

Abstract
N-type calcium channels are essential mediators of spinal nociceptive transmission. The core subunit of the N-type channel is encoded by a single gene, and multiple N-type channel isoforms can be generated by alternate splicing. In particular, cell-specific inclusion of an alternatively spliced exon 37a generates a novel form of the N-type channel that is highly enriched in nociceptive neurons and, as we show here, downregulated in a neuropathic pain model. Splice isoform-specific small interfering RNA silencing in vivo reveals that channels containing exon 37a are specifically required for mediating basal thermal nociception and for developing thermal and mechanical hyperalgesia during inflammatory and neuropathic pain. In contrast, both N-type channel isoforms (e37a- and e37b-containing) contribute to tactile neuropathic allodynia. Hence, exon 37a acts as a molecular switch that tailors the channels toward specific roles in pain.
Keywords: pain, calcium channels, N-type, splice isoforms, siRNA, dorsal root ganglion
Introduction
Detection and transmission of nociceptive stimuli in dorsal root ganglion (DRG) neurons is mediated by several types of ion channels, such as persistent Na+ channels, inwardly rectifying K+ channels, and voltage-gated Ca2+ channels, which regulate either cellular excitability or synaptic transmission (McCleskey and Gold, 1999; Woolf and Salter, 2000; Julius and Basbaum, 2001; Scholz and Woolf, 2002). N-type (CaV2.2) calcium channels are key mediators of nociceptive signaling (Altier and Zamponi, 2004). In the dorsal horn of the spinal cord, these channels control the release of glutamate and neuropeptides such as substance P (Smith et al., 2002), thereby supporting pain transmission via afferent Aβ, Aδ, and C fibers to neurons projecting to the thalamus (Krarup, 2003). Consequently, inhibition of N-type channels via activation of opioid receptors or by N-type channel antagonists mediates analgesia in animals and humans (Hu et al., 1999; Scott et al., 2002; Seko et al., 2002; Staats et al., 2004). Moreover, CaV2.2 channel knock-out mice have decreased pain responses in models of neuropathic and inflammatory pain (Hatakeyama et al., 2001; Kim et al., 2001; Saegusa et al., 2001). N-type channels are now a validated pharmacological target for treating pain (Snutch, 2005).
CaV2.2 RNA undergoes alternative splicing, producing multiple CaV2.2 channel isoforms with distinct electrophysiological properties, subcellular distributions, and likely distinct physiological roles (Lin et al., 1997, 1999; Stea et al., 1999; Pan and Lipscombe, 2000; Kaneko et al., 2002; Maximov and Bezprozvanny, 2002; Bell et al., 2004; Lin et al., 2004; Thaler et al., 2004). One such splicing event occurs through the alternate use of exons e37a and e37b to produce two isoforms with proximal C termini that differ by 14 amino acids (Bell et al., 2004). As a consequence, e37a and e37b isoforms of CaV2.2 have distinct electrophysiological properties, different current densities, and different G-protein sensitivities (Castiglioni et al., 2006; Raingo et al., 2007). Most intriguingly, small, capsaicin receptor-expressing DRG neurons, which participate in the transmission of nociceptive responses to heat stimuli (Tominaga and Caterina, 2004), preferentially express the e37a isoform (Bell et al., 2004). Are the two CaV2.2 splice isoforms equally effective in supporting nociceptive signaling or does one isoform have a preferred role in transmission of thermal or mechanical nociception (Bell et al., 2004; Zamponi and McCleskey, 2004)? To address this question, we used a specific small interfering RNA (siRNA) silencing approach to mediate a selective and localized knockdown of each e37 isoform in DRG neurons in culture and in vivo. We show that knockdown of the e37a- and e37b-containing channels reduces CaV2.2 protein expression and blocks substance P release from cultured DRG neurons, with depletion of the CaV2.2e[37a] isoform preferentially affecting substance P release from capsaicin-sensitive cells. Knockdown of one or the other isoform in vivo differentially affects basal nociception, inflammatory pain, and neuropathic pain, revealing a unique role of e37a-containing CaV2.2 channels in pain pathways. Hence, alternative splicing is a key mechanism that tailors CaV2.2 channels to support specific roles in the processing of pain signals.
Materials and Methods
Animals
Sprague Dawley rats (175–200 g) were purchased from Charles River Laboratories (Montreal, Quebec, Canada) and were housed under constant temperatures and 12 h light/dark cycles. Rats had access to food and water ad libitum. All experiments were approved by the Animal Care Committee of the University of Calgary and were in accordance with the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain (Zimmerman, 1983).
Preparation of siRNAs
6-FAM (6-carboxyfluorescein) siRNA constructs were synthesized at the University of Calgary DNA synthesis facility. siRNAs were dissolved in DEPC water at a stock concentration of 6 μg/μl. For e37a, sense and antisense strands were as follows: siRNA[a] beginning at nucleotide 5285, 6-FAM, 5′-AGAACUGCCCUCGUAGGUUGG-3′ (sense) and 5′-AACCUACGAGGGCAGUUCUUC-3′ (antisense). For e37b, sense and antisense strands were as follows: siRNA[b] beginning at nucleotide 5279, 6-FAM, 5′-GAAAUGCCCGGCUCGAGUUd(TT)-3′ (sense) and 5′-AACUCGAGCCGGGCAUUUCd(TT) (antisense). Control siRNA with approximately the same percentage of GC but with no corresponding sequence was designed as follows: 6-FAM, 5′-CAUGCUAGGUUAGUACUUGd(TT)-3′ (sense); 5′-CAAGUACUAACCUAGCAUGd(TT)-3′ (antisense).
Ideally, one would like to use multiple siRNA constructs, targeted to different channel regions, for knockdown experiments. However, the need for exon-specific knockdown together with exon 37 being short (32 amino acids) and e37a and e37b differing in only 14 amino acid positions precluded the use of multiple siRNA molecules. BLAST (basic local alignment search tool) nucleotide searches based on the siRNA sequences yielded no significant homology with any other known rat sequences.
Electrophysiology on tsA-201 cells
tsA-201 cells plated at high density in 10 cm dishes were transfected with CaV2.2e[37a] or CaV2.2e[37b] (+ β1b + α2–δ1 + eGFP, in which eGFP is enhanced green fluorescent protein) as described in our previous work (Beedle et al., 2004). On the next day, dishes were transfected with 6 μg of siRNA using i-Fect (Neuromics, Minneapolis, MN). Cells were split 24 h after siRNA transfection and subjected to whole-cell patch-clamp analysis on the next day. Electrophysiological recordings were conducted with 20 mm barium as the charge carrier as described in detail in our previous work (Beedle et al.,2004; Altier et al., 2006).
Culturing of DRG neurons
For high-density DRG cultures, 1- to 4-d-old Sprague Dawley rats were killed, and the DRG neurons were removed from all spinal levels and dissociated enzymatically and mechanically. Neurons were plated at high density onto laminin–polyornithine-coated coverslips and transfected on the next day. Three siRNA treatments over 2 d were applied to mimic in vivo experiments. First, siRNAs (3 μg per two coverslips) were transfected using i-Fect. During the second and third treatments, siRNA was simply diluted into the culture medium to reduce transfection toxicity. DRG neurons were used on day 3 after siRNA treatment for the substance P release assay (see below). siRNA localization in DRG cultured neuron was examined after completion of the substance P ELISA. For this, DRG neurons were fixed with 4% paraformaldehyde after KCl depolarization. Coverslips were then mounted for fluorescence imaging on a Zeiss (Oberkochen, Germany) LSM510 microscope.
Substance P release assay
The medium of cultured DRG neurons was removed and immediately frozen for analysis. DRG neurons were then incubated in NaCl-based extracellular solution [in mm: 120 NaCl, 3 CaCl2, 5 KCl, 2 MgCl2, 10 d-glucose, and 10 mm HEPES, pH 7.3 with NaOH, and protease inhibitor cocktail tablet (Roche, Indianapolis, IN)] for 20 min. Then, cells were challenged with 50 mm KCl (containing protease inhibitor) for 3 min. Every step was performed at 37°C in the incubator. Medium and KCl fractions were vacuum dried and used for the substance P ELISA (Assay Designs, Ann Arbor, MI) according to the instructions of the manufacturer. Samples were read with a microplate reader at a wavelength of 405 nm. For capsaicin experiments, DRG neurons were challenged with 10 μm capsaicin at days 2 and 3 before proceeding with the above substance P assay.
Immunoblotting
After the substance P assay, DRG neurons were lysed on ice for 1 h [300 mm NaCl, 50 mm Tris-HCl, pH 7.5, 1% Triton X-100, and protease inhibitor cocktail tablet (Roche)]. Collected supernatant was protein quantified using the DC Protein Assay kit (Bio-Rad, Hercules, CA) according to the instructions of the manufacturer, mixed with 2× SDS loading buffer, and boiled for 5 min. Fifteen micrograms of protein per lane were loaded onto a 6% SDS-PAGE gel; the same amount of sample was loaded in parallel onto a 10% SDS-PAGE gel for an actin control. After transfer, polyvinylidene difluoride (PVDF) membranes were probed with N-type antibody (1:200; Alomone Labs, Jerusalem, Israel) or actin antibody (1:1000; Sigma, St. Louis, MO), respectively. Membranes were subsequently washed and incubated for 1 h with secondary antibody, anti-rabbit Ig, horseradish peroxidase (1:2000; GE Healthcare, Little Chalfont, UK). Membranes were washed three times, and Western blot analysis was performed using ECL detection methods. Band intensity (normalized to actin control) was quantified using AlphaDigiDoc software (Alpha Innotech, San Leandro, CA).
RNase protection assay
Our RNase protection assay (RPA) used the RPA III and the Bright Star Biodetect protocols (Ambion, Austin, TX) essentially as described by the manufacturer. We generated templates for RNA probes by reverse transcription-PCR from DRG mRNA and 3′-deoxyadenosine, 3′-deoxythymidine overhang cloning strategy cloned them into the pCRII vector (Invitrogen, Carlsbad, CA) for in vitro transcription using the Maxiscript kit (Ambion) and biotin-UTP (Roche). The fully protected and partially protected probe sizes are as follows: CaV2.2e[37a] (520 and 429 bp); CaV2.2e[37b] (520 and 429 bp); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (229 bp and fully protected). We performed control hybridizations, containing a fixed amount of probe and increasing amounts of total RNA to ensure that all probes were present in excess compared with their mRNA targets. Nuclease digestion products were separated on 5% acrylamide gels. Following the Biodetect procedure, we collected a time series of exposures to Hyperfilm (GE Healthcare). We scanned the films as 8-bit, 96-dpi, TIFF images, quantified band volumes with ImageQuant software (GE Healthcare), and determined background in each lane, for each band of interest. Hybridizations for CaV2.2e[37b] used GAPDH as an internal control. We normalized CaV2.2e[37b] signals to the GAPDH signal from the same hybridization. However, we report the amount of CaV2.2e[37a] mRNA differently than CaV2.2e[37b] mRNA for the following reasons: CaV2.2e[37a] mRNA is present at ∼10-fold lower levels than CaV2.2e[37b] mRNA, which necessitated using greater amounts of total DRG RNA (∼10 μg) to obtain measurable CaV2.2e[37a] signal. When using this amount of total RNA, we determined that GAPDH mRNA levels were too high to ensure reliably that the GAPDH probe would be in molar excess. Therefore, we calculated percentage of CaV2.2e[37a] mRNA in total CaV2.2 mRNA. This is equivalent to the volume of e37a fully protected probe divided by the volume sum of e37a fully protected probe plus e37a partially protected probe. We obtained these band volumes at different exposure time points because the partially protected probe saturated the linear range of the film at the time point necessary to quantify the fully protected probe. To determine the volume of the partially protected probe, we created a standard curve of volumes obtained from a series of exposures in the linear range of the film and extrapolated the value for partially protected probe volume at the time point used to measure fully protected probe.
In vivo injection of siRNA
Rats were anesthetized with halothane (5%) and received three percutaneous intrathecal injections (L5–L6) of 6-FAM–siRNA (60 μg/rat in 10 μl final volume of DEPC water) over 2 d. To localize siRNA in DRG tissues, rats were anesthetized with sodium pentobarbital (200 mg/kg, i.p.) and transcardially perfused with 4% paraformaldehyde in 100 mm PBS, pH 7.4. DRG (T10–L6) were removed and fixed overnight at 4°C, placed in 25% sucrose in PBS for 24 h at 4°C, embedded in OCT compound (Miles, Elkhart, IN), and sectioned at 25 μm. Confocal images were obtained from fixed DRG slices mounted for visualization. Images were acquired using a Zeiss LSM-510 META confocal microscope using a 20× objective in the inverted configuration. For all confocal images, a regular phase transmission image was obtained. 6-FAM–siRNA was observed using argon laser excitation (488 nm) and emission (505–530 nm) filter sets.
Formalin injection
Two days after intrathecal injection of siRNA, animals received an intraplantar injection of a 5% Formalin solution, diluted in saline (50 μl/paw) under light halothane anesthesia. The evaluation of nociception was performed at various time points after Formalin injection. Number of flinches and duration of licking behaviors were recorded for the first 5 min after intraplantar Formalin injection and then from 15 to 30 min. Nociceptive responses to von Frey filament exposure (see below, Mechanical nociception) and thermal stimulation (see below, Thermal nociception) were measured between 30 min and 4 h after intraplantar Formalin injection. We note that, at early time points (30 and 60 min), analysis may be complicated by spontaneous flinching behavior that occurs in response to the Formalin insult. At time points of 2 h and beyond, spontaneous nociceptive behaviors are not typically observed and thus do not affect the nociceptive scores or paw-withdrawal latencies.
Chronic constriction injury of rat sciatic nerves
Chronic constriction injury (CCI) was performed as described by Bennett and Xie (1988). Rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p.; supplemented as necessary). The common sciatic nerve was exposed at the level of the middle of the thigh by blunt dissection through biceps femoris, proximal to the trifurcation of the sciatic nerve. Seven millimeters of nerve were freed of adhering tissue, and four ligatures (4.0 chromic gut) were tied loosely around the nerve at intervals of ∼1 mm. Pain scores were evaluated on days 3 and 7 after surgery. The siRNA injection was performed on days 8 and 9 after surgery, and the animals were evaluated between days 10 and 14 after surgery.
Spinal nerve ligation
Surgery and behavioral testing were performed as described previously (Matthews and Dickenson, 2001). Male rats (Sprague Dawley; Central Biological Services, University College London, London, UK) initially weighing 130–150 g were used in this study. All experimental procedures were approved by the Home Office and follow the guidelines under the International Association for the Study of Pain (Zimmermann, 1983). The Kim and Chung model of neuropathy was used as first described (Kim and Chung, 1992). Briefly, selective tight ligation of spinal nerves L5 and L6 was performed with 6-0 silk thread under halothane anesthesia (66% N2O and 33% O2), and a sham procedure in which L5 and L6 nerves were isolated but not ligated was performed. Hemostasis was confirmed, the wound was sutured, and the rats were allowed to recover. After confirmation of mechanical allodynia at day 14 after spinal nerve ligation (SNL), we collected dorsal root ganglia L4, L5, and L6 from sides ipsilateral and contralateral to the ligation. We combined ganglia that originated from the same location and prepared total RNA for RNase protection analysis. Of 21 SNL animals, four did not develop mechanical hypersensitivity. We only combined and analyzed ganglia from those 17 animals with a clear behavioral phenotype. We performed sham surgeries on eight additional animals. The RNA analyzed from each ganglion level represents a pool of 17 animals for SNL surgeries and eight animals for sham surgeries.
Nociception measures
Thermal hyperalgesia.
Paw-withdrawal latency to radiant heat stimuli was measured using a Ugo Basile (Comerio, Italy) plantar test essentially as described by Hargreaves et al. (1988). Thermal hyperalgesia was defined as a significant decrease in the withdrawal latency compared with the basal measurement at different times after treatment.
Mechanical nociception.
Allodynia of the hindpaw was assessed as follows. Rats were placed individually into a plastic cage. von Frey filaments with bending forces of 4, 15, and 60 g (4.93, 5.18, and 5.88, respectively) were pressed perpendicularly against the plantar skin and held for 5 s. The stimulation of the same intensity was applied three times to each hindpaw at intervals of several seconds. The responses to these stimuli were ranked as follows: 0, no response; 1, move away from von Frey filament; and 2, immediate flinching or licking of the hindpaw. The nociceptive score was calculated as follows:
 |
The nociceptive score was measured before and after the intraplantar injection of Formalin (5%, 50 μl) and before and after CCI or SNL. Nociceptive tests were performed according to the guidelines published in a Guest Editorial in Pain on ethical standards for investigations of experimental pain in animals (Zimmermann, 1983).
Allodynia for SNL animals.
For 2 weeks after surgery, the rats were housed in groups of four in plastic cages under a 12 h day/night cycle, and their general health was monitored. Successful reproduction of the neuropathic model was confirmed by behavioral testing at postoperative days 2, 6, and 14 assessing the sensitivity of both the ipsilateral and contralateral hindpaws in SNL and sham-operated rats to a series of calibrated normally non-noxious punctate mechanical von Frey filaments (1, 5, and 9 g). Rats were placed in transparent plastic cubicles on a mesh floor and allowed to acclimatize before initiating any tests. Starting with the lowest intensity, the von Frey filament was applied to the plantar surface of the foot 10 times, for no more than 3 s per application, and the number of withdrawals was quantified. This was performed on the other hindpaw and then repeated with von Frey stimuli of increasing intensity. The number of responses per animal to each von Frey filament was quantified.
Statistical analysis
All error bars denote SEs. Statistical analysis was conducted via one-way ANOVA, followed by Tukey's and/or Bonferroni's multiple comparison tests.
Results
Electrophysiological evidence of specific siRNA silencing
The C-terminal regions of CaV2.2e[37a] and CaV2.2e[37b] channels differ by 14 amino acids (Fig. 1A). To explore the role of the two e37 isoforms in the pain pathway, we generated fluorescein-conjugated siRNA molecules targeted against exons 37a and 37b to allow for splice isoform-specific knockdown. To confirm the specificity of these reagents, tsA-201 cells were transfected with either CaV2.2e[37a] or CaV2.2e[37b] (along with ancillary α2–δ1 and β1b subunits), followed by siRNA transfection 24 h later. N-type current densities were then compared via whole-cell patch-clamp recordings in control, siRNA[a], and siRNA[b] treated cells. Treatment with siRNA[b] resulted in a near complete elimination of CaV2.2e[37b] currents (Fig. 1B), whereas currents mediated by the CaV2.2e[37a] channels were not affected (Fig. 1C). Conversely, treatment of cells with siRNA[a] failed to alter CaV2.2e[37b] current densities (Fig. 1B), whereas a significant reduction in CaV2.2e[37a] currents was observed (Fig. 1C). The siRNA-mediated reduction in current densities was accompanied by a twofold to fourfold increase in the percentage of cells that did not yield detectable CaV2.2 currents when treated with the siRNA constructs directed against the expressed splice isoform (Fig. 1B,C). Because we used a transient expression system, in which expression of transfected cDNA varies intrinsically over time, we did not examine the time course of recovery from the siRNA knockdown. Nonetheless, these data collectively indicate that the siRNA constructs effectively reduce CaV2.2 currents and do so in a splice isoform-specific manner.
Figure 1.

Electrophysiological evidence of the specificity of siRNA silencing. A, Sequence alignment between exons 37a and 37b of the rat CaV2.2 calcium channel α1 subunit. B, Whole-cell recordings of peak current densities of tsA-201 cells transfected with the CaV2.2e[37b] (+ β1b and α2–δ) isoform and treated with different types of siRNA (left). Numbers in parentheses are numbers of cells patched, and the asterisks denote statistical significance between untreated, siRNA[a]-treated, and siRNA-treated cells. Right, Portion of cells with or without current for different siRNA treatments. Middle, Current–voltage relationships of CaV2.2e[37b]-transfected cells represented in the left and treated with siRNA[a] or siRNA [b]. C, Same as in B but for CaV2.2e[37a] isoform (+β1b and α2–δ). Note that, in this case, only siRNA[a] decreased CaV2.2e[37a] peak current density and amplitude of currents. The solid lines in the current–voltage relationships were obtained via the Boltzmann equation, and error bars denote SEs.
Role of CaV2.2 splice isoforms in substance P release from cultured DRG neurons
To assess the role of CaV2.2 splice isoforms on neuropeptide release, cultured DRG neurons were subjected to multiple transfections with different types of siRNA constructs before tonic and evoked substance P release was assayed. Measurement of substance P secreted into the culture medium over a 48 h period revealed that there was no significant difference between the various treatments (Fig. 2A, left), indicating that, in culture, without any stimulation, N-type channels do not significantly contribute to tonic substance P release. It is possible that, at this stage of the culture, the synaptic connections are not mature enough to observe autonomous synaptic transmission and neuropeptide release. Moreover, the cumulative measure conducted over a 48 h period may not reflect the contribution of N-type channels to neuropeptide release. However, when cells were depolarized with 50 mm KCl (for 3 min) to induce substance P release, we confirmed substantial levels of substance P in the culture medium for the mismatch control condition. In KCl-stimulated neurons treated with siRNA[a] or siRNA[b], the amount of substance P released into the medium was significantly reduced (Fig. 2A, right). Surprisingly, no additive blockade was observed when cells were treated with both siRNA molecules. This might perhaps reflect the complex dependence of evoked substance P release on the free cytosolic calcium and/or limitations in the sensitivity of the ELISA. Treatment of DRG neurons with the N-type channel inhibitor ω-conotoxin GVIA completely abolished KCl-evoked substance P release (Fig. 2A). Overall, our data show that, in culture, both channel isoforms appear to contribute similarly to the release of the neuropeptide substance P, with acute block of N-type channels being only marginally more effective than concomitant siRNA knockdown of both splice isoforms.
Figure 2.
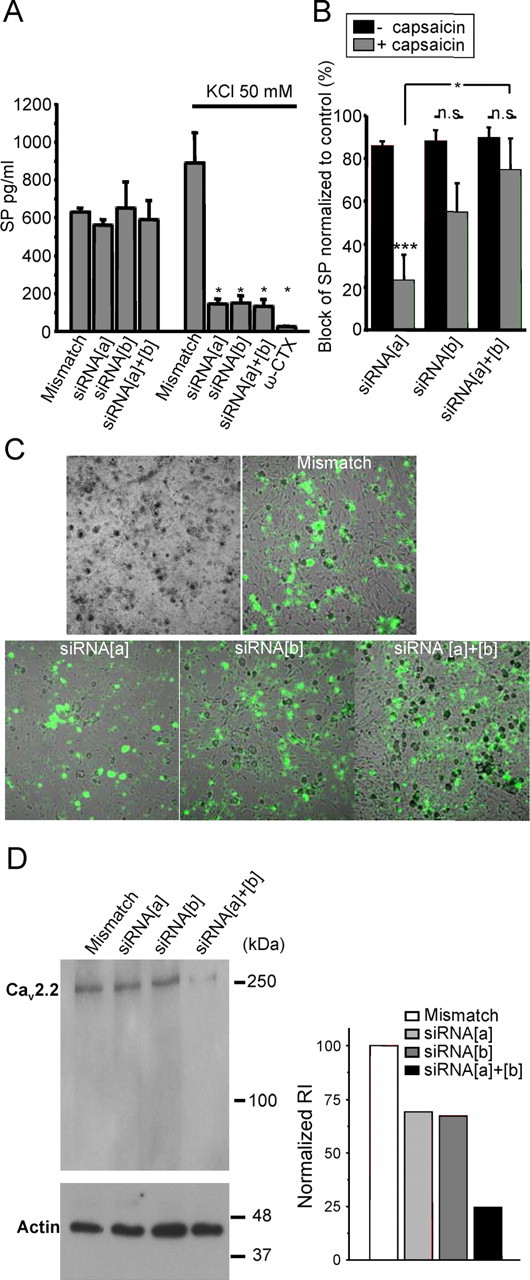
Effects of siRNA treatment on substance P release and CaV2.2 protein expression in cultured DRG neurons. A, Quantification of substance P release by ELISA. DRG neurons were plated at the same density and treated with 6-FAM labeled siRNA constructs (i.e., mismatch, siRNA[a], or siRNA[b]). The amount of substance P secreted by the DRG culture was measured by sampling either the culture medium (reflecting tonic release) or immediately after replacement with medium and subsequent depolarization with 50 mm KCl (reflecting evoked release). ω-Conotoxin GVIA at 1 μm was applied to the cultures to block N-type channels as indicated. In this case, the KCl solution was also supplemented with toxin. B, Inhibition of substance P release normalized to control with or without previous treatment of the neurons with (10 μm) capsaicin to deplete capsaicin-sensitive nociceptors of their neuropeptide contents. *p < 0.05, among the capsaicin-treated groups, significant difference between siRNA[a]- and siRNA[a + b]-injected animals. ***p < 0.01, significant difference among siRNA[a] groups, between capsaicin-treated and untreated DRG. C, Uptake of 6-FAM-tagged siRNA into dorsal root ganglia neurons 2 d after siRNA transfection. D, Western blot analysis of CaV2.2 channel protein in lysate of the DRG neurons used in A. Protein (15 μg) was loaded in every lane. The Western blot was probed with an anti- CaV2.2 calcium channel antibody. As a control, an actin antibody was used on a second gel to verify the amount of protein. The bar chart (right) is a quantification of relative integrated density value from each band. Data from three experiments are included in the bar chart; no statistical analysis was conducted. n.s., Not significant.
Previous findings indicated that ∼55% of neurons that respond to capsaicin express e37a, whereas this isoform is expressed in only ∼20% of the capsaicin-insensitive neurons (Bell et al., 2004). To ascertain to what extent the two CaV2.2 isoforms contributed to the release of substance P from capsaicin-sensitive and -insensitive neurons, substance P release was assayed in cultured neurons exposed previously to capsaicin. Under those conditions, only capsaicin-insensitive neurons can release substance P because capsaicin-sensitive neurons are depleted of their neuropeptide content by capsaicin preexposure. As shown in Figure 2B, after capsaicin treatment (gray bars), the ability of the siRNA[a] construct to inhibit substance P release was significantly reduced compared with untreated cultures (black bars) (from 86 to 23% inhibition; p = 0.006). In contrast, capsaicin treatment affected the ability of siRNA[b] to reduce substance P release to a much smaller extent and had virtually no effect on the inhibition mediated by siRNA[a + b]. As outlined above, it may be difficult to precisely correlate changes in the amount of substance P release with the degree of calcium channel inhibition caused by siRNA treatment. Nonetheless, the reduced efficacy of siRNA[a] at inhibiting substance P release suggests that substance P release from capsaicin-insensitive neurons may be mediated predominantly by CaV2.2e[37b] channels, whereas the CaV2.2e[37a] channel isoform contributes more extensively to substance P release from capsaicin receptor-positive neurons. This finding is consistent with the above mentioned preferential expression of the e37a isoform in these neurons.
DRG neurons used for the substance P assay were fixed and visualized with fluorescence microscopy to establish the transfection efficiency of the siRNA constructs (Fig. 2C). Approximately 90% of the DRG neurons presented with green fluorescence, indicating a high efficiency of siRNA uptake into the cultures, consistent with the large block of evoked substance P release (Fig. 2A). Cells from parallel wells were lysed and used for biochemical analysis via SDS-PAGE and Western blot analysis (Fig. 2D). Quantification of band intensity relative to the actin control reveals that either siRNA[a] or siRNA[b] decreases N-type channel protein expression, with both constructs showing >75% reduction in total N-type channel protein (Fig. 2D, right). Together, our data indicate efficient knockdown of N-type channel protein in neurons and a differential role of e37a and e37b channels in substance P release from capsaicin-sensitive and -insensitive neurons.
In vivo silencing of CaV2.2e[37a] reduces basal thermal and mechanical nociception
To elucidate a putative role of exon 37 splicing in vivo, we intrathecally injected rats with siRNA (L5–L6 level). Proper uptake of the siRNA was verified in lumbar DRG slices obtained 2 d after siRNA injection. As shown in Figure 3A, DRG slices were characterized by pronounced fluorescein epifluorescence, indicating effective uptake of both siRNA[a] and siRNA[b] (Fig. 3A). We verified the efficiency of siRNA knockdown in vivo by conducting Western blot analysis on DRG lysate isolated from siRNA-treated animals (Fig. 3B, left). Quantification of band intensity relative to the actin control (Fig. 3B, right) shows that reduction of protein expression with siRNA[a] is slightly greater than the reduction observed in cultured DRG neurons, but overall there is a remarkable consistency between in vitro and in vivo knockdown, with the combination of siRNA[a + b] reducing N-type channel protein expression by ∼80%. Immunostaining on DRG slices after siRNA treatment from different animals revealed a similar decrease in N-type channel expression after siRNA treatment (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 3.

Effects of CaV2.2 splice isoform-specific siRNA silencing on acute nociception. A, Uptake and distribution of 6-FAM-tagged siRNA into dorsal root ganglia after siRNA injection. Three days after 6-FAM–siRNA intrathecal injection, DRG were harvested, and fluorescence of siRNA[a] and siRNA[b] was visualized in DRG slices. Scale bars, 0.2 mm. B, Western blot analysis of CaV2.2 channel protein from DRG lysate (L4–L6) obtained from rats treated with siRNA. The Western blot was probed with an anti-CaV2.2 calcium channel antibody. An antibody against actin was used on a second gel to verify the amount of protein. The bar chart (right) is a quantification of the relative integrated density value from each band. Collected DRG lysate samples were run blind. Data from three experiments are included in the bar chart; no statistical analysis was conducted. C–F, Kinetics of basal nociceptive responses to mechanical stimuli (von Frey filament 4.93, 4 g bending force; von Frey filament 5.18, 15 g bending force; and filament 5.88, 60 g bending force) as a function of time after siRNA treatment (C–E) or thermal stimulus (F) after intrathecal injection of siRNA. Data are expressed as mean ± SE; data are from seven rats per group. The asterisks and number symbols indicate significant difference from basal conditions (p < 0.05). IB, Immunoblot.
As shown in Figure 3C–F, a differential role of the two channel isoforms in nociceptive signaling was indeed observed. The smallest von Frey filament (filament 4.93) used did not evoke changes in nociceptive responses between groups that received different siRNA treatment (Fig. 3C). The intermediate von Frey filament (filament 5.18) resulted in a greater nociceptive score, which was selectively reduced at days 2–4 by siRNA[a] treatment (Fig. 3D). Similar results (i.e., an siRNA[a]-mediated reduction at days 2–4) were observed with other filaments of intermediate size (5.07 and 5.46) (data not shown). The use of a larger filament (that is expected to mediate a strong noxious mechanical stimulus) resulted in a substantial increase in nociceptive score that, interestingly, was insensitive to siRNA treatment altogether (Fig. 3E). These data indicate that the exon 37a isoform mediates the transmission of mild mechanical stimuli, whereas the 37b isoform did not appear to play a significant role in any of the mechanically evoked basal nociceptive responses. In response to a thermal stimulus (i.e., radiant heat), a significant increase in paw-withdrawal latency was observed in rats injected with siRNA[a] but not siRNA[b] or mismatch. The anti-nociceptive effects of intrathecal siRNA[a] treatment appeared 2 d after intrathecal injection and lasted until day 5 (Fig. 3F). These data show that only e37a is responsible for mediating basal thermal nociception.
It has been shown that pharmacological blockade or gene knock-out of N-type calcium channels does not affect acute pain processing in tail-flick assays (Saegusa et al., 2001; Scott et al., 2002). To confirm the specificity of our siRNA constructs, we conducted tail-flick assays on animals injected with siRNA[a], siRNA[b], or siRNA[a + b]. In agreement with previous work, neither treatment affected tail-flick latencies (data not shown), suggesting that N-type channels do not contribute to acute pain signaling (Saegusa et al., 2001; Scott et al., 2002).
In vivo silencing of CaV2.2e[37a] prevents thermal and mechanical inflammatory hyperalgesia
Injection of Formalin into the rat paw induces inflammatory allodynia and hyperalgesia in response to mechanical or thermal stimuli. Allodynia is characterized by the appearance of a nociceptive response to an innocuous stimulus such as a small-sized von Frey filament (filament 4.93). This is reflected by an increase in nociceptive score 30 min after Formalin injection (Fig. 4A, vehicle). Hyperalgesia is characterized by an increased nociceptive score in response to larger noxious filaments (5.18 and 5.88) compared with basal measurements (Fig. 4B,C, vehicle) and by a decreased withdrawal latency after exposure to a thermal stimulus (Fig. 4D, vehicle).
Figure 4.
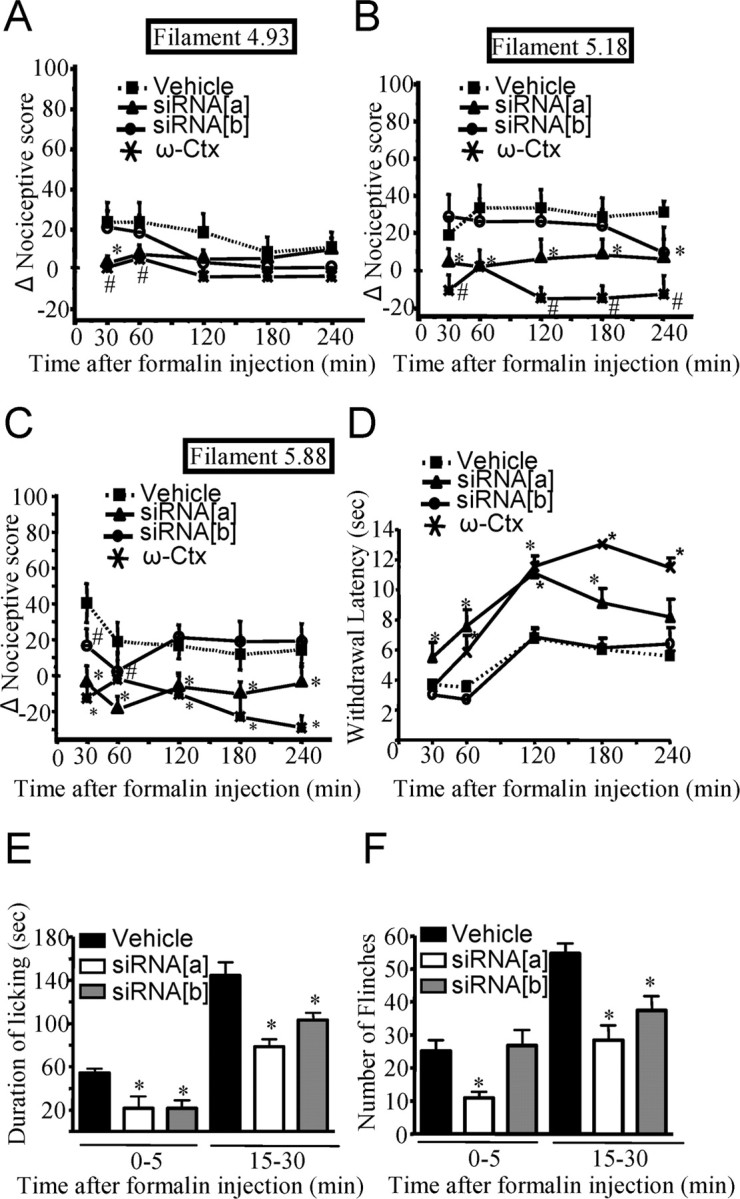
Effect of CaV2.2 splice isoform-specific siRNA silencing on inflammatory allodynia and hyperalgesia induced by plantar injection of Formalin. A–C, Change in nociceptive scores compared with basal measurements in response to a mechanical innocuous stimulus (4.93 von Frey filament of 4 g bending force; A) as a function of time after Formalin injection. Animals were injected with siRNA 2 d earlier. Noxious stimuli (5.18 and 5.88 von Frey filaments of 15 and 60 g bending force, respectively) (B, C) in rats injected intrathecally with siRNA[a], siRNA[b], or vehicle 2 d before the induction of inflammation by intraplantar injection of Formalin. D, Kinetics of the withdrawal latency in response to a thermal stimulus plotted against the time after intraplantar injection of Formalin in rats treated intrathecally with siRNA[a], siRNA[b], or vehicle 2 d before the experiment. E, F, Total time spent during licking (E) and number of flinches (F) for the periods of 0–5 or 15–30 min after Formalin injection in groups of rats injected with siRNA. Data are expressed as mean ± SE; data are from eight rats per group. Asterisks and number symbols denote significant difference from vehicle-treated group (p < 0.05). ω-Cx, ω-Conotoxin.
To determine whether the two CaV2.2 splice isoforms mediate specific roles in inflammatory allodynia and hyperalgesia, we evaluated the responses of siRNA and ω-conotoxin GVIA-injected rats to thermal and mechanical stimuli after Formalin injection (Fig. 4A–F). In Formalin-injected rats, siRNA[a] but not siRNA[b] treatment significantly attenuated the Formalin-induced increase in nociceptive score for the small-diameter (4.93) von Frey filament at the 30 min mark (Fig. 4A). This result was similar to the nociceptive scores obtained after injection of ω-conotoxin GVIA. Although these results might suggest a selective involvement of e37a splice isoform in the generation of tactile inflammatory allodynia, the analysis of data obtained up to 1 h after Formalin injection is complicated by spontaneous flinches caused by the Formalin treatment. When examined with the intermediate-diameter von Frey filament (5.18), Formalin injection induced the development of mechanical hyperalgesia (Fig. 4B). This response was abolished at all time points (including 2 to 4 h after injection at which spontaneous Formalin flinches no longer occur) in animals that were injected with siRNA[a] or ω-conotoxin GVIA but not in those treated with siRNA[b] (Fig. 4B). Treatment with siRNA[b] resulted in a statistically significant reduction in nociceptive score in animals stimulated with the large-diameter von Frey filament (5.88) (Fig. 4C). However, this effect did not persist beyond the two earliest time points (30 and 60 min) and may again be attributable to a contribution from spontaneous Formalin flinches. In contrast, treatment with siRNA[a] or ω-conotoxin GVIA reduced the nociceptive scores across the entire range of time points examined (Fig. 4C), suggesting that the CaV2.2e[37a] channels is the predominant N-type channel isoform involved in the transmission of noxious mechanical stimuli during inflammatory mechanical hyperalgesia. Interestingly, treatment with siRNA[a] molecules or ω-conotoxin GVIA resulted in a net decrease in nociceptive score such that the score dropped below the levels observed before Formalin injection. This is surprising considering that basal mechanical nociception in animals not injected with Formalin did not change with siRNA treatment (Fig. 3D) or ω-conotoxin GVIA (data not shown). It is possible that the inflammatory response to Formalin injection sensitizes nociceptors such that they may rely more strongly on e37a N-type calcium channels, thus responding more readily to siRNA knockdown compared with basal conditions.
Thermal hyperalgesia was also observed after Formalin-induced inflammation (Fig. 4D). In this case, only rats injected with siRNA[a] or ω-conotoxin GVIA presented with an inhibition of thermal hyperalgesia (increased withdrawal latency) compared with vehicle-injected rats at all time points (Fig. 4D). These data suggest that only e37a channels are involved in mediating thermal hyperalgesia. We note that ω-conotoxin GVIA also significantly attenuated basal thermal nociception compared with vehicle control (data not shown). This effect appeared slightly less pronounced than that of CaV2.2e[37a] siRNA treatment, but there was no statistical difference between ω-conotoxin GVIA and CaV2.2e[37a] siRNA-treated groups.
Formalin-induced pain is characterized by two phases: a first phase (phase I) that lasts for the first 5 min after Formalin intraplantar injection and is associated with direct stimulation of nociceptors, and a late phase (phase II) that is observed starting at 10–15 min after Formalin injection, in which inflammatory mediators are released and participate in nociceptor sensitization. Other parameters of pain behavior (duration of licking and number of flinches) were recorded in phases I and II after Formalin intraplantar injection (Fig. 4E,F). During phase II (15–30 min), vehicle RNA interference-injected animals displayed an increased incidence of paw licking and a higher number of flinches compared with both siRNA[a]- and siRNA[b]-injected animals. In contrast, in phase I (0–5 min), only the siRNA[a]-injected animals presented with a reduction in the numbers of flinches, suggesting a major implication for the e37a splice isoform in immediate nociceptor activation (Fig. 4F). In addition to these results, injection of ω-conotoxin GVIA also mediated a decrease in the duration of licking and in the numbers of flinches, and the effect was greater than the reduction observed with siRNA[a] (duration of licking, 17.87 ± 2.2 s, n = 8, and numbers of flinches, 1.0 ± 0.62, n = 8, for phase I).
Together, these results show that the e37a splice isoform is the predominant form of CaV2.2 responsible for thermal and mechanical hyperalgesia in inflammatory and neuropathic models, whereas both isoforms contribute to tactile allodynia during neuropathy.
In vivo silencing of CaV2.2e[37a] prevents thermal hyperalgesia
To examine the relative roles of the two CaV2.2 splice isoforms in neuropathic pain, we induced neuropathy by CCI of the sciatic nerve. This neuropathic pain model induces mechanical and thermal hypersensitivity (Woolf and Mannion, 1999; Whiteside et al., 2004). As shown in Figure 5, CCI induced tactile allodynia (assessed using the 4.93-diameter von Frey filaments) and mechanical hyperalgesia (assessed using larger-sized von Frey filaments, 5.18) as evident from increased nociceptive scores after surgery (which occurred at day 0) (Fig. 5A,B). Moreover, CCI induced thermal hyperalgesia as reflected by a decreased withdrawal latency in response to heat stimulation (Fig. 5C). We then examined the abilities of siRNA[a] or siRNA[b] to reverse the established tactile allodynia and hyperalgesia. siRNA was injected at days 8 and 9, and responses to tactile and thermal stimuli were measured from days 10 to 14. Injection of siRNA[a] reversed tactile allodynia from day 10 to 14, whereas siRNA[b] reversed allodynia only at days 10 and 14. Nonetheless, the nociceptive responses of the siRNA[b]-treated group were consistently below those of the vehicle-treated rats, and, as a consequence, the area under the curve for the siRNA[b]-treated group was significantly lower (p < 0.05) than that of the vehicle control (Fig. 5A). Mechanical hyperalgesia was significantly inhibited only by siRNA[a] treatment (Fig. 5B) and only at day 10. These data are in contrast with what was observed with inflammatory hyperalgesia, in which both channel subtypes appeared to contribute to mechanical hyperalgesia with a larger analgesic effect in response to siRNA[a] silencing. Compared with vehicle-injected animals, only the siRNA[a]-treated group displayed an increase in paw-withdrawal latency, resulting in a complete reversal of thermal hyperalgesia (i.e., return to baseline) between days 12 and 14 (Fig. 5C).
Figure 5.
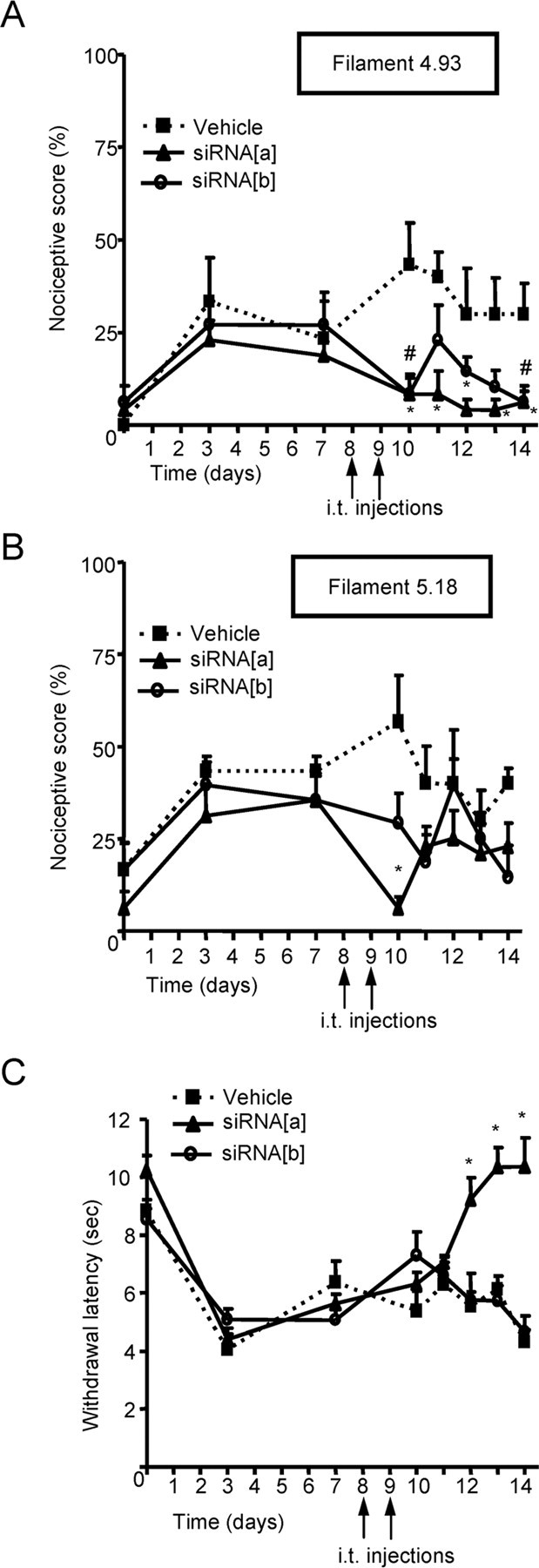
Effect of CaV2.2 splice isoform-specific siRNA silencing on neuropathic allodynia and hyperalgesia induced by sciatic nerve ligature. A–C, Kinetics of nociceptive scores (A, B) and paw-withdrawal latency (C) in response to either a mechanical innocuous stimulus (4.93 von Frey filament of 4 g bending force; A), a noxious stimulus (5.18 von Frey filament of 15 g bending force; B), or a thermal stimulus (C) in rats injected intrathecally with siRNA[a], siRNA[b] or vehicle, 8 and 9 d after the induction of sciatic nerve constriction injury. Data are expressed as mean ± SE; data are from eight rats per group. Asterisks and number symbols denote significant difference from vehicle-treated group (p < 0.05).
Collectively, these in vivo data show that the generation of thermal hyperalgesia in models of inflammatory or neuropathic pain depends exclusively on CaV2.2e[37a] channels. A more complex dependence is seen with mechanical nociception, in which the roles of the two splice isoforms in allodynia and hyperalgesia depend on the type of insult (i.e., inflammation vs neuropathy), with knockdown of the e37a isoform generally resulting in a more prolonged and more robust analgesic effect.
CaV2.2e[37a] is downregulated in rats subjected to spinal nerve ligation
To increase our knowledge about the role of CaV2.2e[37a] and CaV2.2e[37b] channels during neuropathic pain, we quantified mRNA levels of each isoform in whole DRG by RPA. For these experiments, we chose to use the SNL model because the CCI model used in Figure 5 can cause an intermingling of damaged and undamaged fibers, potentially complicating assessment of splice isoform regulation. Furthermore, we chose the SNL model because it involves specific, tight ligation of spinal nerves L5 and L6 but leaves spinal nerve L4 unligated, providing ipsilateral L4 ganglia as an additional control. Fourteen days after surgery, SNL animals presented with mechanical allodynia on the ipsilateral side but not on the contralateral side (Fig. 6A). On the same day, we harvested DRGs from these animals and isolated RNA. We found CaV2.2e[37b] message not altered significantly in ligated ganglia compared with all unligated controls (Fig. 6B,D). In contrast, our RPA demonstrated a significant decrease in CaV2.2e[37a] mRNA only in ipsilateral ganglia L5 and L6, not in L4 ipsilateral ganglia, contralateral ganglia, or sham controls (Fig. 6C,E and data not shown). Collectively, these data suggest that CaV2.2e[37a] mRNA is downregulated during neuropathic pain, possibility reflecting an adaptive response. Importantly, the percentage of CaV2.2e[37a] mRNA is reduced by ∼50% after ligation. Nonetheless, this means that significant levels of CaV2.2e[37a] message remain during neuropathic pain, thus providing a substrate for siRNA knockdown in our experiments.
Figure 6.
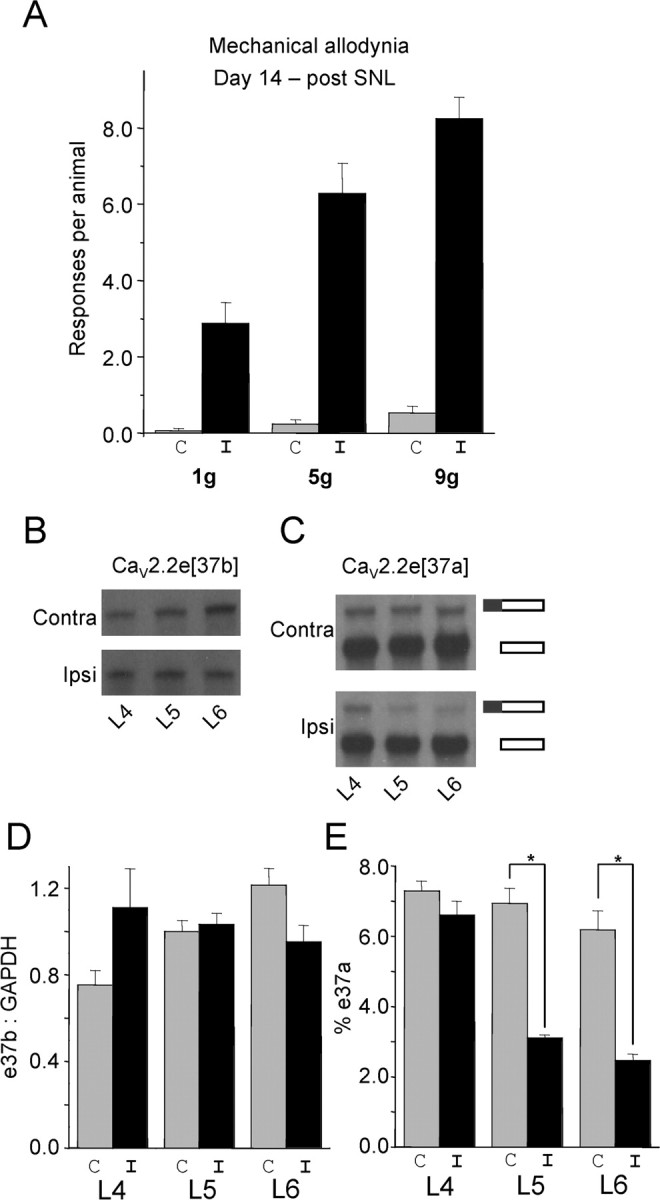
Regulation of CaV2.2e[37a] and CaV2.2e[37b] during neuropathy. A, Mechanical allodynia was evaluated by measuring nociceptive responses using different von Frey filaments 14 d after SNL in the ipsilateral (I; black) and contralateral (C; gray) side. B, RPA analysis using the e37b probe. We pooled total RNA isolated from DRG neurons from the same location (L4, L5, or L6) from animals presenting allodynia (n = 17). C, RPA analysis using the e37a probe. Total RNA is the same as in B. The top band in both contralateral and ipsilateral gels shows “fully protected” e37a probe and indicates the amount of CaV2.2e[37a] mRNA. The bottom band shows “partially protected” e37a probe and indicates non-e37a-containing mRNA (essentially CaV2.2e[37b] mRNA). D, Quantification of e37b mRNA protection levels presented as a ratio of e37b to GAPDH. E, Quantification of e37a mRNA protection levels represented as a percentage of total CaV2.2 mRNA (for calculation, see Materials and Methods). All data reflect the mean ± SE for three independent hybridizations of probe or probe pairs with the pooled DRG RNA.
Discussion
All N-type channels contain a CaV2.2 subunit that is encoded by a single gene with substantial functional diversity in different cell populations. This likely results from the association of CaV2.2 subunits with different ancillary subunits and from cell-dependent alternate splicing of CaV2.2 RNA (Lipscombe et al., 2002; Yasuda et al., 2004). Here we show that different splice isoforms of CaV2.2 mediate distinct physiological functions. Specifically, cell-specific splicing of exon 37 acts as a molecular switch controlling N-type channel coupling to distinct nociceptive pathways, with exon 37a preferentially controlling the transmission of thermal and mechanical stimuli during hyperalgesia.
Acute blockade of N-type channels via intrathecal infusion of ω-conotoxin MVIIA (which does not discriminate among channel splice isoforms) inhibits the early and the late phase of thermal hyperalgesia in Formalin-injected rats and reverses mechanical allodynia after nerve injury (Chaplan et al., 1994; Malmberg and Yaksh, 1995; Bowersox et al., 1996; Diaz and Dickenson, 1997; Sluka, 1998). Our data with ω-conotoxin GVIA are consistent with these previous findings. By using splice isoform-specific gene silencing via in vivo siRNA injection, we show specificity of N-type channel coupling to these pain mechanisms. N-type channels containing exon 37a but not exon 37b appear to be necessary and sufficient for the transmission of thermal nociception both in basal conditions (undamaged tissues) and in inflammatory or neuropathic models. This observation is consistent with the preferential expression of these isoforms in capsaicin-sensitive neurons (Bell et al., 2004) and with our data showing that the effect of the e37a knockdown was diminished on elimination of capsaicin receptor expressing DRG neurons. The CaV2.2e[37a] isoform also appears to be the predominant channel subtype in the transmission of mechanical hyperalgesia, whereas both exon 37 isoforms contribute to tactile allodynia during neuropathy. The latter observation is consistent with previous findings showing that rats treated with ω-conotoxin MVIIA displayed reduced mechanical allodynia in a chronic constriction injury model (Xiao and Bennett, 1995).
The siRNA[a]-mediated inhibition of both phases I and II of the Formalin test are consistent with the overall phenotype of acute N-type channel blockade. Our data are also in general agreement with observations from CaV2.2-deficient mice, in that these mice show reduced mechanical allodynia and thermal hyperalgesia during neuropathy and show reduced thermal hyperalgesia during inflammatory pain (Saegusa et al., 2001). There are, however, some subtle differences between our findings and previously published results with acute block or knock-out of CaV2.2 channels. First, Xiao and Bennett (1995) reported that ω-conotoxin MVIIA application to a normal nerve had no effect on nociceptive responses to thermal or mechanical stimuli, which contrasts with our observation that knockdown of the e37a isoform decreases basal thermal and mechanical nociception (Fig. 3D,F). It is possible that ω-conotoxin MVIIA applied to nerve fibers may not have complete access to all synaptic sites, whereas knockdown of channel expression in DRG neuron cell bodies may be a more effective means of eliminating these channels. In CaV2.2-deficient mice, the responses to such nonpainful thermal stimuli were somewhat ambiguous (Saegusa et al., 2001). Also, in these mice, only phase II of the Formalin test was reduced (Hatakeyama et al., 2001; Kim et al., 2001; Saegusa et al., 2001), which contrasts with our findings and with what is seen after acute block of the channel via ω-conotoxin MVIIA (Bowersox et al., 1996). The difference between long-term gene knock-out and acute block/knockdown of the channels might be attributable to compensatory mechanisms in the knock-out mouse. That said, the bulk of our findings are in line with the overall phenotype expected from removing or inhibiting N-type channels at the DRG level and reveal specific roles of individual N-type calcium channel isoforms in pain pathways. Indeed, intrathecal injection of ω-conotoxin GVIA produced results that were consistent with those obtained with siRNA treatment, such that siRNA treatment was never effective in conditions that were insensitive to ω-conotoxin GVIA, and the magnitudes of the ω-conotoxin GVIA-induced effects were either similar to or slightly enhanced compared with those seen with siRNA treatment. These considerations support the specificity of our siRNA constructs.
Our data indicate that intrathecal siRNA injection is an effective means of selectively removing specific calcium channel splice isoforms in vivo. It has been shown that intrathecally applied antisense oligonucleotides are effectively taken up into dorsal root ganglia, remain localized to the intrathecal sites, and do not cross into the spinal cord (Bourinet et al., 2005). It has also been reported that siRNA is effectively taken up into DRG after intrathecal injection (Luo et al., 2005; Tan et al., 2005). Although we did not examine whether the siRNA constructs cross into the spinal cord, our experiments clearly indicate a similarly efficient uptake of siRNA oligonucleotides into DRG. Both constructs effectively reduced CaV2.2 protein in cultured DRG neurons, with >75% removal of total N-type channel protein in culture. Although we did not specifically examine the extent of channel knockdown in vivo by electrophysiology on the siRNA-positive DRG cells, the effectiveness and selectivity of both constructs is also underscored by their ability to knock down CaV2.2 protein as assayed by Western blotting after in vivo siRNA injection and their distinct actions in capsaicin-treated and untreated neurons. Furthermore, both constructs showed efficacy in vivo (for example tactile allodynia in the CCI model) yet produced distinct effects in thermal hyperalgesia models. In particular, the siRNA[a] construct produced the strongest effects in vivo, although the knockdown in transient expression systems was not as complete as that seen with siRNA[b]. However, considering that tsA-201 cells express much higher levels of N-type channels compared with native cells, this constitutes a robust effect of both siRNA constructs. Overall, our data suggest that the siRNA sequences were effectively taken up and resulted in robust and splice isoform-selective inhibition of N-type channel expression.
Our data showing the selective downregulation of CaV2.2e[37a] mRNA after spinal nerve ligation are intriguing and may reflect an endogenous antinociception mechanism during neuropathic pain. Two other observations are also important. First, we saw no significant changes in CaV2.2e[37b] mRNA, suggesting that there is little compensation from this splice isoform, although a compensatory 3% change in e37b mRNA would be difficult to detect. Second, even in states of neuropathic pain, approximately half of the CaV2.2e[37a] mRNA remains, a key requirement to account for the effects of siRNA[a] injection. It is possible that, during certain types of pain, one splice isoform may become the predominant form, whereas the other could be downregulated, and perhaps so in a neuron subtype-specific manner. A dynamic regulation of CaV2.2 channel splicing could perhaps explain why only siRNA[a] treatment was effective in depressing thermal hyperalgesia, although both channel isoforms are expressed in capsaicin-sensitive neurons under normal conditions (Bell et al., 2004).
It remains unclear as to what makes the exon 37a-containing channels unique in terms of supporting the transmission of specific types of pain stimuli. During transient expression, CaV2.2e[37a] channels yield larger N-type current densities and longer open times (Castiglioni et al., 2006), which would increase the amount of calcium entry during action potentials, thus enhancing neurotransmitter release. However, it was recently shown that e37a renders the channel susceptible to a novel form of G-protein inhibition used by both GABA and opiates (Raingo et al., 2007). The C terminus of CaV2.2 channels therefore mediates important inhibitory effects of G-protein-coupled receptors. It is also a region in which numerous regulatory proteins (Maximov et al., 1999; Liang et al., 2003; Li et al., 2004; Altier et al., 2006) interact with and modulate channel function. Small nociceptive neurons may perhaps contain cellular components that selectively interact with the e37a isoform to modulate channel activity selectively or to target channels to specific subcellular compartments to optimize their role in transmitter release.
The N-type channel is considered an important pharmacological target for the treatment of pain, by either activation of opioid receptors or direct pharmacological inhibition (Altier and Zamponi, 2004; Snutch, 2005). Both avenues have been linked to the occurrence of side effects (Penn and Paice, 2000). This may be attributable to the fact that N-type channels control neurotransmitter release from multiple subtypes (including non-nociceptive) of sensory neurons as well as autonomic neurons (Gruner and Silva, 1994; Meir et al., 1999; Serone and Angus, 1999). The present challenge lies in devising ways to selectively inhibit N-type channels within the pain pathway, eliminating pathological pain but maintaining normal nociception, as well as sensory and autonomic function. In this context, our findings detailing a splice isoform dependence on thermal and mechanical hyperalgesia may provide a potential for treatment. This treatment might take the form of intrathecal administration of siRNA molecules or, alternatively, development of blockers that might discriminate between e37a and e37b.
Footnotes
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) (G.W.Z., N.V.) and National Institutes of Health Grants NS29967 and NS055251 (D.L.). N.V. is a Scholar of the Alberta Heritage Foundation for Medical Research (AHFMR) and a CIHR Young Investigator. G.W.Z. is an AHFMR Scientist and a Canada Research Chair in Molecular Neurobiology. C.A. holds postdoctoral fellowship awards from the AHFMR and the Heart and Stroke Foundation of Canada. C.S.D. was the recipient of a Coordenação de Aperfeiçoamento de Pessoal de Nível Superior studentship from the Brazilian government. R.M.E. is the recipient of an AHFMR fellowship award. A.E.K. holds doctoral studentship awards from AHFMR and the Heart and Stroke Foundation of Canada. E.A.M. is supported by the London Pain Consortium. We thank Dr. Terry Snutch for providing CaV2.2e[37b] cDNA and ancillary calcium channel subunits and Dr. John McRory for technical discussion.
References
- Altier C, Zamponi GW. Targeting Ca2+ channels to treat pain: T-type versus N-type. Trends Pharmacol Sci. 2004;25:465–470. doi: 10.1016/j.tips.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, Chen L, Beedle AM, Ferguson SS, Mezghrani A, Dubel SJ, Bourinet E, McRory JE, Zamponi GW. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7:118–125. doi: 10.1038/nn1180. [DOI] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41:127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J. Silencing of the Ca(v)3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowersox SS, Gadbois T, Singh T, Pettus M, Wang YX, Luther RR. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J Pharmacol Exp Ther. 1996;279:1243–1249. [PubMed] [Google Scholar]
- Castiglioni AJ, Raingo J, Lipscombe D. Alternative splicing in the C-terminus of CaV2.2 controls expression and gating of N-type calcium channels. J Physiol (Lond) 2006;576:119–134. doi: 10.1113/jphysiol.2006.115030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994;269:1117–1123. [PubMed] [Google Scholar]
- Diaz A, Dickenson AH. Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammation. Pain. 1997;69:93–100. doi: 10.1016/s0304-3959(96)03271-x. [DOI] [PubMed] [Google Scholar]
- Gruner W, Silva LR. ω-Conotoxin sensitivity and presynaptic inhibition of glutamatergic sensory neurotransmission in vitro. J Neurosci. 1994;14:2800–2808. doi: 10.1523/JNEUROSCI.14-05-02800.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Wakamori M, Ino M, Miyamoto N, Takahashi E, Yoshinaga T, Sawada K, Imoto K, Tanaka I, Yoshizawa T, Nishizawa Y, Mori Y, Niidome T, Shoji S. Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca2+ channels. NeuroReport. 2001;12:2423–2427. doi: 10.1097/00001756-200108080-00027. [DOI] [PubMed] [Google Scholar]
- Hu LY, Ryder TR, Nikam SS, Millerman E, Szoke BG, Rafferty MF. Synthesis and biological evaluation of substituted 4-(OBz)phenylalanine derivatives as novel N-type calcium channel blockers. Bioorg Med Chem Lett. 1999;9:1121–1126. doi: 10.1016/s0960-894x(99)00160-2. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Cooper CB, Nishioka N, Yamasaki H, Suzuki A, Jarvis SE, Akaike A, Satoh M, Zamponi GW. Identification and characterization of novel human Cav2.2 (α1B) calcium channel isoforms lacking the synaptic protein interaction site. J Neurosci. 2002;22:82–92. doi: 10.1523/JNEUROSCI.22-01-00082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H, Kim HL, Park JM, Kim DK, Jung SJ, Kim J, Shin HS. Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol Cell Neurosci. 2001;18:235–245. doi: 10.1006/mcne.2001.1013. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Krarup C. An update on electrophysiological studies in neuropathy. Curr Opin Neurol. 2003;16:603–612. doi: 10.1097/01.wco.0000093104.34793.94. [DOI] [PubMed] [Google Scholar]
- Li B, Zhong H, Scheuer T, Catterall WA. Functional role of a C-terminal Gbetagamma-binding domain of Ca(v) 2.2 channels. Mol Pharmacol. 2004;66:761–769. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lin Y, McDonough SI, Lipscombe D. Alternative splicing in the voltage-sensing region of N-Type CaV2.2 channels modulates channel kinetics. J Neurophysiol. 2004;92:2820–2830. doi: 10.1152/jn.00048.2004. [DOI] [PubMed] [Google Scholar]
- Lin Z, Haus S, Edgerton J, Lipscombe D. Identification of functionally distinct isoforms of the N-type Ca2+ channel in rat sympathetic ganglia and brain. Neuron. 1997;18:153–166. doi: 10.1016/s0896-6273(01)80054-4. [DOI] [PubMed] [Google Scholar]
- Lin Z, Lin Y, Schorge S, Pan JQ, Beierlein M, Lipscombe D. Alternative splicing of a short cassette exon in α1B generates functionally distinct N-type calcium channels in central and peripheral neurons. J Neurosci. 1999;19:5322–5331. doi: 10.1523/JNEUROSCI.19-13-05322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Pan JQ, Gray AC. Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of Ca(v)alpha1. Mol Neurobiol. 2002;26:21–44. doi: 10.1385/MN:26:1:021. [DOI] [PubMed] [Google Scholar]
- Luo MC, Zhang DQ, Ma SW, Huang YY, Shuster SJ, Porreca F, Lai J. An efficient intrathecal delivery of small interfering RNA to the spinal cord and peripheral neurons. Mol Pain. 2005;1:29. doi: 10.1186/1744-8069-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain. 1995;60:83–90. doi: 10.1016/0304-3959(94)00094-U. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Dickenson AH. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246. doi: 10.1016/s0304-3959(01)00255-x. [DOI] [PubMed] [Google Scholar]
- Maximov A, Bezprozvanny I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J Neurosci. 2002;22:6939–6952. doi: 10.1523/JNEUROSCI.22-16-06939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Sudhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999;274:24453–24456. doi: 10.1074/jbc.274.35.24453. [DOI] [PubMed] [Google Scholar]
- McCleskey EW, Gold MS. Ion channels of nociception. Annu Rev Physiol. 1999;61:835–856. doi: 10.1146/annurev.physiol.61.1.835. [DOI] [PubMed] [Google Scholar]
- Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- Pan JQ, Lipscombe D. Alternative splicing in the cytoplasmic II–III loop of the N-type Ca channel α1B subunit: functional differences are β-subunit specific. J Neurosci. 2000;20:4769–4775. doi: 10.1523/JNEUROSCI.20-13-04769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn RD, Paice JA. Adverse effects associated with the intrathecal administration of ziconotide. Pain. 2000;85:291–296. doi: 10.1016/s0304-3959(99)00254-7. [DOI] [PubMed] [Google Scholar]
- Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T, Han W, Toriyama H, Tanabe T. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 2001;20:2349–2356. doi: 10.1093/emboj/20.10.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci. 2002;5(Suppl):1062–1067. doi: 10.1038/nn942. [DOI] [PubMed] [Google Scholar]
- Scott DA, Wright CE, Angus JA. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur J Pharmacol. 2002;451:279–286. doi: 10.1016/s0014-2999(02)02247-1. [DOI] [PubMed] [Google Scholar]
- Seko T, Kato M, Kohno H, Ono S, Hashimura K, Takenobu Y, Takimizu H, Nakai K, Maegawa H, Katsube N, Toda M. l-Cysteine based N-type calcium channel blockers: structure-activity relationships of the C-terminal lipophilic moiety, and oral analgesic efficacy in rat pain models. Bioorg Med Chem Lett. 2002;12:2267–2269. doi: 10.1016/s0960-894x(02)00456-0. [DOI] [PubMed] [Google Scholar]
- Serone AP, Angus JA. Role of N-type calcium channels in autonomic neurotransmission in guinea-pig isolated left atria. Br J Pharmacol. 1999;127:927–934. doi: 10.1038/sj.bjp.0702629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluka KA. Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammation. J Pharmacol Exp Ther. 1998;287:232–237. [PubMed] [Google Scholar]
- Smith MT, Cabot PJ, Ross FB, Robertson AD, Lewis RJ. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain. 2002;96:119–127. doi: 10.1016/s0304-3959(01)00436-5. [DOI] [PubMed] [Google Scholar]
- Snutch TP. Targeting chronic and neuropathic pain: the N-type calcium channel comes of age. NeuroRx. 2005;2:662–670. doi: 10.1602/neurorx.2.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M, Fisher R, Bryce DA, Mangieri EA, Luther RR, Mayo M, McGuire D, Ellis D. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- Stea A, Dubel SJ, Snutch TP. alpha 1B N-type calcium channel isoforms with distinct biophysical properties. Ann NY Acad Sci. 1999;868:118–130. doi: 10.1111/j.1749-6632.1999.tb11282.x. [DOI] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Thaler C, Gray AC, Lipscombe D. Cumulative inactivation of N-type CaV2.2 calcium channels modified by alternative splicing. Proc Natl Acad Sci USA. 2004;101:5675–5679. doi: 10.1073/pnas.0303402101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ. Thermosensation and pain. J Neurobiol. 2004;61:3–12. doi: 10.1002/neu.20079. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Harrison J, Boulet J, Mark L, Pearson M, Gottshall S, Walker K. Pharmacological characterisation of a rat model of incisional pain. Br J Pharmacol. 2004;141:85–91. doi: 10.1038/sj.bjp.0705568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xiao WH, Bennett GJ. Synthetic omega-conopeptides applied to the site of nerve injury suppress neuropathic pains in rats. J Pharmacol Exp Ther. 1995;274:666–672. [PubMed] [Google Scholar]
- Yasuda T, Chen L, Barr W, McRory JE, Lewis RJ, Adams DJ, Zamponi GW. Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur J Neurosci. 2004;20:1–13. doi: 10.1111/j.1460-9568.2004.03434.x. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, McCleskey EW. Splicing it up: a isoform of the N-type calcium channel specific for pain. Neuron. 2004;41:3–4. doi: 10.1016/s0896-6273(03)00846-8. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]