

Abstract
Postoperative cognitive dysfunction, confusion, and delirium are common after general anesthesia in the elderly, with symptoms persisting for months or years in some patients. Even middle-aged patients are likely to have postoperative cognitive dysfunction for months after surgery, and Alzheimer's disease (AD) patients appear to be particularly at risk of deterioration after anesthesia. Several investigators have thus examined whether general anesthesia is associated with AD, with some studies suggesting that exposure to anesthetics may increase the risk of AD. However, little is known on the biochemical consequences of anesthesia on pathogenic pathways in vivo. Here, we investigated the effect of anesthesia on tau phosphorylation and amyloid precursor protein (APP) metabolism in mouse brain. We found that, regardless of the anesthetic used, anesthesia induced rapid and massive hyperphosphorylation of tau, rapid and prolonged hypothermia, inhibition of Ser/Thr PP2A (protein phosphatase 2A), but no changes in APP metabolism or Aβ (β-amyloid peptide) accumulation. Reestablishing normothermia during anesthesia completely rescued tau phosphorylation to normal levels. Our results indicate that changes in tau phosphorylation were not a result of anesthesia per se, but a consequence of anesthesia-induced hypothermia, which led to inhibition of phosphatase activity and subsequent hyperphosphorylation of tau. These findings call for careful monitoring of core temperature during anesthesia in laboratory animals to avoid artifactual elevation of protein phosphorylation. Furthermore, a thorough examination of the effect of anesthesia-induced hypothermia on the risk and progression of AD is warranted.
Keywords: Alzheimer's disease, anesthesia, tau, phosphorylation, hypothermia, amyloid-β
Introduction
The two histopathological hallmarks of Alzheimer's disease (AD) are senile plaques, composed of extracellular aggregates of the β-amyloid peptide (Aβ) (Selkoe, 2001), and intraneuronal neurofibrillary tangles (NFTs), composed of abnormally hyperphosphorylated tau protein assembled into paired helical filaments (PHFs) (Buee et al., 2000). Only a small proportion of AD is attributable to genetic variants; the large majority of cases (∼95%) is late onset and sporadic in origin. The cause of sporadic AD is likely to be multifactorial, with external factors interacting with biological or genetic susceptibility to accelerate the manifestation of the disease.
Postoperative cognitive dysfunction, confusion, and delirium are common after general anesthesia in the elderly, with symptoms persisting for months or years in some patients (Bedford, 1955; Gustafson et al., 1988; O'Keeffe and Ni Chonchubhair, 1994; Moller et al., 1998; Ancelin et al., 2001). Although the risk of developing symptoms increases with age, even middle-aged patients are likely to have postoperative cognitive dysfunction for months after surgery (Johnson et al., 2002). Some reports suggest that AD patients may be particularly at risk of deterioration after anesthesia (Rees and Gaines, 1985; Bone and Rosen, 2000). Several investigators have thus examined whether general anesthesia is associated with AD. Some studies find no association (French et al., 1985; Amaducci et al., 1986; Broe et al., 1990; Graves et al., 1990; Breteler et al., 1991; Kokmen et al., 1991; Bohnen et al., 1994b; Gasparini et al., 2002), whereas others suggest that exposure to anesthetics may increase the risk of AD (Bohnen et al., 1994a; Johansson and Skoog, 1996; Bone and Rosen, 2000).
Although these clinical studies investigated the relationship between anesthesia and AD, little is known about the impact of anesthesia on two pathological hallmarks of AD: Aβ accumulation and abnormal tau phosphorylation. In the present report, we examine the effect of anesthesia on tau phosphorylation and amyloid precursor protein (APP) metabolism in vivo. Our results indicate that anesthesia induced rapid and massive tau hyperphosphorylation at all the epitopes examined. However, it did not induce any change in APP metabolism or Aβ accumulation. Tau hyperphosphorylation correlated with low body temperature and was completely rescued when the animals were returned to normothermia, demonstrating that anesthesia induces tau hyperphosphorylation through hypothermia. Thus, more thorough studies on the effect of anesthesia-induced hypothermia on risk and progression of AD are warranted.
Materials and Methods
Animals.
Four- to 6-month-old C57BL/6J male mice were used (The Jackson Laboratory, Bar Harbor, ME). Animals were used in full compliance with National Institutes of Health/Institutional Animal Care and Use Committee guidelines.
Anesthesia.
Anesthesia was induced by intraperitoneal injections of chloral hydrate in PBS (500 mg/kg; Sigma, St. Louis, MO), pentobarbital sodium (Nembutal; 100 mg/kg; Abbott Laboratories, North Chicago, IL), or exposure to vapors of isoflurane (AErrane; Baxter Pharmaceutical, Deerfield, IL). Animals were kept at room temperature for 1 h before killing. Control injections included PBS (for chloral hydrate) or vehicle (50% water, 40% propylene glycol, 10% ethanol) for pentobarbital. For the recovery study, 30 min after exposure, the animals were placed in a ventilated incubator at 37°C for another 30 min. Temperature was monitored with a rectal probe (Thermalert TH-5; Physitemp, Clifton, NJ).
Protein extraction and SDS-PAGE.
Mice were killed by cervical dislocation, brains were immediately removed, and tissues were dissected quickly on ice and frozen on dry ice. The whole process took <1 min. Frozen hemispheres (hippocampus and neocortex) were weighed and homogenized without thawing in 5× volume/weight of modified RIPA buffer [50 mm Tris-HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, 10 μl/ml protease inhibitor mixture P8340 (Sigma)], with a mechanical homogenizer (TH; Omni International, Marietta, GA). Samples were then centrifuged for 20 min at 20,000 × g at 4°C, and protein content of the supernatants was determined. The supernatants were diluted 1/10 in O+ buffer [62.5 mm Tris-HCl, pH 6.8, 10% (w/v) glycerol, 5% 2-mercaptoethanol, 2.3% SDS, 1 mm EGTA, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, 10 μl/ml protease inhibitor mixture P8340 (Sigma)] and boiled for 5 min. Depending on the antibody used, 7–21 μg of protein were analyzed as described previously (Planel et al., 2001).
Antibodies.
The following anti-tau monoclonal antibodies (specificity given in parentheses) were a generous gift from Dr. Peter Davies (Albert Einstein University, New York, NY): TG-3, phospho-Ser231 and conformation specific (Jicha et al., 1997); MC-6, phospho-Ser235 (Jicha et al., 1997); and PHF-1, phospho-Ser396/404 (Otvos et al., 1994). Total tau was detected with either Tau T57120 (monoclonal; BD Transduction Laboratories, San Jose, CA) or Tau A0024 (polyclonal; DakoCytomation, Carpinteria, CA). AT8 (Pierce Biotechnology, Rockford, IL) reacts with tau phosphorylated at Ser202 and Thr205 (Goedert et al., 1995), and Tau-1 (Chemicon International, Temecula, CA) recognizes tau dephosphorylated at Ser195, Ser198, Ser199, and Ser202 (Szendrei et al., 1993). Purified rabbit polyclonal anti-tau antibodies anti-tau pS199, pS262, and pS422 were purchased from Biosource International (Camarillo, CA). Changes in tau kinases were investigated with the following antibodies: cyclin-dependent kinase 5 (cdk5), anti-p35C, calcium/calmodulin-dependent protein kinase II (CaMKII), phospho-CaMKII (Santa Cruz Biotechnology, Santa Cruz, CA), glycogen synthase kinase-3β (GSK-3β) (BD Transduction Laboratories), anti-GSK-3α/β [pY219/pY216] (Biosource International), phospho-GSK-3β (Ser9), stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK), phospho-SAPK/JNK (T183/Y185) G9, p44/42 mitogen-activated protein (MAP) kinase, phospho-p44/42 mitogen-activated protein kinase (MAPK) (T202/Y204), phospho-Akt (S473), phospho-p38 MAP kinase (T180/Y182), and p38 MAP kinase (Cell Signaling Technology, Danvers, MA). For Aβ analysis, the following antibodies were used: C1/6.1 (Mathews et al., 2002) [anti-APP full-length and C-terminal fragments (CTFs); generous gift from Dr. Paul Mathews, Nathan S. Kline Institute, Orangeburg, NY], JRF/cAβ 40 and JRF/cAβ 42/26 (murine Aβ 1–40 and Aβ 1–42, respectively; generous gift from Dr. Sonia Jung, Centocor R&D, Radnor, PA).
Western blot analysis and ELISA.
Membrane blocking and antibody incubations were performed as described previously (Noble et al., 2005), with appropriate primary and secondary antibody dilutions. Serial dilutions of brain extracts were loaded in gels to obtain calibration curves for reliable quantification. In some cases (see Fig. 4), tau was measured using a modified sandwich ELISA with ruthenium-labeled detection antibodies (BioVeris, Gaithersburg, MA), using phosphospecific antibody AT8 (Innogenetics, Gent, Belgium; 10 ng/well) and total tau (DakoCytomation; 30 ng/well). For these experiments, mice were killed by focused microwave irradiation, followed by immediate dissection and freezing of the brain at −80°C. ELISA for soluble murine Aβ40 and Aβ42 was performed as described previously (Burns et al., 2003; Schmidt et al., 2005), using diethyl acetate extracts from fresh hemibrains.
Figure 4.

Effects of anesthesia on temperature and tau phosphorylation. A, Rectal temperatures of the mice were recorded after injection of 50, 75, or 100 mg/kg pentobarbital or vehicle (n = 3–5 for each point). B, AT8 and total tau levels were quantified from brains of animals anesthetized with 100 mg/kg pentobarbital, and displayed as percentage of the control animals at different times (n = 3–5 for each point). C, D, Immunoreactive signals of AT8 (C) and total tau (D) from animals anesthetized with 50, 75, or 100 mg/kg pentobarbital were expressed as a percentage of the control mice and plotted against the corresponding individual rectal temperature (n = 47). Data are means ± SD.
Immunohistochemistry.
Mice were killed by cervical dislocation, and brains were immediately removed and frozen in chilled isopentane. Cryostat sections (10 μm) were mounted on Vectabond (Vector Laboratories, Burlingame, CA)-coated slides, air dried, and fixed in 4% paraformaldehyde in phosphate buffer for 15 min at room temperature. Sections were washed in PBS, blocked in 10% normal serum in PBS with 0.1% Triton X-100 and incubated with AT8 (1:500; Innogenetics) or total tau (1:100; DakoCytomation) diluted in blocking buffer overnight at 4°C. After washing, antibodies were visualized with Alexa 488-conjugated goat anti-rabbit or Alexa 594-conjugated goat anti-mouse (Invitrogen, Carlsbad, CA) diluted in blocking buffer for 2 h at room temperature. Cell nuclei were stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) (Invitrogen), and coverslips were mounted with Prolong (Invitrogen). Fluorescence was viewed on a Zeiss (Oberkochen, Germany) Axiovert 100M microscope, and images were captured with a MicroMax camera (Photometrics, Tucson, AZ) using MetaMorph (Molecular Devices, Downington, PA) imaging software.
Phosphatase activity assay.
Phosphatase activity was evaluated by monitoring endogenous tau dephosphorylation according to a modified protocol (Matsuo et al., 1994). Briefly, control or pentobarbital-anesthetized mice were killed by cervical dislocation, brains were immediately removed, and one hemisphere (hippocampus and neocortex) was homogenized in 5× volume/weight of phosphatase sample buffer (50 mm Tris-HCl, pH 7.5, 0.25 m sucrose, 10 mm 2-mercaptoethanol, 0.1 mm EDTA, 1 mm benzamidine, 1 mm PMSF, 10 μl/ml protease inhibitor mixture P8340; Sigma) (Planel et al., 2001). The supernatant was diluted 10 times in phosphatase assay buffer (50 mm Tris-HCl, pH 7.0, 0.1 mm EDTA, 5 mm DTT, 0.01% Brij 35, 1 mm PMSF, 10 μl/ml protease inhibitor mixture P8340; Sigma), incubated at the temperature of the animals at the time of the killing (37°C for control, 26°C for anesthesia), and sampled at different intervals, and endogenous PHF-1 phosphorylation levels were monitored by Western blotting. PHF-1 was chosen because it is an epitope readily dephosphorylated by all of the tau phosphatases (Wang et al., 1996), and it is easily detected in control brains. To verify the results from the above phosphatase assay, we performed a protein phosphatase 2A (PP2A) assay using the Serine/Threonine Phosphatase Assay System from Promega (Madison, WI) (V2460). Briefly, mice were killed by cervical dislocation, the brain was immediately removed, and one hemisphere (hippocampus and neocortex) was homogenized in 5× volume/weight of phosphatase sample buffer and immediately processed as indicated by the manufacturer to remove particulate matter and endogenous free phosphate. For PP2A activity, 2.0 μl of the resulting solution was prepared in duplicate, and the release of phosphate from a chemically synthesized phosphopeptide was assessed over a period of 10 min in PP2A buffer (calibration data not shown). The amount of phosphate released was measured by the absorbance of the molybdate–malachite green–phosphate complex at 630 nm.
Statistical analysis.
Except for Figure 7 (two-way ANOVA followed by Bonferroni's posttests), statistical analysis was performed with a two-tailed t test. Data are means ± SDs, and asterisks indicate significant differences as follows: *p < 0.05 and **p < 0.01.
Figure 7.

Effect of anesthesia on phosphatase activity. A, Kinetic of tau dephosphorylation at PHF-1 epitope in brain extracts, as monitored by Western blotting, with or without 5 nm okadaic acid. Tau dephosphorylation was completely abolished by okadaic acid. B, Phosphatase activity was evaluated by endogenous tau dephosphorylation performed at the temperature of the animals before killing, followed by Western blot analysis. Tau from control brains incubated at 37°C was dephosphorylated faster (n = 3; p = 0.0017; two-way ANOVA followed by Bonferroni's posttests) than tau from animals anesthetized with pentobarbital and incubated at 26°C. C, To confirm these results, PP2A activity was evaluated with the PP2A assay system from Promega. Similarly to the endogenous assay, there was no significant difference in PP2A activity between control and anesthetized mice samples when incubated at 37°C, whereas ∼40% PP2A inhibition was detected when the reaction was performed at 26°C (n = 3; p < 0.05; ANOVA followed by Tukey–Kramer's posttests). Data are means ± SDs. * and ** indicate significant differences with p < 0.05 and 0.01, respectively.
Results
Anesthesia induced tau hyperphosphorylation but did not affect APP metabolism
One hour after the induction of anesthesia by chloral hydrate, a large mobility shift in tau was observed, using a phosphorylation-independent total tau antibody. The prominent normal adult tau band of 56 kDa was diminished, and the phospho-tau 60 kDa band greatly intensified, with the appearance of 64 and 70 kDa bands, but total tau levels did not change (Fig. 1I). This was accompanied by a pronounced enhancement of immunostaining of all phosphorylation-dependent antibodies. PS199 (phospho-Ser199), TG-3 (phospho-Ser231), PS262 (phospho-Ser262), PHF-1 (phospho-Ser396/404), and PS422 (phospho-Ser422) displayed an approximately twofold to fivefold increase in phosphorylation, whereas AT8 (phospho-Ser202/Thr205) and MC-6 (phospho-Ser235) exhibited ∼15–150 times more phosphorylation (Fig. 1). These results were corroborated by a diminished signal for Tau-1 (Fig. 1B), which has optimal immunoreactivity when this phospho-epitope on tau is dephosphorylated. Treatment of the mice with pentobarbital or isoflurane (see Fig. 5B,C, first four lanes) led to similar tau hyperphosphorylation. APP metabolism is affected in AD, with changes in Aβ and C-terminal fragment levels (Sergeant et al., 2002). Here, anesthesia did not affect APP levels (Fig. 2A) or APP CTFs (Fig. 2B). Similarly, the levels of endogenous Aβ1–40 and Aβ1–42 did not change during anesthesia (Fig. 2C). These results demonstrate that, independent of the anesthetic used, 1 h of anesthesia can induce a robust hyperphosphorylation of tau but does not affect APP metabolism in vivo.
Figure 1.

Effect of anesthesia on tau phosphorylation. Proteins from mouse brain hemispheres (hippocampus plus cortex) were separated by SDS-PAGE and identified with the antibodies indicated on top of the boxes. For each antibody, two representative data of four for each condition (Ctl, control; Anes, anesthesia by chloral hydrate) are displayed. Each lane represents an individual mouse. The bands were quantified and the immunoreactivity during anesthesia expressed as a percentage of the control treatment (100%): A, 187 ± 7**; B, 42 ± 3*; C, 1471 ± 177***; D, 326 ± 12*; E, 15683 ± 8059*; F, 475 ± 77**; G, 176 ± 40*; H, 230 ± 23***; I, 116 ± 2. Data are means ± SD. *, **, and *** indicate significant differences with p < 0.05, 0.01, and 0.001, respectively.
Figure 5.

Effect of controlled temperature and different anesthetics on tau phosphorylation. Proteins from mouse brain hemispheres (hippocampus plus cortex) were separated by SDS-PAGE and identified with either AT8 or total tau antibodies. Anesthesia by chloral hydrate (A), pentobarbital (B), or isoflurane (C) induced a robust tau hyperphosphorylation (lanes 3 and 4) that could be totally rescued by returning the core temperature of the animals to normal (lanes 5 and 6). Total tau did not change during the experiment. Ctl, Control; Chlor, chloral hydrate; Pento, pentobarbital; Isofl, isoflurane.
Figure 2.

Effect of anesthesia on APP metabolism. Proteins from mouse brain hemispheres (hippocampus plus cortex) were separated by SDS-PAGE and identified with C1/6.1 antibody, which recognize APP (A) and CTFs (B). A and B display three representative data of five for each condition (Ctl, control; Anes, anesthesia by chloral hydrate). C shows the levels of Aβ1–40 (white bars) and Aβ1–42 (black bars) for control and anesthesia (n = 5), as detected by ELISA. Data are means ± SD.
Hyperphosphorylated tau was not relocalized to somatodendritic region
We then examined the gross anatomical pattern of tau phosphorylation. Anesthesia by pentobarbital led to tau hyperphosphorylation at the AT8 epitope in most anterior areas, such as the neocortex and hippocampus (Fig. 3), whereas total tau immunoreactivity did not change (data not shown). Hyperphosphorylated tau was preferentially localized in the neuropil of cortical regions (Fig. 3C,D), and the hippocampal region (Fig. 3A,B). Tau is an axonal protein in normal adult brain, and axonal localization of hyperphosphorylated tau was confirmed by double immunostaining with AT8 and total tau antibodies in the CA1 region (Fig. 3E–G). In AD brain, hyperphosphorylated tau is found in cell bodies and dendrites of affected neurons. After 1 h of anesthesia, we did not observe somatodendritic relocalization of tau (Fig. 3). These results confirm our Western blot data, which showed significant change in tau phosphorylation at the AT8 epitope (Fig. 1C) in response to anesthesia. Similar results were obtained with chloral hydrate (data not shown). Thus, 1 h of anesthesia leads to tau hyperphosphorylation in the axons and neuropil without detectable somatodendritic relocalization.
Figure 3.
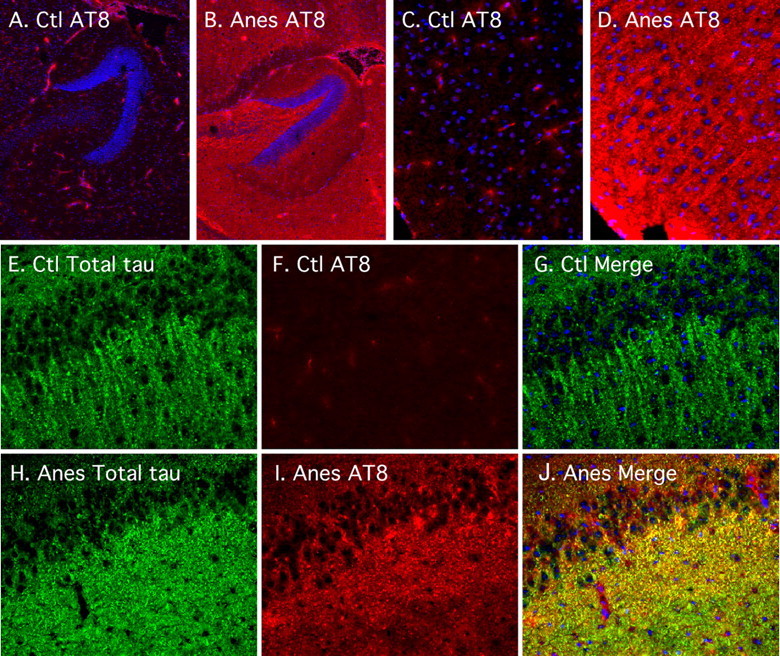
Regional anatomical analysis of tau phosphorylation during anesthesia. Fluorescence photomicrographs of sagittal sections are shown. AT8 (in red) or total tau (in green) immunostaining was performed in control (Ctl) (A, C, E, F, G) and anesthetized (Anes) (pentobarbital; B, D, H, I, J) mice, in the hippocampus (A, B) (magnification, 50×), cortical regions (C, D) (magnification, 200×), or the CA1 (E–J) (magnification, 200×). DAPI nucleus-specific staining is visible in blue.
Anesthesia-induced tau hyperphosphorylation was mediated by hypothermia
We recently demonstrated that hypothermia is a potent inducer of tau hyperphosphorylation. Because anesthesia is known to reduce body temperature (Taguchi and Kurz, 2005), we analyzed the role of body temperature on tau phosphorylation during anesthesia. The rectal temperature of the mice decreased very rapidly to reach ∼26.0°C 1 h after pentobarbital injection (Fig. 4A) (100 mg/kg), ∼27.9°C after chloral hydrate treatment, or ∼26.8°C after isoflurane exposure. We also examined the effects of lower doses of pentobarbital: 75 mg/kg led to rapid hypothermia (30 min, ∼29.4°C; 60 min, ∼27.8°C; 120 min, ∼28.4°C), whereas the effects of 50 mg/kg were less dramatic and the animals regained consciousness quickly (30 min, ∼32.1°C; 60 min, ∼34.8°C; 120 min, ∼36.5°C). Anesthesia led to an immediate enhancement of tau phosphorylation at the AT8 epitope, whereas total tau level did not change (Fig. 4B). In fact, temperature correlated negatively with AT8 phosphorylation (Fig. 4C) (slope, −80%/°C; R2 > 0.907), whereas there was no correlation with total tau (Fig. 4D) (slope, 0.27%/°C; R2 < 0.005). Similar results were obtained with chloral hydrate injection (data not shown). To confirm a causative role for a temperature decrease in the observed tau hyperphosphorylation, we placed the animals in a ventilated incubator at 37°C (for 30 min), 30 min after chloral hydrate treatment at room temperature. This protocol rescued the mouse core temperature to normal (data not shown) and completely restored AT8 phosphorylation to control levels, whereas total tau did not change (Fig. 5A). The effects of pentobarbital and isoflurane on AT8 phosphorylation were also reversed by maintaining the animals at 37°C, whereas total tau did not change (Fig. 5B,C). Similar results were obtained by controlling the animals' core temperature with a rectal probe and a heating blanket (data not shown). These findings demonstrate that the effect of anesthesia on tau phosphorylation is not specific to a given anesthetic but is mediated by hypothermia.
Anesthesia-induced tau hyperphosphorylation was not attributable to kinase activation
We recently demonstrated that tau hyperphosphorylation induced by hypothermia is attributable to inhibition of Ser/Thr protein phosphatases (Planel et al., 2004), suggesting a similar mechanism during anesthesia. However, we could not rule out anesthesia-specific activation of tau kinases that would contribute significantly to the observed tau hyperphosphorylation. Among the kinases able to phosphorylate tau in vitro, GSK-3β, cdk5, MAPK/ERK (extracellular signal-regulated kinase), and JNK are considered to be major physiological and pathological tau kinases (Maccioni et al., 2001; Planel et al., 2002; Zhu et al., 2002). CaMKII is also thought to have a major role in regulating the phosphorylation of tau at epitopes that modulates the binding of tau to microtubules (Litersky et al., 1996; Sironi et al., 1998). We explored the activation patterns of these five kinases with specific antibodies. Phosphorylation of GSK-3β at Tyr216 facilitates substrate binding, whereas phosphorylation at Ser9 leads to partial inhibition (Planel et al., 2002). There was no change at Tyr216 epitope after 1 h of anesthesia (Fig. 6A), but a significant increase of Ser9 phosphorylation could be detected (Fig. 6B). JNK was slightly activated during anesthesia (Fig. 6D), but the activating phosphorylation of MAPK and CaMKII was reduced (Fig. 6F,H, respectively). There were no significant changes in total GSK-3β (Fig. 6C), total JNK (Fig. 6E), or total MAPK (Fig. 6G). Total CaMKII was slightly enhanced (Fig. 6I). In the brain, cdk5 forms a heterodimeric complex with the neuron-specific activator p35 (Maccioni et al., 2001). On cell death, p35 levels rapidly decrease as it is cleaved into p25, which further enhances cdk5 activity toward tau (Taniguchi et al., 2001; Hashiguchi et al., 2002). There was no significant change in cdk5 and p35 during anesthesia (Fig. 6J,K), and there was no detectable p25. Similarly, Akt/PKB (protein kinase B), which was reported to phosphorylate tau (Ksiezak-Reding et al., 2003), did not change during anesthesia (Fig. 6L). We also investigated the activation state of p38, which is known to phosphorylate tau at multiple sites (Reynolds et al., 2000); there was no change in either p38 levels or phosphorylation (Fig. 6M,N). Overall, anesthesia induced inhibitory changes in phosphorylation of GSK-3β, MAPK, and CaMKII. The only kinase activated was JNK, but the slight activation of JNK fails to explain the extent of tau hyperphosphorylation, especially at epitopes not phosphorylated by JNK. Thus, kinase activation is probably not the mechanism leading to tau hyperphosphorylation during anesthesia.
Figure 6.

Effect of anesthesia on tau kinase activation. Proteins from mouse brain hemispheres (hippocampus plus cortex) were separated by SDS-PAGE and identified with the antibodies indicated on top of the boxes. For each antibody, two representative data of four for each condition (Ctl, control; Anes, anesthesia by chloral hydrate) are displayed in the top part of the boxes. Each lane represents an individual mouse. The bands were quantified and the immunoreactivity during anesthesia expressed as a percentage of the control treatment (100%): A, 97 ± 13; B, 808 ± 51***; C, 112 ± 14; D, 128 ± 17*; E, 113 ± 4; F, 24 ± 8**; G, 125 ± 11; H, 72 ± 15*; I, 135 ± 8*; J, 91 ± 14; K, 106 ± 11; L, 119 ± 20; M, 127 ± 33; N, 101 ± 23. Data are means ± SD. *, **, and *** indicate significant differences with p < 0.05, 0.01, and 0.001, respectively.
Anesthesia inhibited PP2A
Based on the result from our previous study (Planel et al., 2004), tau hyperphosphorylation during anesthesia could be attributable to the direct inhibitory effect of hypothermia on Ser/Thr protein phosphatase (PP) activity. To verify this, we evaluated PP activities using an endogenous tau dephosphorylation assay. PP are classified into four types, PP1, PP2A, PP2B, and PP2C, on the basis of their specificity toward certain substrates, and sensitivity to specific activators and inhibitors (Cohen, 1989). Tau can be dephosphorylated by PP1, PP2A, and PP2B, but PP2A shows a much stronger ability to dephosphorylate tau (Goedert et al., 1992; Wang et al., 1996). In vivo and slice culture studies have demonstrated that PP2A is likely to be the main regulator of tau phosphorylation physiologically, whereas PP1 and PP2B are minimally involved (Bennecib et al., 2000; Gong et al., 2000; Planel et al., 2001, 2004). We first confirmed the involvement of PP2A in tau dephosphorylation in mouse brain extract. Dephosphorylation of endogenous tau at the PHF-1 epitope was rapid (Fig. 7A) [0 nm okadaic acid (OA); decay rate constant, 6.18 min; R2 > 0.99]. It was completely inhibited (p = 0.0313) by 5 nm okadaic acid (Fig. 7A) (5 nm OA; slope, 0.23%/min; R2 = 0.48), a concentration inhibitory to PP2A but not PP1 and PP2B (McKintosh, 1993). Thus, PP2A is the main phosphatase driving tau dephosphorylation at the PHF-1 epitope in mouse brain extracts. We then evaluated endogenous tau dephosphorylation in three control and three anesthetized animals. When incubated at 37°C, brain extracts from control and pentobarbital-anesthetized mice did not show any difference in PHF-1 dephosphorylation (data not shown). When extracts from anesthetized mice were incubated at 26°C (temperature of the animals at the time of killing), dephosphorylation of tau (Fig. 7B) (decay rate constant, 16.86 min; R2 = 0.84) was significantly slower (p = 0.0017) than that from the control extracts (Fig. 7B) (decay rate constant, 5.92 min; R2 = 0.92). To verify this endogenous dephosphorylation assay, we performed the same experiment with a commercially available PP2A assay system. Similarly, there was no significant difference in PP2A activity between control and anesthetized mice samples when incubated at 37°C, whereas ∼40% PP2A inhibition was detected when the reaction was performed at 26°C (Fig. 7C). These experiments show that tau hyperphosphorylation during anesthesia is attributable to the direct effect of low temperature on phosphatase activity, because dephosphorylation kinetics of tau in control and anesthetized samples are different only if the temperatures of incubation are different. Together, our results demonstrate that anesthesia-induced hypothermia leads to reduced PP2A activity in the mouse brain.
Discussion
We investigated the impact of anesthesia on tau phosphorylation in vivo. We found that tau was hyperphosphorylated during anesthesia and that this was independent of the anesthetic used. Moreover, hyperphosphorylation was found not to result from anesthesia per se, but from the hypothermia consequent to anesthesia.
Anesthesia by chloral hydrate, sodium pentobarbital, or isoflurane induced a robust hyperphosphorylation of tau, as demonstrated by Western blotting, ELISA, and immunohistochemical analysis. All general anesthetics markedly impair normal autonomic thermoregulatory control (Taguchi and Kurz, 2005), and all of the anesthetics used here induced hypothermia. Restoring normothermia in the mice completely rescued tau phosphorylation to normal levels, demonstrating that tau hyperphosphorylation is not mediated by the anesthetics per se, but by the consequent hypothermia. We found that activation of tau kinases was unable to explain the extent of tau hyperphosphorylation during anesthesia. In fact GSK-3β, MAPK, and CaMKII were markedly inhibited, whereas JNK was only slightly activated. However, PP2A was inhibited by the low temperatures induced by anesthesia. We argue that inhibition of PP2A is sufficient to cause tau hyperphosphorylation despite no kinase activation. This implies that tau hyperphosphorylation can be induced by basal kinase activities, although they are not elevated, simply by a shift in phosphorylation equilibrium as a result of phosphatases inhibition. These results are consistent with our previous work demonstrating that hypothermia mediates tau hyperphosphorylation by direct and exponential inhibition of Ser/Thr protein phosphatase activities that, despite concomitant linear inhibition of tau kinase activities, results in an overall increase in tau phosphorylation in vivo (Planel et al., 2004). We conclude that anesthesia induces hypothermia, which then leads to tau hyperphosphorylation by direct inhibition of phosphatase activities.
These results have important implications for the analysis of phosphoproteins in laboratory animals. Anesthesia without precise control of core body temperature could lead to artifactual elevation of tightly regulated phosphoepitopes as phosphatases are inhibited during hypothermia (Planel et al., 2004). Consistent with our results (Fig. 5B), Li et al. (2005) show that anesthesia enhances the inhibitory phosphorylation of GSK-3β. This enhanced GSK-3β Ser9 phosphorylation is seen in all animal models undergoing hypothermia (Okawa et al., 2003; Planel et al., 2004) and is probably mediated through the inhibition of PP1 and/or PP2A at low temperatures. Anesthesia could also affect transgenic and wild-type animals differently. For example, mice carrying presenilin and APP mutations seem especially vulnerable to hypothermia (Huitron-Resendiz et al., 2002; Iivonen et al., 2003). Therefore, when analyzing phosphorylation of tau or other proteins, the temperature of the animals before killing should be carefully monitored and controlled (in our experience, a simple lamp is not enough).
Some anesthetics have been demonstrated to rapidly accumulate Aβ oligomers in vitro (Eckenhoff et al., 2004) and increase Aβ production in cell cultures (Xie et al., 2006). Here, we could not detect any change in either endogenous APP metabolism or in Aβ1–40 or Aβ1–42 levels after anesthesia. Our data are consistent with a recent report demonstrating that general anesthetics did not change APP protein and mRNA concentration in the rat brain (Palotas et al., 2005). These results exclude any involvement of Aβ in the rapid and reversible tau hyperphosphorylation induced by anesthesia.
Our results show that 1 h of anesthesia led to tau hyperphosphorylation at multiple phosphorylation sites found in AD, including pS422 and TG-3, which are considered pathological epitopes (Jicha et al., 1997; Bussiere et al., 1999), and pS199, which is considered an early marker of tau pathology (Maurage et al., 2003). Tau hyperphosphorylation can lead to PHF formation in vitro (Alonso et al., 2001) and is thought to be a critical event in the pathogenesis of AD (Duff and Planel, 2005; Avila, 2006). Hyperphosphorylated tau was localized in the axons of the neuropil, but we did not observe somatodendritic relocalization. In AD, tau-positive dystrophic axons are widespread, and the earliest detectable hyperphosphorylated tau is preferentially localized in neurites of vulnerable neurons before extending to the soma (Su et al., 1994), where NFTs are formed (Bancher et al., 1989; Braak et al., 1994; Trojanowski and Lee, 1994). Thus, anesthesia can induce a biochemical and histochemical pattern of tau phosphorylation reminiscent of incipient AD.
Anesthesia appears to be associated with long-term cognitive disorder and the acceleration of senile dementia (Ritchie et al., 1997; Xie and Tanzi, 2006). Several reports have suggested that anesthesia could increase the risk of AD (Bohnen et al., 1994a; Johansson and Skoog, 1996; Bone and Rosen, 2000), and hypothermia has also been proposed as a risk factor (Holtzman and Simon, 2000; Avila and Diaz-Nido, 2004). Here, we demonstrated that anesthesia-induced hypothermia led to AD-like tau hyperphosphorylation induced by inhibition of PP2A. In AD, decreased PP2A expression (Vogelsberg-Ragaglia et al., 2001) along with upregulation of its inhibitors (Tanimukai et al., 2005) result in overall inhibition of its activity (Gong et al., 1993, 1995), which might be an important factor in the evolution of the pathology (Tian and Wang, 2002). Thus, during anesthesia-induced hypothermia, inhibition of PP2A and the consequent tau hyperphosphorylation might interact with biological or genetic susceptibilities to increase the vulnerability of the brain to insults associated with AD.
Although recent data show that hypothermia may have advantages in specific critically ill patients, perioperative hypothermia after anesthesia is clearly harmful in certain patients and multiple methods have been developed for thermal management of core temperature (Taguchi and Kurz, 2005). However, most of the studies on anesthesia and AD did not document thermal management methods and body temperatures of patients during and after surgery. Reevaluation of the data with hypothermia as a parameter could lead to a more definitive conclusion on the link between anesthesia and the risk and progression of AD, and possible preventive measures. What remains to be determined is how much impact the transient and reversible tau hyperphosphorylation induced by anesthesia can have on the development of tau pathology in AD. Examination of development of tau pathology in animal models treated with anesthesia may be able to address this question.
Footnotes
This work was supported by National Institutes of Health Pilot Project Grant AG008051 (E.P.) and National Institute of Neurological Disorders and Stroke Grant NS048447 (K.E.D.). We thank Drs. Paul Mathews (Nathan Kline Institute, Orangeburg, NY) and Haung Yu (Columbia University, New York, NY) for helpful discussion. We are grateful to John LaFrançois, Janos Piturca, and Mao Rui Fen for technical help.
References
- Alonso AC, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaducci LA, Fratiglioni L, Rocca WA, Fieschi C, Livrea P, Pedone D, Bracco L, Lippi A, Gandolfo C, Bino G, Prencipe M, Bonatti ML, Girotti F, Carella F, Tavola B, Ferla S, Lenzi GL, Carolei A, Gambi A, Grigoletto F, et al. Risk factors for clinically diagnosed Alzheimer's disease: a case-control study of an Italian population. Neurology. 1986;36:922–931. doi: 10.1212/wnl.36.7.922. [DOI] [PubMed] [Google Scholar]
- Ancelin ML, de Roquefeuil G, Ledesert B, Bonnel F, Cheminal JC, Ritchie K. Exposure to anaesthetic agents, cognitive functioning and depressive symptomatology in the elderly. Br J Psychiatry. 2001;178:360–366. doi: 10.1192/bjp.178.4.360. [DOI] [PubMed] [Google Scholar]
- Avila J. Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett. 2006;580:2922–2927. doi: 10.1016/j.febslet.2006.02.067. [DOI] [PubMed] [Google Scholar]
- Avila J, Diaz-Nido J. Tangling with hypothermia. Nat Med. 2004;10:460–461. doi: 10.1038/nm0504-460. [DOI] [PubMed] [Google Scholar]
- Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, Seitelberger F, Grundke-Iqbal I, Iqbal K, Wisniewski HM. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90–99. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- Bedford PD. Adverse cerebral effects of anaesthesia on old people. Lancet. 1955;269:259–263. doi: 10.1016/s0140-6736(55)92689-1. [DOI] [PubMed] [Google Scholar]
- Bennecib M, Gong C, Grundke-Iqbal I, Iqbal K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000;485:87–93. doi: 10.1016/s0014-5793(00)02203-1. [DOI] [PubMed] [Google Scholar]
- Bohnen N, Warner MA, Kokmen E, Kurland LT. Early and midlife exposure to anesthesia and age of onset of Alzheimer's disease. Int J Neurosci. 1994a;77:181–185. doi: 10.3109/00207459408986029. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Warner MA, Kokmen E, Beard CM, Kurland LT. Alzheimer's disease and cumulative exposure to anesthesia: a case-control study. J Am Geriatr Soc. 1994b;42:198–201. doi: 10.1111/j.1532-5415.1994.tb04952.x. [DOI] [PubMed] [Google Scholar]
- Bone I, Rosen M. Alzheimer's disease and anaesthesia. Anaesthesia. 2000;55:592–593. doi: 10.1046/j.1365-2044.2000.01479-5.x. [DOI] [PubMed] [Google Scholar]
- Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- Breteler MM, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Mortimer JA. Medical history and the risk of Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S36–S42. doi: 10.1093/ije/20.supplement_2.s36. [DOI] [PubMed] [Google Scholar]
- Broe GA, Henderson AS, Creasey H, McCusker E, Korten AE, Jorm AF, Longley W, Anthony JC. A case-control study of Alzheimer's disease in Australia. Neurology. 1990;40:1698–1707. doi: 10.1212/wnl.40.11.1698. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Burns M, Gaynor K, Olm V, Mercken M, LaFrancois J, Wang L, Mathews PM, Noble W, Matsuoka Y, Duff K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J Neurosci. 2003;23:5645–5649. doi: 10.1523/JNEUROSCI.23-13-05645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere T, Hof PR, Mailliot C, Brown CD, Caillet-Boudin ML, Perl DP, Buee L, Delacourte A. Phosphorylated serine422 on tau proteins is a pathological epitope found in several diseases with neurofibrillary degeneration. Acta Neuropathol (Berl) 1999;97:221–230. doi: 10.1007/s004010050978. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Duff K, Planel E. Untangling memory deficits. Nat Med. 2005;11:826–827. doi: 10.1038/nm0805-826. [DOI] [PubMed] [Google Scholar]
- Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101:703–709. doi: 10.1097/00000542-200409000-00019. [DOI] [PubMed] [Google Scholar]
- French LR, Schuman LM, Mortimer JA, Hutton JT, Boatman RA, Christians B. A case-control study of dementia of the Alzheimer type. Am J Epidemiol. 1985;121:414–421. doi: 10.1093/oxfordjournals.aje.a114013. [DOI] [PubMed] [Google Scholar]
- Gasparini M, Vanacore N, Schiaffini C, Brusa L, Panella M, Talarico G, Bruno G, Meco G, Lenzi GL. A case-control study on Alzheimer's disease and exposure to anesthesia. Neurol Sci. 2002;23:11–14. doi: 10.1007/s100720200017. [DOI] [PubMed] [Google Scholar]
- Goedert M, Cohen ES, Jakes R, Cohen P. p42 MAP kinase phosphorylation sites in microtubule-associated protein tau are dephosphorylated by protein phosphatase 2A1. Implications for Alzheimer's disease. FEBS Lett. 1992;312:95–99. doi: 10.1016/0014-5793(92)81418-l. [Erratum (1992) 313:203] [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. J Biol Chem. 2000;275:5535–5544. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, Raskind M. A case-control study of Alzheimer's disease. Ann Neurol. 1990;28:766–774. doi: 10.1002/ana.410280607. [DOI] [PubMed] [Google Scholar]
- Gustafson Y, Berggren D, Brannstrom B, Bucht G, Norberg A, Hansson LI, Winblad B. Acute confusional states in elderly patients treated for femoral neck fracture. J Am Geriatr Soc. 1988;36:525–530. doi: 10.1111/j.1532-5415.1988.tb04023.x. [DOI] [PubMed] [Google Scholar]
- Hashiguchi M, Saito T, Hisanaga S, Hashiguchi T. Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tau. J Biol Chem. 2002;277:44525–44530. doi: 10.1074/jbc.M207426200. [DOI] [PubMed] [Google Scholar]
- Holtzman A, Simon EW. Body temperature as a risk factor for Alzheimer's disease. Med Hypotheses. 2000;55:440–444. doi: 10.1054/mehy.2000.1085. [DOI] [PubMed] [Google Scholar]
- Huitron-Resendiz S, Sanchez-Alavez M, Gallegos R, Berg G, Crawford E, Giacchino JL, Games D, Henriksen SJ, Criado JR. Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Res. 2002;928:126–137. doi: 10.1016/s0006-8993(01)03373-x. [DOI] [PubMed] [Google Scholar]
- Iivonen H, Nurminen L, Harri M, Tanila H, Puolivali J. Hypothermia in mice tested in Morris water maze. Behav Brain Res. 2003;141:207–213. doi: 10.1016/s0166-4328(02)00369-8. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- Johansson C, Skoog I. A population-based study on the association between dementia and hip fractures in 85-year olds. Aging (Milano) 1996;8:189–196. doi: 10.1007/BF03339676. [DOI] [PubMed] [Google Scholar]
- Johnson T, Monk T, Rasmussen LS, Abildstrom H, Houx P, Korttila K, Kuipers HM, Hanning CD, Siersma VD, Kristensen D, Canet J, Ibanaz MT, Moller JT. Postoperative cognitive dysfunction in middle-aged patients. Anesthesiology. 2002;96:1351–1357. doi: 10.1097/00000542-200206000-00014. [DOI] [PubMed] [Google Scholar]
- Kokmen E, Beard CM, Chandra V, Offord KP, Schoenberg BS, Ballard DJ. Clinical risk factors for Alzheimer's disease: a population-based case-control study. Neurology. 1991;41:1393–1397. doi: 10.1212/wnl.41.9.1393. [DOI] [PubMed] [Google Scholar]
- Ksiezak-Reding H, Pyo HK, Feinstein B, Pasinetti GM. Akt/PKB kinase phosphorylates separately Thr212 and Ser214 of tau protein in vitro. Biochim Biophys Acta. 2003;1639:159–168. doi: 10.1016/j.bbadis.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Li X, Friedman AB, Roh MS, Jope RS. Anesthesia and post-mortem interval profoundly influence the regulatory serine phosphorylation of glycogen synthase kinase-3 in mouse brain. J Neurochem. 2005;92:701–704. doi: 10.1111/j.1471-4159.2004.02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litersky JM, Johnson GV, Jakes R, Goedert M, Lee M, Seubert P. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem J. 1996;316:655–660. doi: 10.1042/bj3160655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni RB, Otth C, Concha II, Munoz JP. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem. 2001;268:1518–1527. doi: 10.1046/j.1432-1033.2001.02024.x. [DOI] [PubMed] [Google Scholar]
- Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA. Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of clapains enhances endosomal generation of beta-cleaved C-terminal APP fragments. J Biol Chem. 2002;277:36415–36424. doi: 10.1074/jbc.M205208200. [DOI] [PubMed] [Google Scholar]
- Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O'Connor M, Trojanowski JQ, Lee VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- Maurage CA, Sergeant N, Ruchoux MM, Hauw JJ, Delacourte A. Phosphorylated serine 199 of microtubule-associated protein tau is a neuronal epitope abundantly expressed in youth and an early marker of tau pathology. Acta Neuropathol (Berl) 2003;105:89–97. doi: 10.1007/s00401-002-0608-7. [DOI] [PubMed] [Google Scholar]
- McKintosh C. Assay and purification of protein (serine/threonine) phosphatases. In: Grahame Hardie D, editor. Protein phosphorylation: a practical approach. Oxford: Oxford UP; 1993. pp. 197–230. [Google Scholar]
- Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, Rabbitt P, Jolles J, Larsen K, Hanning CD, Langeron O, Johnson T, Lauven PM, Kristensen PA, Biedler A, van Beem H, Fraidakis O, Silverstein JH, Beneken JE, Gravenstein JS. Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet. 1998;351:857–861. doi: 10.1016/s0140-6736(97)07382-0. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa Y, Ishiguro K, Fujita SC. Stress-induced hyperphosphorylation of tau in the mouse brain. FEBS Lett. 2003;535:183–189. doi: 10.1016/s0014-5793(02)03883-8. [DOI] [PubMed] [Google Scholar]
- O'Keeffe ST, Ni Chonchubhair A. Postoperative delirium in the elderly. Br J Anaesth. 1994;73:673–687. doi: 10.1093/bja/73.5.673. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Palotas M, Palotas A, Bjelik A, Pakaski M, Hugyecz M, Janka Z, Kalman J. Effect of general anesthetics on amyloid precursor protein and mRNA levels in the rat brain. Neurochem Res. 2005;30:1021–1026. doi: 10.1007/s11064-005-6786-7. [DOI] [PubMed] [Google Scholar]
- Planel E, Yasutake K, Fujita SC, Ishiguro K. Inhibition of protein phosphatase 2A overrides Tau protein kinase I/glycogen synthase kinase 3beta and Cyclin-dependant kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 2001;276:34298–34306. doi: 10.1074/jbc.M102780200. [DOI] [PubMed] [Google Scholar]
- Planel E, Sun X, Takashima A. Role of GSK-3 beta in Alzheimer's disease pathology. Drug Dev Res. 2002;56:491–510. [Google Scholar]
- Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer's disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DI, Gaines GY., III Anesthetic considerations for patients with Alzheimer's disease. Tex Med. 1985;81:45–48. [PubMed] [Google Scholar]
- Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- Ritchie K, Polge C, de Roquefeuil G, Djakovic M, Ledesert B. Impact of anesthesia on the cognitive functioning of the elderly. Int Psychogeriatr. 1997;9:309–326. doi: 10.1017/s1041610297004468. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-beta levels. Methods Mol Biol. 2005;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sergeant N, David JP, Champain D, Ghestem A, Wattez A, Delacourte A. Progressive decrease of amyloid precursor protein carboxy terminal fragments (APP-CTFs), associated with tau pathology stages, in Alzheimer's disease. J Neurochem. 2002;81:663–672. doi: 10.1046/j.1471-4159.2002.00901.x. [DOI] [PubMed] [Google Scholar]
- Sironi JJ, Yen SH, Gondal JA, Wu Q, Grundke-Iqbal I, Iqbal K. Ser-262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett. 1998;436:471–475. doi: 10.1016/s0014-5793(98)01185-5. [DOI] [PubMed] [Google Scholar]
- Su JH, Cummings BJ, Cotman CW. Early phosphorylation of tau in Alzheimer's disease occurs at Ser-202 and is preferentially located within neurites. NeuroReport. 1994;5:2358–2362. doi: 10.1097/00001756-199411000-00037. [DOI] [PubMed] [Google Scholar]
- Szendrei GI, Lee VM, Otvos L., Jr Recognition of the minimal epitope of monoclonal antibody Tau-1 depends upon the presence of a phosphate group but not its location. J Neurosci Res. 1993;34:243–249. doi: 10.1002/jnr.490340212. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Kurz A. Thermal management of the patient: where does the patient lose and/or gain temperature? Curr Opin Anaesthesiol. 2005;18:632–639. doi: 10.1097/01.aco.0000191890.36691.cc. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S, Iwatsubo T, Hasegawa M. Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett. 2001;489:46–50. doi: 10.1016/s0014-5793(00)02431-5. [DOI] [PubMed] [Google Scholar]
- Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. 2005;166:1761–1771. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer's disease. Neurosignals. 2002;11:262–269. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Paired helical filament tau in Alzheimer's disease. The kinase connection. Am J Pathol. 1994;144:449–453. [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer's disease hippocampus. Exp Neurol. 2001;168:402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, - 2B and -1. Brain Res. 1996;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- Xie Z, Tanzi RE. Alzheimer's disease and post-operative cognitive dysfunction. Exp Gerontol. 2006;41:346–359. doi: 10.1016/j.exger.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Maeda U, Alfille P, Culley DJ, Crosby G, Tanzi RE. The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid beta protein levels. Anesthesiology. 2006;104:988–994. doi: 10.1097/00000542-200605000-00015. [DOI] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]