

Abstract
Cerebral microvascular amyloid β protein (Aβ) deposition and associated neuroinflammation is increasingly recognized as an important component leading to cognitive impairment in Alzheimer's disease and related cerebral amyloid angiopathy disorders. Transgenic mice expressing the vasculotropic Dutch/Iowa (E693Q/D694N) mutant human Aβ precursor protein in brain (Tg-SwDI) accumulate abundant cerebral microvascular fibrillar amyloid deposits and exhibit robust neuroinflammation. In the present study, we investigated the effect of the anti-inflammatory drug minocycline on Aβ accumulation, neuroinflammation, and behavioral deficits in Tg-SwDI mice. Twelve-month-old mice were treated with saline or minocycline by intraperitoneal injection every other day for a total of 4 weeks. During the final week of treatment, the mice were tested for impaired learning and memory. Brains were then harvested for biochemical and immunohistochemical analysis. Minocycline treatment did not alter the cerebral deposition of Aβ or the restriction of fibrillar amyloid to the cerebral microvasculature. Similarly, minocycline-treated Tg-SwDI mice exhibited no change in the levels of total Aβ, the ratios of Aβ40 and Aβ42, or the amounts of soluble, insoluble, or oligomeric Aβ compared with the saline-treated control Tg-SwDI mice. In contrast, the numbers of activated microglia and levels of interleukin-6 were significantly reduced in minocycline-treated Tg-SwDI mice compared with saline-treated Tg-SwDI mice. In addition, there was a significant improvement in behavioral performance of the minocycline-treated Tg-SwDI mice. These finding suggest that anti-inflammatory treatment targeted for cerebral microvascular amyloid-induced microglial activation can improve cognitive deficits without altering the accumulation and distribution of Aβ.
Keywords: cerebral microvascular amyloid, neuroinflammation, cognitive impairment, microglia, transgenic mice, behavior
Introduction
Characteristic features of Alzheimer's disease (AD) include cognitive decline, amyloid β-peptide (Aβ) deposition, formation of neurofibrillary tangles, and increase of inflammatory cells and molecules (Selkoe, 2001; Walsh and Selkoe, 2004). Aβ peptides are proteolytic cleavage products of the Aβ precursor protein (AβPP) sequentially processed by β- and γ-secretases (Selkoe, 2001). Deposition of Aβ in AD brain occurs in either diffuse or fibrillar forms. In addition to parenchymal plaques, fibrillar Aβ deposition in the cerebral vasculature, a condition known as cerebral amyloid angiopathy (CAA), is commonly found in AD (Jellinger, 2002; Rensink et al., 2003; Zhang-Nunes et al., 2006). Mutations in the Aβ sequence that result in familial forms of CAA, including Dutch type (E22Q) and Iowa type (D23N), cause early-onset and severe cerebral vascular amyloid deposition (Levy et al., 1990; Van Broeckhoven et al., 1990; Grabowski et al., 2001).
Activated microglia and reactive astrocytes, often associated with extracellular Aβ deposits, contribute not only to the production of cytokines, chemokines, reactive oxygen species, and neurotoxic substances, but also to neurotrophic factors (Combs et al., 2001; Mrak and Griffin, 2001; Streit, 2004). The relationship between this cell activation and the cognitive dysfunction in Aβ-depositing diseases is not well understood, partially because of the complex responses generated by activated inflammatory cells. Recent studies have implicated cerebral microvascular Aβ deposition in promoting neuroinflammation and dementia in AD and related familial CAA disorders (Vinters, 2001; Thal et al., 2003; Atterns and Jellinger, 2004; Bailey et al., 2004; Eng et al., 2004; Greenberg et al., 2004). Cerebral microvascular amyloid, especially in familial CAA disorders, is associated with a strong localized neuroinflammatory reaction (Eng et al., 2004; Maat-Schieman et al., 2004; Zhang-Nunes et al., 2006).
Recently, we generated transgenic mice (Tg-SwDI) that express human Swedish/Dutch/Iowa mutant AβPP in brain. The resulting Dutch/Iowa mutant Aβ peptides exhibit highly vasculotropic properties and low efficiency for transporting across the blood–brain barrier, resulting in early-onset and robust accumulation of Aβ in brain, particularly in the cerebral microvasculature (Davis et al., 2004, 2006; Deane et al., 2004; Miao et al., 2005a). In this Tg-SwDI mouse model, we also demonstrated a marked increase of activated microglia and reactive astrocytes in association with the cerebral microvascular amyloid and elevated levels of the inflammatory cytokines interleukin (IL)-1β and IL-6 (Miao et al., 2005a).
Minocycline, a tetracycline derivative with anti-inflammatory properties that crosses the blood–brain barrier, has been shown to suppress the activation of cultured human and mouse microglia stimulated with Aβ (Familian et al., 2006; Seabrook et al., 2006). In animal models of neurological diseases such as Parkinson's disease (Quintero et al., 2006), Huntington's disease (Pattison et al., 2006), amyotrophic lateral sclerosis (Zhu et al., 2002), and Down's syndrome (Hunter et al., 2004), minocycline treatment has been reported to ameliorate symptoms, delay disease progression, prevent cell death, and improve behavioral deficits, all of which closely correlated with its ability to inhibit chronic neuroinflammation associated with these diseases.
In the present study, we investigated the effects of minocycline on Aβ accumulation, inflammatory cell activation, and cognitive performance in Tg-SwDI mice. Minocycline treatment had no effect on cerebral Aβ accumulation, microvascular amyloid load, or soluble oligomeric Aβ levels. Although reactive astrocyte levels were unaffected, there was a significant reduction in the numbers of activated microglial cells in the minocycline-treated Tg-SwDI mice. Moreover, minocycline treatment reduced the levels of proinflammatory IL-6 in Tg-SwDI mice. Last, treatment with minocycline significantly improved the performance of Tg-SwDI mice in spatial learning memory. Together, these findings suggest that cerebral microvascular amyloid-induced activation of microglia is involved in cognitive impairment in Tg-SwDI mice.
Materials and Methods
Animals and treatments.
All work with mice followed National Institutes of Health guidelines and was approved by the Stony Brook University Institutional Animal Care and Use Committee. Generation of Tg-SwDI mice on a pure C57BL/6 background was described recently (Davis et al., 2004). These mice express low levels of human Swedish/Dutch/Iowa mutant AβPP in neurons under control of the mouse Thy1.2 promoter. Homozygous line B Tg-SwDI and nontransgenic C57BL/6 mice at 12 months of age were used in the present study.
Minocycline (Sigma-Aldrich, St. Louis, MO) was prepared as a 5 mg/ml stock solution in saline, pH 7.4, aliquoted, and stored frozen at −80°C. Tg-SwDI mice as well as C57BL/6 mice (n = 9 for each group) were 12 months of age at the beginning of the experiment. Each mouse received intraperitoneal injection of minocycline solution at a dose of 10 ml/kg of body weight (i.e., 50 mg/kg) every other day for the period of 28 d. Another group of Tg-SwDI mice (n = 9) received 10 ml/kg saline injection according to the same schedule. Animal body weights were measured at the beginning of the experiment and every week thereafter. During the last week of injection, all three groups of animals were subjected to behavioral testing as described below.
Tissue preparation.
Mice were killed 24 h after the last injection. After cold-PBS perfusion, brains were removed and dissected through the midsagittal plane. One hemisphere was either immersion fixed with 70% ethanol overnight and subjected to increasing sequential dehydration in ethanol, followed by xylene treatment and embedding in paraffin, or fixed with 4% paraformaldehyde overnight at 4°C and subjected to increasing concentrations (10, 20, or 30%) of sucrose in PBS and then embedded in OCT (optimal cutting temperature) compound (Sakura Finetek, Torrance, CA) and snap frozen in dry ice. Sagittal sections were cut at 10 μm thickness using a Leica RM2135 microtome (Leica Microsystems, Bannockburn, IL), placed in a flotation water bath at 40°C, and then mounted on Colorfrost/Plus slides (Fisher Scientific, Houston, TX). Alternatively, coronal sections were cut at 20 μm thickness from frozen brains using a Leica CM1900 cryostat (Leica Microsystems) and stored in PBS with 0.02% sodium azide at 4°C.
Immunohistochemical analysis.
Immunostainings were performed on deparaffinized sections or free-floating sections. Antigen retrieval was performed by treatment with proteinase K (0.2 mg/ml) for 5 min at room temperature for Aβ and collagen type IV immunostaining, with 1:100 antigen-unmasking solution (Vector Laboratories, Burlingame, CA) for 30 min at 90°C in a water bath for activated microglia immunostaining with 5D4 antibody, or with 10 mm sodium citrate, pH 6.0, for 30 min at 90°C for major histocompatibility complex class II (MHCII) microglial staining. Nonspecific binding was blocked by incubating in PBS containing 0.1% Triton X-100 and 2% bovine serum albumin (Sigma-Aldrich) for 20 min at room temperature. Primary antibodies were incubated with the brain sections overnight at 4°C and detected with horseradish peroxidase-conjugated or alkaline phosphatase-conjugated secondary antibodies. Alternatively, peroxidase-conjugated streptavidin in conjunction with biotinylated secondary antibody was used for detecting microglia. Peroxidase activity was visualized either with a stable diaminobenzidine solution (Invitrogen, Carlsbad, CA) or with the fast red substrate system (Spring Bioscience, Fremont, CA), respectively, as substrate. Thioflavin-S staining for fibrillar amyloid was performed as described previously (Dickson et al., 1990). The following antibodies were used for immunostaining: monoclonal antibody (mAb) 66.1 (1:250), which recognizes residues 1–5 of human Aβ (Deane et al., 2003); rabbit polyclonal antibody to collagen type IV (1:100; Research Diagnostics,, Flanders, NJ); monoclonal antibody to glial fibrillary acidic protein (GFAP) for identification of astrocytes (1:1000; Millipore, Billerica, MA); monoclonal antibody 5D4 to keratan sulfate (1:300; Seikagaku, Tokyo, Japan) and monoclonal antibody to MHC class II (1:200; BD Biosciences PharMingen, San Jose, CA) for identification of activated microglia; and biotinylated goat anti-mouse IgG (1:200) and ABC kit (Vector Laboratories) according to the manufacturer's recommendations.
Quantitative analysis of reactive astrocytes and activated microglia densities.
Total numbers of reactive astrocytes and activated microglia in the frontotemporal cortex, CA1 and CA2 fields of the hippocampus, thalamus, and subiculum regions were estimated using the Stereologer software system (Systems Planning and Analysis, Alexandria, VA) as described previously (Long et al., 1998; Miao et al., 2005a,b). Briefly, every 10th section was selected and generated 10–15 sections per reference space in a systematic-random manner. Immunopositive cells were counted using the optical fractionator method with the dissector principle and unbiased counting rules. Criteria for counting cells required that cells exhibited positive immunostaining (GFAP for astrocytes and mAb to I-A/I-E MHC class II alloantigens or mAb 5D4 to keratan sulfate for activated microglia) and morphological features consistent with each cell type.
Immunochemical analysis of Aβ peptides.
Soluble pools of Aβ40 and Aβ42 were determined by performing ELISA analysis of carbonate-extracted mouse forebrain tissue, and subsequently the insoluble Aβ40 and Aβ42 levels were determined by ELISA of guanidine lysates of the insoluble pellets resulting from the carbonate-extracted brain tissue (Johnson-Wood et al., 1997; DeMattos et al., 2002). The Aβ sandwich ELISA consisted of the capture antibody mAb 21F12 and 2G3 for Aβ42 and Aβ40, respectively, and the biotinylated reporter antibody 3D6. Total Aβ40 and Aβ42 levels were determined by combining the soluble and insoluble levels of each form.
Soluble Aβ oligomers were analyzed in TBS-soluble forebrain fractions using dot blot analysis. Briefly, brain fractions were loaded to nitrocellulose membranes (Schleicher and Schuell BioScience, Hertogen-Bosch, The Netherlands). Blots were washed for 15 min in PBS containing 0.05% Tween 20 (PBST), preincubated with blocking solution (1% milk powder in PBST), washed three times with PBST, and subsequently incubated with primary polyclonal anti-oligomer antibody OC11 and peroxidase-labeled secondary antibodies (GE Healthcare, Piscataway, NJ). Detection was performed by chemiluminescence according to the description of the manufacturer (Bio-Rad, Hercules, CA) and quantitated using Bio-Rad VersaDoc system and the Quantity One software.
Measurement of mouse brain IL-6 levels.
Hemibrains isolated from perfused mice were homogenized in 10 volumes of 50 mm Tris-HCl, 150 mm NaCl, pH 7.5, containing protease inhibitor mixture (SM Biotech, Huntington Station, NY) at 4°C. The samples were centrifuged at 14,000 × g for 50 min at 4°C. The supernatants were collected, and the protein concentrations were determined using the BCA kit (Pierce Biotechnology, Rockford, IL). The levels of IL-6 in the samples were determined using a mouse IL-6 immunoassay kit (Biosource International, Camarillo, CA).
Behavior evaluation.
A modified Barnes maze apparatus (Barnes, 1979) was used to assess ability to learn the location of an escape box over the course of 5 d. The Barnes maze task was chosen because we have shown recently that this test reliably detects differences in Tg-SwDI mice compared with wild-type controls and that these deficits are likely the result of specific damage to the memory- and motivation-related efferents of the hippocampus originating in the subiculum (Xu et al., 2007). The Barnes maze used is a circular table with a diameter of 91 cm, with eight 5-cm-diameter holes located equidistantly around the perimeter. Each hole is 35.5 cm from the center of the table. The Barnes maze was positioned in the center of the test room and 71 cm from the ground. During a trial, the escape box, measuring 11 × 7.5 × 7.5 cm, was placed under the escape hole. The escape hole was in a different position for each mouse but constant for each mouse over the 5 training days. Each mouse was tested twice per day for 5 d, with a 15 min intertrial interval separating each trial. The trial began when the mouse was released from a start box, measuring 16.5 × 9 × 5 cm, attached to a 1 m pole. Each trial lasted up to 2 min or until the mouse entered the escape box. Mice that did not find the escape hole within 2 min were guided by the experimenter to the correct hole. Once the mouse entered the escape box, it was allowed to remain in the box for 1 min.
Statistical analysis.
Histological and biochemical data were analyzed by Student's t test at the 0.05 significance level. The data from the Barnes maze behavioral testing were analyzed by repeated-measures ANOVA. The performance of each subject for each testing day was the average latency of the two trials.
Results
Minocycline treatment does not affect Aβ accumulation in Tg-SwDI mice
Previously, we showed that Tg-SwDI mice selectively accumulate fibrillar amyloid in the cerebral microvasculature and that this was associated with a strong localized neuroinflammatory reaction with robust microglial activation (Davis et al., 2004, Miao et al., 2005a). Therefore, we investigated whether minocycline, a tetracycline derivative with anti-inflammatory properties, was effective in suppressing cerebral microvascular amyloid-induced neuroinflammation in this model. Twelve-month-old mice were administered minocycline or saline via intraperitoneal injection every other day for a period of 4 weeks. After this time, the mice were killed, and the brains were harvested for analysis. The pattern of cerebral Aβ accumulation did not differ between the saline- and minocycline-treated Tg-SwDI mice, with both groups showing the characteristic diffuse parenchymal deposits and fibrillar amyloid being restricted to the cerebral microvasculature (Fig. 1). We next determined whether minocycline treatment affected the amount of cerebral accumulation of different forms of Aβ peptides in Tg-SwDI mice. Quantitative ELISA analysis revealed that there was essentially no difference in the amounts of total Aβ between saline- and minocycline-treated Tg-SwDI mice (29,321 vs 29,295 pg of Aβ/mg of total brain protein, respectively). Furthermore, the ratios of total Aβ40 and Aβ42 or the amounts of soluble and insoluble Aβ between saline- and minocycline-treated Tg-SwDI mice did not differ (Fig. 2 A,B, respectively). Although minocycline did not appear to affect the soluble pools of Aβ in Tg-SwDI mice, we further determined whether there was an effect on the levels of soluble Aβ oligomers. To do this, we performed quantitative dot blot analysis using a polyclonal antibody designated OC11 that is specific for oligomer forms of Aβ. Figure 3 A confirms that the OC11 antibody specifically recognizes soluble oligomeric, but not monomeric, forms of Dutch/Iowa mutant Aβ peptide. Quantitative dot blot analysis using the oligomer-specific antibody showed that there was no difference in the amounts of soluble Aβ oligomers in saline- and minocycline-treated Tg-SwDI mice (Fig. 3 B). The relative levels of Aβ oligomers were 1.0 ± 0.2 versus 1.1 ± 0.1 (mean ± SD; n = 5) in saline- and minocycline-treated Tg-SwDI mice, respectively. Similarly, using a sandwich ELISA composed of mAb 3D6 to capture Aβ and biotinylated mAb 3D6 to detect any size of oligomeric Aβ species as described by DeMattos et al. (2002) failed to show any significant difference in the amounts of soluble Aβ oligomers between the two groups of Tg-SwDI mice (data not shown).
Figure 1.

Minocycline treatment does not alter the spatial accumulation of Aβ in Tg-SwDI mice. A, B, Aβ accumulation in the forebrain of 12-month-old Tg-SwDI mice treated with saline (A) or minocycline (B). Scale bars, 1 mm. C, D, Colocalization of vascular collagen IV immunostaining (red) and thioflavin-S amyloid staining (green) in the thalamus of 12-month-old Tg-SwDI mice treated with saline (C) or minocycline (D). Scale bars, 50 μm.
Figure 2.

Minocycline treatment does not affect the levels of Aβ peptides in Tg-SwDI mouse forebrain. A, ELISA measurements of total Aβ40 (gray bars) and Aβ42 (black bars) in mouse forebrain tissue. B, ELISA measurements of soluble (Sol) Aβ (gray bars) and insoluble (Insol) Aβ (black bars) in mouse forebrain tissue. ELISA data shown are mean ± SD (n = 9 per group).
Figure 3.

Minocycline treatment does not alter the levels of soluble Aβ oligomers in Tg-SwDI mouse forebrain. A, Immunoblot analysis of monomeric and oligomeric Dutch/Iowa Aβ40 using the anti-Aβ monoclonal antibody 6E10 (first lane) and anti-oligomeric Aβ polyclonal antibody OC11 (second lane). B, Representative dot blot analysis of Aβ oligomers in soluble mouse forebrain extracts using polyclonal antibody OC11.
Minocycline reduces microglial, but not astrocyte, activation and lowers IL-6 levels in Tg-SwDI
We reported previously that markedly elevated numbers of reactive astrocytes and activated microglial cells were present in Tg-SwDI mice, especially in the subiculum and thalamus, regions that have extensive fibrillar cerebral microvascular Aβ deposition (Miao et al., 2005a;b). After 4 weeks of treatment, we compared the numbers of GFAP-immunopositive astrocytes in saline- and minocycline-treated Tg-SwDI mice as well as minocycline-treated C57BL/6 mice. Both groups of Tg-SwDI mice showed large increases in the numbers of astrocytes compared with the C57BL/6 mice (Fig. 4). However, there was no effect of minocycline on the increased number of reactive astrocytes in all of the regions measured in Tg-SwDI mice (Fig. 4 D). However, compared with saline-treated animals, the numbers of MHCII-immunopositive activated microglia in the minocycline-treated Tg-SwDI mice were significantly reduced, particularly in the hippocampus, subiculum, and thalamus, the regions with high amounts of cerebral microvascular amyloid (Fig. 5). Wild-type mice exhibited no activated microglia (Fig. 5 C). Furthermore, when immunostaining for activated microglia using another surface marker mAb 5D4, the intensity of staining was demonstrably lower in the minocycline-treated Tg-SwDI mice, further suggesting an altered activation state (Fig. 5 F).
Figure 4.

Minocycline treatment does not affect the numbers of reactive astrocytes in Tg-SwDI mice. A–C, Microvascular-associated reactive astrocytes revealed by GFAP-positive immunostaining (brown) and collagen type IV (red). The thalamic regions of 12-month-old Tg-SwDI mice treated with saline (A) or minocycline (B) or 12-month-old wild-type mice (C) are shown. Scale bars, 50 μm. D, Quantitative stereological estimation of reactive astrocyte densities in different brain regions of wild-type mice (white bars), saline-treated Tg-SwDI mice (black bars), or minocycline-treated Tg-SwDI mice (gray bars). Brain regions measured include frontotemporal cortex (Ctx), hippocampus (Hipp), thalamus (Thal), and subiculum (Sub). Data shown are mean ± SD (n = 5). n.s., Not significant. *p < 0.001.
Figure 5.
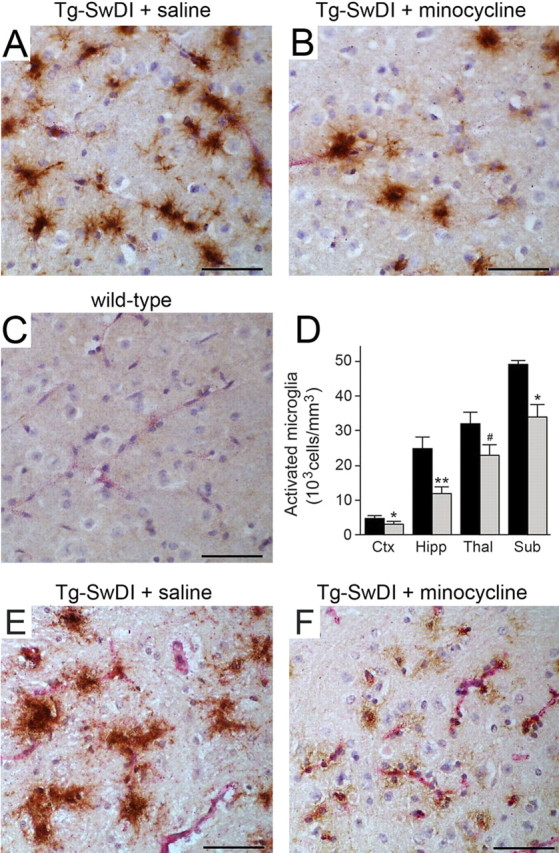
Minocycline treatment reduces microglial activation in Tg-SwDI mice. A–C, Microvascular-associated activated microglia revealed by MHCII-positive immunostaining (brown) and collagen type IV (red). The thalamic regions of 12-month-old Tg-SwDI mice treated with saline (A) or minocycline (B) or 12-month-old wild-type mice (C) are shown. Scale bars, 50 μm. D, Quantitative stereological estimation of activated microglial densities in different brain regions of saline-treated Tg-SwDI mice (black bars) or minocycline-treated Tg-SwDI mice (gray bars). Wild-type mice exhibited no activated microglia. Data shown are mean ± SD (n = 5). *p < 0.02; **p < 0.001; # p < 0.01. Brain regions measured include frontotemporal cortex (Ctx), hippocampus (Hipp), thalamus (Thal), and subiculum (Sub). E, F, Activated microglia revealed by 5D4-positive immunostaining in the thalamic region of 12-month-old Tg-SwDI mice treated with saline (E) or minocycline (F). Scale bars, 50 μm.
We showed previously that IL-6 levels are increased in Tg-SwDI mice exhibiting cerebral microvascular amyloid and microglial activation (Miao et al., 2005b). Here, we show that there was a significant decrease in IL-6 levels in Tg-SwDI mice treated with minocycline compared with saline-treated animals (Fig. 6). Together, these results indicate that minocycline can selectively reduce the numbers and state of activated microglia in response to cerebral microvascular amyloid and lower the levels of proinflammatory IL-6 in Tg-SwDI mice.
Figure 6.

Minocycline reduces elevated levels of IL-6 in Tg-SwDI mice. The levels of IL-6 were measured in soluble forebrain extracts of 12-month-old wild-type and Tg-SwDI mice treated with saline or minocycline by ELISA analysis. Data shown are mean ± SD (n = 5). *p < 0.005; **p < 0.01.
Minocycline improves spatial learning memory performance of Tg-SwDI mice
We used a modified version of the Barnes maze task (Barnes, 1979) to measure deficits in spatial learning memory in Tg-SwDI mice. Figure 7 shows the results for latency to find the escape box of the Barnes maze for 12-month-old animals, demonstrating that saline-treated Tg-SwDI mice took significantly longer to find the escape hole than the age-matched wild-type C57BL/6 mice. However, after 4 weeks of minocycline treatment, the learning memory performance was significantly improved in the Tg-SwDI mice. These observations are supported by both a significant main effect (F (2,23) = 15.1; p < 0.0001) and trial × group interaction (F (8,92) = 2.7; p < 0.01) by repeated-measures ANOVA. These results suggest that the selective reduction of microglial activation by minocycline can reverse cerebral microvascular amyloid-induced behavioral deficits in Tg-SwDI mice.
Figure 7.

Minocycline treatment improves spatial learning memory deficits in Tg-SwDI mice as assessed by performance in the Barnes maze task. Twelve-month-old wild-type mice (squares), saline-treated Tg-SwDI mice (open circles), or minocycline-treated Tg-SwDI mice (closed circles) were measured for their latencies to find the escape box over the course of acquisition. Data shown are mean ± SEM (n = 9 mice per group).
Discussion
In the present study, we demonstrate that treatment of Tg-SwDI mice with the anti-inflammatory drug minocycline significantly reduced the number of activated microglia in thalamus and subiculum, areas that contained extensive cerebral microvascular Aβ deposition. The reduction in microglial activation occurred in the absence of any demonstrable change in the spatial deposition of Aβ or the amounts of soluble or insoluble Aβ, including soluble Aβ oligomers. Furthermore, the reduction in microglial activation was accompanied by a significant improvement in learning memory performance by Tg-SwDI mice.
We showed previously that cerebral microvascular fibrillar amyloid deposition can exclusively induce local neuroinflammation in Tg-SwDI mice in the absence of parenchymal plaque fibrillar amyloid deposition (Miao et al., 2005a,b). Among those inflammatory responses, reactive astrocytes and activated microglia were found to be tightly associated with the cerebral microvascular amyloid deposition. As shown in Tg-SwDI mice, the numbers of these neuroinflammatory cells were highest in the regions with the most extensive cerebral microvascular amyloid (i.e., thalamus and subiculum) and increased in conjunction with the increase in cerebral microvascular Aβ load in all brain regions. The strong association of reactive astrocytes and activated microglia with cerebral microvascular amyloid in Tg-SwDI mice is highly consistent with the cerebral vascular localization of these neuroinflammatory cells in patients with the Dutch- or Iowa-type familial CAA disorders (Grabowski et al., 2001; Maat-Schieman et al., 2004; Zhang-Nunes et al., 2006). In these disorders, as in the Tg-SwDI mice, the induction of a neuroinflammatory reaction appears to mostly correlate with amyloid extending from the vessel wall into the surrounding parenchymal tissues.
Inhibition of chronic neuroinflammation, particularly of microglial activation, has been suggested to be a practical strategy in the treatment of neurodegenerative diseases (Kriz et al., 2002; Zhu et al., 2002; Hirsch et al., 2003). We show here that at 12 months of age, treatment with the anti-inflammatory agent minocycline, which did not affect cerebral Aβ levels or deposition, yet significantly reduced microglial activation and lowered proinflammatory cytokine levels, was able to substantially improve behavioral performance of Tg-SwDI mice. These data further support the hypothesis that microglial activation is involved in promoting cognitive impairment in this transgenic mouse model of cerebral microvascular amyloid. However, the behavioral impairment in Tg-SwDI mice was not fully rescued by minocycline treatment. This may be a result of the incomplete inhibition of microglial activation, because there was still activated microglia well above the wild-type level. In addition, other factors may contribute to the cognitive decline of Tg-SwDI mice at this disease stage. In this regard, Seabrook et al. (2006) recently showed that minocycline treatment reduced the secretion of IL-6, IL-1β, and tumor necrosis factor α from cultured microglia stimulated by Aβ. Consistent with these in vitro findings, we found that minocycline significantly lowered the brain levels of IL-6 in Tg-SwDI mice (Fig. 6). In the future, it will be important to completely define the mediators through which anti-inflammatory treatments prevent/reverse learning memory impairment, by investigating the expression of inflammatory factors by activated microglia in Tg-SwDI mice in the presence or absence of anti-inflammatory drugs.
The present study suggests a significant role for neuroinflammation, in particular microglial activation, in disrupting normal cognitive function in cerebral vascular amyloid-depositing diseases. Because CAA pathology is commonly found in AD, our findings may have additional implications in combined treatment strategies for this neurodegenerative condition as well. There has been significant interest in the use of anti-inflammatory drugs for the treatment of AD and other Aβ-depositing disorders. This interest derives from epidemiological studies that implicate prolonged use of anti-inflammatory drugs in reducing the risk of AD, delaying the age of onset, and slowing the progression of disease and cognitive impairments (In't Veld et al., 2002; Etminan et al., 2003). Despite the potential for anti-inflammatory drugs in the treatment of AD, they have not been successful in slowing disease progression in clinical trials (Aisen, 2002). The lack of success may reflect intervention too late in the pathogenesis of AD. However, recent studies have suggested that treatments aimed at reducing CAA-induced inflammation in afflicted individuals have improved the dementia and the recurrence of cerebral hemorrhage associated with this particular pathology (Hoshi et al., 2000; Eng et al., 2004; Harkness et al., 2004; Oh et al., 2004). However, the bases for these successes in using anti-inflammatory treatment regarding CAA remain unclear. The present results indicate that Tg-SwDI mice provide a unique model to further investigate the role of neuroinflammation and microglial activation associated with cerebral microvascular amyloid leading to cognitive impairment.
Footnotes
This work was supported in part by National Institutes of Health Grants NS55118 and NS36645. Antibody reagents for the Aβ ELISA and detection of soluble Aβ oligomers were generously provided by Lilly Research Laboratories (Indianapolis, IN) and Dr. Charles Glabe (University of California, Irvine, Irvine, CA), respectively.
References
- Aisen PS. The potential of anti-inflammatory drugs for the treatment of Alzheimer's disease. Lancet Neurol. 2002;1:279–284. doi: 10.1016/s1474-4422(02)00133-3. [DOI] [PubMed] [Google Scholar]
- Atterns J, Jellinger KA. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol (Berl) 2004;107:83–90. doi: 10.1007/s00401-003-0796-9. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer's disease. Neurol Res. 2004;26:573–578. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: a neuropsychological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. β-amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Ziegler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Deficient cerebral clearance of vasculotropic Dutch/Iowa double mutant Aβ in human AβPP transgenic mice. Neurobiol Aging. 2006;26:946–954. doi: 10.1016/j.neurobiolaging.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid β-peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Yan SD, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP-amyloid β-peptide (Aβ) interaction regulates differential brain efflux of Aβ isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, O'dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2002;99:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Wertkin A, Mattiace LA, Fier E, Kress Y, Davies P, Yen SH. Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer's disease. Acta Neuropathol (Berl) 1990;79:486–493. doi: 10.1007/BF00296107. [DOI] [PubMed] [Google Scholar]
- Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on the risk of Alzheimer's disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128–132. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004;55:250–256. doi: 10.1002/ana.10810. [DOI] [PubMed] [Google Scholar]
- Familian A, Boshuizen RS, Eikelenboom P, Veerhuis R. Inhibitory effect of minocycline on amyloid beta fibril formation and human microglial activation. Glia. 2006;53:233–240. doi: 10.1002/glia.20268. [DOI] [PubMed] [Google Scholar]
- Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Harkness KA, Coles A, Pohl U, Xuereb JH, Baron JC, Lennox GG. Rapidly reversible dementia in cerebral amyloid inflammatory vasculopathy. Eur J Neurol. 2004;11:59–62. doi: 10.1046/j.1351-5101.2003.00707.x. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson's disease. Ann NY Acad Sci. 2003;991:214–228. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Hoshi K, Yoshida K, Nakamura A, Tada T, Tamaoka A, Ikeda S. Cessation of cerebral hemorrhage recurrence associated with corticosteroid treatment in a patient with cerebral amyloid angiopathy. Amyloid. 2000;7:284–288. doi: 10.3109/13506120009146443. [DOI] [PubMed] [Google Scholar]
- Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann Neurol. 2004;56:675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- In't Veld BA, Launer LJ, Breteler MMB, Hofman A, Stricker BHC. Pharmacologic agents associated with a preventive effect on Alzheimer's disease: a review of the epidemiological evidence. Epidemiol Rev. 2002;24:248–268. doi: 10.1093/epirev/mxf001. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–278. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GTAM, Lyunendijk W, Frangoine B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Long JM, Kalehua AN, Muth NJ, Calhoun ME, Jucker M, Hengemihle JM, Ingram DK, Mouton PR. Stereological estimation of total microglia number in mouse hippocampus. J Neurosci Methods. 1998;84:101–108. doi: 10.1016/s0165-0270(98)00100-9. [DOI] [PubMed] [Google Scholar]
- Maat-Schieman MLC, Yamaguchi H, Hegeman-Kleinn I, Welling-Graafland C, Natte R, Roos RAC, van Duinen SG. Glial reactions and the clearance of amyloid β protein in the brains of patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type. Acta Neuropathol (Berl) 2004;107:389–398. doi: 10.1007/s00401-004-0824-4. [DOI] [PubMed] [Google Scholar]
- Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid β-protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005a;25:6271–6277. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid β-protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β-protein precursor. Am J Pathol. 2005b;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22:915–922. doi: 10.1016/s0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- Oh U, Gupta R, Krakauer JW, Khandji AG, Chin SS, Elkind MSV. Reversible leukoencephalopathy associated with cerebral amyloid angiopathy. Neurology. 2004;62:494–497. doi: 10.1212/01.wnl.0000106951.94624.df. [DOI] [PubMed] [Google Scholar]
- Pattison LR, Kotter MR, Fraga D, Bonelli RM. Apoptotic cascades as possible targets for inhibiting cell death in Huntington's disease. J Neurol. 2006;253:1137–1142. doi: 10.1007/s00415-006-0198-8. [DOI] [PubMed] [Google Scholar]
- Quintero EM, Willis L, Singleton R, Harris N, Huang P, Bhat N, Granholm AC. Behavioral and morphological effects of minocycline in the 6-hydroxydopamine rat model of Parkinson's disease. Brain Res. 2006;1093:198–207. doi: 10.1016/j.brainres.2006.03.104. [DOI] [PubMed] [Google Scholar]
- Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Jiang L, Maier M, Lemere CA. Minocycline affects microglia activation, Aβ deposition, and behavior in APP-tg mice. Glia. 2006;53:776–782. doi: 10.1002/glia.20338. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;8:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and Alzheimer's disease pathogenesis. J Neurosci Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer's disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62:1287–1301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter Van der Vlis M, Roos RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy: a microvascular link between parenchymal and vascular dementia? Ann Neurol. 2001;49:691–692. doi: 10.1002/ana.1055. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Xu F, Grande AM, Robinson JK, Previti ML, Davis J, Van Nostrand WE. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant APP transgenic mice. Neuroscience. 2007 doi: 10.1016/j.neuroscience.2007.01.043. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang-Nunes SX, Maat-Schiemann ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BYS, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]