

Abstract
BDNF contributes to the activity-dependent establishment and refinement of visual connectivity. In Xenopus, BDNF applications in the optic tectum influence retinal ganglion cell (RGC) axon branching and promote synapse formation and stabilization. The expression patterns of BDNF and TrkB suggest that BDNF specifically regulates the maturation of RGC axons at the target. It is possible, however, that BDNF modulates retinotectal synaptic connectivity by differentially influencing presynaptic RGC axons and postsynaptic tectal cells. Here, we combined single-cell expression of a dominant-negative TrkB–enhanced green fluorescent protein (GFP) fusion protein with confocal microscopy imaging in live Xenopus tadpoles to differentiate between presynaptic and postsynaptic actions of BDNF. Disruption of TrkB signaling in individual RGCs influenced the branching and synaptic maturation of presynaptic axon arbors. Specifically, GFP–TrkB.T1 overexpression increased the proportion of axons with immature, growth cone-like morphology, decreased axon branch stability, and increased axon arbor degeneration. In addition, GFP–TrkB.T1 overexpression reduced the number of red fluorescent protein–synaptobrevin-labeled presynaptic specializations per axon terminal. In contrast, overexpression of GFP–TrkB.T1 in tectal neurons did not alter synaptic number or the morphology or dynamic behavior of their dendritic arbors. Electron microscopy analysis revealed a significant decrease in the number of mature synaptic profiles and in the number of docked synaptic vesicles at retinotectal synapses made by RGC axons expressing GFP–TrkB.T1. Together, our results demonstrate that presynaptic TrkB signaling in RGCs is a key determinant in the establishment of visual connectivity and indicate that changes in tectal neuron synaptic connectivity are secondary to the BDNF-elicited enhanced stability and growth of presynaptic RGCs.
Keywords: Xenopus laevis, synapse, axon, tectal neuron, in vivo imaging, transgenic
Introduction
Neuronal connectivity is established through a series of developmental events that involve close communication between presynaptic and postsynaptic neurons. In the developing visual system, neurotrophins have been shown to exert various influences, from guiding the morphological differentiation of neurons to controlling the functional plasticity of visual circuits. BDNF contributes to the establishment and refinement of visual connectivity by acting at multiple levels in the visual pathway, from the retina to the visual cortex (von Bartheld, 1998; Hanover et al., 1999; Huang et al., 1999; Berardi et al., 2003; Cohen-Cory and Lom, 2004; Mandolesi et al., 2005). The spatial and temporal patterns of expression of BDNF and its high-affinity receptor TrkB are consistent with a role for BDNF in modulating visual circuit development. In mammals, BDNF and TrkB are expressed in developing retinal ganglion cells (RGCs), in the lateral geniculate nucleus and superior colliculus, the two retinal axon target areas, as well as in the primary visual cortex (von Bartheld, 1998; Lein and Shatz, 2000; Silver and Stryker, 2001; Vizuete et al., 2001). In nonmammalian vertebrates, both BDNF and TrkB are expressed at peak levels in RGCs and at their target optic tectum at the time of active formation of the retinotectal projection (Cohen-Cory et al., 1996; Garner et al., 1996; Hallbook et al., 1996; Herzog and von Bartheld, 1998).
Evidence suggests that BDNF can act as an anterograde factor to influence postsynaptic neurons or as a target-derived retrograde factor, affecting presynaptic RGCs (Spalding et al., 2002; Menna et al., 2003; Butowt and von Bartheld, 2005). Work in Xenopus has demonstrated that BDNF shapes the morphological differentiation of RGCs and their synaptic connectivity (Lom and Cohen-Cory, 1999; Alsina et al., 2001; Cohen-Cory and Lom, 2004; Du and Poo, 2004; Hu et al., 2005) and to a lesser extent affects the morphological and synaptic differentiation of postsynaptic tectal cells (Du and Poo, 2004; Sanchez et al., 2006). It remained to be determined, however, whether BDNF acts directly on RGCs or whether it influences both presynaptic and postsynaptic neurons to shape their synaptic connectivity. To directly distinguish between presynaptic and postsynaptic actions of BDNF, we expressed a dominant-negative TrkB–green fluorescent protein (GFP) fusion protein (GFP–TrkB.T1) in individual RGCs or tectal neurons of young Xenopus tadpoles and visualized their dynamic growth and differentiation in vivo. TrkB.T1 is a truncated, alternatively spliced receptor isoform that lacks the intracellular kinase domain of TrkB and inhibits full-length TrkB receptor signaling by forming nonfunctional heterodimers and/or by sequestering BDNF ligand (Biffo et al., 1995; Eide et al., 1996; Ninkina et al., 1996; Haapasalo et al., 2001; Offenhauser et al., 2002). Because TrkB.T1 does not induce independent signaling, overexpression of TrkB.T1 has proven useful to inhibit BDNF signaling and to demonstrate both presynaptic and postsynaptic BDNF functions (Li et al., 1998; Gonzalez et al., 1999; Haapasalo et al., 2001; Offenhauser et al., 2002; Elmariah et al., 2004). Here, we combined in vivo time-lapse imaging of fluorescently tagged synaptic specializations with overexpression of GFP–TrkB.T1 in individual RGCs to examine the influence of TrkB signaling on targeting, branching, and synaptic maturation of presynaptic RGC axons. Similarly, expression and in vivo imaging of GFP–TrkB.T1 in tectal neurons was used to determine whether postsynaptic TrkB signaling modulates the morphological and synaptic differentiation of tectal neurons. Electron microscopy analysis of GFP–TrkB.T1-expressing RGC axons provided a direct correlate between changes in synaptic ultrastructure and the dynamic behavior of the axon arbors and synaptic specializations observed in vivo. Together, our results demonstrate that cell-autonomous TrkB signaling in presynaptic RGCs is necessary for the normal development of Xenopus visual connectivity and indicate that the growth and enhanced stability of presynaptic RGC axons affects both structural and functional aspects of retinotectal synaptic connectivity. These in vivo studies, therefore, provide the first direct demonstration of a presynaptic mechanism of BDNF action during visual system development.
Materials and Methods
Xenopus laevis tadpoles were obtained by in vitro fertilization of oocytes from adult females primed with human chorionic gonadotropin. Tadpoles were raised in rearing solution [60 mm NaCl, 0.67 mm KCl, 0.34 mm Ca(NO3)2, 0.83 mm MgSO4 10 mm HEPES, pH 7.4, and 40 mg/L gentamycin] plus 0.001% phenylthiocarbamide to prevent melanocyte pigmentation. Tadpoles were anesthetized during experimental manipulations with 0.05% tricane methanesulfonate (Finquel; Argent Laboratories, Redmond, WA). Staging was done according to Nieuwkoop and Faber (1956). Animal procedures were approved by the University of California, Irvine.
Plasmid constructs.
A cDNA coding for GFP–TrkB.T1 fusion protein was kindly provided by Dr. E. Castren (University of Helsinki, Helsinki, Finland). A t-dimer–red fluorescent protein (RFP)–synaptobrevin plasmid was prepared in our laboratory: a cDNA fragment coding for t-dimer–RFP (kindly provided by Dr. Tsien, University of California, San Diego, La Jolla, CA) (Campbell et al., 2002) was fused in frame to the Xenopus synaptobrevin II cDNA (gift of Dr. Poo, University of California, Berkeley, Berkeley, CA) and cloned under control of cytomegalovirus (CMV) promoter using the pCS2+ expression vector. A pARiCG–signal peptide (SP)–GFP–TrkB.T1 plasmid was prepared and used for generation of transgenic Xenopus embryos. This plasmid encodes the nicotinic acetylcholine receptor β-3 promoter (nARβ3), driving expression of a fusion protein consisting of SP–GFP and rat TrkB.T1. The pARiCG–SP–GFP parent vector was generated as follows. The nARβ3 gene upstream region was PCR amplified from pAR–GFP (Tokuoka et al., 2002) using sense primer 5′-AGCTGTCGACGAGTTGCTGACATGTGATTGGC-3′ and antisense primer 5′-AGCTAAGCTTTTCAGG-AGGGAGTATTAAAAATTGAC-3′, cut with SalI and HindIII, and used to replace the CMV promoter of the Xenopus vector pCS2+ (Turner and Weintraub, 1994), yielding pAR2+. The nARβ3 gene downstream region was amplified from pAR–GFP using sense primer 5′-AGC-TCTCGAGCCTCTAGAGACTCTATTAACACTCATCCACC-3′ and antisense primer 5′-AGCTGCGGCCGCACAATTCACTCATTCTT-CCTGCTG-3′, cut with XhoI and NotI, and used to replace the Simian virus 40 polyadenylation signal of pAR2+, resulting in pAR2+ARpA. To increase the expression efficiency, the chimeric intron of the phosphorylated form of internal ribosomal entry site (Clontech, Palo Alto, CA) was cloned into the BamHI site of pAR2+ARpA, yielding pARi2+. To facilitate identification of transgenic tadpoles, a Cac-GFP–tkpoly(A) cassette, driving expression of the GFP reporter protein from the muscle-specific cardiac actin promoter (Mohun et al., 1986), was cloned into the NotI site of pARi2+, resulting in pARiCG2+. Finally, the SP–GFP cassette (Collin and Martens, 2006) was inserted into the BamHI and EcoRI sites of pARiCG2+, yielding pARiCG–SP–GFP. All constructs were validated by DNA sequence analysis.
Generation of transgenic embryos.
A transgenesis DNA fragment was prepared by digesting pARiCG–SP–GFP–TrkB.T1 plasmid with SalI and NotI restriction enzymes, separating the fragment on 0.8% agarose gel and purifying with QIAquick Gel Extraction kit (Qiagen, Valencia, CA). Ovulation of mature Xenopus females was induced by injection of 800 U of human chorionic gonadotropin (Sigma, St. Louis MO) 1 d before transgenesis. The eggs were dejellied in 2.5% cysteine hydrochloride in 1× MMR (12 mm NaCl, 0.13 mm KCl, 7 μm CaCl2, 0.17 mm MgSO4 1 mm HEPES, and 4 μg/ml gentamycin, pH 8.0) and transferred to a 0.4× MMR, 6% Ficoll solution. The transgenesis DNA fragment (200 ng) was mixed with 1 μl of sperm nuclei (∼150,000 nuclei) and incubated for 5 min at room temperature, and 0.5 μl of egg extract was added as described by Amaya and Kroll (1999). The mixture was diluted with sperm dilution buffer (250 mm sucrose, 75 mm KCl, 0.5 mm spermidine trihydrochloride, and 0.2 mm spermine tetrahydrochloride, pH 7.4) to a concentration of one nucleus per 4.6 nl, and eggs were injected with 4.6 nl of solution per egg using a Drummond Scientific (Broomall, PA) injector. Normally cleaving embryos were selected after 4 h and transferred to 0.1× MMR, 6% Ficoll, and 50 mg/L gentamycin until gastrulation. Embryos that completed gastrulation were transferred to regular rearing solution and handled as described above.
Transfection of RGCs and tectal neurons.
Lipofection of RGC and tectal neurons was performed by pressure injection of 0.1–0.2 nl of DNA (1 μg/μl) mixed with DOTAP liposomal transfection reagent (Roche Diagnostics, Indianapolis, IN) into the eye (for RGCs) or tectal (for tectal neurons) primordium of stage 20–22 tadpoles. In some cases, electroporation of the GFP–TrkB.T1 construct was used to achieve better transfection efficiency in RGCs. Equimolar amounts of GFP–TrkB.T1 and t-dimer–RFP–synaptobrevin (from here on referred to as RFP–synaptobrevin) plasmids were mixed together to the final concentration of 2 μg/μl. The DNA mixture was pressure injected into the eye primordium of stage 20–22 anesthetized tadpoles. Tungsten electrodes (Protech International, San Antonio, TX) were positioned across the injected eye, and a train of 10 40-ms square pulses of 45 V was applied to the animals using a CUY 21 (BEX, Tokyo, Japan) electroporator. After transfection, tadpoles were reared under filtered illumination, in 12 h dark/light cycles, until stage 43–45 when used for experimentation and imaging. Tadpoles with individually labeled RGCs or tectal neurons were selected for imaging.
In vivo time-lapse imaging.
The behavior of individual, fluorescently labeled RGC axons was followed with confocal microscopy in stage 45 tadpoles. Only tadpoles with individual labeled RGC axons that showed specific, punctate RFP–synaptobrevin labeling in their terminals were selected. Tadpoles containing one or two clearly distinguishable double-labeled axons were imaged every 2 h for 6 h and then again at 24 h. Image acquisition was performed as described previously (Hu et al., 2005). Confocal imaging of individually labeled GFP control and GFP–TrkB.T1 tectal neurons was performed similar to the imaging of RGCs. Tectal neurons were imaged at 0, 4, 24, and 48 h as described previously (Sanchez et al., 2006).
Data analysis.
All analysis was performed from raw confocal images without any post-acquisition manipulation or thresholding. Digital three-dimensional reconstructions of GFP (GFP–TrkB.T1)-labeled axonal and dendritic arbors were obtained from individual optical sections through the entire extent of the arbor with the aid of the MetaMorph software (Universal Imaging Corporation, West Chester, PA). To characterize the distribution of RFP–synaptobrevin puncta to particular RGC axonal regions, pixel-by-pixel overlaps from individual optical sections obtained at the two wavelengths were analyzed as described previously (Hu et al., 2005). Only discrete RFP–synaptobrevin puncta with median pixel values 2.0–3.0 times greater than the median pixel values of background nonpunctate RFP within the same axon arbor were counted and included in the analysis. During data analysis, we ensured that similar ratios were maintained for every axon arbor analyzed throughout the entire observation period. Several morphological parameters were measured for the quantitative analysis of axon and dendritic branching: total branch number, the number of first-, second-, and third-order branches, the number of individual branches added, and the number of branches remaining from one observation time point to the next (stable branches). Total arbor length was measured from binarized images of the digitally reconstructed axons or dendritic arbors. A relative measure of cumulative length of all branches per axon terminal or dendritic arbor was obtained by counting total pixel number from the first branch point. Axon or dendrite branch turnover rate was measured as the relative change in branch length within each observation interval and was expressed as percentage of the length in the previous time point. Simple axon projections containing only first-order branches and a motile growth cone structure at the axon tip were defined as growth cone-like axon arbors. The axon complexity index (ACI) of growth cone-like axon arbors was <1.0. Presynaptic specialization density was calculated by dividing the number of RFP–synaptobrevin puncta by the total axon arbor length and is expressed as the number of puncta per 10 μm. A total of 10–17 axonal and dendritic arbors per condition were analyzed, with one RGC axon arbor or one tectal neuron analyzed per tadpole. One- and two-way ANOVA tests (time and treatment) were used for the statistical analysis of data. Results were considered significant as follows: *p < 0.05, **p < 0.005, ***p < 0.0005.
Immunocytochemistry.
For endogenous synaptosome-associated protein of 25 kDa (SNAP-25) colocalization, tadpoles with individual tectal neurons overexpressing GFP control or GFP–TrkB.T1 plasmids were anesthetized and fixed for 2 h by immersion in 2% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4; the brains were removed and postfixed with the same fixative for 1 h. Free-floating brains were preincubated for 1 h in blocking solution (1.5% goat normal serum and 0.1% Triton X-100 in 0.1 m PB) and incubated overnight with rabbit polyclonal anti-SNAP-25 antibody (1:1000 dilution; Stressgen Biotechnologies, Victoria, British Columbia, Canada). Tissues were then rinsed and incubated with Alexa 568 anti-rabbit antibodies (1:200 dilution 0.1 m PB; Invitrogen, Carlsbad, CA). All images were collected with a LSM 5 Pascal confocal microscope using a 63×/1.4 numerical aperture oil immersion objective and 2× electronic zoom. To determine colocalization of fluorescent labels, optical sections were collected at 0.5 μm intervals through the full extent of the GFP-labeled tectal arbor.
Electron microscopy.
Unilateral electroporation of GFP or GFP–TrkB.T1 plasmids into the eye primordium (right eye) of stage 22 tadpoles was used for the ultrastructural analysis. Stage 45 tadpoles with only a few RGCs expressing GFP or GFP–TrkB.T1 in their axon terminals were selected and processed for preembedding immunoelectron microscopy. Tadpoles were anesthetized and fixed in 2% paraformaldehyde and 3.75% acrolein in 0.1 m PB, pH 7.4. Brains were removed, postfixed, and embedded in 1% agarose. Fifty micrometer vibratome sections were collected, incubated in 1% sodium borohydride in PB, cryoprotected, quickly permeabilized in liquid nitrogen, and blocked in 0.5% bovine serum albumin (BSA), 0.1 m Tris-buffered saline (TBS), pH 7.5. Sections were then coded, and all subsequent procedures were performed blind to treatment. Sections were incubated overnight in a primary mouse monoclonal antibody against GFP (1:10 dilution in 0.1% BSA in TBS; Invitrogen), followed by 2 h in a secondary goat anti-mouse IgG coupled to 0.6 nm gold particles (1:50 dilution in 0.5% v/v of 20% fish gelatin, 0.8% BSA in 0.01 m PBS, pH 7.4) (Aurion-EMS, Hatfield, PA). Sections were incubated in 2% glutaraldehyde, and gold particles were enlarged using a British BioCell silver intensification kit (Ted Pella, Redding, CA). Sections were postfixed in 2% osmium tetroxide, dehydrated, and flat embedded in 100% Epon between Aclar sheets. Seventy nanometer thin sections were obtained on copper mesh grids using a Reichert ultramicrotome with a diamond knife (Diatome, Biel, Switzerland) and counterstained with 2% uranyl acetate and Reynolds lead citrate. Ultrastructural analysis was performed using a Philips (Aachen, Germany) CM10 transmission electron microscope. Specificity of the immunostaining was determined by comparing the density of silver-enhanced gold particles within the immunopositive synaptic profile with that of the surrounding neuropil. In addition, to control for specificity, we confirmed that silver-enhanced gold particles would be discretely localized to a few terminals in the unilaterally transfected tadpoles and that it would be absent from the contralateral optic tectum. For the quantitative analysis, the number of synaptic vesicles within the presynaptic terminal area at mature synapses (synapses presenting at least three of the following characteristics: presynaptic and postsynaptic densities, synaptic vesicles, synaptic cleft, and parallel membranes) and the number of docked vesicles per terminal (attached to the presynaptic density at a distance <50 nm) were measured. The length of the presynaptic specialization and the size of the area of the axon terminal were measured using the Image J software (Scion, Frederick, MD). Two-sample unpaired Student's t test was used for the statistical analysis of data. Significance was p < 0.05.
Results
TrkB.T1 overexpression leads to a high proportion of growth cone-like axons among transfected RGC arbors and increases axon degeneration
Overexpression of a GFP–TrkB.T1 fusion protein was used to inhibit TrkB signaling in individual RGCs during the early development of the Xenopus retinotectal projection. Expression of GFP–TrkB.T1 was clearly observed inside the tadpoles' retinas at stage 32, 24 h after transfection. By stage 43, GFP-labeled RGC axons with simple morphology (less than five branches) were observed to project to the optic tectum in both GFP controls and GFP–TrkB.T1-expressing tadpoles. Disruption of TrkB signaling did not appear to cause any errors in RGC targeting because no GFP-labeled RGC axons were observed in any other area of the brain. To further confirm that TrkB signaling is not essential for RGC pathfinding, we created transgenic tadpoles that overexpressed GFP–TrkB.T1 under the control of the nicotinic acetylcholine receptor-3 (nAChR-3) gene promoter. This promoter has been effectively used to drive GFP expression in most RGC axons in zebrafish embryos (Tokuoka et al., 2002). As observed for the single-transfected neurons, RGCs in transgenic tadpoles began to express GFP–TrkB.T1 by stage 32 (data not shown), and the entire population of GFP–TrkB.T1-expressing RGC axons projected appropriately to the optic tectum (Fig. 1 A,B). Thus, our data demonstrate that alterations in TrkB signaling do not impact RGC axon pathfinding or targeting to the tectum.
Figure 1.
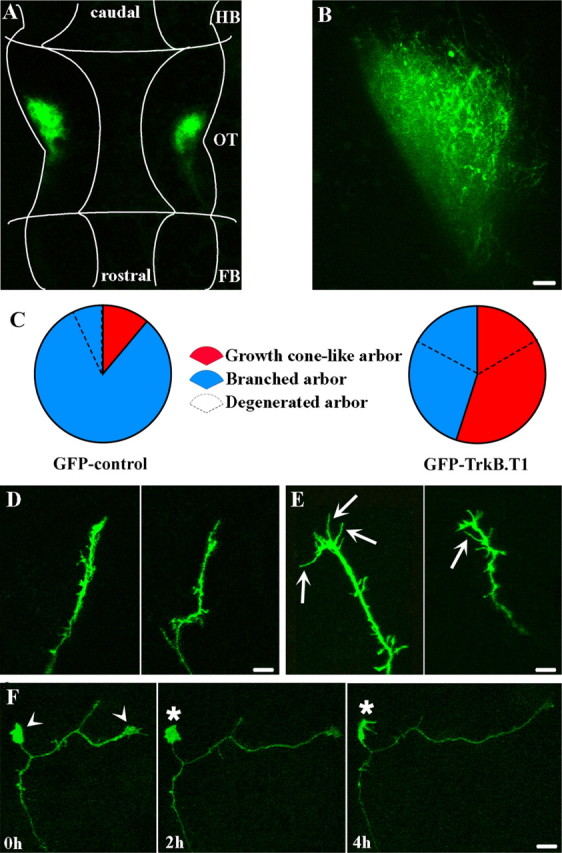
TrkB signaling influences RGC axon growth cone morphology and axon branch initiation. A, B, GFP–TrkB.T1-expressing RGC axons project correctly to the optic tectum. A, Low-magnification confocal image of the brain (outlined by the white tracing) of a stage 45 transgenic tadpole expressing GFP–TrkB.T1 under the control of the nAChR promoter. Fluorescently labeled RGC projections are correctly targeted to the optic tectum (OT). No “mistargeted” RGC axons were observed in other areas of the brain, including forebrain (FB) and hindbrain (HB). B, High-magnification confocal image of the tectal neuropil (right hemisphere of tadpole brain shown in A) better illustrates the GFP–TrkB.T1-expressing RGC axon terminals. Scale bar, 10 μm. C–F, Expression of TrkB.T1 in individual RGC increases the proportion of growth cone-like axon arbors and alters axon morphology. C, Pie charts represent the proportion of GFP control and GFP–TrkB.T1-expressing RGC axons with growth cone-like (red) and branched (blue) morphologies. The broken lines within the pie charts represent the proportion of axons that degenerated within 24 h after the initial imaging session. Note that similar proportions of growth cones and branched arbors degenerated in the GFP–TrkB.T1-expressing RGCs. D–F, Representative RGC axons with terminal growth cone-like structures expressing GFP control (D) or GFP–TrkB.T1 (E, F). Note that TrkB.T1-expressing growth cones possess uncharacteristically long filopodia (arrows) and multiple large lamellipodia (arrowheads). In E, a large, interstitial growth cone-like structure remodeled and persisted throughout at least 4 h of imaging (asterisks). Scale bars, 10 μm.
To investigate the role of TrkB signaling in RGC axon arborization and formation of retinotectal connectivity, we examined the morphology and dynamic behavior of individual GFP–TrkB.T1-expressing RGC axons by in vivo time-lapse confocal microscopy. A significant difference was observed between GFP control and GFP–TrkB.T1-overexpressing axons at stage 45, when most RGC axons have reached the optic tectum and begin to arborize. The majority of GFP–TrkB.T1-expressing RGC axons at this stage possessed an immature, growth cone-like morphology (55%; n = 38), whereas only a few of the GFP control axons were growth cones (11%; n = 28) (Fig. 1 C). In addition, the GFP–TrkB.T1 growth cones had an altered morphology, exhibiting uncharacteristically long filopodia, and often possessed multiple large lamellipodia that persisted over the course of axon growth and were never observed in GFP control growth cones (Fig. 1 D–F). Multiple growth cone-like structures were also observed on branch tips of GFP–TrkB.T1 axons that were branched. Interestingly, more than one-third (34%; n = 38) of the GFP–TrkB.T1-overexpressing axons degenerated during the 24 h observation period. The number of degenerated GFP–TrkB.T1 axons was approximately five times higher than that of controls (7%; n = 28) (Fig. 1 C). Degeneration was observed as a gradual clustering of the GFP label that progressed until discontinuous strings of bright GFP fluorescence formed, indicating disassembly of the entire axon arbor. Axon degeneration was unlikely to be a consequence of potential detrimental effects of accumulated laser irradiation, because degeneration occurred independently of the number of preceding laser exposures and was different between GFP controls and GFP–TrkB.T1 axons imaged in the same manner. Similar proportions of both growth cone-like and branched GFP–TrkB.T1 axon arbors degenerated over time (29% for growth cones and 41% for branched axons) (Fig. 1 C).
RGCs expressing GFP–TrkB.T1 have simpler axon arbors and fail to increase their complexity
Qualitative analysis demonstrates that the majority of the TrkB.T1-expressing RGC axons had immature growth cone-like morphologies. Quantification of total axon branch number in tadpoles at stage 45 showed that the GFP–TrkB.T1 RGC axons had significantly fewer axon branches compared with GFP controls (10.68 ± 1.15 for TrkB.T1; 14.25 ± 1.28 for control; p = 0.0432). After 24 h, the GFP control axons significantly increased their total branch number (18.60 ± 1.66; p = 0.0403), whereas the number of branches in the GFP–TrkB.T1 axons remained unchanged (11.39 ± 1.78; p = 0.7276) (Fig. 2 A). To directly evaluate whether inhibition of TrkB signaling influences RGC axon branch dynamics and axon maturation, we selected for additional analysis only RGC axons that had, at the initial observation time point (0 h), between 10 and 25 branches and an arbor length of at least 120 μm. Based on this selection criteria, both GFP–TrkB.T1 axons (n = 16) and their age-matched GFP controls (n = 17) had similar complexities at the first observation time point (15.94 ± 1.45 and 16.53 ± 1.16 total branches, respectively). We observed that, by 24 h, total branch number in control RGC axons increased by 40% (22.86 ± 1.84; p = 0.0022), but it did not change significantly in the GFP–TrkB.T1 axons (18.45 ± 1.38; p = 0.1544) (Fig. 2 B). Thus, GFP–TrkB.T1-expressing RGCs had a slower axon arborization rate. In addition, we observed that overall axon arbor morphology differed between controls and GFP–TrkB.T1 axons. To evaluate axon arbor complexity, we assigned a topological order to each branch in the individual RGC arbors and calculated ACI as illustrated in Figure 2 C. At 0 h, the ACI value for the GFP–TrkB.T1 axons (1.19 ± 0.05) was significantly lower than that of controls (1.42 ± 0.06; p = 0.0124). Over the 24 h observation period, control GFP-expressing RGCs significantly increased their arbor complexity (ACI, 1.70 ± 0.09; p = 0.0036), whereas GFP–TrkB.T1 axons remained simple and their complexity was not significantly different from that at the 0 h time point (ACI, 1.34 ± 0.07; p = 0.1388) (Fig. 2 D). The branch order distribution analysis revealed that GFP–TrkB.T1 axons had a significantly higher number of first-order branches and significantly lower number of third-order branches than GFP controls at both the 0 and 24 h time points (Fig. 2 E). Consequently, our data demonstrates that interfering with TrkB signaling inhibits the ability of RGC axons to become more complex over time.
Figure 2.

TrkB.T1 overexpression in RGC decreases axon arborization. A, Quantitative analysis of total axon branch number in GFP–TrkB.T1 and GFP control RGCs demonstrates that interfering with TrkB signaling results in axons with significantly fewer branches at stage 45 (0 h) and 24 h later. B, When only axons with similar branch numbers were included in the analysis (axons with 10–25 branches), only control GFP-expressing axons significantly increased their branch number, whereas the GFP–TrkB.T1 axons remained unchanged 24 h after initial imaging. C–E, Branch order analysis and ACI were used for the quantitative analysis of axon arbor morphology of axons with 10–25 initial branches. C, Each branch tip was assigned a number (N, branch order; see schematic diagram) that equals the number of times a primary axon branched to produce that branch tip. D, GFP–TrkB.T1-expressing RGC axons have a lower ACI compared with age-matched controls and fail to significantly increase their complexity over time. E, Branch order distribution shows a significantly higher proportion of first-order branches and lower proportion of third-order branches in GFP–TrkB.T1-expressing RGC axons when compared with age-matched controls. *p < 0.05, **p < 0.005.
Inhibition of TrkB signaling increases RGC axon arbor remodeling
To determine whether the slow growth of TrkB.T1-overexpressing axons was caused by a decreased ability of the neurons to form and maintain branches, we followed branch dynamics of RGC axons by in vivo time-lapse imaging. Analysis of arbor morphology of individual axons at 2 h intervals for a total of 6 h revealed an increased rate of branch remodeling in GFP–TrkB.T1 axons relative to GFP controls (Fig. 3 A). New branch extension in the GFP–TrkB.T1 axons was 1.7 times higher than in control RGCs (50.64 ± 3.81 vs 28.88 ± 2.30% branches added; p < 0.0001) (Fig. 3 B). Similarly, branch elimination was increased in the GFP–TrkB.T1 axons, resulting in only approximately half of the branches (54.25 ± 2.69%) remaining stable over a 2 h time period, a significantly lower number of stable branches than in controls (78.90 ± 1.54%; p < 0.0001) (Fig. 3 B). To obtain a second measure of axon arbor remodeling, we quantified the relative change in length of the axonal arbor within a 2 h period. The GFP–TrkB.T1 axons showed a dramatic increase in turnover rate; the change in GFP–TrkB.T1 axon length was almost twice that of control RGCs (15.87 ± 2.22% per 2 h for GFP–TrkB.T1 vs 8.25 ± 1.21% for controls; p = 0.0029) (Fig. 3 B). Thus, our data demonstrate that GFP–TrkB.T1-overexpressing RGC axons have an increased branch turnover rate and are more motile than their age-matched controls.
Figure 3.
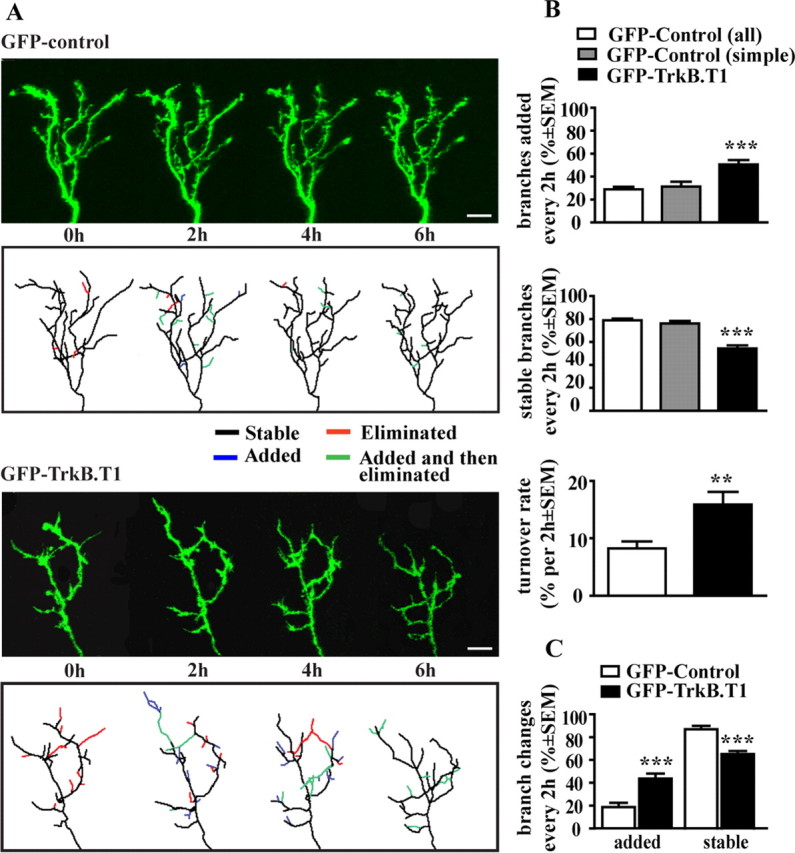
Inhibition of TrkB signaling in RGC increases RGC axon arbor remodeling. A, Time-lapse confocal images of GFP control and GFP–TrkB.T1-expressing RGC axons. Reconstructions of three-dimensional arbors (top) and line tracings (bottom) illustrate the effects of GFP–TrkB.T1 expression on the dynamic remodeling of RGC axonal arbors. Branches that were added are shown in blue, those that were eliminated are in red, and those that were added at one time point and then eliminated in the next are labeled green. Note that, for the GFP–TrkB.T1-expressing RGC axon, a larger portion of the arbor is color coded. Scale bar, 10 μm. B, Quantification of the effects of TrkB.T1 expression on the number of added and stabilized axonal branches and relative length change. Values for added and stable branches are expressed as percentage change from the previous time point. Axon turnover rate represents the relative change in total arbor length within every 2 h observation period (in axons imaged for a total of 6 h) and is expressed as percentage change. Branch addition and stabilization rates of GFP–TrkB.T1-expressing axons were significantly different from age-matched controls (stage 45; white bars) and from GFP controls imaged at younger stages (stage 43 tadpoles; gray bars). C, Branch addition and stabilization rates remain significantly different in GFP–TrkB.T1-expressing arbors even after excluding motile filopodial-like processes (<5 μm) from the analysis. **p < 0.005, ***p < 0.0005.
It is possible that TrkB.T1 overexpression delays axon maturation and that the simpler morphology and more dynamic behavior of the GFP–TrkB.T1-expressing RGC axon arbors reflect the behavior of more immature RGC axons in tadpoles at stages younger than stage 45. Thus, to test whether immature, simple axons exhibit a more dynamic behavior than arborized RGCs, we performed time-lapse imaging of GFP control RGC axons in stage 43 tadpoles, when most RGCs begin to branch and have a simpler arbor complexity. For our analysis, we selected GFP control axons with a similar ACI value to that of GFP–TrkB.T1 RGCs imaged in stage 45 tadpoles. We found that the rates of branch formation and stabilization of simple GFP control axons in stage 43 tadpoles were similar to those of more mature, GFP control branched arbors in stage 45 tadpoles (Fig. 3 B). Thus, these observations indicate that the effect of GFP–TrkB.T1 overexpression on axon branch dynamics is specific and is not caused by a time delay in RGC axon maturation.
Developing axon arbors have transient short filopodia and more stable longer branches. Both types of processes undergo dynamic remodeling during axon arborization (Cohen-Cory and Fraser, 1995; Witte et al., 1996). To determine whether the high rate of branch turnover in GFP–TrkB.T1 RGC axon arbors was attributable to increased filopodial-like dynamics rather than increased turnover of more established branches, we reexamined RGC axon branch dynamics but excluded processes shorter than 5 μm (filopodial-like processes) from the analysis. We found that branch addition and stabilization rates in GFP–TrkB.T1 RGC axons remained significantly different from those in controls (new branches: 18.73 ± 3.71 for control, 43.65 ± 4.50 for GFP–TrkB.T1, p < 0.0001; stable branches: 86.90 ± 1.79 for control, 65.22 ± 2.55 for GFP–TrkB.T1, p < 0.0001) (Fig. 3 C), demonstrating that inhibition of BDNF signaling affects the dynamic behavior of the entire axonal arbor. Furthermore, we found that the proportion of filopodial-like processes (relative to total branch number) remained unaffected by TrkB.T1 overexpression (data not shown).
Intact TrkB signaling is required for presynaptic site formation in developing RGC axons
Our results demonstrate that cell-autonomous TrkB signaling is important for normal arborization of presynaptic RGC axons. Our previous work has shown that BDNF influences both axon branching and synapse formation and that these two processes are related (Alsina et al., 2001; Hu et al., 2005). To investigate whether inhibition of TrkB signaling interferes with RGC synaptic connectivity, we visualized RGC presynaptic specializations by expressing an RFP–synaptobrevin fusion protein together with the GFP control or GFP–TrkB.T1 construct in individual RGCs. The advantage of the t-dimer–RFP fluorophore is that, unlike commercially available Discosoma red protein, it does not require ultimate tetramerization that may cause formation of intracellular aggregates and interfere with normal subcellular localization and function of the red fluorescent chimera (Campbell et al., 2002). Previous studies demonstrated that fluorescently tagged synaptobrevin is correctly targeted to presynaptic specializations in Xenopus RGC axon terminals (Alsina et al., 2001) and localizes to ultrastructurally identified presynaptic terminals as demonstrated by immunoelectron microscopy (Hu et al., 2005). Discrete, punctate distribution of RFP–synaptobrevin was observed in GFP-expressing control RGC axons (Fig. 4 A–D). Although most RGC axons expressing GFP–TrkB.T1 also expressed RFP–synaptobrevin, the GFP–TrkB.T1 axons showed a more diffuse RFP labeling in the growth cone-like tips and fewer less well defined puncta along their branches (Fig. 4 E–H). Quantification of RFP–synaptobrevin puncta revealed that the total number and density of RFP–synaptobrevin-labeled presynaptic specializations in GFP–TrkB.T1 axons was significantly lower than controls at all observation time points (Fig. 4 I), suggesting that inhibition of TrkB signaling in RGCs impairs presynaptic site formation and/or maintenance. These data also suggested that the increased remodeling of TrkB.T1 axon arbors may signify a decreased ability of these axons to form or maintain synapses.
Figure 4.
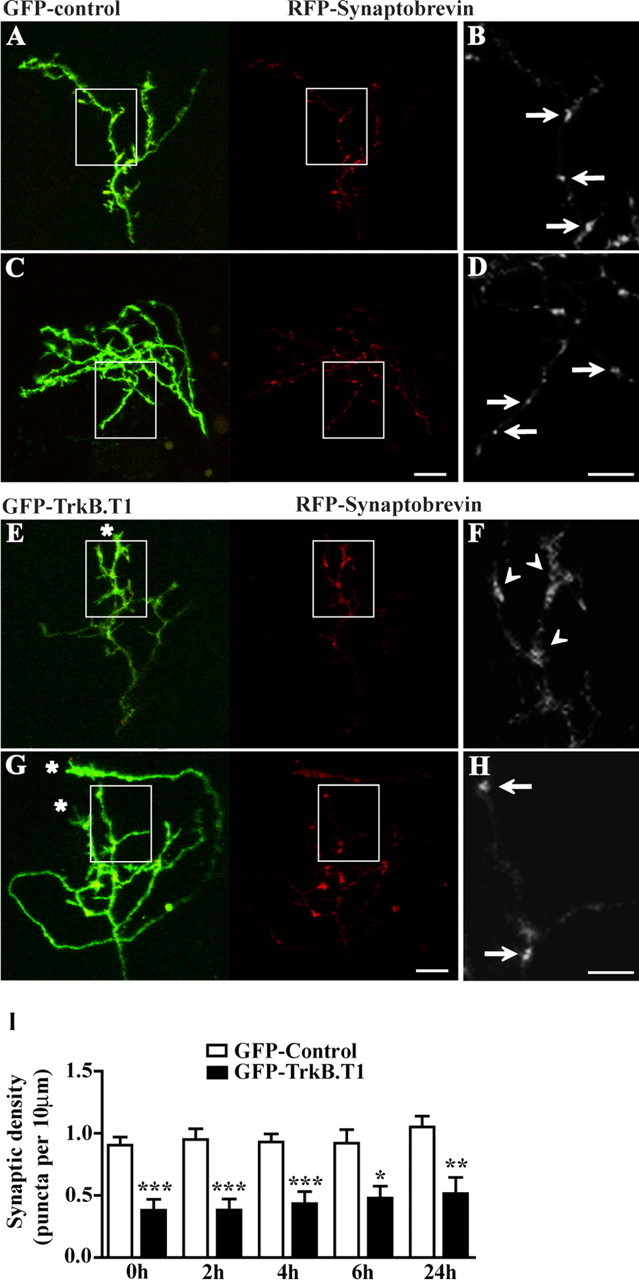
GFP–TrkB.T1-overexpressing axons have a decreased density of presynaptic specializations. A–H, Three-dimensional reconstructions of RGC axon arbors expressing GFP control (A–D) or GFP–TrkB.T1 (E–H) together with RFP–synaptobrevin. Representative examples of relatively simple (A, E) and complex (C, G) axon arbors are shown. Areas of the axon arbor containing isolated single branches are demarcated by the boxes in A, C, E, G and magnified in B, D, F, H, respectively. Note the punctate distribution of the RFP–synaptobrevin presynaptic marker in the GFP controls (arrows in B, D) and the more evenly distributed RFP fluorescence in the GFP–TrkB.T1-expressing axons (arrowheads in F). The branched GFP–TrkB.T1-expressing axons (E, G) have more growth cone-like structures (asterisks) with diffuse RFP fluorescence labeling (arrowheads). The density of RFP–synaptobrevin puncta in the GFP–TrkB.T1-expressing arbors (E–H) is also lower than in GFP controls (A–D). I, Quantification of the effect of GFP–TrkB.T1 expression on the density of presynaptic specializations per axon arbor, expressed as the number of RFP–synaptobrevin puncta per 10 μm. RFP–synaptobrevin puncta (arrows) with diameter of less or equal to 1 μm and fluorescence intensity at least 2.5 times greater than that of background RFP were included in the analysis. *p < 0.05, **p < 0.005, ***p < 0.0005. Scale bars, 10 μm.
Altering TrkB signaling in tectal neurons does not alter dendritic arbor branching or synaptogenesis
Our previous studies demonstrated that manipulations that increase BDNF tectal levels exert significant effects on tectal neuron synaptic connectivity, increasing postsynaptic specialization number but not dendritic branching (Sanchez et al., 2006). Only when BDNF levels were decreased by injection of neutralizing antibodies was dendritic arbor growth prevented. To directly differentiate between cell-autonomous and secondary effects of BDNF on postsynaptic tectal neurons, we overexpressed GFP–TrkB.T1 in tectal neurons of young tadpoles and analyzed dendritic arbor morphology at stage 45. Time-lapse confocal microscopy revealed that GFP–TrkB.T1 expression in individual tectal neurons does not influence dendritic arbor morphology or the rate of dendritic arbor growth (Fig. 5 A). Both GFP–TrkB.T1 and age-matched control tectal neurons had comparable total branch numbers at all observation time points (total branches, 13.55 ± 1.50 for control at 0 h, 20.00 ± 2.32 for control at 48 h, p = 0.0088; total branches, 11.75 ± 0.75 for TrkB.T1 at 0 h, 15.56 ± 1.58 for TrkB.T1 at 48 h, p = 0.0292) (Fig. 5 B). Dendritic complexity index (DCI) of tectal neurons, calculated similarly to the ACI used for analysis of RGC axon complexity, showed that control and GFP–TrkB.T1 tectal neurons possessed similar complexities (DCI values) and significantly increased dendritic arbor complexity within 48 h of observation (DCI for GFP controls: 1.30 ± 0.07 at 0 h, 1.71 ± 0.14 at 48 h, p = 0.0013; DCI for GFP–TrkB.T1 tectal cells: 1.33 ± 0.06 at 0 h, 1.58 ± 0.08 at 48 h, p = 0.0138) (Fig. 5 C). Furthermore, inhibition of TrkB signaling in tectal neurons did not affect the dynamic behavior of the tectal neuron dendritic branches or their growth rate. The rates of dendritic branch addition and stabilization (Fig. 5 D), as well as branch turnover rates were similar in GFP–TrkB.T1 and GFP control tectal neurons (branch turnover rate: 13.88 ± 3.12% for controls, 14.27 ± 4.77% for GFP–TrkB.T1, p = 0.9498; not shown graphically). Moreover, both GFP control and GFP–TrkB.T1 tectal neurons similarly increased their total dendritic length over a 48 h observation period (increase in total arbor length compared with 0 h was, for GFP controls: 108.30 ± 5.49% at 4 h, 132.00 ± 8.94% at 24 h, 172.80 ± 13.23% at 48 h; for GFP–TrkB.T1: 111.60 ± 6.85% at 4 h, 129.00 ± 8.27% at 24 h, 156.20 ± 13.02% at 48 h) (Fig. 5 E). Together, our data demonstrates that inhibition of TrkB signaling in postsynaptic tectal neurons does not alter dendritic arbor morphology or dynamics, further supporting the idea that the effect of neutralizing BDNF on tectal neurons we reported previously is indirect and mediated by the altered growth and maturation of presynaptic RGC axons.
Figure 5.
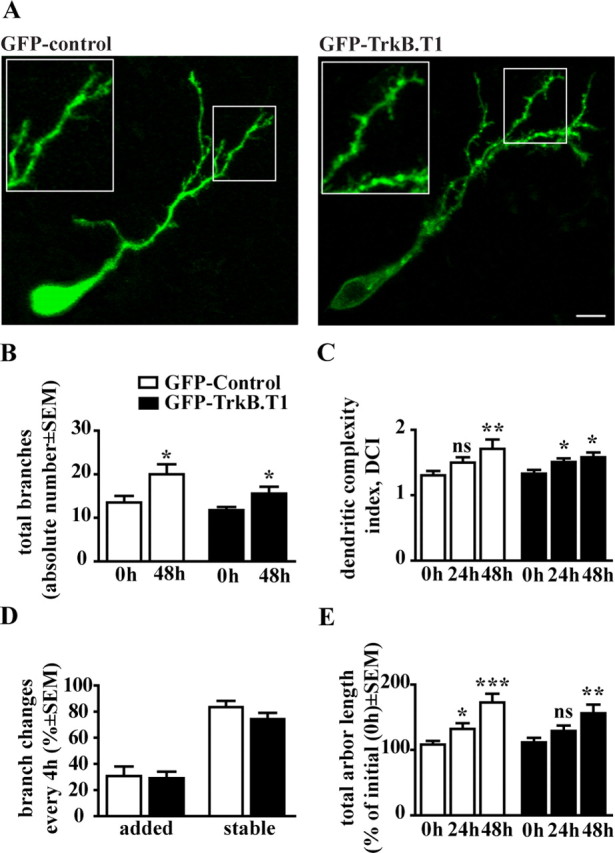
Inhibition of TrkB signaling in postsynaptic tectal neurons does not affect maturation of tectal neuron dendritic arbor. A, Image reconstructions of representative GFP control and GFP–TrkB.T1-labeled tectal neurons. Scale bar, 10 μm. B–E, Quantification of the effects of TrkB.T1 overexpression in postsynaptic tectal neurons on dendritic branch number, dendritic arbor complexity, branch dynamics, and total dendritic arbor length. DCI was calculated similarly to ACI (see Fig. 2 C). Values for new and stable branches are expressed as the percentage change from the previous time point. Total dendritic arbor length is presented as a percentage of total length of the arbor at 0 h time point. *p < 0.05, **p < 0.005, ***p < 0.0005; ns, nonsignificantly different.
Although our previous studies showed that manipulations that increased or decreased tectal BDNF levels had no significant effects on dendritic arbor morphology, these manipulations influenced synapse number in tectal neurons. Specifically, BDNF increased, whereas anti-BDNF decreased, the number of postsynaptic density-95–GFP-labeled postsynaptic specializations present on the tectal neuron dendritic arbors (Sanchez et al., 2006). To determine whether these effects of altered BDNF levels reflect a direct or indirect effect on tectal neuron synaptic connectivity, we performed a quantitative analysis of synaptic sites in tectal neurons overexpressing GFP or GFP–TrkB.T1 in stage 45 tadpoles. Because expression of RFP-tagged postsynaptic proteins interferes with TrkB.T1 expression, for this analysis, we immunostained tadpole brains with antibodies to the presynaptic plasma membrane protein SNAP-25 to identify sites of contact between presynaptic axons and tectal neuron dendritic arbors (Fig. 6 A,B). Confocal z-stacks were collected at a high resolution to resolve individual SNAP-25-immunopositive puncta apposed to the GFP-labeled dendritic branches (Fig. 6 C,D). Our results show that the number of SNAP-25 puncta per unit dendritic arbor length is similar in the GFP–TrkB.T1- and GFP-expressing tectal neurons (2.21 ± 0.31 SNAP-25 puncta per 10 μm for GFP–TrkB.T1 and 2.29 ± 0.25 for GFP control; not shown graphically). Consequently, these results demonstrate that direct alterations in TrkB signaling in tectal neurons do not alter their synaptic connectivity, supporting the idea that BDNF influences the development of retinotectal synaptic connectivity by acting primarily on presynaptic RGCs.
Figure 6.
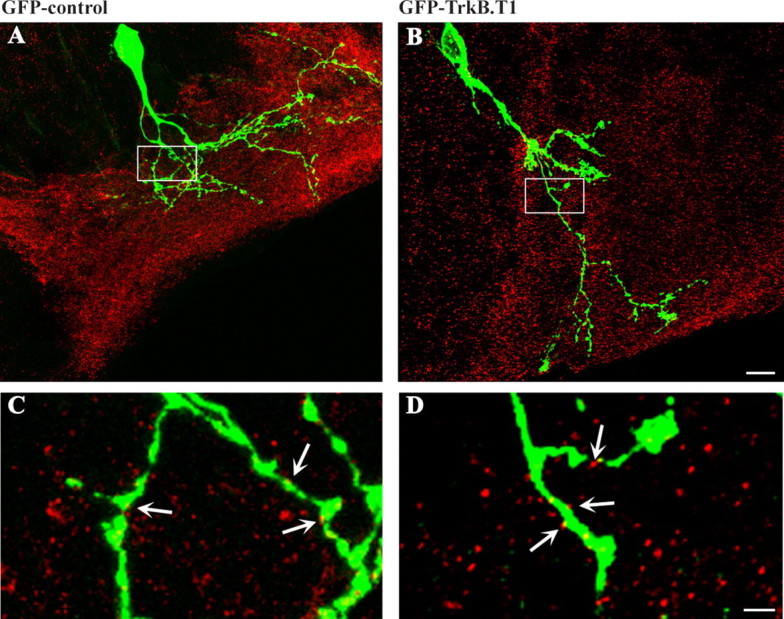
Altering TrkB signaling in postsynaptic tectal neurons does not influence retinotectal synaptic connectivity. A, B, Control (A) and TrkB.T1 (B) overexpressing tectal neurons and the discrete distribution of endogenous SNAP-25 in the tectal neuropil. Tadpole brains with individual tectal neurons expressing GFP or GFP–TrkB.T1 were fixed and immunostained with an antibody to the presynaptic protein SNAP-25 to reveal presynaptic sites apposed to the dendritic arbors. Scale bar, 10 μm. C, D, High-magnification images (projections of 5 sequential confocal planes) show colocalization of single SNAP-25 puncta (arrows) with selected segments of GFP control (C) and GFP–TrkB.T1 (D) dendritic arbor. No significant difference in the number or density of SNAP-25 puncta apposed to GFP–TrkB.T1-expressing tectal neurons versus GFP controls was observed. Scale bar, 2 μm.
Inhibition of TrkB signaling in RGCs influences synaptic vesicle docking at retinotectal synapses
Collectively, our in vivo imaging experiments indicate that cell-autonomous TrkB signaling directly in RGCs mediates the effects of BDNF on axon arborization and presynaptic differentiation. To further evaluate whether TrkB signaling in RGCs is directly involved in synaptic maturation and to correlate in vivo imaging observations with ultrastructural changes at retinotectal synapses, we analyzed brains of stage 45 tadpoles expressing GFP–TrkB.T1 or GFP exclusively in RGCs. Synapses made by the GFP–TrkB.T1- or GFP-expressing RGC axons were specifically identified by preembedding immunoelectron microscopy using an antibody to GFP. Mature and immature retinotectal synapses were observed in both GFP–TrkB.T1- and GFP-expressing tadpoles. Presynaptic profiles analyzed included terminals with synaptic vesicle aggregates but no thickening of presynaptic and postsynaptic membranes (immature synapses), as well as mature synaptic profiles with clearly identifiable presynaptic active zones and postsynaptic densities (Fig. 7 A–C). Almost half of all of the immunopositive profiles with synaptic vesicle aggregates analyzed from RGC terminals expressing GFP in control tadpoles represented mature synapses (46.5 ± 4.46%; n = 154 profiles analyzed from 11 tadpoles expressing GFP in RGCs) (Table 1). In contrast, in the GFP–TrkB.T1-expressing RGC axons, only 21.68 ± 6.6% of profiles with synaptic vesicle aggregates were associated with mature presynaptic and postsynaptic structures (p = 0.0044; n = 188 profiles analyzed from seven tadpoles expressing GFP–TrkB.T1 in RGCs). The significant difference in the number of fully differentiated synaptic profiles in control versus GFP–TrkB.T1-expressing RGC axons suggests that synapse maturation may be affected by the altered TrkB signaling in RGCs. This interpretation is in agreement with our observations that GFP–TrkB.T1-expressing RGC axons have significantly fewer presynaptic specializations than controls, as defined by the RFP–synaptobrevin punctate labeling. Consistent with our observations in single axons imaged in vivo, we also observed that, in addition to mature and immature presynaptic profiles, enlarged growth cone-like presynaptic profiles (data not shown) were prevalent in the GFP–TrkB.T1-expressing RGC axons.
Figure 7.
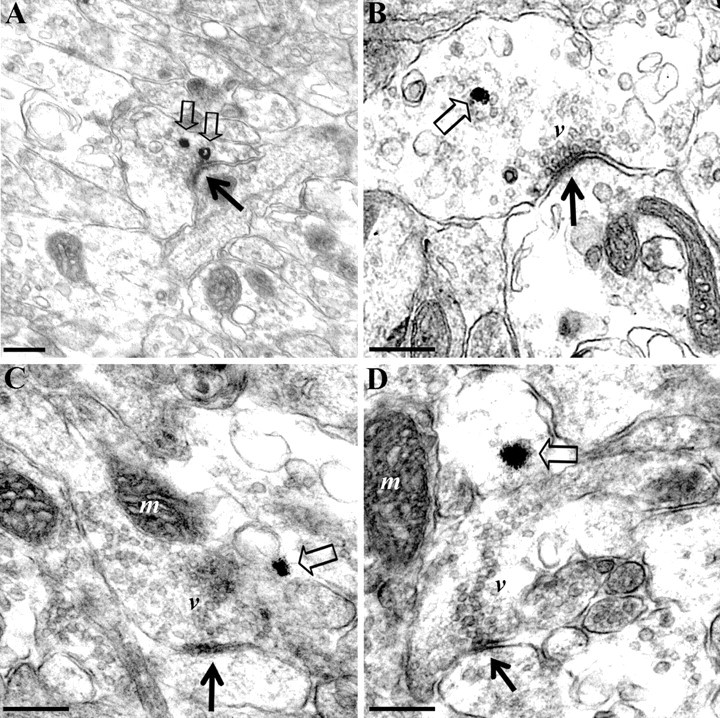
Synapse ultrastructure in the tectal neuropil of stage 45 tadpoles expressing GFP or GFP–TrkB.T1 exclusively in RGCs. A–D, Representative electron photomicrographs of mature synaptic profiles immunopositive for GFP. The silver-enhanced immunogold labeling (open arrows) identifies presynaptic profiles in GFP control (A, B), and GFP–TrkB.T1-expressing RGC terminals (C, D). The lower-magnification micrograph (A) illustrates the specific localization of silver-enhanced gold particles to individual presynaptic terminals and the absence of immunoreactivity in the surrounding neuropil. The number of synaptic vesicles docked at the active zone (black arrows) is higher in GFP control (A, B) than in GFP–TrkB.T1-expressing presynaptic terminals (C, D). v, Synaptic vesicles; m, mitochondria. Scale bars, 0.2 μm.
Table 1.
Ultrastructural analysis of retinotectal synapses with overexpression of GFP or GFP–TrkB.T1 at individual presynaptic RGC terminals
| GFP control |
GFP–TrkB.T1 |
|
|---|---|---|
| Mature synaptic profiles (percentage) | 46.5 ± 4.46 | 21.68 ± 6.6** |
| Synaptic area (μm 2) | 6.2 ± 0.58 | 5.3 ± 0.85 |
| Synaptic vesicle number | 70.8 ± 6.16 | 48.3 ± 5.01* |
| Number of docked vesicles | 6.4 ± 0.46 | 3.9 ± 0.59* |
| Docked vesicle density (per μm) | 39.1 ± 3.67 | 26.9 ± 3.98* |
| Length of active zone (μm) | 0.2 ± 0.013 | 0.165 ± 0.013 |
A total of 188 GFP-immunopositive profiles were analyzed for GFP–TrkB.T1-expressing RGC axons (n = 45 mature synaptic profiles analyzed) and 154 for GFP controls (n = 90 mature synaptic profiles). Two-sample Student's t test were used for the statistical analysis of data.
*p ≤ 0.05;
**p ≤ 0.005.
Detailed morphometric analysis of the tectal neuropil of GFP–TrkB.T1 and GFP-expressing control tadpoles revealed a significant decrease in the number of synaptic vesicles per mature presynaptic profile at synapses made by GFP–TrkB.T1-expressing axons when compared with GFP-expressing controls (Fig. 7 A,B; Table 1). In addition, the number of docked synaptic vesicles, defined as those located up to one-vesicle-diameter distance (∼50 nm) from the active zone, was significantly lower in the GFP–TrkB.T1-immunopositive presynaptic profiles than in GFP controls (6.46 ± 0.47 in GFP–TrkB.T1 vs 3.93 ± 0.59 in GFP-expressing axons; p = 0.015) (Fig. 7 B). Presynaptic bouton area in the GFP–TrkB.T1-positive profiles was not significantly different from controls (Table 1). Thus, our ultrastructural analysis demonstrates that interfering with TrkB signaling selectively in RGCs influences not only synapse number but also structural aspects that influence synaptic function at retinotectal synapses (i.e., docked synaptic vesicle pool).
Discussion
Studies in diverse animal species have established that afferent and efferent neurons as well as interneurons are subject to neurotrophic modulation during their morphological differentiation. The question of where in a specific circuit BDNF acts has been difficult to address because methods that use pharmacologic, transgenic, or knock-out manipulations have not been able to distinguish between presynaptic and postsynaptic events. Our studies directly explored presynaptic and postsynaptic mechanisms that influence the establishment of retinotectal connectivity by specifically interfering with TrkB signaling in presynaptic RGCs or postsynaptic tectal neurons. Real-time, in vivo imaging of individual neurons overexpressing GFP–TrkB.T1 allowed us to demonstrate that cell-autonomous TrkB signaling in presynaptic RGCs is responsible for multiple aspects of retinotectal development that include axon growth cone morphology, axon branch initiation and stabilization, axon arbor maturation and maintenance, synaptogenesis, and synaptic ultrastructure.
TrkB signaling is not required for RGC axon pathfinding
The ability of growth cones to respond to local gradients of neurotrophins in in vitro assays has suggested that BDNF may act as a guidance factor during axon pathfinding or targeting. RGC growth cones in particular are attracted to BDNF gradients when presented during early development in culture (Ming et al., 2001; Zweifel et al., 2005; Chen et al., 2006). Our experiments that interfered with TrkB signaling in RGCs demonstrate, however, that BDNF signaling does not influence RGC axon pathfinding or targeting to the optic tectum, because both individual neurons overexpressing GFP–TrkB.T1 and cohorts of GFP–TrkB.T1 transgenic RGCs projected to their correct targets and no misdirected axons were found. Thus, these results are consistent with previous observations in TrkB and BDNF knock-out mice showing normal eye-specific patterning in the retinorecipient target areas (Rohrer et al., 2001; Lyckman et al., 2005). It is important to note that TrkB signaling, although found to be important for the formation of synaptic connectivity, it is not involved in the guidance of major afferent projections in either the hippocampus or the cerebellum (Martinez et al., 1998; Rico et al., 2002), similar to our observations in developing visual system.
Cell-autonomous TrkB signaling is important for the growth and stabilization of RGC axons
Disruption of intracellular TrkB signaling in presynaptic RGCs caused multiple abnormalities in the growth and maturation of RGC axons that projected to the optic tectum. The TrkB.T1-overexpressing RGCs had much simpler axon arbors than controls, ranging from growth cone-like to relatively complex. The wide variability in arbor complexities may be caused by a difference in the overall ratio of full-length to truncated TrkB isoforms present in the transfected neurons. Indeed, developing RGCs express both full-length and truncated TrkB isoforms endogenously, and the expression level of the different isoforms per RGC may be variable, depending on their maturational state and phenotype (Cohen-Cory and Fraser, 1994; Martin et al., 1995; Cohen-Cory et al., 1996; Garner et al., 1996; Llamosas et al., 1997; Suzuki et al., 1998; Spalding et al., 2005). Consistent with this idea, the responsiveness of Xenopus oocytes to BDNF was found to depend on the ratio of full-length/truncated TrkB on the oocyte membrane (Eide et al., 1996).
Overexpression of TrkB.T1 in RGCs influenced not only the proportion of axons that remained in an immature state but also growth cone morphology. One possible explanation for this effect is that interfering with TrkB signaling delays the outgrowth of RGC axons and therefore the timing at which RGCs reach their final destination, because neurotrophins have been shown to influence axon growth in culture (Zweifel et al., 2005). An alternative, non-exclusive possibility is that interfering with TrkB signaling impairs the ability of RGC axons to branch once they reach the optic tectum by affecting growth cone and axon branch dynamics. Our observations that the first RGC axons in GFP–TrkB.T1 transgenic tadpoles arrive at the optic tectum at the same developmental stage of RGC axons in GFP transgenic tadpoles favor the second possibility. It is possible that altered TrkB signaling impacts RGC growth cone morphology and therefore axon branch initiation by directly influencing signaling mechanisms that regulate microtubule and the actin cytoskeletal dynamics (Gibney and Zheng, 2003; Gehler et al., 2004), because those signaling mechanisms are downstream of TrkB-mediated RhoA and cell division cycle 42 (Cdc42) signaling (Ruchhoeft et al., 1999; Chen et al., 2006). Indeed, the growth cone morphologies observed in TrkB.T1-expressing RGCs are reminiscent of those of Xenopus retinal neurons in vivo with altered Cdc42 signaling (Ruchhoeft et al., 1999).
Our single-cell manipulations and in vivo observations also demonstrated that interfering with TrkB signaling influenced not only axon maturation but also induced an approximately fivefold increase in the number of RGC axons that degenerated after projecting to the optic tectum. Our study did not specifically evaluate whether selective cell death accompanied RGC axon arbor degeneration in RGCs with altered TrkB signaling, because it proved technically difficult to experimentally correlate these two events. The timing of the effect of interfering with TrkB signaling, however, supports the interpretation that BDNF may serve as a target-derived cue that selectively maintains RGC axons that have successfully reached the optic tectum (Isenmann et al., 1999; Menna et al., 2003). It is well known that BDNF can promote neuronal survival of multiple neuronal populations through activation of TrkB receptors (for review, see Kaplan and Miller, 2000). However, it remains controversial whether, and how, BDNF modulates RGC survival in vivo and whether this would be a target-mediated or local effect. BDNF promotes RGC survival in vitro (Rodriguez-Tebar et al., 1989; Cohen-Cory and Fraser, 1994; Meyer-Franke et al., 1995) and in vivo (Ma et al., 1998), but mice lacking all isoforms of the TrkB receptor show no apoptotic RGC death (Rohrer et al., 2001). Interestingly, recent evidence in mammals indicates that BDNF, signaling through the full-length TrkB receptor, influences developmental death dynamics by controlling the timing of programmed cell death but not the final number of RGCs (Pollock et al., 2003). It remains possible, however, that, in the absence of TrkB signaling, BDNF could induce RGC axon degeneration through a p75 receptor-dependent mechanism, similar to the mechanisms described for sympathetic and hippocampal neurons (Kohn et al., 1999; Woo et al., 2005).
TrkB signaling in presynaptic RGCs modulates synaptogenesis and normal maturation of retinotectal synapses
BDNF can influence the development of multiple neuronal populations and neuronal circuits, and, within individual circuits, the effects can occur presynaptically, postsynaptically, or both. In the mammalian visual system, for example, BDNF can act postsynaptically to induce pyramidal cortical neuron dendritic arbor growth and spine maintenance (Horch et al., 1999; Horch and Katz, 2002; Chakravarthy et al., 2006) and can also enhance inhibitory cortical interneuron dendritic maturation (Huang et al., 1999; Jin et al., 2003; Kohara et al., 2003). A postsynaptic mechanism of BDNF action during the development of excitatory and inhibitory circuits has also been favored by studies using hippocampal cultures and knock-out mice as model systems (Elmariah et al., 2004; Elmariah et al., 2005; Luikart et al., 2005; Ohba et al., 2005; Chakravarthy et al., 2006). That BDNF acts presynaptically, however, has only been demonstrated for hippocampal neurons in studies that analyzed changes in synapse number and synaptic function in culture and in conditional TrkB knock-out mice (Li et al., 1998; Luikart et al., 2005; Magby et al., 2006). By directly altering TrkB signaling in single neurons in the living animal, we now demonstrate that presynaptic TrkB signaling is necessary for RGC axon arbor maturation and for the establishment of retinotectal synaptic connectivity because the overexpression of TrkB.T1 exclusively in presynaptic RGCs interfered with both processes.
In previous studies, we used targeted, acute alterations in BDNF levels within the tadpole optic tectum to demonstrate that BDNF influences the establishment of synaptic connectivity between RGC axons and tectal neurons, primarily by influencing RGCs (Alsina et al., 2001; Hu et al., 2005; Sanchez et al., 2006). The present analysis of axon branching and remodeling in RGCs with altered TrkB signaling validates our previous findings and reveals that GFP–TrkB.T1-expressing axons are significantly less stable and have a higher branch turnover rate than control RGC axons expressing GFP. The decreased stability of RGC axons with altered TrkB signaling is in agreement with our recent demonstration that acute neutralization of endogenous tectal BDNF with function-blocking antibodies interferes with RGC axon branch stability (Hu et al., 2005). Longer-term alterations in TrkB signaling, however, influenced RGCs axon branch behavior in a manner that differed from the effect of reducing tectal BDNF levels acutely at the target. TrkB.T1-expressing RGCs possessed higher branch addition and elimination rates, whereas a single treatment with anti-BDNF elicited rapid axon branch elimination without altering branch addition rates (Hu et al., 2005). Thus, the higher axon branch turnover rate in the TrkB.T1-expressing RGCs may reflect an increase in the exploratory behavior of the axon arbors and could represent a mechanism by which RGCs with altered TrkB signaling try to compensate for the impaired ability to stabilize new branches, which interferes with normal growth and maturation of the axonal arbor.
Our in vivo single-cell analysis of tectal neurons also supports the notion that BDNF influences tectal neuron morphology and synaptic connectivity only indirectly, through its effects on presynaptic RGCs. The number and density of presynaptic contact sites per dendritic arbor in the GFP–TrkB.T1-expressing tectal neurons was the same as that of tectal neurons in tadpoles with intact TrkB signaling. Our previous studies demonstrate, however, that presynaptic changes at retinotectal synapses elicited by acute alterations in BDNF levels are paralleled, with a time delay, by synaptic changes in postsynaptic tectal neurons (Sanchez et al., 2006). Thus, presynaptic changes that influence RGC axon arbor shape and synaptic function are then manifested as postsynaptic structural changes at retinotectal synapses.
In addition to influencing axonal architecture and synapse number, presynaptic changes in TrkB signaling impact both the structure and function of retinotectal synapses. Our electron microscopy analysis revealed that a major consequence of interfering with presynaptic TrkB signaling is that synapses established by TrkB.T1-expressing RGC axons are less mature than those made by RGCs with intact TrkB signaling, with TrkB.T1-positive synapses showing significant decreases in the total number of synaptic vesicles and of synaptic vesicles docked at active zones. Because neurotransmitter release probability is proportional to the number of docked synaptic vesicles (Pozzo-Miller et al., 1999; Schikorski and Stevens, 2001; Tyler and Pozzo-Miller, 2001), these structural changes at the level of individual synapses reflect a significant decrease in synaptic function, consistent with electrophysiological studies performed in tadpoles with altered BDNF tectal levels (Du and Poo, 2004).
How does TrkB signaling regulate synaptic structure and function? Most studies have analyzed how BDNF, through its receptor TrkB, modulates the ultrastructural composition of synapses collectively within a circuit, but the specific effects observed can vary slightly among vertebrate species and synapse type. In the hippocampus, deficits in TrkB signaling result in decreased expression of both synaptic vesicle and membrane proteins responsible for synaptic vesicle docking and fusion, a decrease in the number of docked vesicles, and downregulation of neurotransmitter release at synaptic sites (Martinez et al., 1998; Lin and Scheller, 2000; Otal et al., 2005). Cerebellar inhibitory synapses are also modulated by BDNF, in which alterations in TrkB signaling in knock-out mice reduce the numbers of GABAergic boutons and synaptic specializations (Rico et al., 2002), and targeted deletion of the BDNF gene decreases the number of vesicles that are docked (Carter et al., 2002). In the chick visual system, alterations in BDNF levels within the target optic tectum also affect synaptic vesicle pool (Wang et al., 2003). Thus, a commonality between circuits and species is that synaptic vesicle numbers and densities are increased and synapses strengthened by enhanced BDNF signaling, and, conversely, synaptic vesicle numbers are reduced and synapses are weakened if BDNF signaling is impaired (Vicario-Abejon et al., 2002; Shen et al., 2006). Our studies are in agreement with these previous findings and directly demonstrate that alterations in TrkB signaling in individual RGC within an otherwise intact developing visual system are sufficient to affect the synaptic connectivity of its axon terminal in a cell-autonomous way. The decreased number of mature synapses and the decrease in synaptic vesicle release on those mature synapses present in axons with altered TrkB signaling would therefore impact the firing strength of the axons, putting them at a disadvantage compared with other active inputs with intact TrkB signaling. A consequence of that deficit is that the axon terminals destabilize and degenerate, two morphological observations revealed by our imaging studies. Collectively, our in vivo studies demonstrate that cell-autonomous TrkB signaling in presynaptic RGCs is necessary for multiple aspects of RGC differentiation and indicate that the BDNF-mediated growth and enhanced stability of presynaptic RGC axons impacts both structural and functional aspects of retinotectal synaptic connectivity.
Footnotes
This work was supported by National Eye Institute Grant EY119012. We thank Dr. Eero Castren for the GFP–TrkB.T1 construct and Dr. David Bredt for the postsynaptic density-95–GFP construct. We also thank Drs. Ken Cho and Ira Blitz for advice with transgenic techniques and Margarita Meynard for help with multiple aspects of this project.
References
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci. 2001;4:1093–1101. doi: 10.1038/nn735. [DOI] [PubMed] [Google Scholar]
- Amaya E, Kroll KL. A method for generating transgenic frog embryos. Methods Mol Biol. 1999;97:393–414. doi: 10.1385/1-59259-270-8:393. [DOI] [PubMed] [Google Scholar]
- Berardi N, Pizzorusso T, Ratto GM, Maffei L. Molecular basis of plasticity in the visual cortex. Trends Neurosci. 2003;26:369–378. doi: 10.1016/S0166-2236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- Biffo S, Offenhauser N, Carter BD, Barde YA. Selective binding and internalisation by truncated receptors restrict the availability of BDNF during development. Development. 1995;121:2461–2470. doi: 10.1242/dev.121.8.2461. [DOI] [PubMed] [Google Scholar]
- Butowt R, von Bartheld CS. Anterograde axonal transport of BDNF and NT-3 by retinal ganglion cells: roles of neurotrophin receptors. Mol Cell Neurosci. 2005;29:11–25. doi: 10.1016/j.mcn.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AR, Chen C, Schwartz PM, Segal RA. Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J Neurosci. 2002;22:1316–1327. doi: 10.1523/JNEUROSCI.22-04-01316.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ, Mansvelder HD, Levelt CN. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci USA. 2006;103:1071–1076. doi: 10.1073/pnas.0506305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TJ, Gehler S, Shaw AE, Bamburg JR, Letourneau PC. Cdc42 participates in the regulation of ADF/cofilin and retinal growth cone filopodia by brain derived neurotrophic factor. J Neurobiol. 2006;66:103–114. doi: 10.1002/neu.20204. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE. BDNF in the development of the visual system of Xenopus . Neuron. 1994;12:747–761. doi: 10.1016/0896-6273(94)90328-x. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Lom B. Neurotrophic regulation of retinal ganglion cell synaptic connectivity: from axons and dendrites to synapses. Int J Dev Biol. 2004;48:947–956. doi: 10.1387/ijdb.041883sc. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Escandon E, Fraser SE. The cellular patterns of BDNF and trkB expression suggest multiple roles for BDNF during Xenopus visual system development. Dev Biol. 1996;179:102–115. doi: 10.1006/dbio.1996.0244. [DOI] [PubMed] [Google Scholar]
- Collin RW, Martens GJ. The coding sequence of amyloid-beta precursor protein APP contains a neural-specific promoter element. Brain Res. 2006;1087:41–51. doi: 10.1016/j.brainres.2006.02.101. [DOI] [PubMed] [Google Scholar]
- Du JL, Poo MM. Rapid BDNF-induced retrograde synaptic modification in a developing retinotectal system. Nature. 2004;429:878–883. doi: 10.1038/nature02618. [DOI] [PubMed] [Google Scholar]
- Eide FF, Vining ER, Eide BL, Zang K, Wang XY, Reichardt LF. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16:3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Crumling MA, Parsons TD, Balice-Gordon RJ. Postsynaptic TrkB-mediated signaling modulates excitatory and inhibitory neurotransmitter receptor clustering at hippocampal synapses. J Neurosci. 2004;24:2380–2393. doi: 10.1523/JNEUROSCI.4112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AS, Menegay HJ, Boeshore KL, Xie XY, Voci JM, Johnson JE, Large TH. Expression of TrkB receptor isoforms in the developing avian visual system. J Neurosci. 1996;16:1740–1752. doi: 10.1523/JNEUROSCI.16-05-01740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehler S, Shaw AE, Sarmiere PD, Bamburg JR, Letourneau PC. Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J Neurosci. 2004;24:10741–10749. doi: 10.1523/JNEUROSCI.2836-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney J, Zheng JQ. Cytoskeletal dynamics underlying collateral membrane protrusions induced by neurotrophins in cultured Xenopus embryonic neurons. J Neurobiol. 2003;54:393–405. doi: 10.1002/neu.10149. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- Haapasalo A, Koponen E, Hoppe E, Wong G, Castren E. Truncated trkB.T1 is dominant negative inhibitor of trkB.TK+-mediated cell survival. Biochem Biophys Res Commun. 2001;280:1352–1358. doi: 10.1006/bbrc.2001.4296. [DOI] [PubMed] [Google Scholar]
- Hallbook F, Backstrom A, Kullander K, Ebendal T, Carri NG. Expression of neurotrophins and trk receptors in the avian retina. J Comp Neurol. 1996;364:664–676. doi: 10.1002/(SICI)1096-9861(19960122)364:4<664::AID-CNE5>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Hanover JL, Huang ZJ, Tonegawa S, Stryker MP. Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J Neurosci. 1999;19:1–5. doi: 10.1523/JNEUROSCI.19-22-j0003.1999. RC40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog KH, von Bartheld CS. Contributions of the optic tectum and the retina as sources of brain-derived neurotrophic factor for retinal ganglion cells in the chick embryo. J Neurosci. 1998;18:2891–2906. doi: 10.1523/JNEUROSCI.18-08-02891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5:1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- Horch HW, Kruttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–364. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Hu B, Nikolakopoulou AM, Cohen-Cory S. BDNF stabilizes synapses and maintains the structural complexity of optic axons in vivo. Development. 2005;132:4285–4298. doi: 10.1242/dev.02017. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Isenmann S, Cellerino A, Gravel C, Bahr M. Excess target-derived brain-derived neurotrophic factor preserves the transient uncrossed retinal projection to the superior colliculus. Mol Cell Neurosci. 1999;14:52–65. doi: 10.1006/mcne.1999.0763. [DOI] [PubMed] [Google Scholar]
- Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activity-dependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures. J Neurosci. 2003;23:5662–5673. doi: 10.1523/JNEUROSCI.23-13-05662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Adachi N, Nishida M, Itami C, Nakamura S, Tsumoto T. Inhibitory but not excitatory cortical neurons require presynaptic brain-derived neurotrophic factor for dendritic development, as revealed by chimera cell culture. J Neurosci. 2003;23:6123–6131. doi: 10.1523/JNEUROSCI.23-14-06123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD. Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci. 1999;19:5393–5408. doi: 10.1523/JNEUROSCI.19-13-05393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Shatz CJ. Rapid regulation of brain-derived neurotrophic factor mRNA within eye-specific circuits during ocular dominance column formation. J Neurosci. 2000;20:1470–1483. doi: 10.1523/JNEUROSCI.20-04-01470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RC, Scheller RH. Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol. 2000;16:19–49. doi: 10.1146/annurev.cellbio.16.1.19. [DOI] [PubMed] [Google Scholar]
- Llamosas MM, Cernuda-Cernuda R, Huerta JJ, Vega JA, Garcia-Fernandez JM. Neurotrophin receptors expression in the developing mouse retina: an immunohistochemical study. Anat Embryol (Berl) 1997;195:337–344. doi: 10.1007/s004290050053. [DOI] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo . J Neurosci. 1999;19:9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyckman AW, Fan G, Rios M, Jaenisch R, Sur M. Normal eye-specific patterning of retinal inputs to murine subcortical visual nuclei in the absence of brain-derived neurotrophic factor. Vis Neurosci. 2005;22:27–36. doi: 10.1017/S095252380522103X. [DOI] [PubMed] [Google Scholar]
- Ma YT, Hsieh T, Forbes ME, Johnson JE, Frost DO. BDNF injected into the superior colliculus reduces developmental retinal ganglion cell death. J Neurosci. 1998;18:2097–2107. doi: 10.1523/JNEUROSCI.18-06-02097.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magby JP, Bi C, Chen ZY, Lee FS, Plummer MR. Single-cell characterization of retrograde signaling by brain-derived neurotrophic factor. J Neurosci. 2006;26:13531–13536. doi: 10.1523/JNEUROSCI.4576-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandolesi G, Menna E, Harauzov A, von Bartheld CS, Caleo M, Maffei L. A role for retinal brain-derived neurotrophic factor in ocular dominance plasticity. Curr Biol. 2005;15:2119–2124. doi: 10.1016/j.cub.2005.10.045. [DOI] [PubMed] [Google Scholar]
- Martin SC, Marazzi G, Sandell JH, Heinrich G. Five Trk receptors in the zebrafish. Dev Biol. 1995;169:745–758. doi: 10.1006/dbio.1995.1184. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna E, Cenni MC, Naska S, Maffei L. The anterogradely transported BDNF promotes retinal axon remodeling during eye specific segregation within the LGN. Mol Cell Neurosci. 2003;24:972–983. doi: 10.1016/s1044-7431(03)00258-6. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Ming G, Henley J, Tessier-Lavigne M, Song H, Poo M. Electrical activity modulates growth cone guidance by diffusible factors. Neuron. 2001;29:441–452. doi: 10.1016/s0896-6273(01)00217-3. [DOI] [PubMed] [Google Scholar]
- Mohun TJ, Garrett N, Gurdon JB. Upstream sequences required for tissue-specific activation of the cardiac actin gene in Xenopus laevis embryos. EMBO J. 1986;5:3185–3193. doi: 10.1002/j.1460-2075.1986.tb04628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis. The Netherlands: Elsevier North Holland; 1956. [Google Scholar]
- Ninkina N, Adu J, Fischer A, Pinon LG, Buchman VL, Davies AM. Expression and function of TrkB variants in developing sensory neurons. EMBO J. 1996;15:6385–6393. [PMC free article] [PubMed] [Google Scholar]
- Offenhauser N, Muzio V, Biffo S. BDNF binding to truncated trkB.T1 does not affect gene expression. NeuroReport. 2002;13:1189–1193. doi: 10.1097/00001756-200207020-00024. [DOI] [PubMed] [Google Scholar]
- Ohba S, Ikeda T, Ikegaya Y, Nishiyama N, Matsuki N, Yamada MK. BDNF locally potentiates GABAergic presynaptic machineries: target-selective circuit inhibition. Cereb Cortex. 2005;15:291–298. doi: 10.1093/cercor/bhh130. [DOI] [PubMed] [Google Scholar]
- Otal R, Martinez A, Soriano E. Lack of TrkB and TrkC signaling alters the synaptogenesis and maturation of mossy fiber terminals in the hippocampus. Cell Tissue Res. 2005;319:349–358. doi: 10.1007/s00441-004-1020-5. [DOI] [PubMed] [Google Scholar]
- Pollock GS, Robichon R, Boyd KA, Kerkel KA, Kramer M, Lyles J, Ambalavanar R, Khan A, Kaplan DR, Williams RW, Frost DO. TrkB receptor signaling regulates developmental death dynamics, but not final number, of retinal ganglion cells. J Neurosci. 2003;23:10137–10145. doi: 10.1523/JNEUROSCI.23-31-10137.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Xu B, Reichardt LF. TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat Neurosci. 2002;5:225–233. doi: 10.1038/nn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Tebar A, Jeffrey PL, Thoenen H, Barde YA. The survival of chick retinal ganglion cells in response to brain-derived neurotrophic factor depends on their embryonic age. Dev Biol. 1989;136:296–303. doi: 10.1016/0012-1606(89)90256-x. [DOI] [PubMed] [Google Scholar]
- Rohrer B, LaVail MM, Jones KR, Reichardt LF. Neurotrophin receptor TrkB activation is not required for the postnatal survival of retinal ganglion cells in vivo. Exp Neurol. 2001;172:81–91. doi: 10.1006/exnr.2001.7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchhoeft ML, Ohnuma S, McNeill L, Holt CE, Harris WA. The neuronal architecture of Xenopus retinal ganglion cells is sculpted by rho-family GTPases in vivo . J Neurosci. 1999;19:8454–8463. doi: 10.1523/JNEUROSCI.19-19-08454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AL, Matthews BJ, Meynard MM, Hu B, Javed S, Cohen-Cory S. BDNF increases synapse density in dendrites of developing tectal neurons in vivo. Development. 2006;133:2477–2486. doi: 10.1242/dev.02409. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Shen W, Wu B, Zhang Z, Dou Y, Rao ZR, Chen YR, Duan S. Activity-induced rapid synaptic maturation mediated by presynaptic cdc42 signaling. Neuron. 2006;50:401–414. doi: 10.1016/j.neuron.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Silver MA, Stryker MP. TrkB-like immunoreactivity is present on geniculocortical afferents in layer IV of kitten primary visual cortex. J Comp Neurol. 2001;436:391–398. doi: 10.1002/cne.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Tan MM, Hendry IA, Harvey AR. Anterograde transport and trophic actions of BDNF and NT-4/5 in the developing rat visual system. Mol Cell Neurosci. 2002;19:485–500. doi: 10.1006/mcne.2001.1097. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Cui Q, Harvey AR. Retinal ganglion cell neurotrophin receptor levels and trophic requirements following target ablation in the neonatal rat. Neuroscience. 2005;131:387–395. doi: 10.1016/j.neuroscience.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Nomura S, Morii E, Fukuda Y, Kosaka J. Localization of mRNAs for trkB isoforms and p75 in rat retinal ganglion cells. J Neurosci Res. 1998;54:27–37. doi: 10.1002/(SICI)1097-4547(19981001)54:1<27::AID-JNR4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Tokuoka H, Yoshida T, Matsuda N, Mishina M. Regulation by glycogen synthase kinase-3beta of the arborization field and maturation of retinotectal projection in zebrafish. J Neurosci. 2002;22:10324–10332. doi: 10.1523/JNEUROSCI.22-23-10324.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejon C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nat Rev Neurosci. 2002;3:965–974. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- Vizuete ML, Venero JL, Vargas C, Revuelta M, Machado A, Cano J. Potential role of endogenous brain-derived neurotrophic factor in long-term neuronal reorganization of the superior colliculus after bilateral visual deprivation. Neurobiol Dis. 2001;8:866–880. doi: 10.1006/nbdi.2001.0424. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS. Neurotrophins in the developing and regenerating visual system. Histol Histopathol. 1998;13:437–459. doi: 10.14670/HH-13.437. [DOI] [PubMed] [Google Scholar]
- Wang X, Butowt R, von Bartheld CS. Presynaptic neurotrophin-3 increases the number of tectal synapses, vesicle density, and number of docked vesicles in chick embryos. J Comp Neurol. 2003;458:62–77. doi: 10.1002/cne.10558. [DOI] [PubMed] [Google Scholar]
- Witte S, Stier H, Cline HT. In vivo observations of timecourse and distribution of morphological dynamics in Xenopus retinotectal axon arbors. J Neurobiol. 1996;31:219–234. doi: 10.1002/(SICI)1097-4695(199610)31:2<219::AID-NEU7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. 2005;8:1069–1077. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]