

Abstract
Kainate receptors (KARs) are heteromeric ionotropic glutamate receptors that play a variety of functions in the regulation of the activity of synaptic networks. Little is known about the regulation of the function of synaptic KARs in the brain. In the present study, we found that a conditioning activation of synaptic NMDA receptors (NMDARs) induces short-term depression of KAR–EPSCs but not of AMPA receptor–EPSCs at synapses between mossy fibers and CA3 pyramidal cells. Short-term depression of KAR–EPSCs by synaptic NMDARs peaked at 1 s and reversed within 20 s, was likely induced and expressed postsynaptically, and was homosynaptic. It depended on a rise of Ca2+ in the postsynaptic cell and on the activation of the phosphatase calcineurin that likely binds to the GluR6b (glutamate receptor subunit 6b) subunit splice variant allowing the dephosphorylation of KARs and inhibition of activity. Finally, we show in the current-clamp mode that short-term depression of KAR–EPSPs is induced by the coincident discharge of action potentials in the postsynaptic cell together with synaptic stimulation. Hence, this study describes a form of short-term synaptic plasticity that is postsynaptic, depends on the temporal order of presynaptic and postsynaptic spiking, and likely affects the summation properties of mossy fiber EPSPs.
Keywords: kainate receptors, short-term synaptic plasticity, NMDA receptors, mossy fiber, hippocampus
Introduction
Glutamatergic synaptic transmission is mediated by AMPA receptors (AMPARs) and NMDA receptors (NMDARs) that are found colocalized in the postsynaptic density of the vast majority of glutamatergic synapses in the brain and are activated concomitantly after release of glutamate from the presynaptic terminal. Postsynaptic kainate receptors (KARs) show restricted cellular and subcellular expression. Kainate receptor-mediated EPSCs (KAR–EPSCs) have been best characterized at synapses between mossy fibers and CA3 pyramidal cells in the hippocampus (Castillo et al., 1997; Vignes and Collingridge, 1997; Mulle et al., 1998; Contractor et al., 2003), although they have also been observed in a variety of other synapses (Huettner, 2003; Lerma, 2003; Pinheiro and Mulle, 2006). A single stimulation of mossy fibers is sufficient to activate synaptic KARs (Castillo et al., 1997), which only yields EPSCs of small amplitude and slow kinetics. The analysis of KAR mutant mice has further demonstrated that 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine (GYKI 53655)-resistant EPSCs were mediated by KARs composed of the glutamate receptor subunit 6 (GluR6) and KA2 subunits (Mulle et al., 1998; Contractor et al., 2003; Ruiz et al., 2005).
Compared with AMPA and NMDA receptor-mediated synaptic transmission, little is known about the plasticity of KAR–EPSCs. Early during postnatal development, long-term potentiation expression at thalamocortical synapses is associated with a reduction of KAR–EPSCs (Kidd and Isaac, 1999). In perirhinal cortex neurons, activation of PKC by stimulation of mGluR5 enhances KAR–EPSCs (Cho et al., 2003; Park et al., 2006). In these neurons, a train of 200 stimuli at 5 Hz results in long-term depression of KAR–EPSCs via a mechanism involving mGluR5, PKC, and PICK1 (protein interacting with C kinase-1) PDZ (postsynaptic density-95/Discs large/zona occludens-1) domain interactions (Park et al., 2006), and possibly a change in the number of synaptic KARs (Hirbec et al., 2003). At hippocampal mossy fiber synapses, KAR–EPSCs are regulated as a consequence of presynaptic changes in glutamate release properties (Ito et al., 2004), but no form of synaptic plasticity specific for KAR–EPSCs has yet been described. However, the functional properties of recombinant KARs can be regulated on a shorter time scale by direct cAMP-dependent phosphorylation (Traynelis and Wahl, 1997). In cultured hippocampal neurons, a conditioning activation of NMDARs depresses native KAR-mediated responses in a rapid and reversible manner (Ghetti and Heinemann, 2000). This depression depends on Ca2+ influx through NMDA receptor or voltage-gated calcium channels and on the activation of calcineurin (Ghetti and Heinemann, 2000). It also requires the heteromeric assembly of GluR6a and GluR6b and the association of calcineurin with the C-terminal domain of GluR6b in the KAR complex (Coussen et al., 2005). The present experiments were designed to examine whether synaptic KARs at hippocampal mossy fiber synapses were prone to a phos-phorylation-dependent regulation and to define whether this resulted in postsynaptic plasticity of KAR–EPSCs.
Materials and Methods
Transverse hippocampal slices (350 μm thick) were obtained from 15- to 21-d-old C57BL/6 mice. Slices were transferred to a recording chamber and continuously superfused with an oxygenated extracellular medium (95% O2 and 5% CO2) containing (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 0.6 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 16 glucose, pH 7.4. Whole-cell voltage-clamp recordings were made at room temperature from CA3 pyramidal cells. The intracellular solution contained the following (in mm): 140 cesium methanesulfonate, 2 MgC2, 4 NaCl, 5 phospho-creatine, 2 Na2ATP, 1 EGTA, 10 HEPES, 0.33 GTP, pH 7.3. For current-clamp recordings, the following intracellular solution was used (in mm): 130 potassium gluconate, 2 MgCl2, 4 NaCl, 5 phospho-creatine, 2 Na2ATP, 1 EGTA, 10 HEPES, 0.33 GTP, pH 7.3. In some experiments, 1 mm EGTA was replaced by 20 mm BAPTA, and in others, 100 μm peptide corresponding to the last 15 aa of GluR6b or to a 15 aa scramble sequence (Coussen et al., 2005) was added to the intracellular solution together with a cocktail of anti-proteases (pepstatin, aprotinin, leupeptin, and pefabloc, 10 μg/ml each). Neurons were voltage clamped at −80 mV, unless otherwise indicated, and bicuculline (10 μm) was present in the superfusate of all experiments. The access resistance was <20 MΩ, and results were discarded if it changed by >20%.
Mossy fiber (Mf)–EPSCs were evoked by minimal intensity stimulation (Marchal and Mulle, 2004) and were characterized by a marked facilitation after switching stimulus frequency from 0.1 to 1 Hz and by their sensitivity to group 2/3 mGluR agonist LCCG-1 [(2S,1S,2S)-2-(carboxycyclopropyl)glycine] (10 μm). Recordings were made using an EPC 9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) and were filtered at 0.5–1 kHz, digitized at 5 kHz, and stored on a personal computer for additional analysis (IGOR PRO 5.0; WaveMetrics, Lake Oswego, OR). Values are presented as mean ± SEM. Either a paired or unpaired Student's t test was used to define statistical differences between values.
All drugs were obtained from Tocris Bioscience (Ballwin, MO) or Sigma (St. Louis, MO). SR141716A [N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-pyrazole-3-carboxamide] was kindly provided by Olivier Manzoni (INSERM, University of Bordeaux 2).
Results
We examined the possibility that synaptic NMDARs could regulate the function of KARs at synapses between mossy fibers and CA3 pyramidal cells. Mf–EPSCs were evoked in CA3 pyramidal cells at minimal stimulation intensity in the presence of bicuculline (10 μm) to block GABAA receptors, and unless otherwise indicated, GYKI 53655 (50 μm) to block AMPARs. Under these conditions (with 0.6 mm Mg2+ in the extracellular medium), Mf–EPSCs recorded at −80 mV were blocked (>95% inhibition) by CNQX (25 μm; data not shown). In GluR6−/− mice in which KAR–EPSCs are abolished (Mulle et al., 1998), residual Mf–EPSCs of small amplitude could be recorded at −80 mV. These residual EPSCs that corresponded to the NMDA receptor component of the Mf synaptic response only represented 7.5 ± 1.8% (n = 8) of the amplitude of Mf–EPSCs recorded in wild-type mice. Thus, at this membrane potential in the presence of GYKI 53655, Mf–EPSCs are in great part mediated by KARs. In response to a train of five pulses at 20 Hz, the amplitude of KAR–EPSCs during the train increased and remained potentiated 500 ms after the train because of presynaptic facilitation (Salin et al., 1996; Ito et al., 2004) (Fig. 1A).
Figure 1.

Short-term depression of KAR–EPSCs at mossy fiber synapses. A, Representative traces illustrating short-term depression of mossy fiber KAR–EPSCs in CA3 pyramidal cells. A train of five stimulation pulses at 20 Hz was delivered to mossy fibers, followed by a test stimulation (Δt = 500 ms). The train was either given with the cell membrane potential being held at −80 mV [control train (ctr)] or at −40 mV [conditioning train (cond)]. The amplitudes of test KAR–EPSCs after a control (EPSCctr) or a conditioning (EPSCcond) train were compared (enlarged traces are superimposed in inset). B, Average time course of normalized mossy fiber KAR–EPSCs in response to the test stimulus. The KAR–EPSC value was normalized to the average of the amplitudes of KAR–EPSCs obtained in control conditions for the first four KAR–EPSCs in each recording. Arrows indicate the time when the conditioning train was applied. C, Time course of depression of KAR–EPSCs by the conditioning train of mossy fiber stimulation. Time between the end of the train and the control stimulus (Δt) was varied between 0.2 and 20 s.
To examine the consequences of synaptic activation of NMDARs on KAR–EPSCs, we depolarized the membrane to −40 mV while applying the train of five stimulation pulses at 20 Hz (conditioning train). The membrane potential was set back to −80 mV 50 ms after the last stimulus in the train, and a test stimulus was given 500 ms thereafter (Fig. 1A, bottom). At −40 mV, Mf–EPSCs were only inhibited by 25 ± 10% (n = 3) by CNQX, indicating that NMDARs largely contribute to Mf–EPSCs under these conditions. We compared the amplitude of KAR–EPSCs in response to a test stimulus given 500 ms after a control train (membrane potential held at −80 mV) or after a conditioning train. The conditioning train led to the decrease of KAR–EPSC amplitude by 32 ± 3% (n = 11) (Fig. 1A,B). The depression of KAR–EPSCs was transient and highly reproducible during repetition of the conditioning train (Fig. 1B). Depolarization of the cell membrane to −40 mV in the absence of synaptic stimulation did not significantly affect the amplitude of KAR–EPSCs (102 ± 6% of control; n = 8). By varying the interval between the train and the test stimulus, we observed that depression of KAR–EPSCs by a conditioned train developed progressively from almost no effect at 0.2 s to a peak at 1 s (Fig. 1C). KAR–EPSCs returned to control levels with an interval of 20 s. Changing the parameters of the conditioning protocol by increasing the number of stimulation pulses to 10 or increasing the rate of stimulation within the train to 50 Hz did not further increase the extent or time course of KAR–EPSC depression (10 stimulation pulses at 20 Hz, 25 ± 4%, n = 7; 5 stimulation pulses at 50 Hz, 28 ± 2%, n = 4).
Short-term depression of KAR–EPSCs requires changes in the membrane potential of the postsynaptic neuron. This could result in changes in the properties of postsynaptic KARs. Alternatively, depression could be induced postsynaptically and expressed presynaptically through the release of a retrograde messenger, such as what is observed for depolarization-induced suppression of excitation (Diana and Marty, 2004). In this case, depression should affect similarly both AMPAR- and KAR-mediated EPSCs. We thus examined the effect of a conditioning train on AMPAR–EPSCs (in the absence of GYKI 53655) (Fig. 2A). A control train induced post-tetanic potentiation of AMPAR–EPSCs (increase by 434 ± 81%, n = 8) when comparing the first EPSC in the train and the test EPSC (EPSCctr). The amplitude of the test AMPAR–EPSC was not significantly different (p > 0.05) in response to a conditioning train (Fig. 2A,B), indicating that the conditioning protocol selectively affects KAR function and strongly suggesting that the mechanisms of depression do not involve an alteration of glutamate release. In support of this notion, pharmacological manipulations that are known to increase or decrease mossy fiber synaptic transmission at a presynaptic level did not affect the extent of KAR–EPSC depression (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). For instance, activation of adenosine A1 receptors tonically depresses Mf–EPSCs by acting at a presynaptic level (Nicoll and Schmitz, 2005). Accordingly, bath application of the A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (100 nm) increased Mf–EPSC amplitude (153 ± 17% of control Mf–EPSC amplitude; n = 5), without affecting the extent of depression of KAR–EPSCs (29 ± 5%; n = 5). Conversely, the A1 receptor agonist CADO (1 μm) decreased Mf–EPSC amplitude (to 39 ± 9% of control Mf–EPSC amplitude; n = 7) but did not change the depression of KAR–EPSCs (30 ± 3%; n = 7). Finally, forskolin (10 μm), which is known to increase mossy fiber synaptic transmission by acting at a presynaptic level (Nicoll and Schmitz, 2005) (236 ± 15 of control Mf–EPSC amplitude; n = 4), did not affect the depression of KAR–EPSCs (27 ± 5%; n = 7).
Figure 2.
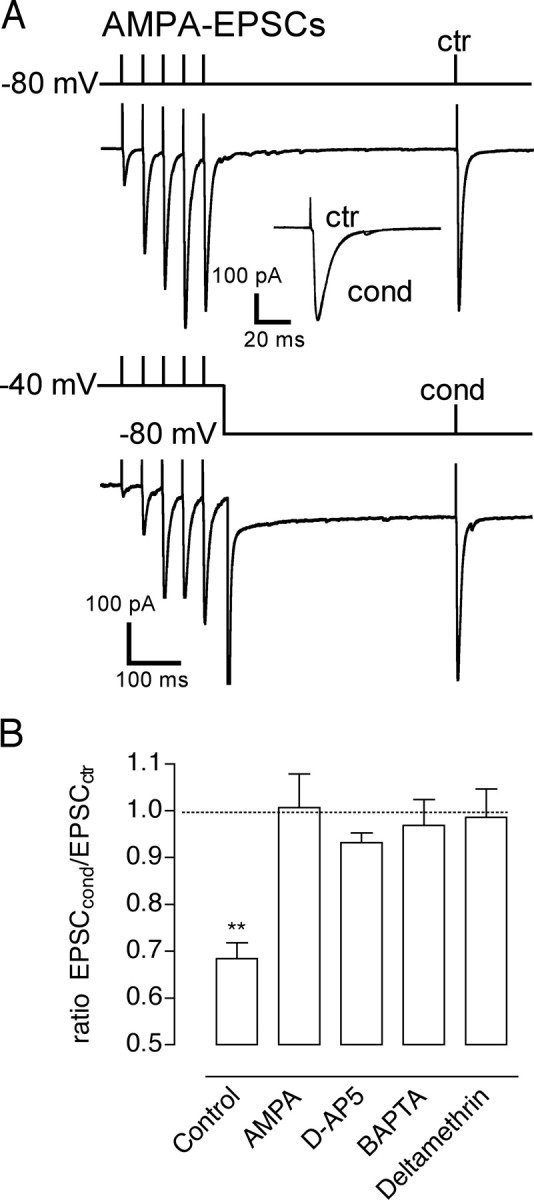
Short-term depression of KAR–EPSCs involves NMDA activation, postsynaptic calcium rise, and calcineurin activation. A, Effects of the conditioning train on AMPAR–EPSCs. Enlarged traces for EPSCctr and EPSCcond are superimposed in inset. ctr, Control train; cond, conditioning train. B, Summary of the effects of the conditioning train in different experimental conditions. The conditioning train depressed KAR–EPSCs (Control), but not AMPAR–EPSCs, and was abolished in presence of the NMDAR antagonist d-AP5 (50 μm), or when BAPTA (20 mm) or the phosphatase calcineurin inhibitor deltamethrin (100 μm) were included in the patch pipette. **p < 0.01.
Short-term depression of KAR–EPSCs was not observed in conditions in which NMDARs were blocked by d-AP5 (50 μm) (depression by 7 ± 2%, n = 7) and in conditions in which intracellular Ca2+ rise in the postsynaptic cell was prevented by the fast Ca2+ chelator BAPTA (20 mm) (depression of KAR–EPSC by 3 ± 6%; n = 4) (Fig. 2B). It was reported that NMDA application on cultured neurons decreases KAR-mediated inward currents by the activation of the phosphatase calcineurin (Ghetti and Heinemann, 2000). When the calcineurin inhibitor deltamethrin (500 nm) was included in the recording pipette, the conditioning protocol did not significantly affect KAR–EPSCs (depression by 1 ± 6%; n = 6) (Fig. 2B). Recovery from depression was strongly reduced when the calcium/calmodulin-dependent protein kinase II (CaMKII) antagonist KN-62 (50 μm), was present in the patch pipette (at Δt = 20 s after the train of stimulation, depression of KAR–EPSC by 21 ± 7%; n = 4). Thus, synaptic activation of NMDARs leads to short-term depression of KAR–EPSCs that appears to depend on the influx of Ca2+ in the postsynaptic pyramidal cell and on the activation of the phosphatase calcineurin.
We recently proposed that binding of calcineurin to GluR6b was necessary for the inhibition of kainate-evoked currents by activation of NMDARs in cultured hippocampal neurons (Coussen et al., 2005). Assuming that postsynaptic KARs at mossy fiber synapses might be composed of both GluR6a and GluR6b, we tested whether GluR6b was also determinant for the NMDAR-induced short-term depression of KAR–EPSCs. We first tested the effects of blocking GluR6b interaction with calcineurin by inclusion of a GluR6b C-terminal peptide (pepR6b) (Coussen et al., 2005) in the recording pipette (Fig. 3A). Dialysis of pepR6b in CA3 pyramidal cells did not affect the average amplitude of KAR–EPSCs during the time of the experiment (Fig. 3C,D). Short-term depression of KAR–EPSCs by a conditioning train was significantly attenuated in the presence of pepR6b (Fig. 3A,E) (depression by 11 ± 3%; n = 8; p < 0.05) but not in the presence of a scramble peptide (see experimental procedures) (depression of 30 ± 2%; n = 6) (Fig. 3E). We also tested the effects of synaptic NMDAR activation on KAR–EPSC depression in mice expressing GluR6a but not GluR6b. For this, we used transgenic mice that express myc-GluR6a in hippocampal neurons on a GluR6−/− background (Coussen et al., 2002). As previously shown, the myc-GluR6a transgene rescued the loss of functional synaptic KARs that is observed in GluR6−/− mice (Fig. 3B). The conditioning train failed to induce a significant depression of KAR–EPSCs in CA3 pyramidal cells from mycGluR6a × GluR6−/− mice (9 ± 9%, n = 6) (Fig. 3B,E). This set of experiments lends additional support to the notion that binding of calcineurin to the C-terminal domain of GluR6b subunit is essential for phosphatase activity on the KAR complex that leads to short-term depression of KAR–EPSCs.
Figure 3.

GluR6b subunit is necessary for NMDA-induced short-term depression of KAR–EPSCs. A, Representative traces illustrating short-term depression of mossy fiber KAR–EPSCs in CA3 pyramidal cells obtained when a GluR6b C-terminal peptide (pepR6b), which blocks the GluR6b interaction with calcineurin, was included in the patch pipette. B, Effects of the conditioning train (cond) in slices from transgenic mice expressing GluR6a but not GluR6b (R6a tg × R6−/−). ctr, Control train. C, Summary plot of the results obtained when interfering with the GluR6b subunit. Blocking GluR6b interaction with calcineurin by inclusion of pepR6b in the recording pipette, but not its scramble version, or working with slices from R6a tg × R6−/− mice greatly attenuated short-term depression of KAR–EPSCs induced by NMDAR activation. **p < 0.01. D, Summary plot of KAR–EPSCs amplitudes obtained in control conditions and in conditions in which the pepR6b was present in the patch pipette. E, Average time course of normalized mossy fiber KAR–EPSCs obtained when the pepR6b was present in the patch pipette. The KAR–EPSC value was normalized to the average of the amplitudes of KAR–EPSCs obtained in control conditions for the first four KAR–EPSCs in each recording. Arrows indicate the time when the conditioning train was applied. The presence of the pepR6b in the patch pipette did not affect the time course of control KAR–EPSCs, but it strongly reduced the depression induced by the conditioning train.
From a functional point of view, it is important to understand whether short-term depression is homosynaptic or heterosynaptic. For this, we stimulated with minimal intensity two independent mossy fiber synaptic pathways by using two electrodes positioned in distinct regions of the dentate granule cell region. This experimental setup provides conditions for maximizing the likelihood that the two mossy fiber pathways were independent. As a confirmation, we checked that there was no cross-potentiation between the two synaptic inputs stimulated (stim 1 or stim 2) as expected for two independent inputs: stimulation of input 1 did not cause any increase in the amplitude of Mf–EPSCs triggered by stimulation of input 2 (and vice versa) (Fig. 4A). A conditioning train to the first stimulating electrode (stim 1) applied at −40 mV did not induce any significant depression of KAR–EPSCs evoked in response to the second stimulating electrode (stim 2) (depression by 1 ± 7%; n = 8) (Fig. 4B, right, C), whereas a similar conditioning protocol in which both the conditioning and the test stimuli were applied to the same mossy fiber pathway led to the depression of KAR–EPSCs (Fig. 4B, left, C). These data indicate that short-term depression of KAR–EPSCs by synaptic NMDARs can be induced homosynaptically and thus requires the close localization of these two receptor populations.
Figure 4.

Short-term depression of KAR–EPSCs is homosynaptic. A, Representative traces of mossy fiber–EPSCs illustrating the protocol used to verify whether two independent mossy fibers were being stimulated. Two glass stimulating electrodes were positioned at different loci in the dentate gyrus [stimulation (stim) 1 and stim 2]. Prominent paired-pulse facilitation is observed for both mossy fiber synaptic inputs applied independently (stim 1 and stim 2). However, no potentiation to stim 1 is observed after stim 2 (and vice versa). The two fibers stimulated by stim 1 and stim 2 were considered to be independent. B, Homosynaptic: the conditioning protocol was applied to the same fiber as the test stimulation leading to short-term depression of KAR–EPSCs. Heterosynaptic: for two independent stimulations, applying the conditioning train to stim 1 was without any effect on KAR–EPSCs evoked in response to stim 2. The corresponding superimposed traces are enlarged in inset. ctr, Control train; cond, conditioning train. C, Summary plot of the results obtained when stimulating two different mossy fibers, testing whether applying the conditioning protocol to one mossy fiber could also induce the short-term depression of KAR–EPSCs recorded from stimulation of a second and independent mossy fiber. *p < 0.05.
To address the physiological relevance of this phenomenon, we performed experiments in the current-clamp mode. Because influx of Ca2+ through NMDARs is needed for short-term depression of KAR–EPSCs, we used a conditioning protocol whereby synaptic NMDARs are relieved from the Mg2+ block through transient depolarization applied in a timely manner with the synaptic release of glutamate. For this, we paired mossy fiber stimulation to a short depolarizing pulse that triggered action potentials (2 ms, 800 pA) given 10 ms after each mossy fiber stimulation in the train. The depolarization resulting from the action potential during activation of NMDARs by synaptically released glutamate will largely favor the opening of NMDAR channels. We recorded a test EPSP 2 s after this conditioning train (at this point the membrane potential was on average −66.7 ± 2.9 mV, n = 7, in control and −66.8 ± 2.8 mV, n = 7, after a conditioning train). This pairing protocol decreased the amplitude of the test KAR–EPSP compared with a control train (no pairing) (depression by 21 ± 5%; n = 7; p < 0.05) (Fig. 5A,C). In contrast, if the short depolarizing pulse was given 10 ms before depolarization, no change in the amplitude of the test KAR–EPSP was observed (Fig. 5B,C). Depression of KAR–EPSPs was prevented when BAPTA (20 mm) was included in the recording pipette (depression by 5 ± 2%; n = 6) and was inhibited by d-AP5 (3 ± 4%, n = 11) (Fig. 5B). Thus, coincidence between action potential discharge and synaptic stimulation at mossy fiber synapses leads to short-term plasticity of KAR–EPSPs through activation of synaptic NMDARs.
Figure 5.

Pairing mossy fiber stimulation with postsynaptic depolarization depresses KAR–EPSPs. A, Current-clamp recordings from CA3 pyramidal cells recorded in the current-clamp mode in the presence of 1.3 mm extracellular Mg+2. Evoking spikes with short depolarizing pulses (2 ms, 800 pA) 10 ms after mossy fiber stimulation (post depol) induced depression of KAR–EPSPs. B, If the depolarizing pulses were each applied 10 ms before mossy fiber stimulation (pre depol), no significant effect of the protocol on KAR–EPSPs was observed. ctr, Control train; cond, conditioning train. C, Summary of the effects of the protocol used in A on KAR–EPSPs in control conditions, in conditions in which 20 mm BAPTA was included in the patch pipette or after inhibition of NMDARs with 50 μm d-AP5, and with the conditions used in B. *p < 0.05
Although a short train of action potentials alone (five spikes) was ineffective in triggering short-term depression of KAR–EPSPs, influx of Ca2+ through voltage-gated channels after a much larger train of action potentials (30 spikes) similarly led to the depression of KAR–EPSPs (depression by 31 ± 2%; n = 3) (Fig. 6B,C) in an NMDA-independent manner (depression by 29 ± 8%; n = 3 in the presence of 50 μm d-AP5). We studied the relevance of this 30% reduction of KAR–EPSPs in more physiological conditions of synaptic transmission, in the absence of the AMPA receptor antagonist GYKI 53655. We stimulated mossy fibers with a train of five stimuli and compared EPSPs obtained in control conditions versus EPSPs obtained after applying the conditioning protocol that consisted of a train of 30 spikes induced by current injection, before synaptic stimulation. This conditioning protocol did not change the amplitude of the first EPSP in the train (Fig. 6D,E), suggesting that the train of 30 spikes did not have any presynaptic effects, nor did it affect AMPA receptors. However, we observed a clear change in the waveform of Mf–EPSPs evoked by a train of five stimuli (Fig. 6D). The kinetic difference is subtle for a single Mf–EPSP, but for subsequent stimuli, a decrease in the decay of Mf–EPSPs is observed that changes summation properties likely attributable to the 30% decrease in KAR–EPSC amplitude. After four stimuli, summation of Mf–EPSPs is significantly decreased (decrease of 37 ± 7%; n = 6) (Fig. 6D, parameter “a,” F), leading in several instances to a difference in the capacity to trigger action potentials (data not shown). This effect was not attributable to the calcium-dependent inactivation of NMDA receptors (Tong et al., 1995) because we could observe the same decrease in Mf–EPSPs summation in the presence of d-AP5 (decrease of 40 ± 5%; n = 5; data not shown).
Figure 6.

Depression of KAR–EPSPs can also be induced by a large train of action potentials and is important for summation properties of mossy fiber EPSPs. A, Applying a train of five spikes without pairing with synaptic stimulation does not change the amplitude of KAR–EPSPs. B, In contrast to what we observed with a train of five spikes, a train of 30 spikes induces a clear reduction in KAR–EPSP amplitude. C, Summary of the effects of trains of 5, 10, or 30 spikes on KAR–EPSPs. The decrease of KAR–EPSPs induced by the train of 30 spikes also occurred in the presence of 50 μm d-AP5. D, Representative traces of Mf–EPSPs obtained without blocking AMPA receptors in control conditions (ctr; black) and after applying a train of 30 spikes (cond; gray). In conditions in which KAR–EPSPs are depressed by ∼30%, the train of EPSPs displayed significantly less summation (measured as a) compared with the control train of EPSPs. E, The train of 30 spikes did not significantly change the amplitude of the first stimulus, suggesting that this protocol did not have any presynaptic effects, nor did it affect AMPA receptors. F, Summary plot of the effect of a train of 30 spikes in the summation (measured in A as indicated in D) of EPSPs obtained after stimulating mossy fiber with a train of five stimuli. *p < 0.05.
Discussion
Short-term changes in synaptic strength are commonly thought to be mediated by presynaptic mechanisms such as Ca2+ control of release probability or presynaptic vesicle replenishment (Zucker and Regehr, 2002). Here we describe a form of short-term plasticity of KAR–EPSCs at mossy fiber synapses that is induced and expressed postsynaptically, in response to the coincident activation of NMDARs during a short burst of afferent mossy fiber stimulation.
Several lines of evidence, such as the requirement for the activity of postsynaptic NMDARs and Ca2+ increase in the postsynaptic CA3 pyramidal cell, argue in favor of a postsynaptic induction mechanism for short-term depression of KAR–EPSCs. Nevertheless, it could be argued that short-term plasticity of KAR–EPSCs is expressed presynaptically. Indeed, postsynaptic depolarization (possibly in conjunction with activation of postsynaptic NMDARs) can release retrograde messengers, such as endocannabinoids, that can act presynaptically to cause short-term depression of transmitter release (Alger, 2002; Diana and Marty, 2004). We do not favor this type of mechanism for several reasons. First, short-term depression does not affect AMPAR–EPSCs, as would be expected if glutamate release was involved in the short-term depression of KAR–EPSCs. In addition, pharmacological manipulations that alter Mf–EPSCs (including KAR–EPSCs) by acting at a presynaptic level are without any effect on the extent of short-term depression of KAR–EPSCs. Finally, short-term depression of KARs by synaptic NMDARs is not observed in transgenic mice with postsynaptic KARs containing GluR6a and not GluR6b, as well as with a postsynaptic infusion of the GluR6b peptide in pyramidal cells. Thus, this form of short-term depression of synaptic transmission is both induced and expressed postsynaptically.
We propose that the mechanism of short-term depression of KAR–EPSCs involves the Ca2+-dependent activation of the phosphatase calcineurin. Previous studies have shown that kainate-activated currents in hippocampal cultured neurons are depressed by NMDARs and by voltage-dependent Ca2+ channels through activation of calcineurin (Ghetti and Heinemann, 2000). We give additional evidence for such a mechanism under physiological conditions of activation of synaptic NMDARs. Our experimental protocol allows to finely detail the time course of inhibition of KAR–EPSCs, which appears to develop progressively over the first second after NMDAR activation and to recover within 20 s. This time course is reminiscent of the calcineurin-dependent regulation of desensitization of synaptic NMDARs (Tong et al., 1995), which can thus regulate their own activity as well as the activity of KARs. It appears that Ca2+ influx through the KAR channel itself is not sufficient to activate the calcium-dependent enzyme calcineurin, possibly because of the low calcium permeability of synaptic KARs that might be predominantly edited (Castillo et al., 1997). As in hippocampal cultured neurons, the recovery from depression of KAR–EPSCs could be prevented by an inhibitor of CaMKII. Overall, these data favor a mechanism whereby synaptic KARs exist, at least partly, in a phosphorylated form and can be dephosphorylated by the calcium-dependent activation of calcineurin, leading to decreased receptor channel activity (Traynelis and Wahl, 1997). It is not yet clear whether one of the KAR subunits of postsynaptic KARs at mossy fiber synapses is prone to phosphorylation by Ca2+-calmodulin kinase II, or whether it is instead a KAR-associated protein. Nevertheless, our data lend further support to the notion that the regulation of KAR function requires the binding of calcineurin to GluR6b (Coussen et al., 2005). Because of its rapid onset and the similarity with studies on recombinant receptors (Traynelis and Wahl, 1997), which implicate a decrease in the opening probability of KAR channels, short-term depression of KAR–EPSCs is likely attributable to a decrease in the function of synaptic KARs, although the removal of KARs from the postsynaptic membrane, a mechanism suggested to occur for long-term depression of synaptic KARs in the perirhinal cortex (Park et al., 2006), cannot be excluded.
Short-term depression of synaptic KARs can occur through a local and synapse-specific mechanism requiring an increase in intracellular Ca2+ by synaptic NMDARs that are likely closely localized with synaptic KARs. However, because mossy fiber synapses have multiple release sites, it remains possible that a bulk increase of Ca2+ in the postsynaptic spine is required for the regulation of KARs. Short-term depression of KAR–EPSPs is triggered by the coordinated firing of action potentials and mossy fiber synaptic stimulation only when action potential fires during the glutamatergic EPSC. It thus represents a novel form of spike-timing-dependent plasticity (Markram et al., 1997; Dan and Poo, 2004).
Although EPSCs mediated by KARs have a much smaller amplitude than that mediated by AMPARs, they also display much slower kinetics and prominent summation properties (Castillo et al., 1997). These features allow KAR–EPSPs to play a prominent role in information transfer in CA1 interneurons by imposing a substantial depolarization during sustained afferent activity (Frerking and Ohliger-Frerking, 2002). In agreement with the important role of kainate receptors for summation properties of EPSPs (Frerking and Ohliger-Frerking, 2002), we observed that a 30% reduction of KAR–EPSPs is sufficient to induce a significant change in the summation of mossy fiber–EPSPs. This indicates that the short-term depression described here might have important physiological consequences mainly during sustained afferent activity controlling CA3 pyramidal cell depolarization. The precise role of kainate receptors in signal integration at mossy fiber–CA3 pyramidal cell synapses is far from being completely understood. Nevertheless, short-term depression of KAR–EPSCs in conditions of high afferent activity leading to spike discharge is expected to exert a feedback inhibition on the sustained depolarization induced by KAR–EPSPs. Interestingly, it was reported that KAR–EPSCs show attenuated forms of presynaptic plasticity compared with AMPAR–EPSCs (Ito et al., 2004). Both mechanisms will concur in preventing the overactivation of synaptic KARs under conditions of sustained network activity and might thus favor information processing in conditions of short bursts of afferent stimulation.
Footnotes
This work was supported by Fundaçao para a Ciência e para a Tecnologia Grant POCI/NEU-SAU/59135/2004, by the Centre National de la Recherche Scientifique, the Ministère de la Recherche of France, the Conseil Régional d'Aquitaine, the Fondation pour la Recherche Médicale (fellowship to S.S.), and the European Commission (EUSynapse, contract number LSH-2004-019055 to C.M.). We thank Paulo Pinheiro for criticial reading of this manuscript.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Cho K, Francis JC, Hirbec H, Dev K, Brown MW, Henley JM, Bashir ZI. Regulation of kainate receptors by protein kinase C and metabotropic glutamate receptors. J Physiol (Lond) 2003;548:723–730. doi: 10.1113/jphysiol.2003.040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Sailer AW, Darstein M, Maron C, Xu J, Swanson GT, Heinemann SF. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2−/− mice. J Neurosci. 2003;23:422–429. doi: 10.1523/JNEUROSCI.23-02-00422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussen F, Normand E, Marchal C, Costet P, Choquet D, Lambert M, Mege RM, Mulle C. Recruitment of the kainate receptor subunit glutamate receptor 6 by cadherin/catenin complexes. J Neurosci. 2002;22:6426–6436. doi: 10.1523/JNEUROSCI.22-15-06426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussen F, Perrais D, Jaskolski F, Sachidhanandam S, Normand E, Bockaert J, Marin P, Mulle C. Co-assembly of two GluR6 kainate receptor splice variants within a functional protein complex. Neuron. 2005;47:555–566. doi: 10.1016/j.neuron.2005.06.033. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Diana MA, Marty A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) Br J Pharmacol. 2004;142:9–19. doi: 10.1038/sj.bjp.0705726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Ohliger-Frerking P. AMPA receptors and kainate receptors encode different features of afferent activity. J Neurosci. 2002;22:7434–7443. doi: 10.1523/JNEUROSCI.22-17-07434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti A, Heinemann SF. NMDA-dependent modulation of hippocampal kainate receptors by calcineurin and Ca(2+)/calmodulin-dependent protein kinase. J Neurosci. 2000;20:2766–2773. doi: 10.1523/JNEUROSCI.20-08-02766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbec H, Francis JC, Lauri SE, Braithwaite SP, Coussen F, Mulle C, Dev KK, Couthino V, Meyer G, Isaac JT, Collingridge GL, Henley JM. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron. 2003;37:625–638. doi: 10.1016/s0896-6273(02)01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE. Kainate receptors and synaptic transmission. Prog Neurobiol. 2003;70:387–407. doi: 10.1016/s0301-0082(03)00122-9. [DOI] [PubMed] [Google Scholar]
- Ito K, Contractor A, Swanson GT. Attenuated plasticity of postsynaptic kainate receptors in hippocampal CA3 pyramidal neurons. J Neurosci. 2004;24:6228–6236. doi: 10.1523/JNEUROSCI.1302-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT. Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature. 1999;400:569–573. doi: 10.1038/23040. [DOI] [PubMed] [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- Marchal C, Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol (Lond) 2004;561:27–37. doi: 10.1113/jphysiol.2004.069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Mulle C, Andreas S, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate induced seizures in GluR6-deficient mice. Nature. 1998;392:601–604. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Park Y, Jo J, Isaac JT, Cho K. Long-term depression of kainate receptor-mediated synaptic transmission. Neuron. 2006;49:95–106. doi: 10.1016/j.neuron.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci. 2005;25:11710–11718. doi: 10.1523/JNEUROSCI.4041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin P, Scanziani M, Malenka R, Nicoll R. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci USA. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wahl P. Control of rat GluR6 glutamate receptor open probability by protein kinase A and calcineurin. J Physiol (Lond) 1997;503:513–531. doi: 10.1111/j.1469-7793.1997.513bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]