.152.9.1372. [DOI] [PubMed] [Google Scholar]
- Rehman HU. Role of CRH in the pathogenesis of dementia of Alzheimer's type and other dementias. Curr Opin Investig Drugs. 2002;3:1637–1642. [PubMed] [Google Scholar]
- Rubenstein R, Kascsak RJ, Merz PA, Wisniewski HM, Carp RI, Iqbal K. Paired helical filaments associated with Alzheimer disease are readily soluble structures. Brain Res. 1986;372:80–88. doi: 10.1016/0006-8993(86)91460-5. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. Prolonged glucocorticoid exposure reduces hippocampal neuron number: implications for aging. J Neurosci. 1985;5:1222–1227. doi: 10.1523/JNEUROSCI.05-05-01222.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Brown ER, Chan RK, Ericsson A, Li HY, Roland BL, Kovacs KJ. The paraventricular nucleus of the hypothalamus and the functional neuroanatomy of visceromotor responses to stress. Prog Brain Res. 1996;107:201–222. doi: 10.1016/s0079-6123(08)61866-x. [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Li HY, Ericsson A. Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog Brain Res. 2000;122:61–78. doi: 10.1016/s0079-6123(08)62131-7. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357:299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- Shu SY, Penny GR, Peterson GM. The ‘marginal division': a new subdivision in the neostriatum of the rat. J Chem Neuroanat. 1988;1:147–163. [PubMed] [Google Scholar]
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev. 2005;4:141–194. doi: 10.1016/j.arr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- Turek VF, Ryabinin AE. Expression of c-Fos in the mouse Edinger-Westphal nucleus following ethanol administration is not secondary to hypothermia or stress. Brain Res. 2005;1063:132–139. doi: 10.1016/j.brainres.2005.09.056. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Watts AG. The impact of physiological stimuli on the expression of corticotropin-releasing hormone (CRH) and other neuropeptide genes. Front Neuroendocrinol. 1996;17:281–326. doi: 10.1006/frne.1996.0008. [DOI] [PubMed] [Google Scholar]
- Webster EL, Lewis DB, Torpy DJ, Zachman EK, Rice KC, Chrousos GP. In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology. 1996;137:5747–5750. doi: 10.1210/endo.137.12.8940412. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer's disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Planel E, Ishiguro K, Fujita SC. Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer's disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Maeda M, Kaku S, Ikeya H, Yamada K, Nakaike S. Lithium inhibits stress-induced changes in tau phosphorylation in the mouse hippocampus. J Neural Transm. 2006;113:1803–1814. doi: 10.1007/s00702-006-0528-0. [DOI] [PubMed] [Google Scholar]