

Abstract
KCNQ2/3 currents are the molecular basis of the neuronal M currents that play a critical role in neuron excitability. Many neurotransmitters modulate M/KCNQ currents through their G-protein-coupled receptors. Membrane PtdIns(4,5)P2 hydrolysis and channel phosphorylation are two mechanisms that have been proposed for modulation of KCNQ2/3 currents. In this study, we studied regulation of KCNQ2/3 currents by the epidermal growth factor (EGF) receptor, a member of another family of membrane receptors, receptor tyrosine kinases. We demonstrate here that EGF induces biphasic inhibition of KCNQ2/3 currents in human embryonic kidney 293 cells and in rat superior cervical ganglia neurons, an initial fast inhibition and a later slow inhibition. Additional studies indicate that the early and late inhibitions resulted from PtdIns(4,5)P2 hydrolysis and tyrosine phosphorylation, respectively. We further demonstrate that these two processes are mutually dependent. This study indicates that EGF is a potent modulator of M/KCNQ currents and provides a new dimension to the understanding of the modulation of these channels.
Keywords: M current, KCNQ current, EGF receptor, modulation, PIP2, phosphorylation
Introduction
M currents play a critical role in the determination of neuronal excitability (Brown and Yu, 2000; Jentsch, 2000). It is now well accepted that KCNQ2/Q3 potassium channels are the molecular basis of M currents (Wang et al., 1998; Shapiro et al., 2000). Mutation of KCNQ2/3 genes leads to the epileptic condition (Biervert et al., 1998). M currents are classically known as being inhibited by activation of the muscarinic acetylcholine receptor (Brown and Adams, 1980). In addition, many neurotransmitters or hormones, such as ATP (Filippov et al., 1998; Ford et al., 2003), bradykinin (Jones et al., 1995; Cruzblanca et al., 1998), angiotensin II (Shapiro et al., 1994), substance P (Adams et al., 1983), and Luteinizing hormone releasing hormone (Adams and Brown, 1980) have also been shown to be able to inhibit M currents.
Since the discovery of the M current 25 years ago, the mechanism of M current inhibition has been debated (Ikeda and Kammermeier, 2002). It is now clear that a variety of neurotransmitters inhibit M currents through Gq/11-coupled receptors (Delmas and Brown, 2005). However, the identities of the molecules downstream of Gq/11 activation are still debated. Ca2+ (Selyanko and Brown, 1996; Gamper and Shapiro, 2003) mediated by calmodulin (Gamper et al., 2005) is responsible for bradykinin-induced inhibition of M currents (Cruzblanca et al., 1998). However, other neurotransmitters (like M receptor agonists) either do not raise intracellular Ca2+ while they inhibit M currents, or only modestly raise intracellular Ca2+ in a manner that is not related to M-current inhibition (Del Río et al., 1999). Protein kinase C (PKC) is another signaling molecule that may participate in the modulation of M currents (Hoshi et al., 2003; Higashida et al., 2005).
As with several other ion channels and membrane transport proteins (Hilgemann et al., 2001; Delmas and Brown, 2005; Suh and Hille, 2005), KCNQ channels require PtdIns(4,5)P2 in the cell membrane to be able to open. It was therefore proposed that channel inhibition by Gq-protein-coupled receptors might result from the depletion of membrane PtdIns(4,5)P2 as a result of its hydrolysis (Suh and Hille, 2002; Ford et al., 2003, 2004; Zhang et al., 2003; Winks et al., 2005).
Evidence has been accumulating which suggests that activation of receptor tyrosine kinases (RTKs) modulates the function of ion channels including K+ channels (Holmes et al., 1996; Bowlby et al., 1997; Fadool et al., 1997; Wischmeyer et al., 1998; Colley et al., 2004; Nesti et al., 2004). As far as the KCNQ channels are concerned, nonreceptor tyrosine kinase Src inhibits M/KCNQ currents through direct channel phosphorylation (Gamper et al., 2003; Li et al., 2004). In light of the above findings and the fact that activation of RTKs would not only activate the tyrosine phosphorylation process but also initiate membrane PtdIns(4,5)P2 hydrolysis through phospholipase C (PLC)γ, it is tempting for us to investigate if RTK activation modulates M/KCNQ channel functions.
We found in the present study that EGF induced a biphasic inhibition of KCNQ2/3 currents expressed in human embryonic kidney 293 (HEK293) cells with an initial fast inhibition and a later slow inhibition. Pharmacological and mutagenesis studies indicate that the early and late inhibitions resulted from PtdIns(4,5)P2 hydrolysis and tyrosine phosphorylation, respectively.
Materials and Methods
Molecular biology.
All cDNA constructs were subcloned into the pCDNA3.0 plasmid vector. Point mutants were produced by Pfu mutagenesis with a Quickchange kit (Stratagene, La Jolla, CA). Sequences were confirmed by DNA sequencing. Recombinant KCNQ channels, M1 receptors, and PLCδ1 pleckstrin homology-green fluorescent protein (GFP) were transfected into HEK293 cells. The GenBank accession numbers for the KCNQ channels are as follows: AF110020 for hKCNQ2 and AF091247 for rKCNQ3.
Cell culture and transfection.
HEK293 cells were seeded in 24-well plates on 12 mm glass coverslips and cultured in 0.3 ml of DMEM supplemented with 10% (V/V) fetal bovine serum (FBS), 100 μg/ml streptomycin, and 100 U/ml penicillin at 5% CO2, 37°C. After reaching 60–70% confluency, the cells were transiently transfected with DNA constructs for 4–6 h with Lipofectamine 2000 (Invitrogen, Eugene, OR) according to the manufacturer's instructions. The total amount of cDNA used for transfection was the same for all control and experimental groups. To visualize responses to EGF, the transfected cells were serum-starved for 12–24 h in the DMEM that lacked FBS.
Primary cultures of neurons were prepared from superior cervical ganglia (SCGs) from 2- to 4-week-old Sprague Dawley rats using a previously described procedure (Tatulian et al., 2001). Briefly, rats were killed by cervical vertebra luxation and then ganglia were rapidly removed from the carotid bifurcation and placed in modified d-Hanks' solution. After being cut into several pieces with iridectomy scissors, ganglia were digested at 37°C with collagenase (1 mg/ml; Worthington, Freehold, NJ) for 30 min, followed by another 30 min digestion with trypsin (2.5 mg/ml; Worthington). They were subsequently suspended at least three times in DMEM plus 10% bovine calf serum (HyClone, Novato, CA) to stop digestion. Ganglia were then dissociated into a suspension of individual cells and planted on poly-d-lysine-coated glass coverslips in 24-well tissue culture plates (Costar, Cambridge, MA). Cells were incubated at 37°C with 5% CO2 and 95% air atmosphere. The medium was changed to neurobasal A medium plus 2% B27 supplement (no NGF; Invitrogen) after 12 h and neurons were used for recording within 48 h.
Confocal microscopy and image analysis.
HEK293 cells were grown on glass coverslips and transfected with PLCδ1-PH-GFP and hM1 receptor expression plasmids. For fluorescence detection, after 1 d after transfection the cells were washed twice with a modified Krebs–Rings buffer [containing the following (in mm): 120 NaCl, 4.7 KCl, 0.7 MgSO4, 1.2 CaCl2, 10 glucose, with 10 HEPES, pH 7.4] and the coverslips were placed into a flow-through chamber and mounted on an inverted microscope. For confocal imaging, a Leica (Nussloch, Germany) DM-IRBE inverted microscope with a 20× objective (numerical aperture, 0.7) and a TCS-SP2 scanhead was used. Excitation of PLCδ1PH-GFP and Fluo 3-AM was accomplished with the 488 nm argon ion laserline, and emission was collected at 500–565 nm. For translocation studies, a series of confocal images was taken at 3–10 s intervals and was stored on a disc. Laser intensity and pinhole settings were kept constant between experiments. Pinhole size was set to scan sections with a thickness of 0.28 μm. Control images were obtained for 1 min before drug application. ACh and EGF were applied for 2 min. Determination of the ratio of membrane to cytosolic fluorescence was accomplished by assigning regions of interest for membrane and cytosol. TCS-SP2 confocal software (Leica) was used to analyze data off line.
Electrophysiology.
Whole-cell and perforated-patch recordings were used in this study. Recordings were made at room temperature (20–25°C). Pipettes were pulled from borosilicate glass capillaries and had a resistance of 4–6 MΩ when filled with internal solution. Currents were recorded using Axon patch 200B amplifier and PClamp 9.0 software (Molecular Devices, Union City, CA) and were filtered at 2 KHz. The voltage protocol used to study M currents of SCG neurons was as follows: the cell was held at −20 mV and a 1 s hyperpolarizing step to −60 mV was applied every 4 s. The amplitude of M currents was defined as the outward current sensitive to 30 μm linopirdine and was measured from deactivation current records at −60 mV as the difference between the average of a 10 ms segment, taken 10–20 ms into the hyperpolarizing step, and the average during the last 10 ms of that step (Suh and Hille, 2002). Solutions were applied through the VM8 fast superfusion system (ALA Instruments, Westbury, NY) or by gravity. The external solution used to record KCNQ2/3 currents in HEK293 cells contained the following (in mm): 150 NaCl, 2 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose, pH 7.2. The internal solution for HEK293 cells contained the following (in mm): 140 KCl, 5 EGTA, 5 NaCl, 10 HEPES, 3 MgATP, and 0.2 Na2GTP, pH 7.2. For the perforated-patch recording, a pipette was first front-filled with the standard internal solution, then backfilled with the same internal solution containing amphotericin B (120 ng/ml). The external solution used to record SCG neuron M current contained the following (in mm): 120 NaCl, 3 KCl, 5 HEPES, 23 NaHCO3, 11 glucose, 1.2 MgCl2, 2.5 CaCl2, and 0.0005 TTX, adjusted to pH 7.4 with NaOH. The internal solution for M-current recording consisted of the following (in mm): 90 K+ acetate, 40 KCl, 20 HEPES, and 3 MgCl2, adjusted to pH 7.3–7.4 with KOH (Tatulian et al., 2001).
Western blot, cell immunofluorescence, and confocal imaging.
Isolated SCGs were cut into small pieces and sonicated in 200 μl of radioimmunoprecipitation assay lysis buffer containing protease inhibitors (pepstatin A, 0.01 μg/μl; leupeptin, 0.025 μg/μl; aprotinin, 0.01 μg/μl; PMSF, 0.03 μg/μl) for 20 min at 0°C. SCG homogenates were centrifuged at 18,000 rpm at 4°C for 20 min and the pellets were discarded. The concentration of protein in the supernatant was assessed by BCA assay (Pierce, Rockford, IL), a loading buffer (10% SDS, 0.5 m Tris-HCl, 50% glycerol, 0.24% bromophenyl blue, 20% ME) was added, and it was incubated at 70°C for 10 min. Proteins were fractionated on 5% SDS-PAGE, transferred to polyvinyl difluoride membranes, and blotted with a rabbit anti-EGF receptor (EGFR) antibody (Ab) (1:200; Santa Cruz Biotechnology, Santa Cruz, CA). Bands were visualized with chemiluminescence from goat anti-rabbit horseradish peroxidase-coupled secondary antibodies (Zhongshan, Beijing, China).
SCG neurons were seeded on poly-lysine coated coverslips and were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked by 3% BSA for 30 min before incubation for labeling. The cells were washed three times with 0.01 m PBS, pH 7.4, before each of above treatments. Subsequently, neurons were incubated with primary rabbit anti-EGFR Ab (1:100; Santa Cruz Biotechnology) in blocking solution overnight at 4°C, followed by three washes with PBS and incubation with goat anti-rabbit IgG-FITC secondary Ab (1:100; Millipore, Temecula, CA) for 30 min at 37°C. Cells were examined on inverted laser-scanning microscope (SP2; Leica). FITC was excited at 488 nm and emitted fluorescence signals at 500 nm.
Chemicals.
The drugs and chemicals used in this experiment were obtained as follows: acetylcholine (ACh), AG1478, daidazin, erbatatin, EGF, genistein, linopirdine, LiCl, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), quinacrine, 1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U73122), 1-6((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)2,5,-pyrrolidinedione (U73343), vanadate, wortmannin, EGTA, K2ATP, NaGTP, HEPES, DMEM, and DMSO were purchased from Sigma (St. Louis, MO). Perorthovanadate (Na3VO4) in the bath solution was prepared by a 20 min incubation with the oxidant H2O2 (0.003%) and subsequent quenching of residual H2O2 with 100 U/ml catalase for 5 min (Holmes et al., 1996).
Statistics
The programs Origin (version 7.0; OriginLab, Northampton, MA) and Excel (Microsoft, Redmond, WA) were used for data analysis. Data were presented as the mean ± SEM. Statistical significance was computed using Student's t test or ANOVA. The differences were considered significant if p < 0.05.
Results
EGF induces distinct fast and slow inhibition of KCNQ2/3 currents in HEK293 cells and M currents in SCG neurons
KCNQ2/3 currents were recorded 24 h after channel DNA transfection of HEK293 cells by using the whole-cell patch-clamp technique. The KCNQ2/3 currents were activated at a depolarizing potential of 0 mV, and the holding currents at −80 mV were recorded as the control current level (Fig. 1A). EGF (100 ng/ml) was applied to the solution bathing the HEK293 cells for a period of 20–30 min. HEK293 cells express endogenous EGF receptors (Kramer et al., 2002, Shah et al., 2006). A fast inhibition of KCNQ2/3 current was seen immediately after EGF application. This inhibition was transient and was able to recover, even in the continuous presence of EGF (Fig. 1A). A second inhibition developed slowly after the fast transient inhibition (Fig. 1A). The slow inhibition started with a delay and took ∼20 min to fully develop. The degree of slow inhibition was much more substantial than the fast one. For comparison, the average fast inhibition was 28 ± 9.6%, and the slow inhibition was 82 ± 13% of the original current amplitudes. Both fast and slow inhibitions were reduced by treatments with either a selective inhibitor of EGF receptor, AG1478 (200 nm) (Levitzki and Gazit, 1995) (Fig. 1B), or a nonspecific tyrosine kinase inhibitor, genistein (100 μm) (Fig. 1C). Figure 1, E and F, summarizes the data for the fast and the slow inhibition of KCNQ2/3 currents, respectively, under different treatment conditions. AG1478 and genistein almost abolished EGF-induced fast inhibition of KCNQ2/3 currents (Fig. 1E); AG1478 reduced the slow inhibition by half, and genistein reduced the slow inhibition to a minimum (Fig. 1F). An inactivate analog of genistein, daidzein (100 μm) was tested and it did not affect the effects of EGF (data not shown). These data indicate that both the fast and the slow inhibition of KCNQ2/3 currents induced by EGF are mediated through tyrosine kinase component of EGF signaling. We also tested the effect of EGF on homomeric KCNQ2 currents (Fig. 1D). Clearly, EGF induced a similar inhibition of KCNQ2 as that of KCNQ2/3 currents. However, an interesting difference was noticed. Whereas the degree of the fast inhibition was similar for both KCNQ2/3 and homomeric KCNQ2 (Fig. 1E), the slow inhibition of KCNQ2 was significantly less than that of KCNQ2/3 (Fig. 1F). In other words, homomeric KCNQ2 was less sensitive than KCNQ2/3 for EGF-induced slow inhibition.
Figure 1.
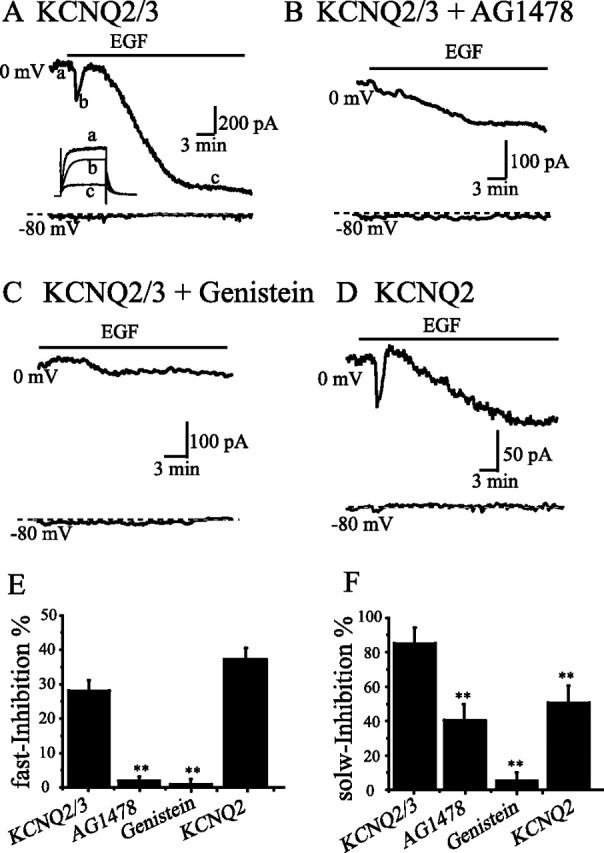
EGF induced a fast and a slow inhibition of KCNQ2/3 and homomeric KCNQ2 expressed in HEK293 cells. Whole-cell KCNQ2/3 currents were activated at 0 mV for 1 s from a holding potential of −80 mV. A–C, The time course of KCNQ2/3 current traces at 0 mV (the top trace) and −80 mV (the bottom trace) was shown in the cell treated with EGF alone (A), with EGF+AG1478 (B), or with EGF+genistein (C). D, EGF also induced a fast and a slow inhibition on homomeric KCNQ2 currents and a summary of these inhibitions is shown in E and F. E, F, The percentage of the fast (E) and the slow (F) inhibition of KCNQ2/3 currents by EGF, and the effects of AG1478 and genistein on these inhibitions are summarized. **p < 0.01 compared with the KCNQ2/3; n = 7–9 for each of the experimental groups. Error bars indicate SEM.
We investigated if EGF could also inhibit native M currents in SCG neurons. M currents from 2- to 4-week-old rats were recorded using perforated patch recordings. M currents were measured from the deactivation current at −60 mV (for details, see Materials and Methods) (Suh and Hille, 2002). EGF (100 ng/ml for 10 min) always induced slow inhibition of M currents in all SCG cells tested. In some cells (7 of 12), EGF (100 ng/ml) induced a small transient fast inhibition of M currents (Fig. 2A, arrow), similar to the fast inhibition of KCNQ2/3 currents seen in HEK 293 cells; an average inhibition of 14.5 ± 1.2% was recorded in these seven cells. Concentration-dependent effects of EGF were also tested (Fig. 2B,C) and a concentration–response curve was established for the effect of EGF (Fig. 2D). EGF began to inhibit M currents at concentration as low as 30 ng/ml and reached a maximal inhibition concentration at 1000 ng/ml; the concentration for the half-maximum inhibition (IC50) is 115 ng/ml (Fig. 2D). EGF (1000 ng/ml) induced a maximal inhibition of 64.5 ± 1.3% (n = 7) of M currents. Genistein (100 μm), PP2 (1 μm), and AG1478 (1 μm) significantly attenuated EGF-induced inhibition of M currents (Fig. 2E). Genistein and PP2 reduced the inhibition to 10.7 ± 2.6% (n = 7) and 17.4 ± 3.2% (n = 7), respectively, from 31.2 ± 2.2% (n = 9), whereas AG1478 totally abolished the inhibition (n = 7). To provide additional evidence that the effects of EGF observed above were through activation of EGFR, Western blot and immunocytochemical studies were performed to confirm the presence of EGFR in SCG neurons. A band with the expected size of EGFR was recognized by anti-EGFR antibody (Fig. 2F) from SCG preparations. In isolated SCG neurons, immunocytochemical studies confirmed the presence of EGFR with clear membrane staining (Fig. 2G).
Figure 2.

EGF inhibited M currents in SCG neurons. Whole-cell currents of rat SCG neurons were recorded using perforated-patch recording. M currents were measured from deactivation currents recorded at −60 mV. A, EGF (100 ng/ml) induced a fast (indicated by the arrow) and a slow inhibition of M currents form SCG neurons. B, EGF of various concentrations (100, 300, 1000 ng/ml) was applied for a period of time (10 min for each concentration) as indicated. C, The current traces elicited by the protocol shown above. The labels a–e indicate the current traces taken at the time shown in B. D, Concentration–response curve for the inhibition of M currents by EGF. The curve was fitted with Hill function. The IC50 for inhibition of the M currents by EGF is 114 ng/ml, and the Hill coefficient is 0.55 (n = 7 for each point). E, Summary data show EGF-induced M current modulation in the absence and presence of genistein (GST), PP2, and AG1478. **p < 0.01 compared with the EGF alone. F, Western blot result shows EGF receptor is present in SCG neurons. G, Immunofluorescence of EGF receptor in SCG neurons. The left panel shows confocal images of SCG neurons probed with anti-EGFR antibody and FITC-conjugated anti-rabbit antibody. The right panel shows phase-contrast microscopy of the same cells. Error bars indicate SEM.
We performed a more detailed study on EGF-induced fast and slow inhibition of KCNQ2/3 currents. KCNQ2/3 currents have distinct characteristics of slow-activation and slow-deactivation. We compared the activation kinetics from the currents activated at 0 mV from −80 mV and the deactivation kinetics from the currents deactivated from 0 to −50 mV (Fig. 3A,B). A single exponential function fit both the activation and the deactivation processes, and the time constants from these fittings are shown in Figure 3C. For the EGF-induced fast inhibition, the activation and the deactivation kinetics were not altered. In contrast, for the EGF-induced slow inhibition, the activation time constant was increased from 227 ± 26 to 641 ± 46 ms, and the deactivation constant was increased from 133 ± 24 to 335 ± 33 ms. We also measured the current–voltage relationship for KCNQ2/3 activation (Fig. 3D). This was done by measuring the tail currents at −60 mV followed by multiple depolarizing steps. The current-voltage relationship was fitted with the Boltzmann function. Again, for the EGF-induced fast inhibition, the activation current–voltage relationship was not altered. For the EGF-induced slow inhibition, the half-maximal activation voltage was right shifted from −17.1 ± 0.7 mV to −9.7 ± 0.3 mV (Fig. 3D). These data suggest that EGF induces distinct fast and slow inhibition of KCNQ2/3 currents. We also studied voltage-dependence and kinetics of M currents from SCG neurons. These analyses were not performed on EGF-induced fast inhibition of M currents because the inhibition was small and not present in all SCG neurons, and these properties were not altered by EGF in the KCNQ2/3 current of HEK cells. For EGF (300 ng/ml)-induced slow inhibition, the half-maximal activation voltage was right shifted from −37.1 ± 1.6 mV to −30.9 ± 1.9 mV (Fig. 3E). However, we found that measurements of activation and deactivation kinetics were quite variable in SCG neurons and, thus, comparable kinetic changes in SCG neurons could not be detected because of experimental limitations.
Figure 3.

EGF-induced fast and slow inhibitions have distinct effects on the kinetics of activation and deactivation of KCNQ2/3 currents. A, B, The kinetics of activation and deactivation of EGF-induced fast (A) and slow (B) inhibition of KCNQ2/3 currents were analyzed from the activating currents at −80 to 0 mV and from the deactivating currents at 0 to −50 mV. The activation and the deactivation of KCNQ2/3 currents can each be fitted with a single exponential function and the time constants from these fittings are shown in C. **p < 0.01 compared with the control; n = 8 for each of the experimental groups. D, The current-voltage relationships of KCNQ2/3 current activation. The tail currents at −60 mV after multiple voltage steps were normalized based on the maximal tail current observed. The data were fitted with the Boltzmann function. The solid and dotted lines show the fitted curves of KCNQ2/3 current activation from the control and from the EGF-induced slow inhibition, respectively. The half-activation voltage of the curves for the control and the EGF-treated cells were −17.1 ± 0.7 and −9.7 ± 0.3 mV, respectively (n = 8). E, The current–voltage relationships of M currents from SCG neurons were also established. The half-activation voltage of the curves for the control and the EGF-treated cells were −37.1 ± 1.6 and −30.9 ± 1.9 mV, respectively (n = 8). Error bars indicate SEM.
EGF-induced fast inhibition of KCNQ2/3 currents is the result of membrane PtdIns(4,5)P2 hydrolysis
It has been shown previously that ACh (through activation of M1 receptor) inhibits KCNQ2/3 currents through hydrolysis of membrane PtdIns(4,5)P2 (Zhang et al. 2003). Activation of the EGF receptor can also hydrolyze PtdIns(4,5)P2 through PLCγ (Schmidt et al., 2000). Thus, we hypothesized that EGF-induced fast inhibition of KCNQ2/3 currents might be the result of PtdIns(4,5)P2 hydrolysis. We tested this hypothesis by comparing ACh and EGF-induced inhibition of KCNQ2/3 currents and observing the effects of pharmacological modulation of these inhibitions. For this study, the M1 receptor was coexpressed with KCNQ2/3 channels in HEK293 cells. Figure 4A shows whole-cell currents of KCNQ2/3 recorded at 0 and −80 mV. A brief application of EGF (100 ng/ml) for 2 min induced a fast and reversible inhibition of the currents. Application of ACh (10 μm) induced a fast and large inhibition of KCNQ2/3 currents; the ACh-induced inhibition was partially reversible after washout. The second round of EGF and ACh application induced similar inhibition (Fig. 4A). On average, EGF inhibited 28 ± 6.1% whereas ACh inhibited 80 ± 12.2% of the KCNQ2/3 currents. We next used U73122 (2 μm) to block PLC activity and observed its effect on ACh- and EGF-induced inhibition. As is shown in Figure 4, B and C, U73122 attenuated both ACh- and EGF-induced inhibition of KCNQ2/3 currents. Figure 4D summarizes the data for EGF- and ACh-induced inhibition in the presence and absence of U73122. U73343 (2 μm), an analog of U73122, did not affect the effects of EGF and ACh (data not shown). These data clearly indicate that EGF, like ACh, induced a fast inhibition of KCNQ2/3 currents through a PLC-dependent mechanism. We noticed an interesting difference between the current treated with ACh (Fig. 4B) and the current treated with EGF (Fig. 4C), namely that the former was much more stable than the latter. All cells treated with EGF (like the one shown in Fig. 4C) demonstrated a delayed run-down of KCNQ2/3 currents after a brief application of EGF. We believe that this delayed current run-down is mostly the result of EGF application, not the result of intrinsic current run-down, because the same slow inhibition was induced by a longer application of EGF (Fig. 1), and only a small (10 ± 5%; n = 12) current run-down was recorded in untreated cell during a same period of time. Thus, even a short application of EGF initiates the delayed slow inhibition of KCNQ2/3 currents.
Figure 4.

A comparative study of EGF- and ACh-induced fast inhibition of KCNQ2/3 currents and translocation of membrane PtdIns(4,5)P2. A, KCNQ2/3 channels and M1 receptors were coexpressed in HEK 293 cells. KCNQ2/3 currents were recorded at 0 mV, and the effects of EGF and ACh were studied in the same cell. B–D, The effects of U73122 on Ach-induced (B) and EGF-induced (C) inhibition of KCNQ2/3 currents are shown and are summarized in D. **p < 0.01 compared with ACh or EGF alone; n = 7. E, HEK293 cells were transfected with PLCδ1PH-GFP and M1 receptors, and fluorescence was monitored using a confocal microscope. Application of ACh (top) and EGF (bottom) led to translocation of PLCδ1PH-GFP to the cytoplasm. Scale bars, 1 μm. F, Effects of ACh and EGF on the kinetics and extent of this translocation are indicated by the ratio of the membrane and the cytoplasm fluorescence intensity (n = 8). Error bars indicate SEM.
We then compared the effects of ACh and EGF on the hydrolysis of membrane PtdIns(4,5)P2. For this, PLCδ1PH-GFP, M1 receptor, and KCNQ2/3 channels were coexpressed in HEK293 cells, and GFP fluorescence was visualized using confocal microscopy. PLCδ1PH-GFP is known to bind PtdIns(4,5)P2 and its translocation between the cytosol and the membrane has been successfully used as an indicator of PtdIns(4,5)P2 breakdown and resynthesis (Stauffer et al., 1998). Figure 4E shows two cells with visible GFP signal that were treated with either ACh or EGF. Figure 4F shows a time course for the ratio of membrane (Fm) and cytosolic (Fc) fluorescence intensity. The intensity ratio was normalized based on the intensity ratio before ACh or EGF application. ACh induced a ∼80% translocation of GFP signal from the membrane to the cytosol. EGF, however, induced a brief translocation of <20% of the GFP signal, which was completely blocked by U73122. Both the amplitudes and time courses of GFP translocation induced by ACh and EGF are compatible with those of the current inhibition shown earlier (Fig. 4A). These results indicate that, similar to ACh, EGF induces a fast inhibition of KCNQ2/3 currents through breakdown of membrane-localized PtdIns(4,5)P2.
We noticed an interesting difference between EGF- and ACh-induced fast inhibition: the onset of action by EGF was significantly slower than that of ACh; EGF-induced fast inhibition started at 35 ± 8 s (n = 16) whereas ACh-induced fast inhibition started at 18 ± 7 s (n = 20). This difference may reflex a slower process of the RTK–PLCγ pathway compared with that Gq–PLCβ pathway.
One key piece of evidence that supports the notion that PtdIns(4,5)P2 is the molecular determinant responsible for M1-mediated inhibition of M/KCNQ currents came from experiments showing that a block of PtdIns(4,5)P2 resynthesis after hydrolysis leads to a block in the recovery of the inhibited M/KCNQ currents (Suh and Hille, 2002; Zhang et al. 2003). We proceeded to test if the recovery of the EGF-induced fast inhibition of KCNQ2/3 currents depends on the resynthesis of PtdIns(4,5)P2. Wortmannin (10 μm) was used in this study to block PtdIns(4)P kinase, leading to a block in the resynthesis of PtdIns(4,5)P2, as has been described previously (Suh and Hille, 2002; Zhang et al., 2003). For comparison, ACh-induced inhibition was also tested in this part of the study. Figure 5A shows the different recovery rates of KCNQ2/3 currents from the inhibition induced by ACh in the absence or presence of wortmannin. It is clear that wortmannin greatly slowed the recovery of currents after inhibition, a result that is consistent with a previous study (Zhang et al., 2003). Similarly, wortmannin also blocked recovery of EGF-induced inhibition of KCNQ2/3 currents (Fig. 5B). In this case, the current continued to decay even after washout of wortmannin, a result that is in contrast with the ACh-induced inhibition shown in Figure 5A. This slow continuous run-down of the currents, as we discussed earlier, is most likely a delayed response to EGF.
Figure 5.
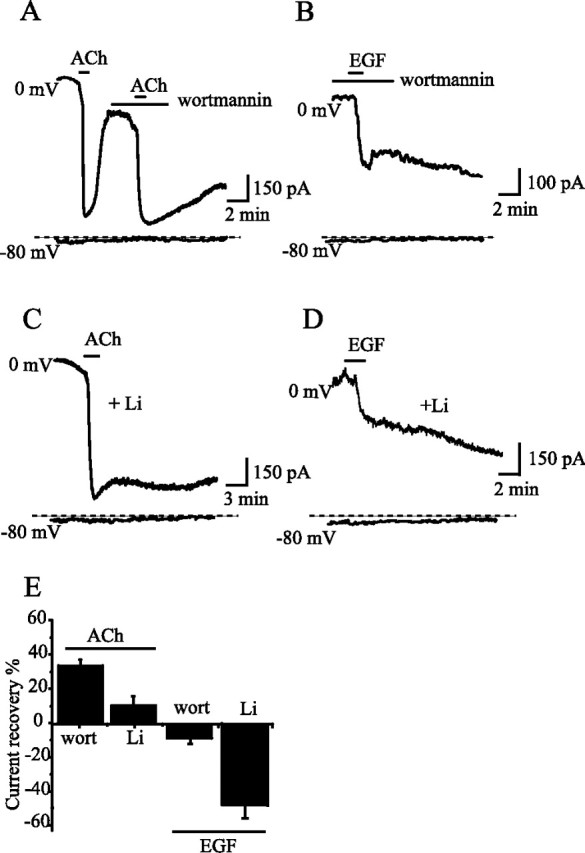
Wortmannin and lithium blocked the recovery of EGF- and ACh-induced fast inhibition of KCNQ2/3 currents. KCNQ2/3 channels and M1 receptors were coexpressed in HEK293 cells. A, B, The effects of wortmannin (10 μm) on Ach-induced (A) or EGF-induced (B) inhibition of KCNQ2/3 currents were studied. In both cases, wortmannin blocked the recovery of the currents after inhibition. C, D, Similarly, the effects of lithium on Ach-induced (C) or EGF-induced (D) inhibition of KCNQ2/3 currents were studied. In this case, the cells were treated with lithium (100 μm) for 6 h before subjecting them to ACh or EGF. The percentage recovery of the currents is summarized in E (n = 7–9). The current amplitude before application of either ACh or EGF was used as the normalization factor. Error bars indicate SEM.
We also used Li+ to block the resynthesis of PtdIns(4,5)P2. Li+ inhibits inositol monophosphate phosphatase and inositol polyphosphate 1-phosphatase and blocks conversion of inositol phosphate to inositol, resulting in inositol depletion. Li+ treatment eventually affects PtdIns(4,5)P2 synthesis. HEK293 cells expressing M1 receptor and KCNQ2/3 channels were incubated with Li+ (100 μm) for 6 h to deplete the cellular pool of inositol. After inositol depletion, ACh and EGF were applied and the inhibition and recovery of KCNQ2/3 currents was observed (Fig. 5C,D). In Li+ treated cells, the initial current amplitudes of KCNQ2/3 were reduced by 23 ± 8% (n = 8; p < 0.01) compared with nontreated cells. ACh inhibited KCNQ2/3 currents with an amplitude that was compatible with non-Li+ treated cells (Fig. 5, compare A, C). However, in Li+-treated cells, only a negligible recovery of the currents was seen after inhibition (Fig. 5C). Similar results were observed for EGF-induced inhibition (Fig. 5D). Again, in this case, not only was recovery not observed, but the currents continued to run-down after wash out of EGF (Fig. 5D) likely resulting from a delayed response to EGF as we discussed previously. The percentage of recovery of the inhibited currents is summarized in Figure 5E. It is apparent that Li+ is more efficient than wortmannin at blocking current recovery, probably reflecting the difference between the effects of substrate depletion and kinase inhibition on PtdIns(4,5)P2 synthesis. These results strongly suggest that, like activation of M1 receptor, activation of EGF receptor induces a fast inhibition of KCNQ2/3 currents by a mechanism that involves PtdIns(4,5)P2 hydrolysis.
EGF-induced slow inhibition is the result of channel tyrosine phosphorylation
The delayed slow inhibition of KCNQ2/3 currents induced by EGF may not be related to PtdIns(4,5)P2 hydrolysis, because the inhibition was not prevented by U73122 (Fig. 4C) or recovered after withdrawal of wortmannin (Fig. 5B), and the amplitude and the time course of inhibition were not compatible with PtdIns(4,5)P2 hydrolysis (Fig. 4F). We hypothesize that the slow inhibition of KCNQ2/3 currents induced by EGF is the result of channel phosphorylation. We proceeded to test this hypothesis in the following experiments. We tested the effect of vanadate, an inhibitor of protein tyrosine phosphatase. If the slow inhibition of KCNQ2/3 current by EGF is the result of channel tyrosine phosphorylation, then vanadate, which inhibits protein dephosphorylation, would be expected to have an effect on KCNQ2/3 currents that is similar to that of EGF. In cells expressing KCNQ2/3 channels, vanadate (200 μm) inhibited KCNQ2/3 currents with characteristics similar to those of EGF-induced inhibition. After a 20 min incubation with vanadate, the current amplitudes of KCNQ2/3 were reduced to 45 ± 13% of the original values. The activation time constant of KCNQ2/3 currents was increased by vanadate from 238 ± 35 ms to 535 ± 42 ms, and the deactivation constant was increased from 136 ± 26 ms to 358 ± 39 ms (Fig. 6A,B). These effects are similar to those of EGF as described in Figure 3, B and C.
Figure 6.
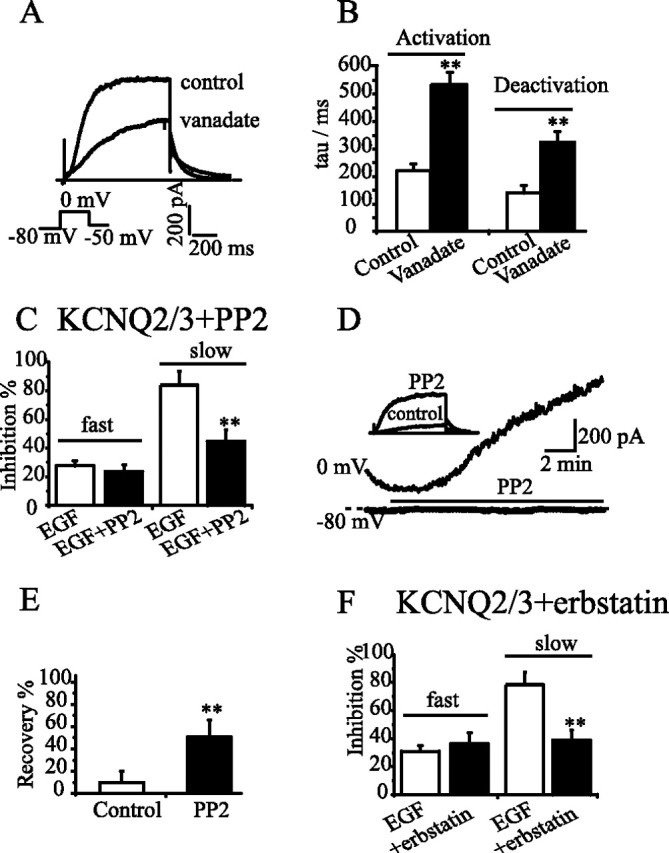
EGF-induced slow inhibition of KCNQ2/3 currents was mimicked by vanadate and attenuated by PP2 and erbstatin. A, B, A long (30 min) incubation of the cell expressing KCNQ2/3 channels with vanadate (200 μm) suppressed the current amplitude (A) and slowed the activation and deactivation of the currents, (B). KCNQ2/3 currents were activated and deactivated with the protocol shown in A. A single exponential function was used to fit both activation and deactivation of KCNQ2/3 currents, and the time constants are summarized in B. **p < 0.01, compared with the control; n = 7–8. C, PP2 selectively antagonized the slow inhibition without affecting the fast inhibition of KCNQ2/3 currents by EGF. **p < 0.01, compared with EGF alone; n = 7. D, E, PP2 (200 nm) reversed EGF-induced irreversible inhibition of KCNQ2/3 currents (D), and the summarized data are shown in E. In these experiments, the cells were first treated with EGF for 30 min to reach a maximal inhibition of the currents (**p < 0.01, compared with the control; n = 10 for the control and n = 7 for PP2). F, Erbstatin (10 μm) also selectively suppressed EGF-induced slow inhibition (**p < 0.01, compared with EGF alone, n = 6). Error bars indicate SEM.
We also observed the effects of PP2, a specific Src tyrosine kinase inhibitor, on EGF-induced inhibition of KCNQ2/3 currents. PP2 did not greatly affect the fast inhibition, but did reduce the slow inhibition (Fig. 6C). Furthermore, the irreversible inhibition of KCNQ2/3 currents induced by EGF was partially reversed to 52 ± 15% of the pre-EGF level by PP2 (Fig. 6D,E). The effect of erbstatin, another Src tyrosine kinase inhibitor was also tested. Similar to PP2, erbstatin significantly reduced EGF-induced slow inhibition but did not affect the fast inhibition (Fig. 6F). These results suggest EGF-induced slow inhibition is possibly mediated by Src tyrosine kinase.
These results indicate that tyrosine phosphorylation is most likely the underlying mechanism for EGF-induced slow inhibition of KCNQ2/3 currents. A previous study demonstrated that over-expression of Src kinase inhibited KCNQ2/3 currents through phosphorylation of two tyrosine residues, Y67 and Y349, of KCNQ3 (Gamper et al., 2003). In the present study, we investigated the consequences of these two mutations on EGF-induced slow inhibition of KCNQ2/3 currents. For this, KCNQ2 was coexpressed with either KCNQ3, KCNQ3(Y67F), KCNQ3(Y349F), or KCNQ3(Y67F,Y349F) in HEK293 cells, and the effects of EGF on these channel currents were observed at the end of an ∼25 min EGF application. Figure 7, A and B, shows two representative recordings of EGF-induced inhibition of KCNQ2/KCNQ3(Y67F) and KCNQ2/KCNQ3(Y67F/Y349F). Figure 7C summarizes the data for EGF-induced inhibition of the KCNQ currents. It is apparent that a single mutation of either Y67F or Y349F of KCNQ3 reduced EGF-induced slow inhibition of the KCNQ currents, and simultaneous mutation of both residues had an additional effect. EGF induced only 20 ± 10% inhibition of KCNQ2/KCNQ3(Y67F, Y349F), as compared with 82 ± 20% inhibition of KCNQ2/3. These results strongly suggest that channel tyrosine phosphorylation is largely responsible for EGF-induced slow inhibition of KCNQ2/3 currents.
Figure 7.
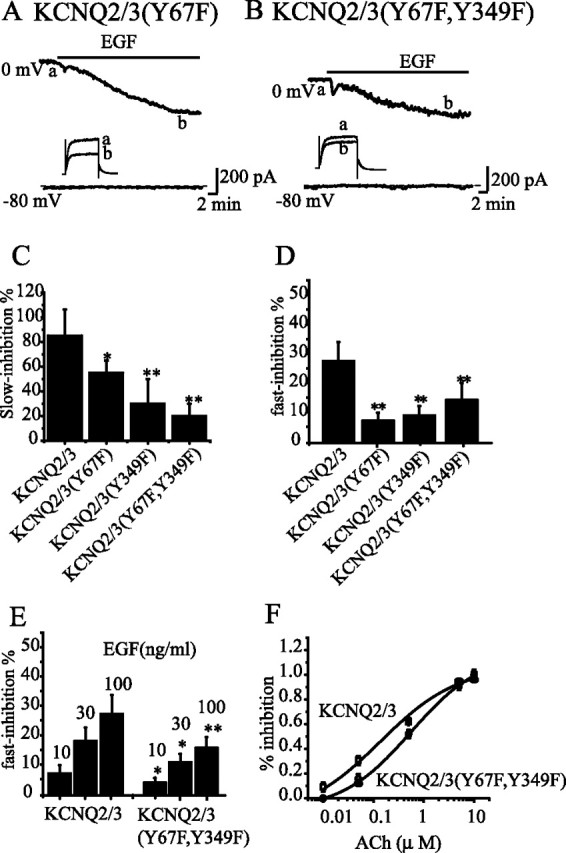
Mutation of two tyrosine residues in KCNQ3 attenuated EGF- and ACh-induced inhibition of KCNQ2/3 currents. Wild-type KCNQ2/3, KCNQ2/3(Y67F), KCNQ2/3(Y349F), and KCNQ2/3(Y67F, Y349F) were each expressed in different HEK293 cells, and EGF and ACh actions on these channel currents were studied. Two representative cells expressing either KCNQ2/3(Y67F) or KCNQ2/3(Y67F, Y349F) are shown in A and B, respectively. C, D, The relative EGF-induced slow (C) and fast (D) inhibitions of the wild-type and the mutant channel currents are summarized. **p < 0.01; *p < 0.05, compared with KCNQ2/3; n = 5–7. E, Concentration-dependent effects of EGF on fast inhibition of wild-type and mutant KCNQ2/3 channels. **p < 0.01; *p < 0.05, compared with KCNQ2/3, n = 5–9. F, Concentration–response relationship for ACh-induced inhibition of wild-type and mutant KCNQ2/3 channels. Each point represents the summary of seven to nine repeated experiments. Sensitivity of KCNQ currents to ACh were described by the Hill equation Y = A/1 + (IC50/X)n, where A is the maximum inhibition, X is ACh concentration, and n is the slope. IC50 and slope are 0.15 and 0.86 μm, respectively, for KCNQ2/3, and 0.58 and 0.75 μm, respectively, for KCNQ2/3(Y67F, Y349F). Error bars indicate SEM.
Phosphorylation affects PtdIns(4,5)P2-dependent channel modulation
To our surprise, these tyrosine mutations not only reduced EGF-induced slow inhibition of KCNQ currents, but also reduced the EGF-induced fast inhibition, as can be seen in Figure 7, A and B. Summary data shown in Figure 7D clearly demonstrated that states of channel tyrosine phosphorylation indeed affected PtdIns(4,5)P2-dependent fast inhibition of KCNQ currents. In the case of KCNQ2/KCNQ3(Y67F, Y349F), the percentage of fast inhibition was reduced to 13 ± 8%, as opposed to 28 ± 10% in the case of KCNQ2/3. To get a more complete assessment of the effect of channel phosphorylation on KCNQ2/3 sensitivity to PtdIns(4,5)P2 depletion, we compared the effects of the KCNQ mutations on fast inhibitions by EGF and ACh by completing concentration-response curves to assess sensitivity changes. Figure 7E shows that at three concentrations tested, mutation indeed reduced EGF-induced fast inhibition at each concentration. We were unable to get reliable results from higher concentrations of EGF because at these concentrations, EGF-induced slow inhibition appeared sooner and followed fast inhibition immediately, thus, making it difficult to make a reliable judgment. Complete concentration-response curves were established for the effect of ACh and were presented in Figure 7F; mutation indeed shifted the response of the channels to ACh to the right (IC50 from 0.15 to 0.58 μm). We went further to test the effect of vanadate on EGF-induced fast inhibition. As we showed previously (Fig. 6A,B), vanadate mimicked EGF-induced slow inhibition of KCNQ currents. We reasoned that reduced channel dephosphorylation by vanadate would enhance EGF-induced fast inhibition of KCNQ2/3 currents, in contrast to the effects of mutation of KCNQ3 tyrosine residues. Figure 8A shows a representative recording of EGF-induced inhibition of KCNQ2/3 currents from a cell pretreated (∼20 min) with 0.1 mm vanadate (Huang et al., 1998). From the summary data shown in Figure 8B, it is clear that vanadate treatment significantly enhanced EGF-induced inhibition of KCNQ2/3 currents (from 28 ± 10% to 45 ± 8%). Another interesting observation is that the recovery of the currents from inhibition was slowed by vanadate and the inhibition became irreversible in most instances (Fig. 8A).
Figure 8.

Vanadate enhanced the EGF-induced fast inhibition of KCNQ2/3 currents. Pretreatment of HEK293 cells expressing KCNQ2/3 channels with vanadate (100 μm) for 20 min enhanced EGF-induced fast inhibition of KCNQ2/3 currents. A, B, The representative record (A) was from six similar experiments (B). **p < 0.01, compared with EGF alone; n = 4 for EGF and n = 6 for vanadate + EGF. Error bars indicate SEM.
Discussion
In the present study, we demonstrate that activation of EGF receptor inhibits KCNQ2/3 and M currents with two distinct characteristics: an initial transient fast inhibition followed by a delayed slow inhibition. We further demonstrate that the fast inhibition is most likely the result of EGF-induced PtdIns(4,5)P2 hydrolysis and that the slow inhibition is caused by channel phosphorylation. In addition, these two processes are interconnected because mutation of tyrosine phosphorylation sites that reduce the slow inhibition also attenuate the fast inhibition.
It is well established that functional EGF receptors are expressed in HEK293 cells. These cells have been extensively used to study EGF-induced signaling (Schmidt et al., 2000; Stope et al., 2004; Shah et al., 2006). It is clear that EGF induced two types of characteristic inhibition of KCNQ2/3 currents through the activation of EGF receptors, because AG1478, a specific EGF receptor inhibitor, significantly attenuated the inhibitions. Initiation and propagation of EGF signaling include many phosphorylation steps, including phosphorylation of the EGF receptor itself, and many down stream signaling molecules and many adaptor and effector proteins (Schlessinger, 2000). Thus, it is not surprising that EGF-induced inhibition of KCNQ2/3 was almost abolished by the tyrosine kinase inhibitor genistein in the present study.
In native SCG neurons, EGF inhibited M currents in a concentration-dependent manner. At a maximal inhibition concentration (1000 ng/ml), EGF induced a final 65% inhibition of M currents. In some of the cells tested (7 of 12), a transient fast and sustained slow inhibition could also be distinguished (Fig. 2A,B). Although the fast inhibition was small and not present in all cells, we believe it is true and is not an artifact. Similar to KCNQ currents, EGF-induced inhibition of M currents was also reduced by pharmacological inhibition of EGF signaling pathway. Although EGF receptor is believed to be ubiquitously expressed in many tissues and has been found to be expressed in the CNS (Tucker et al., 1993a,b; Yamada et al., 1997), there was no direct evidence that EGFR was present in SCG. We show, by the means of Western blot and immunocytochemical studies, that EGFR was indeed present in the SCG. Thus, EGF modulated both KCNQ2/3 currents and native M currents in a similar manner.
It seems clear that EGF-induced fast and slow inhibitions involve distinct mechanisms. First, the fast inhibition was small in amplitude and transient whereas the slow inhibition started with a delay and the inhibition was much more profound and irreversible. Second, the fast inhibition only affected the amplitude of the currents whereas the slow inhibition affected both the amplitude and the kinetics of current activation and deactivation (Fig. 3).
Several lines of evidence suggest that EGF-induced fast inhibition of KCNQ2/3 current is the result of PtdIns(4,5)P2 hydrolysis: (1) the inhibition was significantly reduced by the PLC inhibitor U73122; (2) treatments with wortmannin or Li+, which block the resynthesis of PtdIns(4,5)P2, make the transient fast inhibition irreversible, indicating that recovery of the currents from inhibition is the result of PtdIns(4,5)P2 resynthesis; (3) the time course and amplitude of PtdIns(4,5)P2 hydrolysis and resynthesis indicated by PLCδ1PH-GFP translocation was consistent with that of the current inhibition. Finally, EGF-induced fast inhibition is similar to M1-mediated inhibition of KCNQ2/3 currents, which has been shown to be mediated by PtdIns(4,5)P2 hydrolysis (Zhang et al., 2003). Then why does EGF induce a fast transient inhibition of KCNQ2/3 currents when activation of M1 and other G-protein-coupled receptors induces a persistent inhibition of the currents? It is not easy to give a definite answer to this question from data presented in this study. It is evident that EGF receptor (and other RTKs) signaling is more complicated than the G-protein-coupled receptor pathway. It is clear that EGF is much less potent than M1 activation at activating hydrolysis of PtdIns(4,5)P2 hydrolysis (Fig. 4E,F) in HEK293 cells. This probably reflects the distinct natures of the EGF receptor-PLCγ-PtdIns(4,5)P2 pathway and the M1-PLCβ-PtdIns(4,5)P2 pathway, but likely does not reflect a difference in the amount of endogenously expressed EGF receptor and over-expressed M1 receptor, because we observed a similar result from cells over-expressing EGF receptor (data not shown). A EGF-induced activation of the phospholipase A2 (PLA2)–arachidonic acid pathway, may provide an explanation for the above question. Such a mechanism has been proposed for somatostatin-induced increase of M currents (Schweitzer et al., 1990). Ca2+-dependent activation of PLA2 is also responsible for the enhancement of M currents that generates the over-recovery that is observed after the removal of a muscarinic receptor agonist (Marrion et al., 1991; Villarroel, 1994). Indeed, EGF is able to activate the PLA2–arachidonic acid pathway (Schalkwijk et al., 1996). We used a PLA2 inhibitor, quinacrine, to see if the recovery of the fast inhibition under continuous presence of EGF would be affected. In eight cells tested, the EGF-induced fast inhibition recovered to 73 ± 9% of the pre-EGF level in the presence of quinacrine, as compared with a 102 ± 9% (n = 8; p < 0.01) recovery rate in the absence of quinacrine. Thus, at least partially, the PLA2–arachidonic acid pathway may responsible for the transient nature of EGF-induced fast inhibition.
Several lines of evidence suggest that EGF-induced slow inhibition of KCNQ2/3 currents is mediated through tyrosine phosphorylation: (1) the tyrosine phosphatase inhibitor vanadate fully mimicked all of the effects of EGF; (2) tyrosine kinase inhibitor PP2 reduced and reversed the slow inhibition, but did not affect the fast inhibition; (3) mutations of tyrosine residues on KCNQ3 significantly reduced EGF-induced inhibition of KCNQ2/3 currents. Gamper et al. (2003) showed that overexpression of Src tyrosine kinase inhibited KCNQ2/3 currents in Chinese hamster ovary cells. They later showed that Src directly phosphorylates two tyrosines (Tyr-67 and Tyr-349) on KCNQ3 (Li et al., 2004). Our present work is consistent with their findings. Mutation of either Tyr-67 or Tyr-349 significantly reduced EGF-induced slow inhibition of KCNQ2/3 currents; mutation of both residues reduced the inhibition to ∼20% (compared with >80% inhibition with wild types of channels). Together, our data strongly suggest that EGF-induced slow inhibition is mediated through channel tyrosine phosphorylation. Src tyrosine kinase may participate, at least partially, in EGF-induced slow inhibition, because PP2, a specific Src tyrosine kinase inhibitor, reduced the slow inhibition (Fig. 6C). We found that EGF also induced slow inhibition of homomeric KCNQ2 currents, but the degree of the inhibition was less than that of heteromeric KCNQ2/3 currents (Fig. 1F). This result ensured two suggestions: EGF induces phosphorylation of KCNQ2 as well as KCNQ3, and EGF-induced slow inhibition of KCNQ2/3 currents is the results of combined KCNQ2 and KCNQ3 phosphorylations.
To our surprise, mutation of the phosphorylation sites on the KCNQ3 channel not only reduced EGF-induced slow inhibition of KCNQ2/3 currents, but also reduced the fast inhibition. Furthermore, as it is shown in Figure 7, E and F, the mutation not only reduced EGF-induced fast inhibition but also reduced ACh-induced inhibition. One attractive hypothesis is that the states of channel phosphorylation affect PtdIns(4,5)P2-mediated modulation of KCNQ2/3 currents, and channel phosphorylation makes the channel more sensitive to PtdIns(4,5)P2 depletion. At least one more line of evidence from the present study supports this hypothesis. Pretreatment with vanadate, which presumably increases the level of channel phosphorylation by inhibiting the activity of protein phosphatase, greatly increased EGF-induced fast inhibition of KCNQ2/3 currents (Fig. 8). A similar mechanism has been proposed for the M3-mediated modulation of Kir3.1/3.2 channel currents. In this case, PKC phosphorylation of Kir3.1/Kir3.2 sensitizes the response of the channels to PtdIns(4,5)P2 reduction, thus resulting in a greater inhibition of Kir3.1/3.2 currents (Brown et al., 2005). Similarly, A-kinase anchoring protein-PKC mediated phosphorylation of M/KCNQ channels increases muscarinic receptor-induced inhibition of M/KCNQ currents (Hoshi et al., 2003). This may be related to the channels' increased sensitivity to PtdIns(4,5)P2 depletion (Shen et al., 2005). Furthermore, a mutation that supposedly increases the channel-PtdIns(4,5)P2 interaction not only reduces M1, but also PKC-mediated inhibition of Kir2.3 channel currents (Du et al., 2004). In light of these findings we propose the following working hypothesis: there is ongoing tyrosine phosphorylation of KCNQ2/3 channels opposed by tyrosine phosphatases in resting cells; if the tyrosine dephosphorylation is blocked, phosphorylated tyrosine accumulates; the accumulated resting amount of phosphorylated tyrosine on KCNQ determines how sensitive the channel will be to PtdIns(4,5)P2 depletion. This same phosphorylation is also the inhibitory phosphorylation, which may reflect the very slow phosphorylation once receptors are stimulated.
There is growing evidence that RTKs are important modulators of ion channels. Neurotrophins, including NGF, regulate neuronal ion channels and membrane electrical properties in both central and peripheral neurons (Holm et al., 1997; Baldelli et al., 2000; Adamson et al., 2002; Zhang et al., 2002; Luther and Birren, 2006). These RTKs, including EGFR and also Src kinase may launch tyrosine phosphorylation which may converge (much as many Gq receptor actions converge) to change PtdIns(4,5)P2 sensitivity and gating of channels like KCNQ. The modulation of M/KCNQ2/3 currents by EGF or by activation of other RTKs will generally increase cell excitability and oppose any hyperpolarization-induced decrease in excitability. Thus, complex integration of signals at the level of individual ion channels could lead to changes in growth rates, calcium influx, and a host of other cellular phenomena.
Footnotes
This work was supported by National Natural Science Foundation of China Grant 30270361, Ministry of Science and Technology of China Grant 2003CCA00300, and Hebei Nature Science Foundation Grant 303464 (H.Z.). H.Z. is a beneficiary of the National Science Fund for Distinguished Young Scholars of China (No. 30325038). We are grateful for Prof. B Hille for all of his suggestions.
References
- Adams PR, Brown DA. Luteinizing hormone-releasing factor and muscarinic agonists act on the same voltage-sensitive K+-current in bullfrog sympathetic neurons. Br J Pharmacol. 1980;68:353–355. doi: 10.1111/j.1476-5381.1980.tb14547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PR, Brown DA, Jones SW. Substance P inhibits the M-current in bullfrog sympathetic neurons. Br J Pharmacol. 1983;79:330–333. doi: 10.1111/j.1476-5381.1983.tb11004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson CL, Reid MA, Davis RL. Opposite actions of brain-derived neurotrophic factor and neurotrophin-3 on firing features and ion channel composition of murine spiral ganglion neurons. J Neurosci. 2002;22:1385–1396. doi: 10.1523/JNEUROSCI.22-04-01385.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli P, Forni PE, Carbone E. BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. Eur J Neurosci. 2000;12:4017–4032. doi: 10.1046/j.1460-9568.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Bowlby MR, Fadool DA, Holmes TC, Levitan IB. Modulation of the Kv1.3 potassium channel by receptor tyrosine kinases. J Gen Physiol. 1997;110:601–610. doi: 10.1085/jgp.110.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BS, Yu SP. Modulation and genetic identification of the M channel. Prog Biophys Mol Biol. 2000;73:135–166. doi: 10.1016/s0079-6107(00)00004-3. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown SG, Thomas A, Dekker LV, Tinker A, Leaney JL. PKC-delta sensitizes Kir3.1/3.2 channels to changes in membrane phospholipid levels after M3 receptor activation in HEK-293 cells. Am J Physiol Cell Physiol. 2005;289:C543–C556. doi: 10.1152/ajpcell.00025.2005. [DOI] [PubMed] [Google Scholar]
- Colley B, Tucker K, Fadool DA. Comparison of modulation of Kv1.3 channel by two receptor tyrosine kinases in olfactory bulb neurons of rodents. Receptors Channels. 2004;10:25–36. [PMC free article] [PubMed] [Google Scholar]
- Cruzblanca H, Koh DS, Hille B. Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA. 1998;95:7151–7156. doi: 10.1073/pnas.95.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Del Río E, Bevilacqua JA, Marsh SJ, Halley P, Caulfield MP. Muscarinic M1 receptors activate phosphoinositide turnover and Ca2+ mobilisation in rat sympathetic neurones, but this signalling pathway does not mediate M-current inhibition. J Physiol (Lond) 1999;520:101–111. doi: 10.1111/j.1469-7793.1999.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of kir channels by diverse modulators. J Biol Chem. 2004;279:37271–37281. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- Fadool DA, Holmes TC, Berman K, Dagan D, Levitan IB. Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA. P2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents. J Neurosci. 1998;18:5170–5179. doi: 10.1523/JNEUROSCI.18-14-05170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Stemkowski PL, Light PE, Smith PA. Experiments to test the role of phosphatidylinositol 4,5-bisphosphate in neurotransmitter-induced M-channel closure in bullfrog sympathetic neurons. J Neurosci. 2003;23:4931–4941. doi: 10.1523/JNEUROSCI.23-12-04931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Stemkowski PL, Smith PA. Possible role of phosphatidylinositol 4,5 bisphosphate in luteinizing hormone releasing hormone-mediated M-current inhibition in bullfrog sympathetic neurons. Eur J Neurosci. 2004;20:2990–2998. doi: 10.1111/j.1460-9568.2004.03786.x. [DOI] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol. 2003;122:17–31. doi: 10.1085/jgp.200208783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Stockand JD, Shapiro MS. Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J Neurosci. 2003;23:84–95. doi: 10.1523/JNEUROSCI.23-01-00084.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Li Y, Shapiro MS. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell. 2005;16:3538–3551. doi: 10.1091/mbc.E04-09-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida H, Hoshi N, Zhang JS, Yokoyama S, Hashii M, Jin D, Noda M, Robbins J. Protein kinase C bound with A-kinase anchoring protein is involved in muscarinic receptor-activated modulation of M-type KCNQ potassium channels. Neurosci Res. 2005;51:231–234. doi: 10.1016/j.neures.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001;111:RE19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- Holm NR, Christophersen P, Olesen SP, Gammeltoft S. Activation of calcium dependent potassium channels in mouse [correction of rat] brain neurons by neurotrophin-3 and nerve growth factor. Proc Natl Acad Sci USA. 1997;94:1002–1006. doi: 10.1073/pnas.94.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Levitan IB. Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci. 1996;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H. AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Kammermeier PJM. Current mystery messenger revealed? Neuron. 2002;35:411–412. doi: 10.1016/s0896-6273(02)00792-4. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and G alpha q/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- Kramer HK, Onoprishvili I, Andria ML, Hanna K, Sheinkman K, Haddad LB, Simon EJ. Delta opioid activation of the mitogen-activated protein kinase cascade does not require transphosphorylation of receptor tyrosine kinases. BMC Pharmacol. 2002;2:5. doi: 10.1186/1471-2210-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Li Y, Langlais P, Gamper N, Liu F, Shapiro MS. Dual phosphorylations underlie modulation of unitary KCNQ K+ channels by Src tyrosine kinase. J Biol Chem. 2004;279:399–407. doi: 10.1074/jbc.M408410200. [DOI] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol. 2006;96:946–958. doi: 10.1152/jn.01078.2005. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Zucker RS, Marsh SJ, Adams PR. Modulation of M-current by intracellular Ca2+ Neuron. 1991;6:533–545. doi: 10.1016/0896-6273(91)90056-6. [DOI] [PubMed] [Google Scholar]
- Nesti E, Everill B, Morielli AD. Endocytosis as a mechanism for tyrosine kinase-dependent suppression of a voltage-gated potassium channel. Mol Biol Cell. 2004;15:4073–4088. doi: 10.1091/mbc.E03-11-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalkwijk CG, van der Heijden MA, Bunt G, Maas R, Tertoolen LG, van Bergen en Henegouwen PM, Verkleij AJ, van den Bosch H, Boonstra J. Maximal epidermal growth-factor-induced cytosolic phospholipase A2 activation in vivo requires phosphorylation followed by an increased intracellular calcium concentration. Biochem J. 1996;313:91–96. doi: 10.1042/bj3130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Frings M, Mono ML, Guo Y, Weernink PA, Evellin S, Han L, Jakobs KH. G protein-coupled receptor-induced sensitization of phospholipase C stimulation by receptor tyrosine kinases. J Biol Chem. 2000;275:32603–32610. doi: 10.1074/jbc.M004784200. [DOI] [PubMed] [Google Scholar]
- Schweitzer P, Madamba S, Siggins GR. Arachidonic acid metabolites as mediators of somatostatin-induced increase of neuronal M-current. Nature. 1990;346:464–467. doi: 10.1038/346464a0. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Brown DA. Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron. 1996;16:151–162. doi: 10.1016/s0896-6273(00)80032-x. [DOI] [PubMed] [Google Scholar]
- Shah BH, Neithardt A, Chu DB, Shah FB, Catt KJ. Role of EGF receptor transactivation in phosphoinositide 3-kinase-dependent activation of MAP kinase by GPCRs. J Cell Physiol. 2006;206:47–57. doi: 10.1002/jcp.20423. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron. 1994;12:1319–1329. doi: 10.1016/0896-6273(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Stope MB, Vom Dorp F, Szatkowski D, Bohm A, Keiper M, Nolte J, Oude Weernink PA, Rosskopf D, Evellin S, Jakobs KH, Schmidt M. Rap2B-dependent stimulation of phospholipase C-epsilon by epidermal growth factor receptor mediated by c-Src phosphorylation of RasGRP3. Mol Cell Biol. 2004;24:4664–4676. doi: 10.1128/MCB.24.11.4664-4676.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker MS, Eves EM, Wainer BH, Rosner MR. Activation of mitogen-activated protein kinase by epidermal growth factor in hippocampal neurons and neuronal cell lines. J Neurochem. 1993a;61:1376–1387. doi: 10.1111/j.1471-4159.1993.tb13631.x. [DOI] [PubMed] [Google Scholar]
- Tucker MS, Khan I, Fuchs-Young R, Price S, Steininger TL, Greene G, Wainer BH, Rosner MR. Localization of immunoreactive epidermal growth factor receptor in neonatal and adult rat hippocampus. Brain Res. 1993b;631:65–71. doi: 10.1016/0006-8993(93)91187-w. [DOI] [PubMed] [Google Scholar]
- Villarroel A. On the role of arachidonic acid in M-current modulation by muscarine in bullfrog sympathetic neurons. J Neurosci. 1994;14:7053–7066. doi: 10.1523/JNEUROSCI.14-11-07053.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ. Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischmeyer E, Doring F, Karschin A. Acute suppression of inwardly rectifying Kir2.1 channels by direct tyrosine kinase phosphorylation. J Biol Chem. 1998;273:34063–34068. doi: 10.1074/jbc.273.51.34063. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ikeuchi T, Hatanaka H. The neurotrophic action and signalling of epidermal growth factor. Prog Neurobiol. 1997;51:19–37. doi: 10.1016/s0301-0082(96)00046-9. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na(+) current and delayed rectifier K(+) current in rat sensory neurons. J Physiol (Lond) 2002;544:385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]