

Abstract
Growing evidence implicates microglia in the loss of dopaminergic neurons in Parkinson's disease (PD). However, factors mediating microglial activation in PD are poorly understood. Proinflammatory cytokines, such as interferon-γ (IFN-γ), orchestrate the actions of microglia. We report here that PD patients express significantly elevated levels of IFN-γ in their blood plasma. After this initial finding, we found that IFN-γ-deficient mice displayed attenuated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced substantia nigra pars compacta dopaminergic cell loss along with reduced loss of striatal tyrosine hydroxylase and dopamine transporter fiber density. MPTP-induced depletion of striatal dopamine and its metabolite DOPAC (3,4-dihydroxyphenylacetic acid), as well as ΔFosB, a marker of postsynaptic dysfunction, were also attenuated in these knock-out mice. Consistent with the role for IFN-γ in microglial activation, MPTP-induced morphological activation of microglia was abrogated compared with wild-type mice. To examine more mechanistically the role of IFN-γ in microglial activation, we evaluated the interactions between microglia and dopaminergic neurons in an in vitro mixed microglia/midbrain neuron rotenone-induced death paradigm. In this in vitro paradigm, dopaminergic neurons are selectively damaged by rotenone. Exogenous IFN-γ ligand alone and without rotenone resulted in dopaminergic cell loss, but only in the presence of microglia. The addition of an IFN-γ neutralizing antibody attenuated neuronal loss as a result of rotenone treatment. The presence of only wild-type microglia and not those deficient in IFN-γ receptor elicited significant dopaminergic cell loss when exposed to rotenone. Neurons deficient in IFN-γ receptor, however, did not display increased resistance to death. Finally, levels of IFN-γ message increased in microglia in response to rotenone. Together, these data suggest that IFN-γ participates in death of dopaminergic neurons by regulating microglial activity.
Keywords: interferon-γ (IFN-γ); Parkinson's disease; neurodegeneration; dopamine; cytokine; microglia; 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
Introduction
Parkinson's disease (PD) is a neurodegenerative condition characterized by progressive motor deficits including tremor, bradykinesia, rigidity, and postural instabilities (Przedborski et al., 2001). Many of the cardinal symptoms of PD are attributed to depletion of dopamine (DA) in the brain resulting from the loss of dopaminergic neurons of the substantia nigra pars compacta (SNc) (Smeyne and Jackson-Lewis, 2005). Current therapies, such as l-DOPA (L-3,4-dihydroxyphenylalanine), are mainly directed toward replacing DA levels in the brain and, as such, provide only symptomatic relief. These drugs do not modify the progressive neurodegenerative cell loss process associated with PD that, in many cases, results in debilitating side effects (Kostic et al., 1991). Attenuation of the underlying degeneration in PD may provide opportunities for more effective therapeutic intervention. However, the mechanism(s) controlling dopaminergic loss is not completely clear.
Administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) produces a syndrome in mice and primates resembling that of clinical PD (Langston et al., 2000; Fukuda, 2001). Similar to the histopathological features of PD, MPTP treatment provokes a selective loss of dopaminergic neurons in the SNc coupled with a profound reduction in striatal DA with little alteration in other monoamine neurotransmitter systems (Smeyne and Jackson-Lewis, 2005). This model is frequently used to examine dopaminergic cell loss in an in vivo setting.
A common characteristic of PD and the MPTP model of degeneration is increased microglial activation (Czlonkowska et al., 1996; Gao et al., 2002b; Barcia et al., 2004). Several lines of evidence support the importance of this activation in the deterioration of dopaminergic neurons. The use of the pharmacological anti-inflammatory agent minocycline attenuates activation of microglia as well as MPTP-induced dopaminergic cell loss (Du et al., 2001; Wu et al., 2002). Direct administration of lipopolysaccharide (LPS) induces microgliosis and has been correlated with the loss of dopaminergic neurons in vivo (Arai et al., 2004; De Pablos et al., 2005). Activation of this innate immunity has also been implicated in neurodegeneration with the specific triggering of Toll-like receptor-4-dependent pathways (Lehnardt et al., 2003). Furthermore, low concentrations of rotenone require microglia to induce death of dopaminergic neurons (Gao et al., 2002a). One potential mechanism underlying the pro-death effects of microglia on dopaminergic neurons involves increased oxidative insult. For example, microglia are known to activate proteins that increase oxidative load, including NADPH oxidase and inducible nitric oxide synthase (iNOS) (Almer et al., 1999; Liberatore et al., 1999; Wu et al., 2003). Indeed, mice deficient in both of these signals display attenuated MPTP-induced loss of dopaminergic neurons. Although this evidence implicates microglial activation, the functional relevance of microgliosis is still controversial. Specifically, it is unclear, particularly in vivo, whether microglial activation is causative or correlative to dopaminergic neuron loss. In addition, other types of glial activation (i.e., astrocytes) might also be important in dopaminergic cell death (Suzumura et al., 2006). Accordingly, questions of (1) whether microglia may only be a secondary effect to initial dopaminergic degeneration and (2) how important microglial activation may be to dopaminergic degeneration in vivo remain. For example, agents such as minocycline may protect by means other than microglial activation (Wu et al., 2002; Yang et al., 2003).
Inflammatory mediators are central to microglial activation (McGeer and McGeer, 2002). However, the required nature of the inflammatory response, as it relates to microglia and dopaminergic death, is not completely clear. Various cytokines including interleukin (IL)-1β, IL-2, IL-4, IL-6, transforming growth factor-β, and tumor necrosis factor-α (TNF-α) are increased in the brains of PD patients (Nagatsu et al., 2000). Likewise, cDNA microarray revealed that MPTP-treated mice displayed alterations in genes for numerous proinflammatory cytokines (IL-1, IL-6, and TNF-α) as well as the anti-inflammatory cytokine IL-10 (Mandel et al., 2000). However, the function of these cytokines in dopaminergic neuronal loss is less clear.
There is recent evidence that another critical inflammatory cytokine, interferon-γ (IFN-γ), may be important in PD. IFN-γ is believed to play an important role in early immunological responses to viral and tumor insults, as well as slow developing adaptive responses to infections (Mamane et al., 1999). In the brain, IFN-γ is produced by microglia (De Simone et al., 1998) and can be a strong activator of microglia in vitro (Badie et al., 2000). IFN-γ, through the Janus kinase/signal transducers and activators of transcription-1 (Jak/STAT1) pathway, mediates the induction of iNOS (Delgado, 2003). Recently, Ciesielska et al. (2003) have shown IFN-γ transcripts increase after MPTP treatment in mice. Evidence for a relationship between IFN-γ and PD is suggested by the fact that allelic differences between early- and late-onset PD patients were reported for the IFN-γ gene (Mizuta et al., 2001). However, to date, there is no clear evidence to indicate that IFN-γ is central to dopaminergic cell loss. Consequently, we examined whether IFN-γ may be required in the loss of dopaminergic neurons in both in vivo and in vitro models of PD. We present evidence for the importance of IFN-γ in the death of dopaminergic neurons induced by MPTP (in vivo) and rotenone (in vitro). We also provide evidence that IFN-γ can act through microglia to promote dopaminergic loss and suggest that this may be critical in the pathogenesis of PD.
Materials and Methods
Patient IFN-γ plasma analysis.
Human plasma was examined using a multiplex bead-based immunoassaying system as described previously (Hulse et al., 2004). The patients' sera were diluted fourfold in PBS containing 1% BSA and 0.05% NaN3. Fifty microliters of each sample were incubated for 2 h at room temperature with the antibody-coupled beads in a filter-bottomed 96-well plate (Nunc, Naperville, IL; VWR International, West Chester, PA). The biotinylated anti-cytokine antibody conjugate (1:500) was added to the well and incubated for 1 h at room temperature. Bound antibodies were revealed with a streptavidin–phycoerythrin conjugate. After washing and resuspension in 2% paraformaldehyde in PBS, the beads were analyzed on a luminex-100 system (Luminex, Austin, TX) equipped with Luminex Data Collector software, version 1.7, using the median fluorescence intensity as the read out. Standard curves were established using recombinant IFN-γ, and sample IFN-γ concentrations were derived from the linear portion of the curve. Patient diagnosis was assessed by David Grimes after clinical history and examination. Blood samples from 13 PD patients and 7 disease control patients were analyzed in this study (supplemental Table 1, available at www.jneurosci.org as supplemental material). The Ottawa Hospital Research Ethics Board sanctioned approval for this study. Samples were drawn and immediately centrifuged with serum drawn off and frozen. Patient confidentiality was maintained using number coding, and samples were blinded to condition. The mean age of the PD patients was 66 years (range, 44–77 years), the duration of the disease was 7.5 ± 1.3 years, and all patients had been medicated as specified (supplemental Table 1, available at www.jneurosci.org as supplemental material). The mean age of the disease control patients was 66 years (range, 61–76 years).
Experimental animals.
Male IFN-γ −/− knock-out (KO), IFN-γ receptor −/− KO, and +/+ wild-type (WT) mice (all on a C57BK/6J background; 12 generations of KO mice) were obtained from The Jackson Laboratory (Bar Harbor, ME). The animals were maintained on a 12 h light/dark cycle with lights on at 6:00 A.M., the room temperature was maintained at 21°C, and the mice were permitted an ad libitum diet of Ralston Purina (St. Louis, MO) mouse chow. For the in vivo experiments, two separate groups of animals were analyzed. In the first, IFN-γ KO and WT mice were treated with both MPTP and saline. To corroborate data from these findings, the IFN-γ KO and WT mice were interbred for two generations to produce littermate controls, which were then used in a second set of experiments. All experimental procedures were approved by the University of Ottawa Committee for Animal Care and met the guidelines set out by the Canadian Council on Animal Care.
MPTP administration.
N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP-HCl; 25 mg/kg, i.p., measured as free base; Sigma, St. Louis, MO) was administered to mice (8–10 weeks old) intraperitoneally once per day for 5 consecutive days (Crocker et al., 2003). Mice used as controls received an equivalent volume of saline (0.9%). Mice were killed 14 d after initial MPTP/saline treatment.
Immunohistochemistry.
After overnight postfixation in 4% paraformaldehyde, brains were cryoprotected in 10% sucrose before cryosectioning into 14 or 40 μm (used for stereology) free-floating sections, obtained as described previously (Smith et al., 2003). Immunostaining was performed as described previously using mouse anti-tyrosine hydroxylase (TH; 1:10,000; ImmunoStar, Hudson, WI), rat anti-DA transporter (DAT; 1:2000; Millipore, Bedford, MA), rat anti-CD11b (1:200; Serotec, Oxford, UK), or rabbit anti-ΔFosB (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). Primary antibodies were visualized using diaminobenzidine, as described previously (Crocker et al., 2003).
Assessment of neuronal survival.
Dopaminergic neurons in the SNc of treated mice were assessed for survival using the dopaminergic cell marker TH. Assessment of survival was performed using two different counting methodologies. In the first set of animals, dopaminergic neurons in the SNc of WT and IFN-γ KO mice were determined by serial section analysis of the total number of TH-positive (TH+) neurons as described previously (Crocker et al., 2003; Smith et al., 2003) at 14 d after initial MPTP treatment. Briefly, midbrain tissue sections were stained for TH. At least two sections were obtained from each animal representing each of five levels from −2.92 to −3.52 relative to bregma. Sections from each level and animal were averaged for the number of TH+ neurons. A total of six to eight animals per group were analyzed. Estimates of total TH+-stained neurons in the SNc were calculated using Abercrombie's correction for the previously stated bregma levels, centering on the central region of the SNc, the medial terminal nucleus (MTN) (Hayley et al., 2004). Adjacent tissue was also stained using anatomical stain, cresyl violet, to validate survival as assessed by TH analysis. These results were subsequently corroborated, by examining a second group of animals interbred to produce littermate controls, as described above. The SNc region of this second group of animals was analyzed by unbiased stereological estimates. The total number of TH+ neurons in the SNc was obtained by applying optical fractionation (Gundersen et al., 1988) using Stereo Investigator (version 6; MicroBrightField, Williston, VT), as described previously (Rymar et al., 2004). In brief, 40 μm brain sections were examined within the rostal and caudal limits of the SNc (bregma −2.54 to −3.88) (Paxinos and Franklin, 2001). For each brain, six coronal sections were examined. After immunohistochemistry, mounting, defatting, and coverslipping, the mean section thickness, as measured with a z-axis microcator, was 16 μm. Sections were analyzed using a 100× lens. Total number of TH+ neurons was determined using the optical fractionator.
Quantification of striatal immunohistochemistry.
Quantification of striatal dopaminergic density fiber staining and striatal ΔFosB-positive nuclei were analyzed using Eclipse densitometry analysis (Crocker et al., 2003). In all cases of immunohistochemical quantification, analyses were performed by an individual unaware of the experimental treatments. Each tissue quantified was compared relative to its own nonstained background.
N-methyl-4-phenylpyridinium ion measurements. Striatal concentrations of N-methyl-4-phenylpyridinium ion (MPP+) were measured 90 min after a single dose of MPTP using HPLC measurements, as described previously (Crocker et al., 2003).
Neurochemical analyses.
After decapitation, brains were sectioned into a series of coronal slices using a dissecting block with adjacent slots spaced 2 mm apart. The SNc and striatum were obtained by micropunch using a 1-mm-diameter biopsy needle. Brain punches were taken according to the mouse brain atlas of Paxinos and Franklin (Paxinos and Franklin, 2001; Smith et al., 2003). HPLC analysis was performed 14 d after initial MPTP treatment. Levels of DA and its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) were determined by HPLC using methods described previously (Seegal et al., 1986; Smith et al., 2003).
Primary neuron-enriched mesencephalic cultures.
The entire midbrain, without meninges and blood vessels, was collected from the brains of embryonic day 13.5 (E13.5) mice, as described previously (Liu et al., 2000). The midbrain tissue was removed and subjected to trypsin (T-4549; 1 mg/ml; Sigma) for 25 min at 37°C. After dissociation, the cells were treated with trypsin inhibitor (0.92 mg/ml dissolved in 1 mm HCl; catalog #10137100; Roche, Indianapolis, IN) and DNase (1 mg/ml; catalog #1284-932; Roche) and were counted using a hemacytometer (Bright-Line; Hausser Scientific, Horsham, PA). Midbrain cells were seeded (5 × 105 per well) in 24-well culture plates precoated for 1 h with poly-d-lysine (20 μg/ml). Cultures were maintained in 0.5 ml per well of Neurobasal medium supplemented with 2% B27, 1% N2, 416 μm l-glutamine, 41.6 U/ml penicillin, and 41.6 μg/ml streptomycin and kept in a 37°C incubator-humidified atmosphere of 5% CO2 and 95% air.
Microglia harvest.
Postnatal day 2–3 mice were used for mixed glia cultures. Brains were dissociated using a tissue grinder kit and plated in minimum essential medium supplemented with 5% heat-inactivated fetal bovine plasma, 5% heat-inactivated horse plasma, and 0.05 mg/ml gentamycin. Two days later, the medium was replenished, and the culture reached confluency ∼8 d later. Confluent mixed-glia cultures were shaken for 5 h at 180 rpm at 37°C to separate microglia from astrocytes. This produces a yield of >94% enriched microglia and is comparable with similar harvesting protocols (Gao et al., 2003a).
Coculturing.
Harvested microglia were added (5 × 104 per well) to 7-d-old neuron-enriched mesencephalic cultures. The following day, rotenone was added (20–25 nm), and 7 d later the cultures were fixed and stained for TH (1:2500) and Hoescht (0.24 μg/ml; Sigma). TH immunoreactivity was quantified by counting the total number of all live cells (assessed by Hoescht staining) that stained positive for TH. All live cells typically appear intact with uncondensed nuclei, whereas dead cells displayed fragmented with a condensed morphology. Rotenone treatment has been shown previously to diminish the total number of TH+ live cells in this culture model (Gao et al., 2002a). The results were performed in triplicate and show the average total number of live TH-immunoreactive cells per well. This was compared with untreated control wells that were plated identically and obtained from the same neuronal culture preparation as the experimental wells. For neutralizing antibody studies, IFN-γ-neutralizing IgG (rIFN-γ-purified goat IgG; R & D Systems, Minneapolis, MN) or control nonspecific IgG (normal goat IgG; R & D Systems) was added (15 μg/ml) 30 min before rotenone application. In our ligand application studies, IFN-γ ligand (recombinant mouse IFN-y; R & D Systems) was added 24 h after coculturing, and the cultures were fixed 7 d later. IFN-γ receptor KO neurons and microglia were cultured in the same way as WT cells.
Detection of IFN-γ mRNA using reverse transcription-PCR.
One day after plating, WT microglia-enriched cultures were treated with rotenone (20 nm) for 0, 1, or 3 d. RNA was extracted using Trizol (Invitrogen, San Diego, CA). After RNA purification and determination of RNA concentration, reverse transcription (RT)-PCR analysis for IFN-γ mRNA was conducted using the following primers: forward, ACT GGC AAA AGG ATG GTG AC; reverse, GAC CTG TGG GTT GTT GAC CT. As an internal control, S12 RNA was amplified using the following primers: forward, GGA AGG CAT AGC TGC TGG; reverse, CCT CGA TGA CAT CCT TGG. Cycle conditions were as follows: 94°C (30 s), 59°C (30 s), and 72°C (1 min). Positive control samples were extracted from CD4+ T-cells after being stimulated in the presence of TH1 (T helper cell 1) cytokines for 6 d. After amplification, the PCR products were run on a 2.25% agarose electrophoretic gel in 1× Tris–acetate–EDTA buffer for 30 min and visualized by ethidium bromide staining. Densitometry analysis revealed an increase in IFN-γ mRNA after exposure with rotenone for 1 and 3 d.
Statistical analysis.
All data analyses were performed by using one-way ANOVA, followed by the Newman–Keuls post hoc test (unless otherwise stated).
Results
Elevated levels of plasma IFN-γ in PD patients
We initially examined the levels of several cytokines [IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, and TNF-α] by multiplex bead-based immunoassaying analyses in blood plasma from 13 PD patients compared with 7 normal control individuals. As shown in the Figure 1 dot plot, immunoassay analysis revealed a significant elevation of IFN-γ in blood plasma from PD patients over that in control subjects (33.0 ± 4.1% increase; p < 0.01), as was revealed previously by Gribova et al. (2003). Other cytokines, including GM-CSF, TNF-α, IL-2, IL-4, and IL-5, were not detectable, whereas no significant differences in IL-6 and IL-10 were detected (supplemental Table 2, available at www.jneurosci.org as supplemental material). Accordingly, combined with results by Ciesielska et al. (2003), displaying elevated IFN-γ transcripts in the SNc of mice treated with MPTP, we hypothesized that IFN-γ may be central to dopaminergic loss occurring in PD and MPTP-induced neurotoxicity. Accordingly, we proceeded to examine whether IFN-γ may be important in an in vivo animal model of dopaminergic loss.
Figure 1.

PD patients display elevated IFN-γ plasma levels relative to non-PD patients. Blood plasma from 13 PD and 7 control patients was analyzed by a multiplex bead assay system for several cytokines. Results for IFN-γ concentrations are provided here in dot plot representation (two-tailed t test, **p < 0.01).
IFN-γ-deficient mice exhibit attenuated MPTP-induced degeneration of dopaminergic cell bodies in the SNc
We first assessed dopaminergic neuron survival after saline or MPTP treatment of separate groups of WT and IFN-γ KO mice on a C57BK/6J background (Fig. 2A–C). No statistical difference in the number of TH+ neurons in the SNc could be detected between saline-treated WT and IFN-γ KO mice (Fig. 2B). Similarly, no statistical difference in striatal TH or DAT fiber density was detected by immunohistochemistry (Fig. 2D–G). DAT is critical for the reuptake of DA and MPP+, the active MPTP metabolite in dopaminergic neurons (Meissner et al., 2003). Finally, no differences in levels of striatal DA or its metabolite DOPAC were detected in these mutant mice under basal conditions (Fig. 3C,D). After MPTP administration, there was a significant reduction (42%; p < 0.01) in TH+ SNc neurons in WT mice (Fig. 2E). Interestingly, IFN-γ-deficient mice displayed significantly more TH+ cells than WT mice treated with MPTP (p < 0.05).
Figure 2.

IFN-γ-deficient mice display protection of dopaminergic cell bodies and striatal terminals of MPTP-induced degeneration of SNc dopaminergic neurons. A, Representative photomicrographs illustrating TH immunoreactivity in the ventral midbrain SNc of the indicated non-littermate treatment groups, as indicated in Materials and Methods. B, Quantification of TH+ neurons using Abercrombie correction, as described in Materials and Methods. C, Quantification of cresyl violet-stained cells of the SNc (MTN level). D, Representative photomicrographs of striatal sections stained with TH from the animal groups as described in A. Magnified images are included in the insets of D–J. E, Quantification of optical density of TH striatal fibers. F, Representative photomicrographs illustrating DAT immunoreactivity in the striatum of indicated treatment groups as in A. G, Quantification of optical density of striatal DAT-stained fiber density. H, Representative photomicrographs illustrating TH immunoreactivity in the SNc of the indicated littermate treatment animal groups. Staining is more intense here than in A because of increased section thickness required for stereological analyses. I, Quantification of TH+ neurons, in littermate animals by stereology, as described in Materials and Methods. J, Representative photomicrographs of striatal TH fiber density of indicated littermate treatment groups. K, Quantification of OD of striatal TH-stained fiber density of indicated littermates. Error bars represent mean ± SEM. ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001; n = 6–8 animals per group. OD, Optical density; Sal, saline.
Figure 3.

IFN-γ KO mice display reduced striatal ΔFosB, a marker for postsynaptic changes in the denervated striatum, as well as attenuated DA and DOPAC. A, Representative photomicrographs showing ΔFosB staining in the striatum of mice treated as indicated. Sal, Saline. B, Quantification of ΔFosB-positive cells/nuclei. Str, striatum. C, D, Levels of DA (C) and its metabolite DOPAC (D) in the striatum were analyzed using HPLC on 14-d-old tissue after saline/MPTP injection. Error bars represent mean ± SEM. ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001; n = 6 animals per group.
To further substantiate these findings, we repeated the experiment using WT and IFN-γ KO mice interbred to produce littermate controls. These animals were analyzed by stereological assessment for surviving TH+ neurons over the entire nigral region. Identical results were obtained in comparison to the initial dopaminergic cell survival experiment (Fig. 2A–C). MPTP-treated IFN-γ KO mice exhibited a significant dopaminergic cell-body survival (p < 0.001) compared with WT littermate control animals also treated with MPTP (Fig. 2H,I).
To corroborate TH analysis of dopaminergic neuron survival, the number of neurons at the level of the MTN (bregma −3.16) within the SNc of all groups was determined using cresyl violet staining (Fig. 2C). Analysis was consistent with that of TH survival, showing no difference between saline-treated groups, a significant loss of TH+ neurons in WT MPTP-treated mice (p < 0.001), and a significant reduction in cells lost in IFN-γ-deficient MPTP-treated animals compared with WT mice (p < 0.001).
MPTP derives its toxic effects once it crosses the blood–brain barrier (BBB) and is converted to MPP+, the active dopaminergic neural toxin, by monoamine oxidase-B. The protective effects observed in IFN-γ-deficient animals may be attributed to altered MPTP metabolism and therefore reduced MPP+. To examine this possibility, striatal tissue was evaluated from mice 90 min after a single injection of MPTP, and levels of MPP+ were determined using HPLC. It was observed that MPP+ levels did not significantly differ between WT and IFN-γ-deficient animals (data not shown), suggesting that the protection afforded by IFN-γ deficiency was not because of impaired MPTP metabolism.
IFN-γ-deficient mice exhibit attenuated MPTP-induced degeneration of the DA axon terminal nigrostriatal dopaminergic pathway
Previous results indicated that MPTP-induced loss of dopaminergic cell bodies in the SNc is attenuated by IFN-γ deficiency. To determine whether the striatal projections of the SNc neurons were also protected in IFN-γ-deficient mice, striatal dopaminergic terminal fiber density was assessed initially in non-littermate animals. This analysis is important because this is the region where dopaminergic neurons express their activity through DA release. Densitometry analysis of TH striatal fiber staining showed a significant reduction (68%; p < 0.001) in WT MPTP-treated animals compared with saline controls (Fig. 2D,E). Similar to that observed in the SNc, there was no statistical difference in the TH striatal density of saline-treated WT or IFN-γ-deficient mice. IFN-γ KO animals exhibited an attenuation of MPTP-induced reduction in striatal TH density compared with MPTP-treated WT animals (32 vs 68%). This suggests striatal terminal fibers were protected by IFN-γ deficiency similar to the dopaminergic cell bodies. Results using littermate control animals paralleled these findings, with IFN-γ KO mice displaying attenuation in TH-fiber density (p < 0.01) (Fig. 2J,K).
Additional corroboration of these findings included examining density levels of striatal DAT. Densitometric analysis of DAT staining of striatal fibers showed a significant reduction (38%; p < 0.001) in WT MPTP-treated animals compared with saline controls (Fig. 2F,G). IFN-γ KO animals exhibited an attenuation of MPTP-induced reduction in striatal DAT density compared with WT treated mice (p < 0.01).
Postsynaptic functional effects of IFN-γ deficiency
Treatment with MPTP has been demonstrated previously to induce expression of the transcription factor ΔFosB within the striatum, which is thought to reflect postsynaptic changes (Smith et al., 2003). ΔFosB is suggested to mediate the supersensitivity of striatal DA receptors after denervation (Dragunow et al., 1995). Consistent with these reports, WT MPTP-treated mice displayed a significant increase in the number of ΔFosB-positive cells (Fig. 3A,B) compared with saline controls. Importantly, MPTP-treated IFN-γ-deficient mice showed a significantly reduced increase in FosB staining.
Effect of IFN-γ deficiency on amine levels
Finally, we examined how DA levels are affected in IFN-γ-deficient mice after MPTP treatment. Previous analyses report diminished DA and its metabolite, DOPAC, in the striatum 14 d after MPTP treatment (Crocker et al., 2003). The effects of IFN-γ deficiency parallel the dopaminergic cell survival results observed previously (Fig. 3C,D). In both the striatum and the SNc, MPTP significantly reduced the levels of DA and DOPAC in WT mice. However, there was a significant decrease in the loss of DA in the MPTP-treated IFN-γ KO mice.
IFN-γ-deficient mice display attenuated microglial activation in the SNc in response to MPTP
In WT mice, MPTP has been shown to initiate microgliosis, the morphological and pathological activation of microglia, within the SNc where the highest concentration of microglia resides (Czlonkowska et al., 1996, 2002). Because IFN-γ has also been reported to activate microglia, we examined whether microgliosis was altered in IFN-γ-deficient mice treated with MPTP compared with WT animals. Microglia were stained for CD11b, the α-subunit of the αMβ2 integrin (CD11b/CD18, complement 3bi receptor) that plays an important role in leukocyte adhesion and whose expression in the brain is confined to microglial cells (Zeng et al., 2005). Activated microglia appear more compact and rounded with cellular thickening and complexing (Fig. 4A, WT/MPTP, arrow) (Soltys et al., 2001). Although normal resident microglia express basal levels of CD11b, they appear smaller and have a ramified shape with thin processes (Fig. 4A, WT/Sal, arrow). Quantification of microgliosis yielded a significant difference (p < 0.01) with twice the number of activated microglia in MPTP WT mice than in saline-treated mice (Fig. 4B). Basal activity of microglia in IFN-γ KO mice treated with saline displayed no difference in microgliosis compared with WT saline-treated mice (Fig. 4B). The increase in microgliosis exhibited with MPTP treatment, as seen in WT mice, was not observed in IFN-γ-deficient mice. There was no significant difference in active microglia between IFN-γ-deficient saline- versus MPTP-treated mice. However, there was a significant difference between WT and IFN-γ KO mice treated with MPTP (p < 0.01) (Fig. 4B).
Figure 4.

Analysis of active microglia using anti-CD11b. A, Representative photomicrographs illustrating CD11b immunoreactivity in the SNc (MTN) of WT and KO animals treated with either saline (Sal) or MPTP. Arrows indicate resting CD11b-positive microglia (WT/Sal) and activated microglia (WT/MPTP). B, Quantification of morphologically active microglia. Magnified images of each are provided in the insets of A (WT/Sal and WT/MPTP). Error bars represent mean ± SEM (ANOVA, **p < 0.01; n = 6 animals per group).
Role of IFN-γ in dopaminergic cell loss in an in vitro microglia/neuron coculture system
Our in vivo results indicate that IFN-γ is important in MPTP-induced dopaminergic cell death. The protection associated with IFN-γ deficiency is accompanied with reduced microglial activation. However, the mechanism by which IFN-γ deficiency may be protective and whether it involves microglial activation is difficult to assess in an in vivo setting. This is particularly true in this present situation in which reduced microglial activation could be a secondary effect of reduced neuronal loss. Accordingly, we used an in vitro model of dopaminergic cell loss in which effects of microglia on dopaminergic neurons can be studied more directly. Therefore, to address whether IFN-γ may promote dopaminergic loss through activation of microglia and to more closely study the interaction between these two populations of cells, we used a microglia/midbrain neuron coculture system, similar to that reported previously (Gao et al., 2002a). In this system, midbrain cultures enriched for neurons, obtained from E13.5 embryos, are cultured in defined Neurobasal medium. In these cultures, dopaminergic neurons constitute <1% of all the neurons in culture. All neurons, including TH+ cells, in these cultures are resistant to 20–25 nm rotenone, another complex I inhibitor similar to MPP+ (Kotake and Ohta, 2003). No dopaminergic cell death is observed even after 7 d of rotenone treatment in the absence of microglia (Fig. 5A). The addition of microglia to these cultures alone, in the absence of rotenone, does not significantly affect survival of total neurons or the more sensitive dopaminergic neurons. However, with the addition of 20–25 nm rotenone, a significant loss of TH+ neurons is observed only in the presence of microglia (Fig. 5A). These findings indicate that death of TH+ neurons in this system, at a defined rotenone concentration, is dependent on microglia. It is important to note that this effect is concentration specific; at higher concentrations, rotenone will begin to kill nondopaminergic cell types as well, even in the absence of microglia (data not shown).
Figure 5.
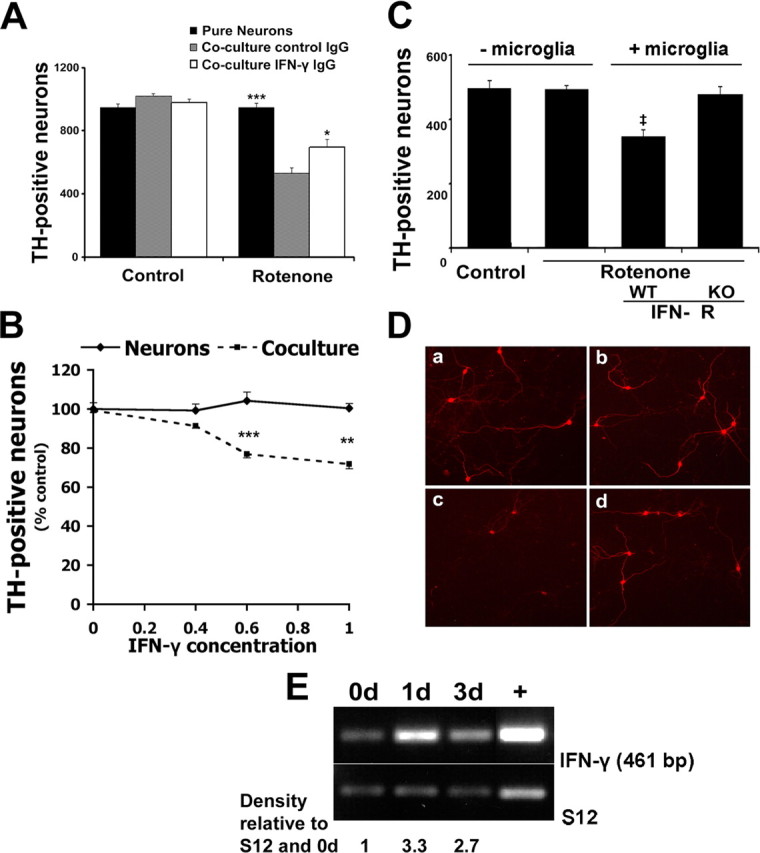
IFN-γ requirement in a coculture model of dopaminergic cell loss induced by rotenone in vitro. A, Neutralizing IFN-γ antibody protects dopaminergic neurons from rotenone-induced death in the presence of microglia. Seven-day-old WT neuron-enriched midbrain cultures were cocultured with and without WT microglia in the presence or absence of rotenone and with control or IFN-γ neutralizing antibody, as described in Materials and Methods and as shown. The number of live TH+ neurons was assessed as described in Materials and Methods. *p < 0.05, ***p < 0.001 relative to coculture with control IgG (two-tailed t test). B, Seven-day-old neuron-enriched midbrain cultures supplemented with or without microglia were subjected to IFN-γ ligand (nanograms per milliliter) applications. The cultures were fixed 7 d later and stained with TH and Hoechst. ***p < 0.001 compared with control, **p < 0.01 (two-tailed t test; n = 3). C, Dopaminergic neurons treated with rotenone are resistant to death in the presence of microglia lacking IFN-γ receptor. Dopaminergic neurons used were similar to those in A, except WT neurons were cultured in the presence of either WT or IFN-γ receptor (IFN- R)-deficient microglia. ‡p < 0.05 relative to IFN-γ receptor KO (two-tailed t test; n = 3). D, Representative immunofluorescence of cocultures quantified in C, stained with TH antibody are as follows: a, WT mesencephalic neurons; b, WT mesencephalic neurons treated with rotenone; c, WT mesencephalic neurons and microglia treated with rotenone; d, WT mesencephalic neurons and IFN-γ receptor KO microglia treated with rotenone. E, WT microglia-enriched cultures were treated with rotenone for 0, 1, and 3 d. One week after plating, microglia were collected, RNA was extracted, and RT-PCR was performed for IFN-γ and S12 control. Densitometric analysis was performed with values normalized to each respective S12 signal and further normalized to 0 d value. +, IFN-γ-positive RNA control sample obtained from CD4+ T-cells. Error bars represent mean ± SEM.
Using this system, we first asked whether inhibition of IFN-γ would affect rotenone-induced dopaminergic death as was observed in vivo. As shown in Figure 5A, a neutralizing antibody to IFN-γ but not control IgG significantly improved dopaminergic survival. This indicates that IFN-γ can modulate the rotenone-induced dopaminergic cell death, which occurs only in the presence of microglia. To further support the importance of IFN-γ in microglia-induced sensitivity of dopaminergic neurons, we exposed midbrain neurons with and without cocultured microglia to varying concentrations of exogenous IFN-γ ligand. As shown in Figure 5B, 0.6 and 1 ng/ml concentrations of exogenous IFN-γ caused significant loss of dopaminergic neurons. This, however, only occurred in the presence of microglia and not in their absence. This indicates that IFN-γ is an important mediator of dopaminergic neuron loss, but only in the presence of microglia.
To determine more clearly whether IFN-γ may act on microglia to control rotenone-induced dopaminergic cell loss in this system, we cultured microglia from IFN-γ receptor KO or WT control mice. These WT or IFN-γ receptor-deficient microglia were then cocultured with normal neurons with or without rotenone treatment. Death of dopaminergic neurons only occurred in the presence of WT microglia with rotenone treatment (Fig. 5C,D). Cultures treated with rotenone but containing IFN-γ receptor-deficient microglia displayed no death. Conversely, we also examined whether midbrain neurons cultured from IFN-γ receptor-deficient animals were resistant to rotenone in the presence of WT microglia. There was no difference in death of IFN-γ receptor-deficient neurons compared with WT neurons exposed to rotenone in the presence of WT microglia (data not shown). Finally, if rotenone-mediated death of dopaminergic neurons occurs through IFN-γ, we would predict that IFN-γ should be present in microglia. Accordingly, we isolated microglia, exposed them to rotenone, and determined the IFN-γ message at certain times after treatment by RT-PCR. As shown in Figure 5E, there was a basal level of IFN-γ message present in untreated microglia that increased on exposure to rotenone. Together, these results indicate that IFN-γ plays a crucial role in modulating microglial responses in vitro. In addition, it suggests that IFN-γ may promote MPTP-induced dopaminergic loss in vivo, at least partially through activation of microglia.
Discussion
Previous work has suggested the importance of the inflammatory response and microglial activation in dopaminergic loss in animal models of PD as well as in PD patients (McGeer et al., 1988; Banati et al., 1998; Wu et al., 2002; Barcia et al., 2004). However, the required nature of the inflammatory mediators or their mechanisms of action in PD is not fully understood. Our findings are of significance because they directly demonstrate that a central inflammatory mediator, IFN-γ, plays a critical role in dopaminergic loss, both in vitro and in vivo. We found that (1) levels of IFN-γ are elevated in the serum of PD patients, (2) IFN-γ deficiency attenuates the degeneration of the nigrostriatal system and significantly reduces microgliosis in a model of in vivo dopaminergic loss, and (3) IFN-γ is involved in microglial-mediating dopaminergic neurodegeneration.
Our analysis of human blood serum for possible elevated cytokines yielded results with only levels of IFN-γ displaying a significant increase from those of control patients. It must be noted that cytokine detection in this assay may be hampered because of a lack in cytokine stability (Noguchi et al., 1996). In addition, it should also be stressed that cytokine blood levels do not necessarily reflect actions in the CNS. However, systemic IFN-γ-producing inflammatory cells have been reported to infiltrate the brain (Perry, 2004). In addition, Banks (2005) has recently suggested that cytokines circulating in the blood may cross into the CNS through direct cytokine transport across the BBB. Therefore, we also examined the role of IFN-γ in dopaminergic neuronal loss.
Our results, in an in vivo mouse model of dopaminergic neurodegeneration, reveal that mice deficient in IFN-γ show reduced loss of dopaminergic cell bodies and striatal terminals compared with WT animals treated with MPTP. Germline loss of IFN-γ did not appear to grossly perturb the dopaminergic system, including possible postsynaptic signaling changes (FosB), as assessed in saline-treated animals. In addition, DA and DOPAC levels, as well as markers of postsynaptic function (FosB), are significantly attenuated in IFN-γ-deficient animals treated with MPTP. Interestingly, microglial activation is also reduced in these deficient mice.
Consistent with our in vivo findings, we observed that IFN-γ is required for the death of dopaminergic neurons cultured in the presence of microglia with rotenone. Importantly, reduced cell loss was only observed when microglia deficient in IFN-γ receptor, but not WT microglia, were added to the dopaminergic neurons. In contrast, deficiency of IFN-γ receptors in dopaminergic neurons did not result in protection in our coculture death paradigm. IFN-γ was also detected in cultured microglia that increased on rotenone exposure. Together, the data not only provide evidence for direct relevance of the microglial response in dopaminergic cell loss induced by mitochondrial toxins but also define IFN-γ as a critical inflammatory mediator in this process.
Our results are consistent with the view that IFN-γ is a primary mediator of microglial activation (Moss and Bates, 2001; Platten et al., 2001; Abbas et al., 2002; Delgado, 2003). There are a number of possible effects of IFN-γ on microglia that may contribute to dopaminergic loss, both in vivo and in vitro. First, IFN-γ, through binding to its receptor and activation of the Jak/STAT pathway, can activate iNOS. Alternatively, IFN-γ has also been shown to upregulate the death receptor Fas and its ligand, FasL (Badie et al., 2000). Accordingly, IFN-γ could mediate its effects on microglia through direct activation of the death receptor on dopaminergic neurons. Indeed, Fas has been shown to be localized to dopaminergic neurons and is implicated in MPTP-induced dopaminergic cell death (Hayley et al., 2004).
Our model that IFN-γ promotes dopaminergic cell death through the activation of microglia is consistent with growing evidence that the inflammatory pathway is central to dopaminergic loss in in vivo and in in vitro models. Indeed, microglial activation has been noted in PD animal models and postmortem brains (McGeer et al., 1988; Banati et al., 1998). Direct activation of microglia through administration of the endotoxin LPS was found to induce loss of dopaminergic neurons in mice (Arai et al., 2004), while also stimulating the production of iNOS and subsequent reactive oxygen species (ROS) (Li et al., 2005). In fact, the nigra appears to be exquisitely sensitive to microglia activation because equivalent LPS injections in alternate brain regions (hippocampus, cortex) did not have the same pattern of degeneration (Xie et al., 2003). Finally, LPS and MPTP can synergistically increase dopaminergic death (Gao et al., 2003b).
However, most functional evidence implicating microglia as a direct cause of dopaminergic cell degeneration is controversial. Minocycline, a tetracycline derivative known to inhibit microglia activation/inflammation and protect dopaminergic neurons in an MPTP model of PD, can also inhibit classic death molecules, such as caspases (Chen et al., 2000; Wei et al., 2005). Moreover, others have reported that minocycline can exacerbate MPTP-induced toxicity (Yang et al., 2003). In addition, the role of microglia may also be dependent on the mode of activation. For example, activation of microglia in the hippocampus by IL-4- and IFN-γ-expressing T-cells is neuroprotective (Butovsky et al., 2005). Our evidence presented here is important because it provides clear evidence for the central death-promoting nature of the inflammatory response in dopaminergic neuronal loss. Finally, it is important to note that microglial activation is likely a graded response, and the degree of activation may depend on the type and severity of insult.
Although our evidence indicates the important nature of IFN-γ activation of microglia in vitro, the exact mechanism by which IFN-γ promotes dopaminergic death in vivo and whether this is strictly reliant on microglial activation is unclear. Indeed, IFN-γ receptors are expressed ubiquitously (Blanck, 2002). In addition, it is important to note that our results do not imply that inflammatory processes are the exclusive mediators of neuronal damage. Indeed, although IFN-γ deficiency effectively blocks microglial activation, it does not completely eliminate the reduction in striatal DA/DOPAC. This is an interesting observation that suggests that microglial activation, although important, is not the only mechanisms by which dopaminergic neuron damage occurs. This notion is supported by the fact that MPTP (MPP+ metabolite) can directly target mitochondria in neurons and lead to increased ROS and damage (Liberatore et al., 1999; Dehmer et al., 2000; Gonzalez-Polo et al., 2004; Chen et al., 2006).
There are some intriguing links with our present IFN-γ studies to that of human PD. First, microglial activation in PD patients is well characterized (Ouchi et al., 2005). Second, we and others have shown that plasma levels of IFN-γ are elevated in PD patients (Nakamura et al., 1992). The exact significance of this is unknown, particularly because the relationship, if any, between peripheral events and events in the CNS is unclear. However, increased plasma IFN-γ levels could reflect a systemic increase in IFN-γ production by lymphocytes and infiltrates capable of entering the CNS. In support of this, T-cells are known to circulate in the brain with lymphocyte infiltration, and release of proinflammatory cytokines, such as TNF-α and IFN-γ, have been shown at the site of injury (Raivich et al., 2003). Third, IFN-γ levels have also been shown to be increased in CSF and in the substantia nigra of PD patients (Widner et al., 2002). Fourth, IFN-γ has been shown to upregulate iNOS, by binding the IFN-γ receptor and by upregulating expression of astrocyte CD23 (Okuno et al., 2005). Interestingly, CD23 is found in glia from the SNc of PD patients, yet it is not detected in control subjects (Hunot et al., 1999). Finally, genetic studies have linked a dinucleotide CA repeat polymorphism within the first intron of the IFN-γ gene of patients with late onset of PD (Hakansson et al., 2005). This polymorphism leads to a higher production of IFN-γ. Together, this evidence suggests the potential importance of IFN-γ in PD pathogenesis and provides a link to our animal studies presented here.
In conclusion, we have shown that mice deficient in IFN-γ display an attenuation of MPTP-induced dopaminergic cell death and maintenance of DA functional activity. Our data suggest that one mechanism by which this is observed involves the activation of microglia, aiding in the damage to dopaminergic neurons. Accordingly, targeting microgliosis by modulating levels of IFN-γ may be a candidate therapeutic strategy for treatment of PD.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (D.S.P., H.A.), Parkinson's Society Canada, the Parkinson's Disease Foundation (USA), the United States Department of National Defense, the Michael J. Fox Foundation and Parkinson's Research Consortium, Heart and Stroke Foundation Ontario (D.S.P.), and the Natural Sciences and Engineering Research Council (S.H.). H.A. and S.H. have Canadian Research Chairs in Neuroscience. We thank the technical staff at Roger Guindon Hall Animal Care Veterinary Services (University of Ottawa, Ottawa, Ontario, Canada) for their assistance with our animals, Fraser Scott and David Lefebvre for reagents, Abbas Sadikot and Vladimir Rymar for their assistance with stereological analysis, and Yasmilde Rodriguez Gonzalez for her technical assistance.
References
- Abbas N, Bednar I, Mix E, Marie S, Paterson D, Ljungberg A, Morris C, Winblad B, Nordberg A, Zhu J. Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J Neuroimmunol. 2002;126:50–57. doi: 10.1016/s0165-5728(02)00050-4. [DOI] [PubMed] [Google Scholar]
- Almer G, Vukosavic S, Romero N, Przedborski S. Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H. Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1beta, and expression of caspase-11 in mice. J Biol Chem. 2004;279:51647–51653. doi: 10.1074/jbc.M407328200. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner J, Vorpahl J, Preston K. Interferon-gamma induces apoptosis and augments the expression of Fas and Fas ligand by microglia in vitro. Exp Neurol. 2000;162:290–296. doi: 10.1006/exnr.1999.7345. [DOI] [PubMed] [Google Scholar]
- Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson's disease. Mov Disord. 1998;13:221–227. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- Banks WA. Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr Pharm Des. 2005;11:973–984. doi: 10.2174/1381612053381684. [DOI] [PubMed] [Google Scholar]
- Barcia C, Sanchez Bahillo A, Fernandez-Villalba E, Bautista V, Poza YPM, Fernandez-Barreiro A, Hirsch EC, Herrero MT. Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia. 2004;46:402–409. doi: 10.1002/glia.20015. [DOI] [PubMed] [Google Scholar]
- Blanck G. Components of the IFN-gamma signaling pathway in tumorigenesis. Arch Immunol Ther Exp (Warsz) 2002;50:151–158. [PubMed] [Google Scholar]
- Butovsky O, Talpalar AE, Ben-Yaakov K, Schwartz M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci. 2005;29:381–393. doi: 10.1016/j.mcn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Chen J, Tang XQ, Zhi JL, Cui Y, Yu HM, Tang EH, Sun SN, Feng JQ, Chen PX. Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis. 2006;11:943–953. doi: 10.1007/s10495-006-6715-5. [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- Ciesielska A, Joniec I, Przybylkowski A, Gromadzka G, Kurkowska-Jastrzebska I, Czlonkowska A, Czlonkowski A. Dynamics of expression of the mRNA for cytokines and inducible nitric synthase in a murine model of the Parkinson's disease. Acta Neurobiol Exp (Wars) 2003;63:117–126. doi: 10.55782/ane-2003-1461. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, Callaghan SM, Slack RS, Melloni E, Przedborski S, Robertson GS, Anisman H, Merali Z, Park DS. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson's disease. J Neurosci. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration. 1996;5:137–143. doi: 10.1006/neur.1996.0020. [DOI] [PubMed] [Google Scholar]
- Czlonkowska A, Kurkowska-Jastrzebska I, Czlonkowski A, Peter D, Stefano GB. Immune processes in the pathogenesis of Parkinson's disease—a potential role for microglia and nitric oxide. Med Sci Monit. 2002;8:RA165–RA177. [PubMed] [Google Scholar]
- Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem. 2000;74:2213–2216. doi: 10.1046/j.1471-4159.2000.0742213.x. [DOI] [PubMed] [Google Scholar]
- Delgado M. Inhibition of interferon (IFN) gamma-induced Jak-STAT1 activation in microglia by vasoactive intestinal peptide: inhibitory effect on CD40, IFN-induced protein-10, and inducible nitric-oxide synthase expression. J Biol Chem. 2003;278:27620–27629. doi: 10.1074/jbc.M303199200. [DOI] [PubMed] [Google Scholar]
- De Pablos RM, Herrera AJ, Villaran RF, Cano J, Machado A. Dopamine-dependent neurotoxicity of lipopolysaccharide in substantia nigra. FASEB J. 2005;19:407–409. doi: 10.1096/fj.04-2153fje. [DOI] [PubMed] [Google Scholar]
- De Simone R, Levi G, Aloisi F. Interferon gamma gene expression in rat central nervous system glial cells. Cytokine. 1998;10:418–422. doi: 10.1006/cyto.1997.0314. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Butterworth N, Waldvogel H, Faull RL, Nicholson LF. Prolonged expression of Fos-related antigens, Jun B and TrkB in dopamine-denervated striatal neurons. Brain Res Mol Brain Res. 1995;30:393–396. doi: 10.1016/0169-328x(95)00037-s. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T. Neurotoxicity of MPTP. Neuropathology. 2001;21:323–332. doi: 10.1046/j.1440-1789.2001.00402.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002a;22:782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. 2002b;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. J Neurosci. 2003a;23:1228–1236. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Synergistic dopaminergic neurotoxicity of MPTP and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. FASEB J. 2003b;17:1957–1959. doi: 10.1096/fj.03-0203fje. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Soler G, Rodriguezmartin A, Moran JM, Fuentes JM. Protection against MPP+ neurotoxicity in cerebellar granule cells by antioxidants. Cell Biol Int. 2004;28:373–380. doi: 10.1016/j.cellbi.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Gribova IE, Gnedenko BB, Poleshchuk VV, Morozov SG. [Level of interferon-gamma, tumor necrosis factor alpha, and antibodies to them in blood serum from Parkinson disease patients] Biomed Khim. 2003;49:208–212. [PubMed] [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ. The new stereological tools: disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Hakansson A, Westberg L, Nilsson S, Buervenich S, Carmine A, Holmberg B, Sydow O, Olson L, Johnels B, Eriksson E, Nissbrandt H. Investigation of genes coding for inflammatory components in Parkinson's disease. Mov Disord. 2005;20:569–573. doi: 10.1002/mds.20378. [DOI] [PubMed] [Google Scholar]
- Hayley S, Crocker SJ, Smith PD, Shree T, Jackson-Lewis V, Przedborski S, Mount M, Slack R, Anisman H, Park DS. Regulation of dopaminergic loss by Fas in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2004;24:2045–2053. doi: 10.1523/JNEUROSCI.4564-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse RE, Kunkler PE, Fedynyshyn JP, Kraig RP. Optimization of multiplexed bead-based cytokine immunoassays for rat serum and brain tissue. J Neurosci Methods. 2004;136:87–98. doi: 10.1016/j.jneumeth.2003.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch EC. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic V, Przedborski S, Flaster E, Sternic N. Early development of levodopa-induced dyskinesias and response fluctuations in young-onset Parkinson's disease. Neurology. 1991;41:202–205. doi: 10.1212/wnl.41.2_part_1.202. [DOI] [PubMed] [Google Scholar]
- Kotake Y, Ohta S. MPP+ analogs acting on mitochondria and inducing neuro-degeneration. Curr Med Chem. 2003;10:2507–2516. doi: 10.2174/0929867033456558. [DOI] [PubMed] [Google Scholar]
- Langston JW, Quik M, Petzinger G, Jakowec M, Di Monte DA. Investigating levodopa-induced dyskinesias in the parkinsonian primate. Ann Neurol. 2000;47:S79–89. [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci USA. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Wang T, Pei Z, Liu B, Hong JS. Inhibition of microglial activation by the herbal flavonoid baicalein attenuates inflammation-mediated degeneration of dopaminergic neurons. J Neural Transm. 2005;112:331–347. doi: 10.1007/s00702-004-0213-0. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- Mamane Y, Heylbroeck C, Genin P, Algarte M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R, Hiscott J. Interferon regulatory factors: the next generation. Gene. 1999;237:1–14. doi: 10.1016/s0378-1119(99)00262-0. [DOI] [PubMed] [Google Scholar]
- Mandel S, Grunblatt E, Youdim M. cDNA microarray to study gene expression of dopaminergic neurodegeneration and neuroprotection in MPTP and 6-hydroxydopamine models: implications for idiopathic Parkinson's disease. J Neural Transm Suppl. 2000;60:117–124. doi: 10.1007/978-3-7091-6301-6_7. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Meissner W, Prunier C, Guilloteau D, Chalon S, Gross CE, Bezard E. Time-course of nigrostriatal degeneration in a progressive MPTP-lesioned macaque model of Parkinson's disease. Mol Neurobiol. 2003;28:209–218. doi: 10.1385/MN:28:3:209. [DOI] [PubMed] [Google Scholar]
- Mizuta I, Nishimura M, Mizuta E, Yamasaki S, Ohta M, Kuno S, Ota M. Relation between the high production related allele of the interferon-gamma (IFN-gamma) gene and age at onset of idiopathic Parkinson's disease in Japan. J Neurol Neurosurg Psychiatry. 2001;71:818–819. doi: 10.1136/jnnp.71.6.818a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss DW, Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci. 2001;13:529–538. doi: 10.1046/j.1460-9568.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson's disease. J Neural Transm Suppl. 2000;58:143–151. [PubMed] [Google Scholar]
- Nakamura S, Nagano I, Yoshioka M, Onodera J, Kogure K. [Detection of interferon-gamma and IL-6 producing cells in cerebrospinal fluid cells in the central nervous system infectious diseases using immunocytochemistry] Rinsho Shinkeigaku. 1992;32:143–147. [PubMed] [Google Scholar]
- Noguchi Y, Yoshikawa T, Matsumoto A, Svaninger G, Gelin J. Are cytokines possible mediators of cancer cachexia? Surg Today. 1996;26:467–475. doi: 10.1007/BF00311551. [DOI] [PubMed] [Google Scholar]
- Okuno T, Nakatsuji Y, Kumanogoh A, Moriya M, Ichinose H, Sumi H, Fujimura H, Kikutani H, Sakoda S. Loss of dopaminergic neurons by the induction of inducible nitric oxide synthase and cyclooxygenase-2 via CD40: relevance to Parkinson's disease. J Neurosci Res. 2005;81:874–882. doi: 10.1002/jnr.20599. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Platten M, Wick W, Wischhusen J, Weller M. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) suppresses microglial inducible nitric oxide synthase (iNOS) expression and activity induced by interferon-gamma (IFN-gamma) Br J Pharmacol. 2001;134:1279–1284. doi: 10.1038/sj.bjp.0704373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 2001;76:1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Werner A, Jones LL, Galiano M, Kloss CU, Zhu XZ, Pfeffer K, Liu ZQ. Lymphocyte infiltration in the injured brain: role of proinflammatory cytokines. J Neurosci Res. 2003;72:726–733. doi: 10.1002/jnr.10621. [DOI] [PubMed] [Google Scholar]
- Rymar VV, Sasseville R, Luk KC, Sadikot AF. Neurogenesis and stereological morphometry of calretinin-immunoreactive GABAergic interneurons of the neostriatum. J Comp Neurol. 2004;469:325–339. doi: 10.1002/cne.11008. [DOI] [PubMed] [Google Scholar]
- Seegal RF, Brosch KO, Bush B. High-performance liquid chromatography of biogenic amines and metabolites in brain, cerebrospinal fluid, urine and plasma. J Chromatogr. 1986;377:131–144. doi: 10.1016/s0378-4347(00)80768-9. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson's disease. Brain Res Mol Brain Res. 2005;134:57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci USA. 2003;100:13650–13655. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltys Z, Ziaja M, Pawlinski R, Setkowicz Z, Janeczko K. Morphology of reactive microglia in the injured cerebral cortex. Fractal analysis and complementary quantitative methods. J Neurosci Res. 2001;63:90–97. doi: 10.1002/1097-4547(20010101)63:1<90::AID-JNR11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Suzumura A, Takeuchi H, Zhang G, Kuno R, Mizuno T. Roles of glia-derived cytokines on neuronal degeneration and regeneration. Ann NY Acad Sci. 2006;1088:219–229. doi: 10.1196/annals.1366.012. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhao L, Liu J, Dodel RC, Farlow MR, Du Y. Minocycline prevents gentamicin-induced ototoxicity by inhibiting p38 MAP kinase phosphorylation and caspase 3 activation. Neuroscience. 2005;131:513–521. doi: 10.1016/j.neuroscience.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Widner B, Leblhuber F, Fuchs D. Increased neopterin production and tryptophan degradation in advanced Parkinson's disease. J Neural Transm. 2002;109:181–189. doi: 10.1007/s007020200014. [DOI] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci USA. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Morgan TE, Rozovsky I, Finch CE. Aging and glial responses to lipopolysaccharide in vitro: greater induction of IL-1 and IL-6, but smaller induction of neurotoxicity. Exp Neurol. 2003;182:135–141. doi: 10.1016/s0014-4886(03)00057-8. [DOI] [PubMed] [Google Scholar]
- Yang L, Sugama S, Chirichigno JW, Gregorio J, Lorenzl S, Shin DH, Browne SE, Shimizu Y, Joh TH, Beal MF, Albers DS. Minocycline enhances MPTP toxicity to dopaminergic neurons. J Neurosci Res. 2003;74:278–285. doi: 10.1002/jnr.10709. [DOI] [PubMed] [Google Scholar]
- Zeng HY, Zhu XA, Zhang C, Yang LP, Wu LM, Tso MO. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Invest Ophthalmol Vis Sci. 2005;46:2992–2999. doi: 10.1167/iovs.05-0118. [DOI] [PubMed] [Google Scholar]