

Abstract
Alzheimer's disease (AD) is a senile dementia characterized by amyloid plaques, neurofibrillary tangles, and synaptic and cell loss. The “amyloid cascade” hypothesis suggests that amyloid-β (Aβ), the peptide deposited as amyloid plaques, is the primary insult in AD. However, debate continues over the mechanism of Aβ toxicity and whether fibrillar or oligomeric Aβ is the active species of the peptide that ultimately causes the synaptic loss and dementia associated with AD. Brain-derived neurotrophic factor (BDNF) is required for survival and function of cells compromised in AD. Decreased BDNF causes defects in long-term potentiation and memory and correlates with cognitive decline. We previously demonstrated that BDNF reduction occurs early in the course of AD, suggesting that decreased BDNF may promote neuronal dysfunction in AD. We also demonstrated that three of seven human BDNF transcripts are specifically downregulated in AD. What pathological feature(s) of AD leads to the decreased BDNF is unknown.
In this study, we administered both fibrillar and oligomeric conformations of Aβ1–42 to differentiated SH-SY5Y, a human neuroblastoma cell line, and measured both phosphorylated cAMP response element-binding protein (CREB), a regulator of BDNF transcription, and BDNF total mRNA. We found that oligomeric but not fibrillar preparations of Aβ1–42 significantly decrease both phosphorylated CREB and total BDNF mRNA. Furthermore, oligomeric Aβ1–42 decreases BDNF transcripts IV and V in these cells, demonstrating that Aβ1–42 downregulates the major BDNF transcript decreased in vivo in the AD brain. Thus, oligomeric Aβ1–42 could compromise neuronal function, causing memory loss and cognitive dysfunction by downregulation of BDNF in AD.
Keywords: Alzheimer, neurotrophin, exons, real-time RT-PCR, cell culture, CREB
Introduction
Alzheimer's disease (AD) is marked by progressive decline in cognitive functions and distinct pathology including senile plaques, neurofibrillary tangles, and synaptic degeneration (Glenner and Wong, 1984; Grundke-Iqbal et al., 1986; Geula, 1998; Coleman and Yao, 2003). The amyloid cascade hypothesis identifies amyloid-β (Aβ) as the precipitating insult in AD (Selkoe, 1994), although how Aβ produces synaptic degeneration is uncertain. Although fibrillar amyloid deposited as plaques may contribute to AD pathogenesis, oligomeric amyloid is now recognized as the major toxic species (Lambert et al., 1998) and correlates with neurodegeneration in AD (Lue et al., 1999; McLean et al., 1999; Gong et al., 2003).
Brain-derived neurotrophic factor (BDNF) has an established role in neuronal survival (Yuan and Yankner, 2000) and in long-term potentiation (LTP), a form of synaptic plasticity associated with memory formation and consolidation (McAllister et al., 1999; Lu, 2003; Bramham and Messaoudi, 2005). Transgenic mice with reduced BDNF levels exhibit defects in synaptic transmission, LTP, and memory tests that are rescued by exogenous BDNF administration (Korte et al., 1995; Patterson et al., 1996; Olofsdotter et al., 2000). BDNF mRNA and protein are significantly decreased in AD and mildly cognitive impaired (MCI) subjects compared with controls (Phillips et al., 1991; Connor et al., 1997; Ferrer et al., 1999; Hock et al., 2000; Holsinger et al., 2000). Moreover, the decrease in BDNF correlates with the severity of cognitive impairment (Peng et al., 2005), identifying BDNF downregulation as an early and possibly underlying event in AD. In rodents, 42 aa amyloid-β (Aβ1–42) compromises BDNF production and signaling (Tong et al., 2001, 2004; Wu et al., 2006). Although BDNF downregulation by Aβ1–42 has been demonstrated in rodent models, there have been no investigations of human BDNF regulation by Aβ1–42.
To investigate the effects of Aβ1–42 on human BDNF expression, we used the retinoic acid (RA) differentiated human neuroblastoma cell line SH-SY5Y (SY5Y). Differentiated SY5Y cells express BDNF and TrkB, the high-affinity BDNF receptor, and become dependent on BDNF for survival (Kaplan et al., 1993; Encinas et al., 2000; Feng et al., 2001). Furthermore, differentiated SY5Y cells develop a neuronal appearance, with long cell processes and expression of neuron-specific markers (Pahlman et al., 1984, 1990; Encinas et al., 2000) and, like human cortical neurons, are sensitive to Aβ (Lambert et al., 1994; Datki et al., 2004; Deshpande et al., 2006).
The human BDNF gene consists of seven upstream noncoding exons differentially spliced to the downstream coding exon, producing multiple transcripts (Aoyama et al., 2001; Garzon et al., 2002; Liu et al., 2005) (see Fig. 1A). Upstream of the noncoding exons are promoters governing differential mechanisms of activation and tissue-specific transcript expression in the CNS (Ohara et al., 1992; Metsis et al., 1993; Kokaia et al., 1994; Timmusk et al., 1995). In AD, BDNF transcripts I, II, and IV are specifically decreased (Garzon et al., 2002). However, the mechanism responsible for the observed decrease in these BDNF transcripts is unknown. The aim of this study was to determine whether fibrillar or oligomeric Aβ1–42 downregulates BDNF, and if so, whether it downregulates the transcripts that are decreased in cortical tissue of subjects with AD.
Figure 1.

Human BDNF gene with mRNA transcripts. A, Diagram showing the entire BDNF gene with the noncoding exons in white spliced to the coding exon in black. The asterisk (*) indicates splice variants, of which three are known, two on exon II and one on exon VI for a total of 10 transcripts. B, PCR products for all seven BDNF transcripts from retinoic acid-differentiated SH-SY5Y cells at 9 d in vitro. Lanes numbered I–VII correspond to the transcripts in A. PCR primer sequences and size of fragments are found in Table 1. The splice variants, although present, are not significant products in lanes 2 and 6. A 100 bp ladder is shown on the left.
Materials and Methods
SH-SY5Y cells.
SH-SY5Y cells were a generous gift from Dr. L. T. Young (Centre for Addiction and Mental Health, Toronto, Ontario, Canada). Cells were incubated in DMEM (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS) (Invitrogen), 2 mm l-glutamine (l-Gln) (Invitrogen), and 1% penicillin/streptomycin (Pen/Strep) (Invitrogen). Cells were kept at 37°C and 5% CO2 and split 1:5 every 5 d. For cell differentiation, 3.25 × 105 cells per well were seeded in six-well plates (Sarstedt, Montreal, Quebec, Canada) in DMEM, 10% FBS, 2 mm l-Gln, and 1% Pen/Strep for 24 h, after which medium was replaced with DMEM, 1% N-2 supplements (Invitrogen), 2 mm l-Gln, 1% Pen/Strep, and 10 μm all-trans RA (Sigma, St. Louis, MO). One-half of the medium was replaced every other day for 9 d. After 9 d of RA differentiation, 5 μm Aβ1–42 was added in fresh medium and incubated for 48 h.
Amyloid preparation.
Amyloid-β was purchased from American Peptide Company (Sunnyvale, CA) or rPeptide (Athens, GA). For initial experiments, Aβ1–42 was dissolved in 0.05 m Tris buffer, pH 10.5, and either Aβ1–42 or vehicle was added to fresh medium (DMEM, 1% N-2 Supplements, 2 mm l-Gln, 1% Pen/Strep, and 10 μm all-trans RA) to a final concentration of 5 μm.
Preparation of fibrillar or oligomeric amyloid was from Dahlgren et al. (2002). Briefly, both Aβ1–42 and scrambled Aβ1–42 (with the same amino acid composition as Aβ1–42 but in random sequence) (rPeptide) were dissolved to 1 mm in 100% 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). HFIP was dried in the fume hood and resuspended to 5 mm in anhydrous DMSO. For fibril-forming conditions, amyloid or scrambled peptide was brought to a final concentration of 100 μm with 10 mm HCl and incubated for 24 h at 37°C. For oligomer-forming conditions, amyloid or scrambled peptide was diluted to a final concentration of 100 μm with Ham's F-12 (phenol red free; BioSource, Camarillo, CA) and incubated at 4°C for 24 h. After incubation, fibrillar or oligomeric amyloid or vehicle was added to fresh medium (DMEM, N-2 supplements, l-Gln, Pen/Strep, and all-trans RA) and incubated with cells at 37°C for 48 h. Amyloid administration was conducted in two independent experiments with three separate samples per group.
RNA isolation, DNase treatment, reverse transcription, and absolute quantitative PCR.
After 48 h incubation in the presence of Aβ1–42, scrambled peptide, or vehicle, cells were lysed with 1 ml of Trizol (Invitrogen), and RNA was isolated with RNeasy Spin columns (Qiagen, Mississauga, Ontario, Canada). The procedure for RNA isolation with Trizol and RNeasy spin columns was followed as specified by Qiagen. RNA purity was confirmed by spectrophotometry (A260/A280 > 1.7) and RNA integrity was visualized by agarose gel electrophoresis.
One microgram of RNA was treated with 2 U of Turbo DNase (Ambion, Austin, TX). For reverse transcription (RT), the protocol and reagents for Superscript II of Invitrogen were used. The final volume of 20 μl contained 250 ng of random primers, 0.5 mm dNTPs (0.5 mm each of dATP, dTTP, dCTP, and dGTP), 1× first-strand buffer, 0.05 mm DTT, 2 U of RNaseOUT, and 200 U of Superscript II (Moloney murine leukemia virus RT). As a control, 1 μg of RNA was treated according to the same protocol with addition of water instead of the RT enzyme (“no RT” control).
Real-time PCR was performed in the Stratagene (La Jolla, CA) MX3000P using the DNA binding dye SYBR green (Platinum SYBR Green qPCR SuperMix UDG; Invitrogen). The 20 μl PCR mix contained 1× qPCR SuperMix, forward and reverse primers, 30 nm ROX reference dye (Stratagene), and cDNA from 50 ng of RNA or reference standard for absolute quantification. A “no template” control lacking RNA was included. For BDNF detection, 300 nm forward and reverse primers were used (forward primer, 5′-AAA CAT CCG AGG ACA AGG TG; reverse primer, 5′-AGA AGA GGA GGC TCC AAA GG; product, 249 bp), and 150 nm forward and reverse primers were used for β-actin (forward primer, 5′-CTC TTC CAG CCT TCC TTC; reverse primer, 5′-TGT TGG CGT ACA GGT CTT; product, 109 bp). Levels of BDNF and β-actin were determined using absolute quantification. Standards for BDNF were made in our laboratory from a pGEM plasmid containing the entire BDNF coding region of 755 bp (National Center for Biotechnology Information accession no. AY890649) and were run in triplicate with six 10-fold dilutions starting at 2.4 × 106 copies of pGEM plasmid. β-Actin standards were from a plasmid obtained from Invitrogen and were run in duplicate with six 10-fold dilutions starting at 1.0 × 107 copies of plasmid. Standards for individual BDNF transcripts were run in triplicate and obtained from purified PCR products using the primers listed in Table 1. Only experiments with R2 > 0.990 and a PCR efficiency between 90 and 100% were used for analysis. All unknowns, no RT, and no template controls were run in triplicate. The following thermal profile was used for total BDNF and β-actin: 2 min at 50°C, 2 min at 95°C followed by 40 cycles of 95°C for 15 s, 58°C for 30 s, and 72°C for 30 s. For individual BDNF transcripts, the 40 cycles had the following thermal profile: 95°C for 30 s, 58°C for 30 s, and 72°C for 45 s. After PCR, a dissociation curve was added to verify that no secondary products had formed. Results were expressed as copies per nanogram of total RNA.
Table 1.
PCR primer sequences for human BDNF transcripts
| Transcript | Primer sequence | Product (bp) |
|---|---|---|
| BDNF transcript I | (F) GCG GAT ATT GCA AAG GGT TA | 287 |
| (R) ACC TTG TCC TCG GAT GTT TG | ||
| BDNF transcript IIa | (F) GCG GTG ATA GGC TGG AAT AG | 232 |
| (R) ACC TTG TCC TCG GAT GTT TG | ||
| BDNF transcript III | (F) GCG AGT TTC GGG CGC TGG CTT AGA | 269 |
| (R) ATT CAC GCT CTC CAG AGT CCC ATG | ||
| BDNF transcript IV | (F) GAG TAT TAC CTC CGC CAT GC | 381 |
| (R) ATT CAC GCT CTC CAG AGT CC | ||
| BDNF transcript V | (F) AAC CAC GAT GTG ACT CCG C | 216 |
| (R) ATT CAC GCT CTC CAG AGT CC | ||
| BDNF transcript VI | (F) AGT GGA CTT ACA AGT CCG AAG C | 313 |
| (R) ATT CAC GCT CTC CAG AGT CC | ||
| BDNF transcript VII | (F) TTC TGC TGA CAG CAT GAG C | 515 |
| (R) ATT CAC GCT CTC CAG AGT CC |
F, Forward primer; R, reverse primer.
aPrimers for BDNF transcript II encompass two of the three splice variants reported by Liu et al. (2005). We redesigned new primers to detect all three splice variants using the new forward sequence CAT TCA GCA CCT TGG ACA GA and the same reverse sequence from Table 1. PCR analysis with the new primers revealed splice variant 2 continued to be the most predominantly expressed transcript, and we were unable to detect the third splice variant in SH-SY5Y cells.
Western blotting.
To confirm fibrillar and oligomeric speciation of Aβ1–42, Western blotting was performed following the protocols of Dahlgren et al. (2002) and Stine et al. (2003) with the following modifications: Aβ1–42 samples were diluted in 4× loading buffer (0.25 m Tris-HCl, pH 6.8, 8% SDS, 40% glycerol, 20% β-mercaptoethanol, 0.004% bromophenol blue) and separated by SDS-PAGE in a 12% Tris/glycine gel.
For determination of cAMP response element-binding protein (CREB), phosphorylated CREB (P-CREB), caspases-3 and -7, and β-actin, RA-differentiated SY5Y cells subjected to different treatments were lysed in buffer containing 50 mm Tris, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EGTA, pH 8, 1 mm sodium orthovanadate, 1 mm NaF, 1 mm PMSF, and 1 μg/ml each of aprotinin, leupeptin, and pepstatin A. Samples were centrifuged for 5 min at 300 × g. Protein concentration of the supernatant was determined using a DC protein assay (Bio-Rad, Hercules, CA). Twenty micrograms of total protein in 4× loading buffer was separated by SDS-PAGE in a 12% Tris/glycine gel.
After transfer to polyvinylidene fluoride membranes and blocking for 1 h at room temperature in 1× Tris buffered saline (TBS)–0.1% Tween 20 (TBS/T) with 5% nonfat dry milk (MLK) [or PBS containing 3% nonfat dry milk (PBS-MLK) for CREB and P-CREB], blots were incubated overnight at 4°C with primary antibodies for Aβ1–42 (6E10; Signet Laboratories, Dedham, MA; diluted 1:3000 in TBS/T), β-actin (Sigma; diluted 1:5000 in TBS/T), caspases-3 and -7 (Cell Signaling Technology, Danvers, MA; diluted 1:1000 in TBS/T-MLK), or CREB and P-CREB (Upstate, Lake Placid, NY; diluted 1:1000 in PBS-MLK). Secondary antibodies anti-mouse IgG HRP (GE Healthcare, Buckinghamshire, UK) diluted 1:5000 for Aβ1–42, 1:5000 for β-actin, and 1:2000 for caspase-7, or anti-rabbit IgG HRP (Cell Signaling) diluted 1:2000 for caspase-3 and 1:5000 for CREB and P-CREB, were incubated for 1 h at room temperature. Blots were developed using enhanced chemiluminescence (GE Healthcare) and visualized using CL-X Posure film (Pierce Biotechnology, Rockford, IL). Pixel density was determined using Scion Image software (Scion, Frederick, MD).
Cell viability assay.
Amyloid-induced cytotoxicity was determined by measuring release of cytosolic lactate dehydrogenase (LDH) into the medium (Cytotox 96 nonradioactive cytotoxicity assay; Promega, Madison, WI). The protocol was followed as specified by the manufacturer. Each sample was tested in triplicate, and the LDH release was measured at 492 nm using a Titertek Multiscan Plus (MP Biomedicals, Irvine, CA) plate reader.
Quantitative and statistical analysis.
Samples for BDNF mRNA exposed to different species of amyloid-β and LDH cytotoxicity were assayed in triplicate in each of two independent experiments. Data from Western blots for total CREB (T-CREB), P-CREB, and caspases-3 and -7 were obtained from three independent experiments. The data were analyzed by one-way ANOVA with post hoc Tukey's test for pairwise group comparison or Student's t test where indicated (SPSS, version 13, software; SPSS, Chicago, IL).
Results
Retinoic acid-differentiated SH-SY5Y cells express all BDNF transcripts
RA-differentiated SY5Y cells after 9 d in vitro express all seven BDNF transcripts (Fig. 1B). The BDNF transcript expression pattern in SH-SY5Y cells is very similar to that of human cortical tissue. A comparison of levels of each BDNF transcript demonstrates that BDNF transcript IV is the most highly expressed transcript in both the human cortex and SY5Y cells, representing over one-half of the total BDNF (Table 2). Both the human cortex and SY5Y cells predominantly express BDNF transcripts I, IV, and V, whereas transcripts II, VI, and VII are minor contributors to the total, and transcript III is not detectable (Table 2).
Table 2.
BDNF transcript abundance in human parietal cortex and RA-differentiated human neuroblastoma SH-SY5Y cells
| Human parietal cortex |
Human neuroblastoma SH-SY5Y |
||
|---|---|---|---|
| BDNF transcript order | Copiesa | BDNF transcript order | Copiesb |
| Transcript IV | 625 | Transcript IV | 643 |
| Transcript I | 332 | Transcript V | 215 |
| Transcript V | 130 | Transcript I | 121 |
| Transcript VI | 70 | Transcript II | 95 |
| Transcript VII | 16 | Transcript VI | N/D |
| Transcript II | 15 | Transcript VII | N/D |
| Transcript III | N/D | Transcript III | N/D |
aDetermination of BDNF transcript copy number for human parietal cortex was done using relative quantitative RT-PCR with incorporation of 33P (Garzon et al. 2002). The 33P isotope was detected by pixelation intensity on a phosphorimage cassette, and pixel values were averaged for all control subjects for each BDNF transcript. One pixel was assigned a value of 65,000 and was used as the denominator in the following ratio: total averaged pixel value/65,000. The value of the ratio appears in the ‘Copies’ column (under ‘Human parietal cortex’). BDNF transcript III was not detectable (N/D) in human parietal cortex.
bDetermination of BDNF transcript copies after 9 d of retinoic acid differentiation of SH-SY5Y cells, using absolute quantitative real-time PCR. Three samples per group were assayed in triplicate in each of two independent experiments. The value of ‘Copies’ (under ‘Human neuroblastoma SH-SY5Y’) is copies of BDNF per 50 ng of total input RNA. BDNF transcripts III, VI, and VII were below quantifiable levels (N/D, <40 copies/50 ng total RNA).
Retinoic acid induces BDNF mRNA expression
BDNF mRNA levels increased significantly over time when cells were grown in serum free medium with N-2 supplements and 10 μm RA (Fig. 2). BDNF mRNA was significantly increased at 3 d (one-way ANOVA and post hoc Tukey's test, p < 0.01), 5 d (p < 0.001), and 9 d (p < 0.001) of growth in N-2 and RA compared with serum free medium with N-2 supplements alone or with medium with 10% FBS (Fig. 2).
Figure 2.

Measurement of total BDNF mRNA after 3, 5, and 9 d of growth in different media. BDNF is significantly increased in N-2 supplements and 10 μm RA compared with N-2 supplements alone or 10% FBS at 3, 5, and 9 d (*p < 0.01; **p < 0.001). In addition, comparing BDNF mRNA levels for N-2 and RA across the time points, there is a significant increase in BDNF mRNA between days 3 and 9 (*p = 0.036) and between days 5 and 9 (*p = 0.022). Error bars represent SEM; n = 3.
Aβ1–42 downregulates BDNF mRNA in human neuroblastoma SH-SY5Y cells
The effect of 5 μm Aβ1–42 administration on BDNF mRNA in SY5Y cells was examined after incubation for 6, 12, 24, and 48 h. Five micromolar Aβ1–42 is a concentration previously reported to interfere with cellular signaling without induction of apoptosis (Tong et al., 2001, 2004). Although total BDNF mRNA (exon VIII) was decreased at all time points, the greatest decrease occurred at 48 h (data not shown). The 48 h time point was subsequently used for all additional experiments. Administration of 5 μm Aβ1–42 for 48 h to SY5Y cells resulted in a 33% decrease in total BDNF mRNA compared with vehicle-treated cells (Student's t test, p = 0.002). In contrast, amyloid-β did not downregulate BDNF in undifferentiated SY5Y cells (data not shown).
Aβ1–42 (5 μm) in fibrillar or oligomeric conformations is not cytotoxic to SH-SY5Y cells
To confirm that Aβ1–42 is not cytotoxic to SY5Y cells under these conditions, a colorimetric assay measuring the release of cytoplasmic LDH and Western blots measuring levels of caspases-3 and -7 were used. There was no change in levels of inactivated caspases-3 and -7 or in LDH release from cultures exposed to fibrillar or oligomeric Aβ1–42 for 48 h in comparison with the scrambled peptide or no peptide controls (one-way ANOVA, p > 0.05) (Fig. 3, Table 3).
Figure 3.

Western blotting of inactivated caspase-3 and -7. A, No difference in levels of inactivated caspase-3 and -7 among differently treated RA-differentiated SY5Y cells (one-way ANOVA, p = 0.354 for caspase-3, and p = 0.921 for caspase-7). Analysis is based on three independent experiments; n = 9. Error bars represent SEM. B, Western blot shows only inactivated caspase-3 and -7 were detected after treatment with Aβ1–42. As a positive control, SY5Y cells were treated with 25 μm etoposide, an apoptosis-inducing reagent. After 24 h, no inactivated caspase-3 or -7 was detected.
Table 3.
Percentage cytotoxicity after 48 h 5 μm Aβ1–42 administration, determined by LDH assay
| Condition | Percentage cytotoxicitya |
|---|---|
| Control (no peptide) | 17.82 |
| Fibrillar Aβ1–42 | 11.77 |
| Scrambled fibrillar Aβ1–42 | 17.92 |
| Oligomeric Aβ1–42 | 17.36 |
| Scrambled oligomeric Aβ1–42 | 17.17 |
aOne-way ANOVA, p = 0.84.
Oligomeric but not fibrillar Aβ1–42 significantly decreases BDNF mRNA
The 5 μm Aβ1–42 applied for 48 h was an undefined, heterogeneous mixture of fibrillar and oligomeric amyloid. To determine which amyloid species was responsible for BDNF downregulation, homogeneous fibrillar and oligomeric amyloid samples were prepared. Western blotting with an antibody to Aβ confirmed the efficacy of the Aβ1–42 oligomeric and fibrillar speciation procedure (Fig. 4). The oligomeric preparation (lane 1) contained monomers (4 kDa), small oligomeric trimers (∼12 kDa), and the larger oligomeric amyloid (40–98 kDa), but no fibrils. The fibrillar amyloid (lane 2) contained both monomeric amyloid and fibrillar amyloid (>250 kDa), but no detectable oligomers. The procedures for amyloid speciation were also performed on the scrambled peptide, containing the same amino acid composition as Aβ1–42, which served as a negative control and did not cross-react with the amyloid antibody (lanes 3 and 4). After confirmation of the amyloid species, oligomeric or fibrillar Aβ1–42 was added to the SY5Y cells. Scrambled Aβ1–42 treated identically to oligomeric and fibrillar preparations and vehicle without added peptide served as negative controls. Total BDNF mRNA was decreased 62% (ANOVA and post hoc Tukey's test, p < 0.001) in cultures exposed to oligomeric preparations of Aβ1–42 compared with scrambled oligomeric peptide and control without peptide (Fig. 5). SY5Y cells treated with fibrillar and oligomeric scrambled peptide controls showed no change in BDNF levels compared with controls without added peptide (p > 0.05) (Fig. 5). Furthermore, cells treated with fibrillar Aβ1–42 showed no change in BDNF levels compared with scrambled controls or controls without added peptide (p > 0.05) (Fig. 5).
Figure 4.
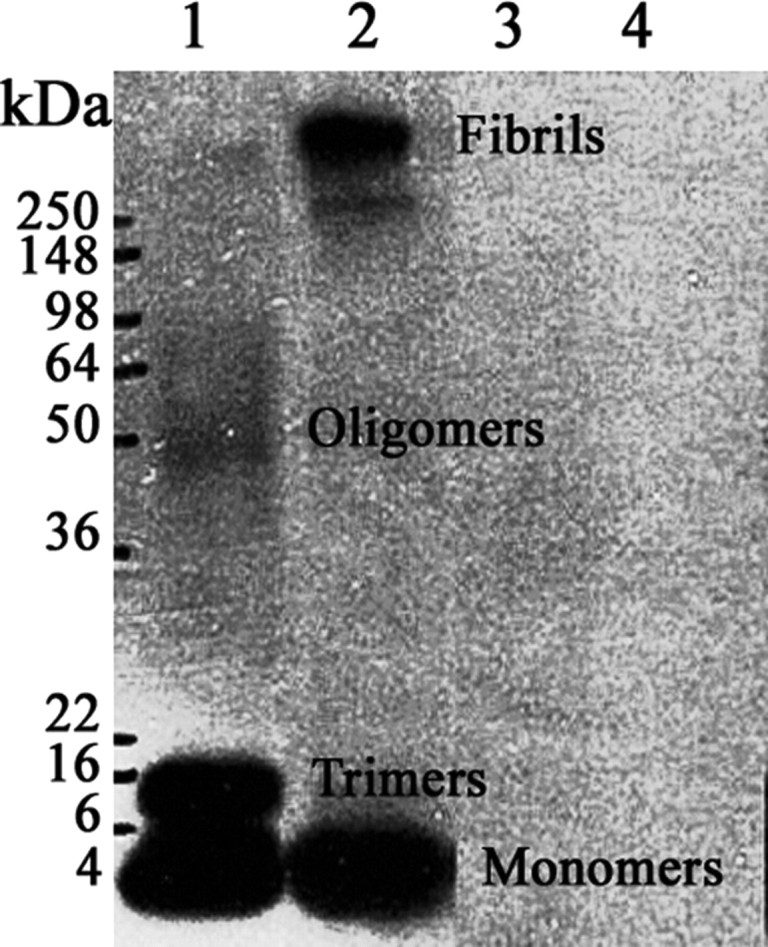
Western blot of fibrillar and oligomeric Aβ1–42 speciation. The oligomeric preparation (lane 1) contains monomers (4 kDa), small oligomeric trimers (∼12 kDa), and the larger oligomeric amyloid (40–98 kDa), but no fibrils. The fibrillar amyloid (lane 2) contains both monomeric amyloid and fibrillar amyloid (>250 kDa), but no oligomers. The procedures for amyloid speciation were also performed on the scrambled peptide, containing the same amino acid composition as Aβ1–42, which served as a negative control and did not cross-react with the amyloid antibody (lanes 3 and 4).
Figure 5.

BDNF mRNA levels in RA-differentiated SY5Y cells exposed to oligomeric or fibrillar Aβ. Five micromolar Aβ1–42, treated to form either oligomeric or fibrillar species, was added to RA-differentiated SH-SY5Y cells for 48 h. Only oligomeric amyloid caused a significant decrease (**p < 0.001) in BDNF mRNA (exon VIII) compared with no peptide and scrambled oligomeric amyloid controls. There was no change in BDNF exposed to fibrillar amyloid compared with either no peptide or scrambled fibril controls. The analysis is based on two independent experiments; n = 6. Error bars represent SEM.
Oligomeric Aβ1–42 decreases BDNF transcripts 4 and 5
Examination of the levels of each of the seven BDNF transcripts demonstrated oligomeric Aβ1–42 decreased BDNF transcripts IV and V by 45% (ANOVA and post hoc Tukey's test, p = 0.001) and 54% (p = 0.015), respectively (Fig. 6C,D), increased BDNF transcript I by 70% (p = 0.034) (Fig. 6A), and did not change BDNF transcript II levels (Fig. 6B). BDNF transcripts III, VI, and VII were below the level of the standard curve both before and after Aβ1–42 administration (<40 copies/50 ng total RNA).
Figure 6.

Measurement of BDNF transcripts I, II, IV, and V in RA-differentiated SY5Y cells exposed to oligomeric or fibrillar Aβ. A, A significant increase (*p = 0.034) in BDNF transcript I after oligomeric amyloid administration compared with both control conditions. B, No change in BDNF transcript II levels among the five experimental conditions. C, D, Significant decreases in BDNF transcripts IV and V (**p = 0.001 and *p = 0.015, respectively) after oligomeric amyloid administration compared with both controls. There was no change in any BDNF transcript level after fibrillar amyloid administration compared with either control. Transcripts III, VI, and VII were below the levels of detection. The analysis is based on two independent experiments; n = 6. Error bars represent SEM.
Oligomeric but not fibrillar Aβ1–42 significantly decreases levels of phosphorylated CREB
Levels of phosphorylated CREB were decreased by 38% in samples treated with oligomeric amyloid compared with all other groups (one-way ANOVA and post hoc Tukey's test, p = 0.005) (Fig. 7). The levels of total CREB protein did not differ among amyloid and control groups (one-way ANOVA, p > 0.05) (Fig. 7).
Figure 7.

Western blots of T-CREB and P-CREB levels in RA-differentiated SY5Y cells treated with oligomeric or fibrillar Aβ. RA-differentiated SY5Y cells administered oligomeric amyloid demonstrate significantly decreased phosphorylated CREB levels (*p = 0.005). Levels of total CREB were unchanged among all five conditions (one-way ANOVA, p = 0.237). The analysis is based on three independent experiments; n = 9. Error bars represent SEM.
Discussion
In this study, we showed that Aβ1–42 significantly decreases BDNF mRNA and P-CREB, a transcriptional regulator of BDNF. We demonstrated that oligomeric, not fibrillar, species of Aβ1–42 are responsible for the observed effects. Furthermore, oligomeric Aβ1–42 significantly decreases BDNF transcripts IV and V. Transcript IV is the most abundantly expressed BDNF transcript in human cortical tissue and in SY5Y cells and is decreased in AD (Garzon et al., 2002). Therefore, our results suggest that Aβ1–42 is responsible for the majority of BDNF downregulation in AD.
RA-differentiated SY5Y cells demonstrate human neuronal characteristics, making their use as a model system ideal. They exhibit neuronal morphology and express neuron-specific markers including neurofilaments, neuron-specific enolase, and growth-associated protein-43 as well as the neuronal polarity markers tau and microtubule-associated protein 2 (Pahlman et al., 1984, 1990; Encinas et al., 2000). We show that, unlike previously reported human and murine cell culture systems, RA-differentiated SY5Y cells produce endogenous BDNF, increasing BDNF expression over the course of RA administration. A previous study in rat demonstrated that membrane depolarization-induced BDNF upregulation is disrupted by Aβ1–42 administration (Tong et al., 2001). Our findings are the first to report that Aβ administration significantly decreases basal levels of BDNF mRNA and to identify oligomeric amyloid as responsible for the observed decrease.
In addition, we report here that the expression of BDNF transcripts in RA-differentiated SY5Y cells correlates closely with the expression pattern of BDNF transcripts in postmitotic human CNS tissue. Furthermore, RA-differentiated SY5Y cells closely resemble human cortical neurons in their response to Aβ (Deshpande et al., 2006). Primary murine neuronal cultures were not used in this study, because the number and the expression patterns of BDNF transcripts in the brain differ between human and mouse (Garzon et al., 2002; Liu et al., 2005, 2006).
The amyloid cascade hypothesis proposes that Aβ production and toxic action on neurons and tissues cause AD. SY5Y cells have been used extensively to study mechanisms of Aβ toxicity. Aβ binds to the neurites of differentiated SY5Y cells, promoting neurite degeneration and induction of tau hyperphosphorylation (Datki et al., 2004). Similar data are reported for rat and human cultured neurons (Gong et al., 2003; Lacor et al., 2004; Deshpande et al., 2006).
Acceptance of the original amyloid cascade hypothesis is not pervasive, because data support no correlation between plaque deposition and neurodegeneration (Cummings and Cotman, 1995; Irizarry et al., 1997; Chui et al., 1999; Naslund et al., 2000). However, oligomeric Aβ1–42 is a potent neurotoxin at nanomolar concentrations and inhibits LTP and synaptic plasticity (Lambert et al., 1998; Walsh et al., 2002). Oligomeric Aβ1–42 levels are increased in areas of the brain compromised in AD and are correlated with areas of neurodegeneration; oligomeric Aβ1–42 is more neurotoxic than fibrillar amyloid and is found intracellularly, in the absence of amyloid plaques and neurofibrillary tangles, suggesting the presence of oligomeric Aβ1–42 early in the progression of AD (Funato et al., 1999; Lue et al., 1999; McLean et al., 1999; Walsh et al., 2000; Dahlgren et al., 2002; Gong et al., 2003). Our findings of BDNF downregulation by oligomeric Aβ1–42 support the growing literature suggesting oligomeric Aβ1–42 is the first neurotoxic insult in AD. Together with our recent findings of decreased BDNF in prodromal AD subjects (Peng et al., 2005), we propose that oligomeric amyloid-induced downregulation of BDNF in the brain is one of the primary and earliest mechanisms of neurodegeneration in AD.
It was previously unknown which species of Aβ1–42 downregulates BDNF. Here, we demonstrate that oligomeric but not fibrillar Aβ1–42 significantly decreases BDNF mRNA levels in differentiated SY5Y cells. Furthermore, undifferentiated, amyloid-treated SY5Y cells do not downregulate BDNF, consistent with previous reports that undifferentiated SY5Y cells are not responsive to Aβ (Lambert et al., 1994).
To confirm that the effects of amyloid administration on BDNF expression occur before any cytotoxic effects, we demonstrated the absence of LDH release and of caspase activation, a proapoptotic marker, in amyloid-treated RA-differentiated SY5Y cells during the time course of BDNF mRNA measurement. Our results are in agreement with recent data demonstrating that Aβ-derived diffusible ligands (of similar molecular weight to our oligomeric amyloid) induce subtle mitochondrial changes, but not overt cell death, in human cortical neurons over a period of 1 week (Deshpande et al., 2006). Therefore, oligomeric amyloid-induced BDNF downregulation is not a result of cell death.
In our previous study, we reported BDNF transcripts I, II, and III were significantly decreased in the parietal cortex of AD compared with control subjects (Garzon et al., 2002). Recently, there has been a revision to both the human and the rodent BDNF genes with the discovery of an additional exon between exons II and III (Liu et al., 2005, 2006). The newly discovered exon is called exon III and the previous exons III and IV are renamed exons IV and V, respectively. Based on this revision, our previous data show a significant decrease in human BDNF transcripts I, II, and IV in the parietal cortex of AD compared with control subjects. In this study, consistent with Alzheimer's subjects, RA-differentiated SY5Y cells exposed to oligomeric Aβ1–42 exhibit significantly decreased transcript IV (Garzon et al., 2002). BDNF transcript IV is the most abundantly expressed transcript in SY5Y cells and in human cortical tissue, exhibiting a 2- to 16-fold greater level of expression than the other transcripts (Table 2) and accounting for more than one-half of the total BDNF mRNA. Therefore, downregulation of transcript IV is likely the primary driver behind downregulation of total BDNF mRNA by Aβ1–42 in both SY5Y cells and in human cortical tissue.
In differentiated SY5Y cells, BDNF transcript V is decreased 54% by oligomeric Aβ1–42. In human cortical tissue, BDNF transcript V showed a similar trend in AD subjects compared with controls, but the finding was not statistically significant (p = 0.062) (Garzon et al., 2002). The discrepancy between these findings is likely a result of insufficient sample numbers in our human cortical tissue analysis, diminished accuracy based on our previous use of relative RT-PCR versus real-time RT-PCR in this study, and intersubject variability in the human cortical tissue samples.
Interestingly, in differentiated SY5Ys, oligomeric amyloid increases BDNF transcript I and does not change BDNF transcript II, whereas in AD, transcripts I and II are both decreased. The discrepancy between amyloid-treated SY5Y cells and human AD cortical samples suggests that factor(s) other than Aβ are responsible for BDNF transcript I and II downregulation in AD. BDNF transcripts III, VI, and VII are minor contributors to total BDNF mRNA levels; in SY5Y cells, they are expressed at levels below our standard curve (<40 copies/50 ng total RNA) and at least 16-fold lower than transcript IV.
The effect of Aβ on LTP and memory is postulated to be mediated by inhibition of CREB phosphorylation. Activation of CREB by phosphorylation is essential for learning and memory (Silva et al., 1998; Tully, 1998); P-CREB levels are decreased in AD hippocampus (Yamamoto-Sasaki et al., 1999) and in Aβ-treated rat primary cortical neurons (Tong et al., 2001). Aβ reduces adenylate cyclase and protein kinase A (PKA) activity and hence P-CREB levels in hippocampal neurons (Vitolo et al., 2002). Consistent with the role of P-CREB as a transcription factor for activity-dependent regulation of BDNF transcript IV (Shieh et al., 1998; Tao et al., 1998; Tong et al., 2001, 2004) and with our results demonstrating oligomeric amyloid-specific downregulation of BDNF transcript IV, we find only oligomeric, not fibrillar amyloid decreases the levels of P-CREB in SY5Y cells. Our results suggest that decreased CREB phosphorylation induced by oligomeric Aβ is responsible for downregulation of BDNF transcript IV in SY5Y cells and in AD.
BDNF is a well known mediator of LTP and memory. Transgenic mice with reduced BDNF levels exhibit defects in synaptic transmission, LTP, and memory tests that are rescued by exogenous BDNF administration (Korte et al., 1995; Patterson et al., 1996; Olofsdotter et al., 2000), and reduced BDNF protein levels are correlated with cognitive impairment in MCI and AD subjects (Peng et al., 2005). Therefore, we propose a mechanism for the effects of Aβ on cognitive and memory function in AD, which is that oligomeric Aβ inhibits adenylate cyclase activity and reduces PKA activation, reducing CREB phosphorylation and subsequent BDNF transcript IV expression. Because transcript IV represents over one-half of the total BDNF mRNA, this mechanism may explain reduced BDNF in both RA-differentiated SY5Y cells and in human neurons exposed to Aβ, and identifies BDNF downregulation as an underlying event in AD. Furthermore, because oligomeric amyloid appears before plaque deposition and because BDNF reduction occurs early in the course of AD and is correlated with cognitive dysfunction, oligomeric amyloid may produce synaptic dysfunction and learning and memory deficits by downregulation of BDNF before the clinical onset of Alzheimer's disease.
Footnotes
This work was supported by grants from the Canadian Institutes for Health Research and the Scottish Rite Charitable Foundation of Canada (M.F.). D.J.G. is supported by studentships from the Alzheimer Society of Canada and the Scottish Rite Charitable Foundation of Canada. We thank Dr. Charlie Goldsmith for his advice on statistical analysis and Dr. Jane Foster for her advice on real-time PCR.
References
- Aoyama M, Asai K, Shishikura T, Kawamoto T, Miyachi T, Yokoi T, Togari H, Wada Y, Kato T, Nakagawara A. Human neuroblastomas with unfavorable biologies express high levels of brain-derived neurotrophic factor mRNA and a variety of its variants. Cancer Lett. 2001;164:51–60. doi: 10.1016/s0304-3835(00)00715-1. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res. 1997;49:71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Cotman CW. Image analysis of beta-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- Datki Z, Papp R, Zadori D, Soos K, Fulop L, Juhasz A, Laskay G, Hetenyi C, Mihalik E, Zarandi M, Penke B. In vitro model of neurotoxicity of Abeta 1–42 and neuroprotection by a pentapeptide: irreversible events during the first hour. Neurobiol Dis. 2004;17:507–515. doi: 10.1016/j.nbd.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75:991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- Feng X, Jiang H, Baik JC, Edgar C, Eide FF. BDNF dependence in neuroblastoma. J Neurosci Res. 2001;64:355–363. doi: 10.1002/jnr.1086. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–739. doi: 10.1097/00005072-199907000-00007. [DOI] [PubMed] [Google Scholar]
- Funato H, Enya M, Yoshimura M, Morishima-Kawashima M, Ihara Y. Presence of sodium dodecyl sulfate-stable amyloid beta-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am J Pathol. 1999;155:23–28. doi: 10.1016/s0002-9440(10)65094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon D, Yu G, Fahnestock M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer's disease parietal cortex. J Neurochem. 2002;82:1058–1064. doi: 10.1046/j.1471-4159.2002.01030.x. [DOI] [PubMed] [Google Scholar]
- Geula C. Abnormalities of neural circuitry in Alzheimer's disease: hippocampus and cortical cholinergic innervation. Neurology. 1998;51:S18–S29. doi: 10.1212/wnl.51.1_suppl_1.s18. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, Heese K, Hulette C, Rosenberg C, Otten U. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol. 2000;57:846–851. doi: 10.1001/archneur.57.6.846. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res. 2000;76:347–354. doi: 10.1016/s0169-328x(00)00023-1. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Matsumoto K, Lucarelli E, Thiele CJ. Induction of TrkB by retinoic acid mediates biologic responsiveness to BDNF and differentiation of human neuroblastoma cells. Eukaryotic Signal Transduction Group. Neuron. 1993;11:321–331. doi: 10.1016/0896-6273(93)90187-v. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Metsis M, Kokaia M, Bengzon J, Elmer E, Smith ML, Timmusk T, Siesjo BK, Persson H, Lindvall O. Brain insults in rats induce increased expression of the BDNF gene through differential use of multiple promoters. Eur J Neurosci. 1994;6:587–596. doi: 10.1111/j.1460-9568.1994.tb00303.x. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Stevens G, Sabo S, Barber K, Wang G, Wade W, Krafft G, Snyder S, Holzman TF, Klein WL. Beta/A4-evoked degeneration of differentiated SH-SY5Y human neuroblastoma cells. J Neurosci Res. 1994;39:377–385. doi: 10.1002/jnr.490390404. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Walther D, Drgon T, Polesskaya O, Lesnick TG, Strain KJ, de Andrade M, Bower JH, Maraganore DM, Uhl GR. Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson's Disease. Am J Med Genet B Neuropsychiatr Genet. 2005;134:93–103. doi: 10.1002/ajmg.b.30109. [DOI] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Metsis M, Timmusk T, Arenas E, Persson H. Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc Natl Acad Sci USA. 1993;90:8802–8806. doi: 10.1073/pnas.90.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Ohara O, Gahara Y, Teraoka H, Kitamura T. A rat brain-derived neurotrophic factor-encoding gene generates multiple transcripts through alternative use of 5′ exons and polyadenylation sites. Gene. 1992;121:383–386. doi: 10.1016/0378-1119(92)90148-i. [DOI] [PubMed] [Google Scholar]
- Olofsdotter K, Lindvall O, Asztely F. Increased synaptic inhibition in dentate gyrus of mice with reduced levels of endogenous brain-derived neurotrophic factor. Neuroscience. 2000;101:531–539. doi: 10.1016/s0306-4522(00)00428-0. [DOI] [PubMed] [Google Scholar]
- Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Pahlman S, Mamaeva S, Meyerson G, Mattsson ME, Bjelfman C, Ortoft E, Hammerling U. Human neuroblastoma cells in culture: a model for neuronal cell differentiation and function. Acta Physiol Scand Suppl. 1990;592:25–37. [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol. 1994;53:438–447. doi: 10.1097/00005072-199409000-00003. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Lendahl U, Funakoshi H, Arenas E, Persson H, Metsis M. Identification of brain-derived neurotrophic factor promoter regions mediating tissue-specific, axotomy-, and neuronal activity-induced expression in transgenic mice. J Cell Biol. 1995;128:185–199. doi: 10.1083/jcb.128.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. Beta-amyloid-(1–42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival Is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T. Toward a molecular biology of memory: the light's coming on! Nat Neurosci. 1998;1:543–545. doi: 10.1038/2780. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wu ZL, Ciallella JR, Flood DG, O'kane TM, Bozyczko-Coyne D, Savage MJ. Comparative analysis of cortical gene expression in mouse models of Alzheimer's disease. Neurobiol Aging. 2006;27:377–386. doi: 10.1016/j.neurobiolaging.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Sasaki M, Ozawa H, Saito T, Rosler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]