

Abstract
ClC-2 is a broadly expressed plasma membrane chloride channel that is modulated by voltage, cell swelling, and pH. A human mutation leading to a heterozygous loss of ClC-2 has previously been reported to be associated with epilepsy, whereas the disruption of Clcn2 in mice led to testicular and retinal degeneration. We now show that the white matter of the brain and spinal cord of ClC-2 knock-out mice developed widespread vacuolation that progressed with age. Fluid-filled spaces appeared between myelin sheaths of the central but not the peripheral nervous system. Neuronal morphology, in contrast, seemed normal. Except for the previously reported blindness, neurological deficits were mild and included a decreased conduction velocity in neurons of the central auditory pathway. The heterozygous loss of ClC-2 had no detectable functional or morphological consequences. Neither heterozygous nor homozygous ClC-2 knock-out mice had lowered seizure thresholds. Sequencing of a large collection of human DNA and electrophysiological analysis showed that several ClC-2 sequence abnormalities previously found in patients with epilepsy most likely represent innocuous polymorphisms.
Keywords: leukodystrophy, oligodendrocyte, glia, knock-out, anion transport, ion siphoning
Introduction
The transport of chloride (Cl−) serves many purposes, including the transport of salt and fluid across epithelia, control of neuronal and muscular excitability, the regulation of cell volume, and the acidification of intracellular organelles. Translocation of Cl− across biological membranes occurs through coupled transport processes and by diffusion through Cl− channels. ClC-2 is a broadly expressed Cl− channel of the CLC family of Cl− channels and transporters. In mammals, this gene family comprises nine members that can be grouped into three branches (Jentsch et al., 2002). The first branch, which includes ClC-2, comprises plasma membrane Cl− channels. Their importance is illustrated by genetic diseases such as myotonia congenita and Bartter syndrome, which are caused by mutations in ClC-1 and ClC-Kb or its β-subunit barttin, respectively (Steinmeyer et al., 1991; Simon et al., 1997; Estévez et al., 2001). ClC-2 is activated by negative membrane voltage, cell swelling, a rise in intracellular Cl− concentration, or mild extracellular acidification (Gründer et al., 1992; Thiemann et al., 1992; Jordt and Jentsch, 1997; Zuñiga et al., 2004). Its mRNA was found in all tissues and cell lines investigated (Thiemann et al., 1992). It was most abundant in brain and in epithelia (Thiemann et al., 1992; Cid et al., 1995). In mice, the disruption of ClC-2 led to testicular and retinal degeneration (Bösl et al., 2001; Nehrke et al., 2002). Germ cells and photoreceptors depend on the transepithelial transport mediated by Sertoli cells and the retinal pigment epithelium (RPE), respectively. It was hypothesized that the lack of ClC-2 impairs this transport, thereby changing the ionic environment of germ cells and photoreceptors (Bösl et al., 2001). Indeed, Ussing chamber experiments revealed a reduced voltage and current across the RPE of ClC-2 knock-out (KO) mice (Bösl et al., 2001).
ClC-2 is abundantly expressed in brain. It was proposed to participate in lowering the cytoplasmic Cl− concentration of neurons, a process that establishes an inhibitory response to the neurotransmitters GABA and glycine (Staley et al., 1996). However, the disruption of ClC-2 in mice did not entail epilepsy, as might have been expected from this hypothesis (Bösl et al., 2001; Nehrke et al., 2002). On the other hand, heterozygous mutations in CLCN2 (the gene encoding ClC-2) were recently reported in a few patients with three clinically distinct forms of epilepsy (Haug et al., 2003). That work prompted us to investigate the CNS of ClC-2 KO mice in detail.
Surprisingly, we now discovered a severe and widespread, spongiform vacuolation of the white matter in the brain and spinal cord of old ClC-2 KO mice. No changes were observed in heterozygous animals. The myelin of central but not of peripheral axons was abnormal, with fluid-filled vacuoles between myelin sheets. In contrast, the morphology of neurons appeared normal, and the neurological phenotype was subtle. No differences in seizure thresholds between wild-type (WT), heterozygous, and homozygous ClC-2 KO mice were detected.
Materials and Methods
Mice.
The generation of ClC-2 KO mice was described by Bösl et al. (2001).
Histology.
Detailed procedures of histochemistry, immunohistochemistry, and electron microscopy are found in the supplemental material (available at www.jneurosci.org). Primary antibodies used were CC1 (1:20; Calbiochem, La Jolla, CA), ClC-2 (1:300) (Bösl et al., 2001), connexin47 (Cx47; 1:250; Chemicon, Temecula, CA), F4/80 (1:200; Serotec, Oxford, UK), glial fibrillary acidic protein (GFAP; 1:500; Sigma, St. Louis, MO), myelin basic protein (MBP; 1:500; Chemicon), NeuN (1:500; Chemicon), and proteolipid protein (PLP; 1:300; Chemicon). Activated microglia was detected with biotinylated Griffonia simplicifolia agglutinin (GSA) (1:50; Vector Laboratories, Burlingame, CA).
Preparation of myelin and Western blot analysis.
Myelin from 8- to 12-month-old animals was prepared as described previously (Norton and Poduslo, 1973). Twenty micrograms of purified myelin and the same amount of the initial brain homogenate were used for SDS-PAGE and Western blot analysis. For detailed protocols, see supplemental material (available at www.jneurosci.org).
Gene expression profiling.
For microarray analysis, RNA was extracted from cerebelli of 2-week-old (n = 3, each genotype) and 5-week-old (n = 4, each genotype) male ClC-2 KO mice and control littermates, converted into labeled cDNA, and hybridized to Affymetrix (Santa Clara, CA) murine genome MOE430A/B microarrays. For quantitative PCR, starting from total RNA from cerebella of 5-week-old animals (n = 4, each genotype) used for the microarray analysis and additionally prepared from 5-week-old (n = 3, each genotype) and 24-week-old (n = 3, each genotype) animals, cDNA was prepared using the Invitrogen (San Diego, CA) cDNA kit and analyzed with the Applied Biosystems (Foster City, CA) Prism 7700 system using SYBR green. For detailed protocols, see supplemental material (available at www.jneurosci.org).
Patients.
DNA samples of 150 unrelated patients with clinical suspicion of leukodystrophy, as well as control patients, were analyzed for CLCN2 mutations as detailed in the supplemental material (available at www.jneurosci.org).
Behavioral tests.
Activity of mice was monitored quantitatively as the overall motion of the animal using the Infrared E Motion System (Infra E-Motion, Hamburg, Germany). Singly caged animals were observed for ≥1 week, and overall activity and resting times were calculated per 2 min. Motor-coordination activity was tested with an accelerating rotarod (TSE Systems, Bad Homburg, Germany). The time mice remained on the roller was measured (up to 240 s). Three trials per mouse were performed on 3 consecutive days.
Electrophysiology of mutant ClC-2 channels.
Two-electrode voltage-clamp analysis of Xenopus oocytes was done as described by Jordt and Jentsch (1997) and as detailed in the supplemental material (available at www.jneurosci.org).
Auditory brainstem response.
Auditory brainstem response (ABR) was performed as in (Kharkovets et al., 2006) and as detailed in supplemental material (available at www.jneurosci.org).
Results
ClC-2 KO mice display progressive spongiform vacuolation in the CNS
Brain sections of 22-month-old ClC-2 KO (Clcn2 −/−) mice and control WT and heterozygous littermates were stained with hematoxylin–eosin. In ClC-2 KO mice, this analysis revealed vacuole-like holes throughout the white matter of the brain and in large fiber tracts of the brainstem (Fig. 1 A) and spinal cord (Fig. 1 I). Such changes were absent from WT (Fig. 1 B) or heterozygous (data not shown) mice. The regions displaying these morphological alterations costained for myelin with Luxol-blue (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Vacuolation was most prominent in fiber tracts of the cerebellum (Fig. 1 A, a), corpus callosum (Fig. 1 A, b), internal capsule (Fig. 1 A, c), and brainstem (Fig. 1 A, d). Myelin vacuolation was also visible in the spinal cord of ClC-2 KO mice (Fig. 1 I). The gray matter did not show myelin-associated vacuoles, and neuronal cell loss was not observed (Fig. 1 A).
Figure 1.

Spongiform vacuolation in white matter tracts of ClC-2 KO mice. A, Hematoxylin–eosin-stained brain section of a 22-month-old Clcn2 −/− mouse shows vacuolation in white matter tracts of the cerebellum (a), corpus callosum (b), internal capsule (c), and brainstem (d). B, An age-matched WT brain shows much less vacuolation, which is mostly attributable to capillaries. C–F, Progressive vacuolation in Clcn2 −/− cerebellum from P14 (C) (no vacuolation) to P28 (D), 4 months (E), and 22 months (F). G, Semithin sections of the middle cerebellar peduncle of 2-, 5-, and 14-month-old WT and ClC-2 KO mice used to quantify the area of vacuolation. Abundant vacuoles (asterisks) were visible in cerebellar white matter of 5- and 14-month-old KO but not WT animals, in which capillaries (c) are indicated. H, The white area of randomly taken sections was determined by automated image analysis and shown as the percentage of the total area [5.2 ± 0.6% (KO) vs 1.4 ± 1.3% (WT) at 2 months, 17.8 ± 0.9% (KO) vs 2.1 ± 0.5% (WT) at 5 months, and 21.3 ± 1.2% (KO) vs 3.3 ± 1.2% (WT) at 14 months; 10 sections per mouse, 3 mice each]. Error bars indicate SEM. I, Prominent vacuolation in the white matter of the spinal cord of a 14-month-old KO but not WT mouse. Scale bars: A, B, 1 mm; C–F, 0.25 mm; G, 20 μm; I, 0.5 mm.
The morphological alterations in Clcn2 −/− brain increased with age. At P14, the KO cerebellum appeared normal (Fig. 1 C). At postnatal day 28 (P28), vacuole-like holes became apparent in the white matter of the cerebellum (Fig. 1 D) and in fiber tracts of the brainstem (data not shown). The vacuolation progressed further with age, as shown for Clcn2 −/− cerebellum at 4 months of age (Fig. 1 E) and 22 months of age (Fig. 1 F). To quantify this progression, semithin sections of the middle cerebellar peduncle of 2-, 5-, and 14-month-old mice (Fig. 1 G) were examined by automated image analysis. It yielded the percentage of the white area as a measure of vacuole-like structures (asterisk) but also includes capillaries (Fig. 1 G, c). In 2-month-old Clcn2 −/− mice, this relative area was already larger than in the wild type. It was dramatically increased at 5 months and showed a modest further expansion at 14 months of age (Fig. 1 H). A qualitative morphological analysis suggested that this increase in the white area primarily reflected an enlargement in vacuole size rather than an increase in their number.
Unlike the CNS, peripheral nerves of Clcn2 −/− mice appeared unaffected. No pathological alterations were visible in semithin sections of sciatic nerves from 14-month-old KO animals (Fig. 2 A), a result that was confirmed by ultrastructural analysis (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The selective presence of myelin pathology in the CNS might be explained by the differential expression pattern of ClC-2, which was detected in brain but not in sciatic nerve (Fig. 2 B). However, the optic nerve, although being part of the CNS, did not show vacuolation either (Fig. 2 H and supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 2.

Ultrastructural analysis of vacuoles and myelin. A, Semithin sections of the sciatic nerve show no difference between the genotypes at 14 months of age (14m). B, Western blot analysis detects the ClC-2 protein in brain but not in sciatic nerve (sciatic n.). KO lysates showed the specificity of the ClC-2 antibody. Actin served as a loading control. C–E, Electron micrographs showing typical vacuoles in white matter tracts of ClC-2 KO cerebellum. These vacuoles (asterisks) contained aberrant myelin sheets (arrows) and were surrounded by thin myelin sheaths (arrowheads). a, Axon. D, Myelinated axon surrounded by a vacuole and a normal-appearing oligodendrocyte (n). F, Part of WT white matter cerebellum. G, The structure of myelin sheaths in the area of best compaction revealed no difference in myelin compaction. Distances between MDLs were unchanged in the KO [10.7 ± 0.1 nm (WT) vs 10.8 ± 0.1 nm (KO); n < 90, 3 animals each]. 7w, 7 weeks old. H, Semithin sections of the optic nerve of 7-week-old (7w) WT and Clcn2 −/− mice revealed no vacuolation in the KO (asterisks indicate capillary). Higher magnification is shown in supplemental Fig. S3 (available at www.jneurosci.org as supplemental material). Scale bars: A, 20 μm; C–F, 1 μm; G, 20 nm; H, 10 μm.
ClC-2 KO and WT brains were analyzed in more detail by electron microscopy (Fig. 2 C–F). Numerous vacuoles (asterisk) of different sizes and shapes were found in the white matter of Clcn2 −/− (Fig. 2 C–E) but not of WT (Fig. 2 F) cerebella. These vacuoles contained abberant myelin sheets (arrows) and were surrounded by thin myelin sheaths (arrowheads), suggesting that they are most likely formed within the compact myelin of axons. Cell bodies of oligodendrocytes (n) did not show aberrant morphology near vacuoles (Fig. 2 D). Axons (a) lacked detectable morphological changes even when close to vacuoles (Fig. 2 C–E). To determine whether there was a general problem in myelin compaction, we measured distances between the dark major dense lines (MDLs) in myelin areas not directly affected by vacuolation, as exemplified in Figure 2 G. There were no obvious differences in these distances between WT and Clcn2 −/− littermates [10.7 ± 0.1 nm (WT) vs 10.8 ± 0.1 nm (KO); mean ± SEM]. We also evaluated the diameters of cerebellar myelin sheaths that were not directly affected by vacuolation at the level of the section. There was no difference, either [WT, 112.2 ± 3.7 nm (123 axons); KO, 110.3 ± 4.1 nm (116 axons); three 7-week-old animals each].
Normal expression and localization of myelin proteins in ClC-2 KO mice
Immunohistochemistry of the CNS myelin proteins PLP and MBP revealed no difference between KO and WT brains at 5 weeks or 8 months of age, except for the presence of vacuoles (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The expression levels of either protein and of 2′–3′ cyclic nucleotide phosphodiesterase (CNP), a marker of oligodendrocytes, were followed by Western blot analysis of whole-brain lysates during myelination. In rodents, this process starts at the end of the first postnatal week and is completed within 2 months. Whereas ClC-2 expression remained constant, MBP, PLP, and CNP levels increased during this time, with no difference between Clcn2 −/− and WT mice (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). There were neither differences in the abundance of MBP, PLP, and CNP in lysates from cerebellum or whole brain at 1 and 12 months of age nor in the expression levels of axonal or neuronal markers (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material).
Upregulation of inflammation-associated genes in ClC-2 KO brain
Comparative microarray expression profiling of whole cerebellar RNA did not reveal differences between the genotypes when performed at P14, a time when Clcn2 −/− brain is still devoid of overt morphological changes (deposited at MIAMExpress with the accession number E-MEXP-1028). In 5-week-old KO mice, however, transcripts of several genes involved in inflammatory processes were increased, whereas those of myelin-specific genes were slightly decreased (supplemental Table 1, available at www.jneurosci.org as supplemental material). These results were confirmed in part by quantitative PCR (supplemental Table 2, available at www.jneurosci.org as supplemental material). We next asked whether the further progression of CNS vacuolation was paralleled by an increased expression of genes that are upregulated during inflammation, such as those encoding the astrocytic GFAP or microglial F4/80 proteins (Eng and Ghirnikar, 1994; Raivich et al., 1999). Quantitative reverse transcription-PCR analysis of cerebellar RNA from 5- and 24-week-old animals (Fig. 3 A) revealed that, except for glutathione peroxidase, their expression relative to WT increased in Clcn2 −/− cerebella from 5 weeks (filled bars) to 24 weeks (open bars). Microglial activation in Clcn2 −/− but not in WT brain was also evident from an increased binding of GSA lectin at 8 months of age (Fig. 3 C) and by an increased immunostaining for F4/80 (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). Such an activation of microglia was not yet seen in 5-week-old KO mice (data not shown). During the same time period, the expression of GFAP increased prominently in Clcn2 −/− brains (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material), indicative of reactive astrogliosis.
Figure 3.

Inflammation and impaired blood–brain barrier in ClC-2 KO mice and functional tests of motor coordination and central nerve conductance. A, Quantitative PCR (qPCR) analysis of selected transcripts from cerebella of 5-week-old (n = 6, each genotype) and 24-week-old (n = 3, each genotype) animals. Expression levels are given as a percentage of wild type. Genes analyzed by qPCR are as follows: 1, cathepsin-S (accession #NM021281); 2, osteopontin (NM009263); 3, lysozyme (NM013590); 4, F4/80 (NM010130); 5, hexosaminidase (NM010422); 6, leukocyte-derived chemotaxin (NM010701); 7, complement component 1 (NM007572); 8, glutathion peroxidase (NM008161); 9, GFAP (NM010277). Except for GFAP and F4/80, these genes were also found to be upregulated in expression profiling (supplemental Table 1, available at www.jneurosci.org as supplemental material). Rel., Relative. B, C, Red staining by labeled GSA showed microglia activation in 8-month-old Clcn2 −/− (C) but not WT (B) cerebella. D, E, Diaminobenzidine staining (brown) for HRP that was previously injected intravenously into 8-month-old (8m) Clcn2 −/− (E) or WT (D) mice revealed extravasation (arrows), indicative of a disrupted blood–brain barrier only in the KO. Nuclei were counterstained with methylgreen. F, In rotarod tests, ClC-2 KO mice (n = 5) performed as well as WT (n = 4) mice. Motor skills improved with the number of trials in both groups. G, IPIs between waves I and III of auditory brain stem responses were significantly increased in older animals, indicating a slowed nerve conductance in the KO [5 weeks (5w), ***p < 0.1%; 12 weeks (12w), **p < 1%; n = 5–18 mice, same as in supplemental Fig. S7, available at www.jneurosci.org as supplemental material]. 3w, 3 weeks. Error bars indicate SEM. Scale bars: B, C, 0.05 mm; D, E, 0.1 mm.
Impaired blood–brain barrier in Clcn2 −/− mice
Because inflammatory processes may disrupt the blood–brain barrier, we tested its integrity by injecting the tracer horseradish peroxidase (HRP) intravenously into 5-week-old and 8-month-old animals of either genotype. Brains were dissected 10 min after injection and fixed. HRP was detected by diaminobenzidine staining. At 5 weeks of age, HRP passed the blood–brain barrier neither in WT nor in Clcn2 −/− mice (data not shown). However, in 8-month-old mice, HRP extravasation was evident in KO but not in WT cerebellum (Fig. 3 D,E, arrows).
Except for blindness, Clcn2 −/− mice have only mild neurological deficits
In adult Clcn2 −/− mice, the vacuolation severely affected the cerebellum, a structure involved in motor learning and coordination. To assess these skills, we performed rotarod tests on 6- to 8-month-old mice. Surprisingly, Clcn2 −/− mice performed as well as WT mice and learned motor skills with a similar time course (Fig. 3 F). The basal motor activity of KO mice as assessed by infrared sensors was also undistinguishable from wild type (data not shown). ABRs were used to explore the conduction and processing velocity of the peripheral and central auditory pathways. Electric responses to acoustic clicks were recorded in anesthetized mice by subcutaneous electrodes at both mastoids and the vertex. The time-averaged electrical response reflects the synchronized electrical activity of the auditory pathway and consists of five well defined peaks (I–V) in the range of <10 μs. Hearing thresholds of 3- to 24-week-old WT and KO animals did not differ significantly (∼64 dB per sound pressure level), and mice of both genotypes displayed similar presbyacusis at 24 weeks (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). Whereas the latency from the acoustic click to the first peak is dominated by cochlear processes, interpeak intervals (IPIs) between peaks I and III are primarily proportional to the nerve conduction velocity in the central auditory pathway. In all age groups, the latency of wave I was not changed in the KO, indicating normal cochlear function (data not shown). Whereas not being significantly different at 3 weeks of age, the I–III latency was increased in KO at 5 and 12 weeks of age (Fig. 3 G). Hence, the progressive myelin vacuolation of Clcn2 −/− mice is reflected in a reduction in nerve conduction velocity.
Localization of ClC-2 in the nervous system
To potentially correlate the CNS phenotype of Clcn2 −/− mice with the cellular and subcellular distribution of ClC-2, brain sections of 5-week-old mice of either genotype were stained with an antibody against ClC-2 (Bösl et al., 2001). The absence of staining in Clcn2 −/− brain showed the specificity of the antibody (Fig. 4 A–C). In the CA1 and CA3 region of the hippocampus, as described previously (Sik et al., 2000), ClC-2 localized to pyramidal neurons (data not shown) and to endfeet of GFAP-positive astrocytes (arrows) surrounding blood vessels (asterisks) (Fig. 4 A). In the cerebellum, ClC-2 was detected in GFAP-positive Bergmann glia of the cortex (Fig. 4 B) and in myelinated fiber tracts of the medulla (Fig. 4 C–F). Because ClC-2 staining did not overlap with the astrocytic GFAP in the medulla (Fig. 4 C), such sections were costained for other marker proteins (Fig. 4 D–F). Whereas the neuronal marker NeuN did not overlap significantly with ClC-2 (Fig. 4 D), ClC-2-positive puncta surrounded cell bodies that showed cytoplasmic labeling for the oligodendrocytic marker CC1 (Bhat et al., 1996) (Fig. 4 E, arrows). Costaining with Cx47, a gap junction protein exclusively expressed on oligodendrocytes (Altevogt and Paul, 2004), showed a similar localization of both proteins along the circumference of oligodendrocytic cell bodies (Fig. 4 F). Possibly reflecting the limited sensitivity of our antibody, immunohistochemistry did not detect ClC-2 in myelinated fiber tracts of other brain regions. Consistent with the cerebellar immunofluorescence, however, Western blots detected ClC-2 in myelin fractions purified from whole mouse brain at roughly similar levels as in whole brain (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). In myelin preparations from Clcn2 −/− mice, the abundance of myelin proteins was similar to wild type. Additional support for the expression of ClC-2 in oligodendrocytes and astrocytes comes from in situ hybridization (supplemental Fig. S9, available at www.jneurosci.org as supplemental material). In addition to brightly labeled neuronal cell bodies (e.g., in the hippocampus), it revealed staining of the white matter. This included the cerebellum and the corpus callosum, structures that show prominent vacuolation in Clcn2 −/− mice.
Figure 4.
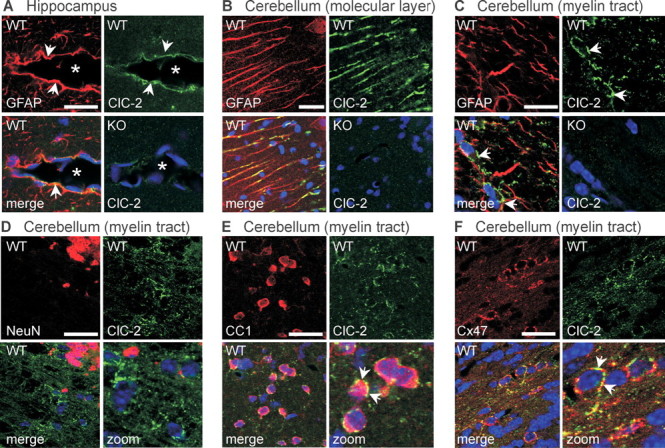
Subcellular localization of ClC-2 in brain. Confocal images of brain sections of 5-week-old WT and ClC-2 KO mice double stained for ClC-2 (green) and marker proteins (red) including GFAP (A–C), NeuN (D), CC1 (E), and Cx47 (F). A, In the hippocampus, ClC-2 colocalized with GFAP in astrocytic endfeet (arrows) surrounding blood vessels (asterisks). B, C, In the cerebellum, ClC-2 was found in GFAP-positive Bergman glia of the molecular layer (B) and in myelinated fiber tracts (C). C, D, In the latter region, ClC-2 did not significantly overlap with either GFAP (C) or NeuN (D). E, Cells with numerous ClC-2 puncta around their cell bodies were identified as oligodendrocytes by staining for CC1. F, Many of these ClC-2-positive puncta also stained for Cx47. Scale bars, 20 μm. Blue indicates TOTO staining of nuclei.
CLCN2 as a candidate gene for leukodystrophy and epilepsy
The observed vacuolation of CNS myelin strongly resembles human leukodystrophy, a clinically and genetically heterogenous group of human pathologies in which several causative genes remain to be identified (Schiffmann and van der Knaap, 2004). We examined the CLCN2 gene in a heterogeneous group of 150 patients with leukodystrophy. In six patients, leukodystrophy had been classified as cystic leukodystrophy, but they did not carry a mutation in MLC1. Fourteen patients were diagnosed to suffer from hypomyelination. In addition to several noncoding single nucleotide polymorphisms, some heterozygous missense mutations were identified (supplemental Table 3, available at www.jneurosci.org as supplemental material). Two of these, E718D and R688Q, are identical to those described by D'Agostino et al. (2004) in epileptic patients, but the leukodystrophy patients carrying these variants lacked a history of epilepsy. This suggests that these sequence variants represent innocuous polymorphisms that are neither associated with leukodystrophy nor epilepsy.
Leukodystrophies are often associated with epilepsy, and mutations in CLCN2 were recently reported to underlie diverse clinical forms of epilepsy in a few families (Haug et al., 2003). We therefore explored the functional impact of the CLCN2 sequence abnormalities that were associated with epilepsy by Haug et al. (2003). Although these authors reported that a truncating CLCN2 mutation (Gins597) strongly suppressed currents of coexpressed WT ClC-2, the truncated protein lacked dominant-negative effects when coexpressed in Xenopus oocytes with WT ClC-2 at a 1:1 ratio that closely mimics the situation in heterozygous patients (Fig. 5 A). This supports similar results from Sepúlveda's group (Niemeyer et al., 2004). We extended our electrophysiological analysis to two sequence variants identified more recently by D'Agostino et al. (2004) in a heterozygous state in patients with epilepsy and that were also present in some of our patients with leukodystrophy (E718D and R688Q; see above). These sequence variants had no detectable influence on functional properties (Fig. 5 B).
Figure 5.

Potential role of ClC-2 in epilepsy. A, Two-electrode voltage-clamp analysis of Xenopus oocytes previously injected with 10 ng of WT ClC-2 cRNA (■) or with a construct modeled on the truncating Gins597 mutation (○) from a family with epilepsy (Haug et al., 2003). Injecting half the amount (5 ng) of WT ClC-2 cRNA gave about half the current amplitude (□). Coinjection of 5 ng of WT and 5 ng of Gins597 cRNA (•) yielded similar current amplitudes, indicating that the mutant lacks a dominant-negative effect. Currents were obtained from >15 oocytes from three batches, averaged, and normalized to wild type. B, Two-electrode voltage-clamp analysis of two sequence variants identified in epileptic patients (D'Agostino et al., 2004) in the present screen of patients with leukodystrophy and by Stogmann et al. (2006). Currents from ClC-2_E718D (▾; n = 21 oocytes) and ClC-2_R688Q (○; n = 23) were indistinguishable from WT ClC-2 currents (■; n = 19). Averaged and normalized currents from three batches of oocytes are shown. C, D, Seizure susceptibility of WT, Clcn2 +/− (HET), and Clcn2 −/− (KO) mice as determined by exposure to fluorethyl (applied at 10 ml/min to the chamber; C) or pentylenetetrazol (PTZ; injected at 50 mg/kg body weight; D). C, The apparent slight decrease in threshold in Clcn2 −/− mice (2.34 ± 0.13 min; n = 8 mice) was not statistically different (p = 0.072) from those of WT (2.95 ± 0.25; n = 12) or Clcn2 +/− mice (2.91 ± 0.21 min; n = 3). D, There was no difference between seizure thresholds of WT and KO animals on exposure to PTZ (3.70 ± 1.41 vs 4.04 ± 1.25 min; n = 6 mice each). Error bars indicate SEM.
We also determined seizure thresholds of WT, Clcn2 −/−, and Clcn2 +/− mice. The latter mice more closely mimic patients heterozygous for the truncating CLCN2 mutation Gins597. Seizure susceptibilities of homozygous and heterozygous KO mice, as well as of WT mice, were determined by exposure to the pro-convulsants fluorethyl (Bösl et al., 2001) (Fig. 5 C) or pentylenetetrazol (Hentschke et al., 2006) (Fig. 5 D). The delay between the onset of exposure and the appearance of seizures did not differ significantly between the three genotypes. Hence, neither homozygous nor heterozygous ClC-2 KO mice have reduced seizure thresholds.
Discussion
We found that the disruption of the ClC-2 Cl− channel not only leads to the previously described blindness and male infertility (Bösl et al., 2001; Nehrke et al., 2002) but also to a widespread, progressive spongiform vacuolation of the white matter of the brain and spinal cord, which was accompanied by glial activation. The CNS vacuolation was found within fiber tracts and developed during the period of myelination. Although new vacuoles may form after that time, our data are compatible with the subsequent increase in vacuolar space being attributable to an expansion of existing vacuoles. Electron microscopy revealed that the vacuoles were surrounded by thin myelin layers and contained aberrant myelin sheets, hence suggesting a formation of vacuoles within myelin sheaths. Despite the widespread and extensive vacuolation of the white matter in the brain of ClC-2 KO mice, these animals lacked overt neurological deficits. This may be explained by the apparent lack of neurodegeneration. Fluoro-Jade, which stains degenerating CNS neurons (Schmued et al., 2005), labeled neurons in the degenerating hippocampus of ClC-3 KO mice (Stobrawa et al., 2001) but did not stain cells in brain sections of Clcn2 −/− mice severely affected by myelin vacuolation (supplemental Fig. S10, available at www.jneurosci.org as supplemental material). Furthermore, axons were still ensheathed by multiple layers of myelin. A slight decrease in central nerve conduction velocity was, however, evident from increased latencies in ABRs. Similar increases were found in demyelinating mouse models (Roncagliolo et al., 2000) and in patients with subclinical forms of CNS myelopathies (Robinson and Rudge, 1975; Starr, 1978; Elidan et al., 1982).
The vacuolation affected only central but not peripheral myelin. This may be because of intrinsic differences between oligodendrocytes and Schwann cells, which enwrap axons in the CNS and PNS, respectively. This is suggested by the detection of ClC-2 in CNS tissue but not in sciatic nerve. However, this difference might also be owed to a dependence of oligodendrocytes on astrocytes, which are absent from the PNS. In the brain, the ClC-2 Cl− channel is expressed in neurons and glia (Sik et al., 2000). This probably includes oligodendrocytes, as suggested by Western blots of myelin preparations (supplemental Fig. S8, available at www.jneurosci.org as supplemental material) and by a punctate ClC-2 staining surrounding cerebellar oligodendrocytic somata (Fig. 4 E,F). Some of these puncta were adjacent to Cx47, a gap junction protein exclusively expressed on oligodendrocytic perikarya (Li et al., 2004) that connects these cells with astrocytes. Light microscopy cannot resolve whether ClC-2 is expressed on the membrane of oligodendrocytic or astrocytic membrane at such contact sites. Because ClC-2 was detected (albeit not enriched) in myelin preparations and because subtypes of astrocytes are known to express ClC-2 (Ferroni et al., 1997; Sik et al., 2000; Makara et al., 2003) (Fig. 4 A), either localization is plausible.
An attractive hypothesis that bears resemblance to that proposed for the testicular and retinal degeneration of Clcn2 −/− mice (Bösl et al., 2001) puts ClC-2 into the context of extracellular ion homeostasis by the astrocytic–oligodendrocytic syncytium. Similar to our hypothesis to explain the testicular and retinal degeneration of Clcn2 −/− mice (Bösl et al., 2001), ClC-2 may be needed to regulate the pH in narrow extracellular spaces (e.g., by recycling Cl− for Cl−/HCO 3 − exchangers). It may also be needed for regulating the K+ concentration in these clefts. To prevent detrimental changes in the concentration of K+ in the narrow extracellular spaces between neurons and glia during neuronal activity, glial cells buffer extracellular potassium by K+ uptake and release (“potassium siphoning”). Together with various connexins such as Cx47 and Cx32 (Kleopa et al., 2004) and the water channel aquaporin 4, the inwardly rectifying K+ channel Kir4.1 (Butt and Kalsi, 2006; Neusch et al., 2006) is important for K+ buffering. Depending on its heteromerization, localization, and the electrochemical potential for K+, Kir4.1 is thought to mediate both K+ uptake and release. Interestingly, and in this respect similar to ClC-2, Kir4.1 is expressed in astrocytic endfeet and on oligodendrocytic somata (Neusch et al., 2001, 2006; Butt and Kalsi, 2006). Importantly, the disruption of Kir4.1, as well as the double-KO of Cx32 and Cx47, entailed myelin-associated vacuoles in the CNS (Neusch et al., 2001; Menichella et al., 2003) that resemble the observed myelin vacuolation in our KO mouse model. The disruption of Cx47 in mice had only minor effects (Odermatt et al., 2003), but mutations in the human ortholog GJA12 underlie the demyelinating Pelizaeus-Merzbacher-like disease (Uhlenberg et al., 2004). Unlike Clcn2 −/− mice, however, Kir.4.1 KO mice had neuronal cell loss that resulted in severe neurological deficits. Recent experiments suggest that Kir4.1 and Cx32 and Cx47 function in a common pathway and that the vacuolization of myelin is enhanced by neuronal activity (Menichella et al., 2006). The vacuolation of the optic nerve of Cx47/32 double-KO mice began after eye opening and could be suppressed by inhibiting optic nerve activity with tetrodotoxin. Clcn2 −/− mice display an early, severe retinal degeneration that results in almost complete blindness from birth on (Bösl et al., 2001). Hence, the selective absence of vacuolation in their optic nerves suggests that the need of ClC-2 for oligodendrocyte integrity is particularly high when neuronal activity leads to dynamic changes of extracellular ion concentrations.
The similarities in the vacuolation and expression patterns mentioned above indicate that ClC-2 disruption might cause myelin vacuolation by disturbing the ion homeostasis of the oligodendrocytic/astrocytic network. Newly formed, immature myelin sheaths may be particularly vulnerable, explaining the early emergence of vacuoles that later expand. ClC-2 might be required to buffer extracellular Cl− close to GABAergic synapses (Sik et al., 2000). However, we expect less stringent requirements for the extracellular buffering of Cl− than for K+, because the >20-fold higher extracellular concentration of Cl− implies that relative concentration changes are much larger for K+. We, rather, suggest that ClC-2 is needed to neutralize the electrical currents that are associated with bulk K+ uptake or release by glial cells. The direction of passive Cl− transport depends on the electrochemical potential for Cl−. Several studies indicate that it is above equilibrium in astrocytes (Walz, 2002). Thus, it is plausible that ClC-2 mediates Cl− efflux in astrocytic endfeet, in particular because the parallel efflux of K+ (via channels containing Kir4.1) will increase the open probability of ClC-2 by hyperpolarizing the membrane (Thiemann et al., 1992). An influx of Cl− via ClC-2 is more easily imaginable in oligodendrocytes, in which the electrochemical potential for Cl− is probably at, or slightly above, equilibrium (Walz, 2002). We propose that ClC-2 might serve both as an influx and efflux pathway for Cl−, influenced by the activity-dependent electrochemical gradients present at specific subcellular sites.
The newly discovered CNS pathology of Clcn2 −/− mice strongly resembles mild forms of human leukodystrophy. Some types of human leukodystrophy are associated with only mild neurological deficits (van der Knaap et al., 1995, 1996), and probably several genes for leukodystrophy remain to be identified. However, we did not find convincing CLCN2 mutations in any of the 150 leukodystrophy patients we have analyzed. We are currently not aware of patients with mild leukodystrophy and retinal degeneration, which would more closely resemble our mouse model. In contrast, mutations in CLCN2 were described to be associated with several distinct forms of human epilepsies (Haug et al., 2003; D'Agostino et al., 2004). However, the coding sequence abnormalities found in epileptic patients by D'Agostino et al. (2004) were also found in some of our patients who lacked a record of epilepsy and were recently found on control chromosomes in another study (Stogmann et al., 2006). Because neither variant changed ClC-2 currents (Fig. 5 B), we conclude that they represent rare innocuous polymorphisms. Our electrophysiological analysis of CLCN2 sequence abnormalities described in patients with epilepsy (Haug et al., 2003) did not provide evidence for them being epileptogenic, thereby confirming data from Sepúlveda's group (Niemeyer et al., 2004). Additionally, neither homozygous nor heterozygous ClC-2 KO mice showed spontaneous epileptic activities or reduced seizure thresholds. However, marked effects of the genetic background of mice on seizure susceptibility (D'Agostino et al., 2004) have to be considered.
This work has, rather, revealed a pivotal role of ClC-2 in glial function, putting it into a functional relationship to other ion and water transport proteins that are involved in the ionic homeostasis in the CNS. Together with the previously reported retinal and testicular degeneration of Clcn2 −/− mice (Bösl et al., 2001), the present analysis of their newly discovered leukodystrophy leads us to suggest a more general role of ClC-2 in regulating ion concentrations in small extracellular spaces.
Footnotes
This work was supported in part by Deutsche Forschungsgemeinschaft Grant SFB 444 and the NGFN2 program of the Bundesministerium für Bildung und Forschung. M.A. was supported by a fellowship from the Swiss National Science Foundation. We thank A. Gal and J. Gärtner for providing access to their collection of DNA from patients with leukodystrophy, R. Martini and H. Goebel for helpful discussions, A. Senning for sequence analysis in Hamburg, and B. Merz and E. Orthey for technical assistance.
References
- Altevogt BM, Paul DL. Four classes of intercellular channels between glial cells in the CNS. J Neurosci. 2004;24:4313–4323. doi: 10.1523/JNEUROSCI.3303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bösl MR, Stein V, Hübner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, Davidoff MS, Holstein AF, Jentsch TJ. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl− channel disruption. EMBO J. 2001;20:1289–1299. doi: 10.1093/emboj/20.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid LP, Montrose-Rafizadeh C, Smith DI, Guggino WB, Cutting GR. Cloning of a putative human voltage-gated chloride channel (ClC-2) cDNA widely expressed in human tissues. Hum Mol Genet. 1995;4:407–413. doi: 10.1093/hmg/4.3.407. [DOI] [PubMed] [Google Scholar]
- D'Agostino D, Bertelli M, Gallo S, Cecchin S, Albiero E, Garofalo PG, Gambardella A, St Hilaire JM, Kwiecinski H, Andermann E, Pandolfo M. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology. 2004;63:1500–1502. doi: 10.1212/01.wnl.0000142093.94998.1a. [DOI] [PubMed] [Google Scholar]
- Elidan J, Sohmer H, Gafni M, Kahana E. Contribution of changes in click rate and intensity on diagnosis of multiple sclerosis by brainstem auditory evoked potentials. Acta Neurol Scand. 1982;65:570–585. doi: 10.1111/j.1600-0404.1982.tb03111.x. [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229–237. doi: 10.1111/j.1750-3639.1994.tb00838.x. [DOI] [PubMed] [Google Scholar]
- Estévez R, Boettger T, Stein V, Birkenhäger R, Otto E, Hildebrandt F, Jentsch TJ. Barttin is a Cl− channel β-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- Ferroni S, Marchini C, Nobile M, Rapisarda C. Characterization of an inwardly rectifying chloride conductance expressed by cultured rat cortical astrocytes. Glia. 1997;21:217–227. doi: 10.1002/(sici)1098-1136(199710)21:2<217::aid-glia5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Gründer S, Thiemann A, Pusch M, Jentsch TJ. Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature. 1992;360:759–762. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- Haug K, Warnstedt M, Alekov AK, Sander T, Ramirez A, Poser B, Maljevic S, Hebeisen S, Kubisch C, Rebstock J, Horvath S, Hallmann K, Dullinger JS, Rau B, Haverkamp F, Beyenburg S, Schulz H, Janz D, Giese B, Müller-Newen G, et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003;33:527–532. doi: 10.1038/ng1121. [DOI] [PubMed] [Google Scholar]
- Hentschke M, Wiemann M, Hentschke S, Kurth I, Hermans-Borgmeyer I, Seidenbecher T, Jentsch TJ, Gal A, Hübner CA. Mice with a targeted disruption of the Cl−/HCO3 − exchanger AE3 display a reduced seizure threshold. Mol Cell Biol. 2006;26:182–191. doi: 10.1128/MCB.26.1.182-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Jentsch TJ. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997;16:1582–1592. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006;25:642–652. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa KA, Orthmann JL, Enriquez A, Paul DL, Scherer SS. Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia. 2004;47:346–357. doi: 10.1002/glia.20043. [DOI] [PubMed] [Google Scholar]
- Li X, Ionescu AV, Lynn BD, Lu S, Kamasawa N, Morita M, Davidson KG, Yasumura T, Rash JE, Nagy JI. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Rappert A, Matthias K, Steinhäuser C, Spät A, Kettenmann H. Astrocytes from mouse brain slices express ClC-2-mediated Cl− currents regulated during development and after injury. Mol Cell Neurosci. 2003;23:521–530. doi: 10.1016/s1044-7431(03)00080-0. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, Baggs R, Shull GE, Melvin JE. Loss of hyperpolarization-activated Cl− current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem. 2002;277:23604–23611. doi: 10.1074/jbc.M202900200. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Müller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bähr M, Richter DW, Kirchhoff F, Hülsmann S. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–1852. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Yusef YR, Cornejo I, Flores CA, Sepúlveda FV, Cid LP. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol Genomics. 2004;19:74–83. doi: 10.1152/physiolgenomics.00070.2004. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Büssow H, Schilling K, Steinhäuser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Robinson K, Rudge P. Auditory evoked responses in multiple sclerosis. Lancet. 1975;1:1164–1166. doi: 10.1016/s0140-6736(75)93138-4. [DOI] [PubMed] [Google Scholar]
- Roncagliolo M, Benitez J, Eguibar JR. Progressive deterioration of central components of auditory brainstem responses during postnatal development of the myelin mutant taiep rat. Audiol Neurootol. 2000;5:267–275. doi: 10.1159/000013891. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, van der Knaap MS. The latest on leukodystrophies. Curr Opin Neurol. 2004;17:187–192. doi: 10.1097/00019052-200404000-00017. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Sik A, Smith RL, Freund TF. Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience. 2000;101:51–65. doi: 10.1016/s0306-4522(00)00360-2. [DOI] [PubMed] [Google Scholar]
- Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- Staley K, Smith R, Schaack J, Wilcox C, Jentsch TJ. Alteration of GABAA receptor function following gene transfer of the CLC-2 chloride channel. Neuron. 1996;17:543–551. doi: 10.1016/s0896-6273(00)80186-5. [DOI] [PubMed] [Google Scholar]
- Starr A. Sensory evoked potentials in clinical disorders of the nervous system. Annu Rev Neurosci. 1978;1:103–127. doi: 10.1146/annurev.ne.01.030178.000535. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch TJ. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. 1991;354:304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/s0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Stogmann E, Lichtner P, Baumgärtner C, Schmied M, Hotzy C, Asmus F, Leutmezer F, Bonelli S, Assem-Hilger E, Vass K, Hatala K, Strom TM, Meitinger T, Zimprich F, Zimprich A. Mutations in the CLCN2 gene are a rare cause of idiopathic generalized epilepsy syndromes. Neurogenetics. 2006;7:265–268. doi: 10.1007/s10048-006-0057-x. [DOI] [PubMed] [Google Scholar]
- Thiemann A, Gründer S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- Uhlenberg B, Schuelke M, Rüschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloglu H, Nürnberg P, Hübner C, Weschke B, Gartner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Valk J, Barth PG, Smit LM, van Engelen BG, Tortori Donati P. Leukoencephalopathy with swelling in children and adolescents: MRI patterns and differential diagnosis. Neuroradiology. 1995;37:679–686. doi: 10.1007/BF00593394. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS, Barth PG, Vrensen GF, Valk J. Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol (Berl) 1996;92:206–212. doi: 10.1007/s004010050510. [DOI] [PubMed] [Google Scholar]
- Walz W. Chloride/anion channels in glial cell membranes. Glia. 2002;40:1–10. doi: 10.1002/glia.10125. [DOI] [PubMed] [Google Scholar]
- Zuñiga L, Niemeyer MI, Varela D, Catalán M, Cid LP, Sepúlveda FV. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J Physiol (Lond) 2004;555:671–682. doi: 10.1113/jphysiol.2003.060046. [DOI] [PMC free article] [PubMed] [Google Scholar]