

Abstract
The homeostasis of amyloid-β (Aβ) in the brain is critical to the pathogenesis of Alzheimer's disease (AD). Aβ is a fragment of amyloid-β precursor protein (APP) generated in neurons by two proteases, β- and γ-secretases. APP and β-secretase, both present on cell surface, are endocytosed into endosomes to produce Aβ. The molecular mechanism by which neurons trigger the production of Aβ is poorly understood. We describe here evidence that the binding of lipid-carrying apolipoprotein E (ApoE) to receptor apolipoprotein E receptor 2 (ApoER2) triggers the endocytosis of APP, β-secretase, and ApoER2 in neuroblastoma cells, leading to the production of Aβ. This mechanism, mediated by adaptor protein X11α or X11β (X11α/β), whose PTB (phosphotyrosine-binding) domain binds to APP and a newly recognized motif in the cytosolic domain of ApoER2. Isomorphic form ApoE4 triggers the production of more Aβ than by ApoE2 or ApoE3; thus, it may play a role in the genetic risk of ApoE4 for the sporadic AD. The mechanism, which functions independently from Reelin–ApoER2 interaction, also provides a link between lipid uptake and Aβ production, which may be important for the regulation of neuronal activity.
Keywords: amyloid-β precursor protein, apolipoprotein E, amyloid-β, β-secretase, endocytosis, Alzheimer's disease
Introduction
An excess brain level of amyloid-β (Aβ) is a major factor in the pathogenesis of Alzheimer's disease (AD) (Selkoe, 2001). Aβ is formed from proteolysis of amyloid-β precursor protein (APP) by β-secretase [BACE1 (β-site APP-cleaving enzyme 1); memapsin 2] (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Lin et al., 2000) and γ-secretase. APP and β-secretase, an aspartic protease with an optimal activity around pH 4 are both present on the cell surface, where at pH 7, β-secretase has only minimal activity (Lin et al., 2000). Both proteins are endocytosed into endosomes, a major cellular site for APP cleavage and Aβ production (Koo and Squazzo, 1994; Cook et al., 1997; Hartmann et al., 1997). β-Secretase is then recycled back to the cell surface (Huse et al., 2000; Walter et al., 2001). Therefore, β-secretase traffics between an active and a relatively inactive compartment to facilitate the production of Aβ. The mechanism and regulation for the endocytosis of both proteins may influence cellular Aβ production and pathogenesis of AD.
Cellular trafficking of APP and β-secretase are mediated by interactions of motifs in their cytosolic domains with adaptor proteins. The endocytosis of APP is facilitated by the interaction of a YENPTY motif with several adaptor proteins including X11α/β and Fe65 (for review, see King and Scott Turner, 2004), both of which are specifically present in the brain. β-Secretase has a “dileucine acid cluster” motif (He et al., 2002, 2003, 2005), which interacts with GGA proteins to facilitate the transport of the protease in the recycling but not the endocytic pathway (He et al., 2005; Wahle et al., 2005), although the two leucines in the motif are required for the latter (Huse et al., 2000; Pastorino et al., 2002). APP has also been shown to be a “carrier” for the endocytosis of the ectodomain of β-secretase, most likely by the interaction of the proteins in the assembly of endocytic vesicles on the cell surface (X. P. Huang et al., 2004).
Apolipoprotein E (ApoE) polymorphism is associated with the sporadic form of AD for which ApoE4, but not ApoE2 or ApoE3, is a risk factor (for review, see Huang, 2006). The molecular mechanism underlying this association is unclear. One of the possibilities is that ApoE4 supports a high level of Aβ production through a brain-specific receptor. This hypothesis led us to examine the influence of ApoE and the role of ApoE receptor 2 (ApoER2) in Aβ production in neuronal cells. ApoER2, a member of the low-density lipoprotein (LDL) receptor family, is specifically expressed in the brain. Its physiological function is to bind ApoE-containing lipoprotein particles to import cholesterol and lipids into brain cells. This process is essential for remodeling of neuronal membrane and mediating synaptic plasticity. Here we report that ApoE stimulated Aβ production through a coordinated endocytosis of ApoE, ApoER2, APP, and β-secretase. This mechanism involves adaptor protein X11α/β, which facilitates the formation of a complex among ApoER2, X11, and APP. In this system, ApoE4 produces more Aβ than does ApoE2 or ApoE3.
Materials and Methods
cDNA vectors.
Vectors for human β-secretase and Swedish mutant of APP were described previously (Lin et al., 2000). Vectors for human X11α, X11β, human LDL receptor-related protein (LRP), murine ApoER2, Reelin, and bovine ADAM-10 were kindly provided by Drs. Chistopher C. J. Miller (Kings College London, London, UK), Ben Margolis (University of Michigan, Ann Arbor, MI), Guojun Bu (Washington University, St. Louis, MO), Johannes Nimpf (University of Vienna, Vienna, Austria), Tom Currant (St. Jude Children's Research Hospital, Memphis, TN), and Falk Fahrenholz (Johannes Gutenberg University, Mainz, Germany), respectively. PCR was used for transferring murine ApoER2 gene into pcDNA6 with V5-His-Tag at the C terminus and for creating mutants of murine ApoER2 in pcDNA6. Phosphotyrosine-binding (PTB) domain of X11α (residue 457–643) was amplified by PCR and inserted into pcDNA6. GST constructs of GST–PTB (457–643), GST–postsynaptic density-95/Discs large/zona occludens-1 (PDZ) (650–837) of X11α, and GST constructs of ApoER2 cytosolic domain GST–ApoC1 (881–996), GST–ApoC2 (881–925), GST–ApoC3 (926–996), and GST–APPc (650–695) were amplified by PCR and inserted into plasmid pGEX6.1 (GE Healthcare, Uppsala, Sweden). All constructs were verified by sequencing. GST fusion proteins were produced in Escherichia coli BL21 strain and purified as described previously (He et al., 2003).
Antibodies.
Goat polyclonal antibodies against recombinant pro-β-secretase were affinity-purified using Affigel (Bio-Rad, Hercules, CA)-immobilized recombinant β-secretase. Rabbit polyclonal APP antibody AB5228, AB5352, monoclonal APP antibody MAB1560, monoclonal Reelin antibody MAB5364, and control mouse IgG were obtained from Chemicon (Temecula, CA). Mouse monoclonal antibody for X11α was purchased from BD Biosciences (San Jose, CA). Mouse monoclonal antibody for LRP was purchased from Calbiochem (La Jolla, CA). Monoclonal V5 antibody, hemagglutinin (HA)-tag antibody, Myc antibody, and polyclonal antibody for ApoER2 were purchased from Invitrogen (Carlsbad, CA). Rabbit polyclonal anti-β-actin was purchased from Novus Biologicals (Littleton, CO). Monoclonal apoE and apoE4 antibodies were purchased from MBL (Naka-ku Nagoya, Japan).
Lipoproteins.
ApoE and ApoE4 were from Biodesign (Saco, ME); ApoE, ApoE2, ApoE3, and ApoE4 were from Alpha Diagnostic International (San Antonio, TX). Human plasma lipoprotein fractions were prepared as follows: out-dated plasma was obtained from the Oklahoma Blood Bank and was supplemented with antibiotics, preservatives, and phenylmethylsulfonyl fluoride to prevent oxidative and proteolytic damage (Manchekar et al., 2004). Very low-density lipoprotein (VLDL) [density (d) < 1.006 g/ml], LDL (d 1.006–1.063 g/ml), and high-density lipoprotein (HDL) (d 1.063–1.21 g/ml) were prepared by sequential ultracentrifugation and dialyzed against PBS. The contents of ApoE4 and total ApoE were analyzed for individual VLDL samples using Western blots with monoclonal antibodies. Quantity of bands was determined by optical scanning using STORM Scanner (GE Healthcare) and ImageQuant (Molecular Dynamics, Sunnyvale, CA).
Immunoprecipitation and Western blots.
Human embryonic kidney 293 (HEK293) cells were transfected as described previously (He et al., 2005) with the following combinations of plasmids: ApoER2/X11α, ApoER2/X11β, or ApoER2/X11α/APPsw695. Cells were harvested 36 h after transfection and lysed on ice in lysate buffer [PBS, pH 7.4, with 1% NP-40 and 1% saponin with protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN)]. Cell lysates were cleared by centrifugation and incubated with indicated antibody for 4 h at 4°C. Immune complexes were isolated using protein G-agarose beads (Sigma, St. Louis, MO) and subjected to SDS-PAGE and Western blotting using specific antibodies as indicated.
Pull-down assays.
For GST pull-down, GST fusion proteins were retrieved as described previously (He et al., 2003). The amount of protein recovered with glutathione beads was determined using Bio-Rad Protein Assay and diluted to yield a concentration of 1 mg of protein per milliliter of beads. Cell lysates from HEK293 cells transfected with X11α or X11β plasmids were cleared by centrifugation and aliquots of the supernatant were incubated with the same amount of GST fusion protein bound to glutathione beads in PBS buffer for 4 h at 4°C. For nickel-nitrilotriacetic acid (NI-NTA) pull-down experiments, lysates from HEK 293 cells overexpressed X11α, X11α/ApoER2, X11β, or X11β/ApoER2 were individually incubated with same amount NI-NTA beads (Qiagen, Valencia, CA) in PBS for 4 h at 4°C. The bound proteins on both types of pull-down beads were recovered with SDS sample buffer at boiling temperature and subjected to Western blot.
Cellular Aβ40/Aβ42 production.
Neuroblastoma N2a-APPsw cells (from Dr. Riqiang Yan, Lerner Research Institute, Cleveland, OH) were cultured in 24-well plates in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) with 80 μg/ml G418 1 d before use. Transient transfections were done using FuGENE6 (Roche Diagnostics) and fresh Optimum medium (Invitrogen) was supplied 5 h after transfection. Apolipoproteins were added in the fresh medium at 10 μg/ml. Conditioned medium was collected 24 h later. Aβ40 or Aβ42 was determined in triplicate using Aβ40 or Aβ42 ELISA Kit (Biosource International, Camarillo, CA). For studying the effect of PTB domain on Aβ production, N2a-APPsw cells were transfected with (0.4 μg/per well for 24 well plate) PTB-containing vector before being subjected to the above procedure.
Internalization of cell surface proteins.
N2a-APPsw cells were transiently transfected with ApoER2 and β-secretase or with ApoER2 and ADAM-10 using Lipofectamine 2000. Twenty-four hours after transfection, 10 μg/ml ApoE or ApoE4 was added into medium and incubated for 2 h. Cells were then placed on ice, rinsed in cold PBS, and incubated in PBS containing 1.5 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL) for 20 min at 4°C. Cells were rinsed twice in PBS and lysed in 800 μl of PBS containing 0.1%SDS, 1% NP40, and a complete protease inhibitor mixture. Total protein concentration was determined by immunoblotting from an aliquot of 50 μl of cell lysate. Biotinylated proteins were recovered with NeutrAvidin agarose (50 μl; Pierce), rinsed three times, eluted in 20 μl of SDS sample buffer at boiling temperature, and used for Western blot. Quantity of bands was determined by optical scanning instruments specified above. For the determination of internalized proteins, cells were prepared as above, rinsed in PBS, and then incubated with cold 1.5 mg/ml cleavable biotin reagent in PBS (EZ-Link Sulfo-NHS-SS biotin; Pierce) for 20 min at 4°C. Cells were then rinsed quickly with medium at room temperature, incubated for 15 min at 37°C in control medium or in medium containing 10 μg/ml ApoE or ApoE4, placed on ice, and rinsed in cold stripping buffer (50 mm glutathione, 75 mm NaCl, 75 mm NaOH, 10% FBS, pH 8.5–9.0). Cells were then lysed in 800 μl of PBS containing 0.1% SDS, 1% NP40, and a complete protease inhibitor mixture. After centrifugation, biotinylated proteins were retrieved by incubation with NeutrAvidin agarose (50 μl; Pierce). Isolated proteins were rinsed in buffer three times and boiled in 20 μl of sample buffer for Western blots.
Immunofluorescence studies of cellular proteins.
HeLa cells were seeded onto six-well plates with glass coverslips and expressed the ApoER2/X11α, ApoER2/X11β, or ApoER2/APP constructs for 36 h after transfection. Cells were gently fixed in 4% paraformaldehyde, in PBS, pH 7.4, at room temperature for 15 min. Coverslips were then washed twice for 10 min each in PBS and incubated for 1 h at room temperature with the indicated combinations of primary antibodies diluted in 0.1% BSA, 0.1% saponin, and 0.02% sodium azide in PBS (immunofluorescence buffer). At the end of this period, coverslips were washed twice with PBS followed by incubation for 30 min with the indicated combinations of secondary antibodies diluted in immunofluorescence buffer. Coverslips were again washed twice with PBS and mounted on slides using Vectashield (Vector Laboratories, Burlingame, CA). APP was immunolabeled with Rabbit polyclonal antibody 5352 (Chemicon) and followed by cyanine 3 (Cy3)-conjugated sheep anti-rabbit secondary antibody (Sigma). X11α and X11β were blotted by monoclonal antibodies against them (BD Biosciences) and then recognized by Alexa 488-conjugated donkey anti-mouse secondary antibody (Invitrogen). ApoER2 was labeled either by rabbit polyclonal antibody against His-tag (colocalized with X11) or monoclonal antibody against V5 (colocalized with APP) and then was labeled by either Cy3 or Alexa 488-conjugated secondary antibodies. Images were obtained in an inverted confocal laser-scanning microscope (LSM 510; Zeiss, Oberkochen, Germany).
Small interfering RNA interference of ApoER2.
Double-stranded small interfering RNA (siRNA) specific for the mouse ApoER2 (ON-TARGETplus SMARTpool, catalog number L-046407-00) was chemically synthesized by Dharmacon (Chicago, IL). N2a-APPsw cells grown in 24-well plates for 24 h were transfected with either ApoER2 siRNA or control siRNA (NO-TARGETplus siRNA; Dharmacon) with Oligofectamine (Invitrogen). The medium, Opti-MEM, was replaced 4 h later with ApoE or ApoE4 and cultured for 24 h. Aβ40 in the medium samples were assayed as described above.
Studies on Reelin and Reelin effect.
N2a-APPsw cells grown in a T25 flask were transfected with 5 μg of expression vector pCrl containing the entire mouse Reelin gene using Lipofectamine 2000. Control cells were transfected with blank vector. Cells were conditioned in Opti-MEM for 36 h, and the medium was collected and concentrated. Western blot was performed on concentrated medium and cell lysate using anti-Reelin antibody MAB5364 (Chemicon). To determine the effect of Reelin antibody on Aβ production, N2a-APPsw cells were cultured for 24 h with 10 μg/ml ApoE and either with or without antibody MAB5364. Aβ40 in the medium was determined by ELISA.
Results
ApoE increases Aβ in N2a cells
We observed that Aβ production in Neuro-2a cells stably transfected with human APPswedish (N2a-APPsw) increased up to twofold in the presence of purified human VLDL (p < 0.01), whereas LDL and HDL had no effect (Fig. 1a). Because VLDL is the main lipoprotein fraction containing ApoE (Fig. 1a, inset), we tested the effect of each of the isomorphic ApoE forms and found that the addition of recombinant ApoE2, ApoE3, and ApoE4 to the cell culture at 10 μg/ml increased Aβ production (Fig. 1b) with the highest response by ApoE4. Because these ApoE samples contained no lipids, the differential response may be influenced by their non-native physical properties. We therefore investigated whether ApoE4 from native human VLDL stimulates more Aβ production than either ApoE2 or ApoE3. VLDL was prepared from individual human plasma and the content of total ApoE and ApoE4 was determined. VLDL samples containing different levels of ApoE were chosen for measuring the production of Aβ40 and Aβ42 in N2a-APPsw cells. We observed a linear correlation of ApoE4 content and the production of both Aβ species (Fig. 1c) with similar correlation coefficients. These results indicate that ApoE4 in native lipoprotein particles also stimulates higher Aβ production in N2a cells than those for ApoE2 and ApoE3.
Figure 1.

ApoE increases Aβ production in neuroblastoma N2a-APPsw cells. a, Human VLDL (15 μg/ml) increased Aβ production in N2a-APPsw cells. HDL and LDL (same concentration) had no effect. Aβ in the medium was analyzed after 24 h of culture. Inset, Western blots of ApoE in three lipoprotein fractions. b, Purified human ApoE, recombinant ApoE2, ApoE3, and ApoE4 (each at 10 μg/ml) increased Aβ production in N2a-APPsw cells. Experiment conditions are the same as in a. *p < 0.05; **p < 0.01. c, Correlation of ApoE4 content of human VLDL with Aβ40 and Aβ42 production in N2a-APPsw cells. VLDL samples purified from individuals were analyzed for ApoE4 and total ApoE contents and measured with ELISA for their effect on Aβ40 and Aβ42 production in N2a-APPsw cells. d, Transfect of ApoER2 into N2a-APPsw cells increased ApoE or VLDL induced increase of Aβ production (black columns) over cells transfected with blank vectors (gray columns). The experimental conditions were the same as above. e, ApoER2 siRNA knockdown abolished ApoE- or ApoE4-induced increase of Aβ production (left). Western blots (right) show that the endogenous ApoER2 band was greatly reduced in ApoER2 siRNA knockdown, whereas the control β-actin band was not significantly changed.
ApoER2 mediates Aβ increase by ApoE
Because ApoER2 is a brain-specific receptor for ApoE, we asked whether the ApoE effect on Aβ is mediated by ApoER2. In the first set of experiments, the influence of ApoE and VLDL on Aβ production was compared for N2a-APPsw cells with or without transfection of ApoER2. Cells transfected with ApoER2 produced more Aβ in response to ApoE, recombinant ApoE4, or purified VLDL (Fig. 1d). These results suggest that ApoER2 mediated the increase of Aβ production in response to ApoE. ApoE also caused Aβ to increase in cells transfected by blank vector (Fig. 1d), which provided an opportunity to study whether the increase was mediated by endogenous ApoER2. In the second series of experiments, we used siRNA to knock down ApoER2 and examined Aβ production in response to the various ApoE species. Figure 1e shows that only residual expression of endogenous ApoER2 remained in knockdown cells, which lost approximately one-fourth of their Aβ production and which did not increase after the addition of ApoE or ApoE4 to the media (Fig. 1d). These observations indicate that ApoER2 mediates ApoE-stimulated Aβ production in N2a-APPsw cells.
ApoE triggers the endocytosis of ApoER2, APP, and β-secretase
A possible explanation for ApoE stimulation of Aβ production is that ApoE triggers the internalization of not only ApoER2, but also APP and β-secretase from cell surface to endosomes. We examined whether the addition of ApoE causes the reduction of these proteins from the cell surface. N2a-APPsw cells were treated with ApoE or ApoE4 for 2 h and then biotinylated to label the cell-surface proteins. Biotinylated ApoER2, APP, and β-secretase were recovered and revealed in Western blot. Compared with controls, the remaining cell-surface ApoER2, APP, and β-secretase decreased by ∼25, 25, and 7%, respectively, when ApoE was added and 50, 50, and 35%, respectively, when ApoE4 was present (Fig. 2a). The total amounts of the three proteins in the cell lysates remained approximately the same. Under identical conditions, cell-surface ADAM-10 (known to be an α-secretase) was unchanged in the presence of either ApoE or ApoE4 (Fig. 2b), indicating the specificity of ApoE induced changes. We then determined whether ApoEs increased these three proteins inside the cells. Cell-surface proteins were pulse labeled by biotinylation and permitted to internalize for 15 min. Biotinyl groups remained at cell surface were stripped, and the biotinylated ApoER2, APP, and β-secretase inside the cells were retrieved and visualized in Western blot. We observed small increases of ApoER2 and β-secretase in response to ApoE and ApoE4 (Fig. 2c), whereas the increase of APP was much greater, ∼2.5 and fourfold in response to ApoE and ApoE4, respectively (Fig. 2c). Together, the depletion of cell-surface ApoER2, APP, and β-secretase and the increase of the intracellular pool of these proteins in response to the addition of ApoE are consistent with the mechanism that the binding of ApoE to ApoER2 triggered the endocytosis of all three proteins. To further substantiate that the endocytosed APP was subjected to proteolysis by β-secretase, we also determined in the above experiments the APP C-terminal fragment resulting from β-secretase cut (C99). We observed that C99 was significantly increased in the presence of ApoE and ApoE4 over the control (Fig. 2d). Again, ApoE4 generated more C99 than did ApoE. An increased endocytosis and decreased cell-surface APP stimulated by ApoE should be accompanied by a decrease of APP processing by α-secretase. Western blots of the APP ectodomain generated by α-secretase, sα-APP, confirmed a reduction of sα-APP by ApoE and ApoE4 (Fig. 2e).
Figure 2.

ApoE or ApoE4 induced the internalization of ApoER2, APP, and β-secretase from the cell surface. a, ApoE reduced cell surface ApoER2, APP, and β-secretase. N2a-APPsw cells transfected with ApoER2 and β-secretase were incubated with ApoE or ApoE4 for 2 h. Cell-surface proteins were biotinylated, retrieved with avidin–agarose gel, and subjected to Western blots for ApoER2, APP, and β-secretase (top right). Parallel experiments performed without biotinylation from total cell lysates were subjected to Western blots (top left). Quantitation of the bands was done by scanning densitometer and displayed here as relative intensity as the ratios of the surface protein to the total amount proteins (bottom). Controls (white columns) were set as 1.0 (average of 2 experiments). b, ApoE did not change cell-surface α-secretase (ADAM-10). N2a-APPsw cells transfected with bovine ADAM-10 and then cell-surface ADAM-10 was determined in the absence and presence of ApoE or ApoE4 using the same procedure and experimental conditions as described in a. An example of Western blots shows that neither ADAM-10 (HA-tag; right) nor the total cell lysates (left) was significantly changed in the presence of ApoE or ApoE4. c, ApoE increased intracellular ApoER2, APP, and β-secretase. Cells were prepared as in a, and then the cell-surface proteins were labeled with a cleavable biotinylation reagent at 4°C. The internalization was affected at 37°C for 15 min. After stripping the biotinyl group on cell surface, the cells were lysed, and the biotinylated proteins were retrieved for Western blots as above. The displayed panels are the same as in a except that at the bottom, the amounts of ApoE (black columns) were set as 1.0. d, Western blot of intracellular APP fragment C99 from N2a-APPsw cells in the absence and presence of ApoE. The samples were those described in d, blotted with antibody MAB1560. e, Western blot of soluble APP ectodomain generated by α-secretase (s-APPα) from N2a-APPsw cells in the absence and presence of ApoE. The condition medium from a was immunoprecipitated with MAB1560 and Western blotted with the same antibody.
Role of X11α/β in the ApoE and Aβ relationship
A hypothesis that may explain the ApoE triggered endocytosis of ApoER2 and APP is that their cytosolic domains may interact with a same adaptor protein that regulates cellular transport. The cytosolic domain of APP contains a YENPTY motif known to bind brain-specific adaptor proteins X11α or X11β (Fig. 3a). We seek to determine whether X11α/β also interacts with ApoER2. When ApoER2 was immunoprecipitated from lysates of HEK293 cells transiently expressing ApoER2 and either X11α or X11β, the adaptor proteins were seen in the Western blots of the precipitate (Fig. 3b). Substitution of ApoER2 with LRP did not produce coimmunoprecipitation with X11α or the ApoER2-binding PTB domain of X11α (see below) (Fig. 3c), indicating that X11 binding to ApoER2 is specific rather than a general phenomenon with LDL receptor family. X11α and X11β gave essentially identical results. X11γ was not tested because it is not expressed in the brain. In reverse pull-down, ApoER2 containing a His6 tag in the lysate collected with Ni-affinity gel also recovered X11α/β (Fig. 3d). These results suggest that X11α/β binds ApoER2 with high affinity. Because X11α/β binds both ApoER2 and APP, we tested whether ApoER2 and APP form a complex in non-neuronal HEK293 cells devoid of these endogenous proteins of interest. When the lysates of cells expressing ApoER2, APP, and X11α/β were immunoprecipitated with anti-APP antibody 5228, both APP and X11 were found in the Western blot of the precipitates (Fig. 3e). Again, both X11α and X11β supported the coprecipitation. Cells transfected with ApoER2 and APP but without X11 had virtually no coprecipitation of ApoER2 and APP (Fig. 3e). These results indicate that X11α/β is required for the complex formation involving ApoER2, APP, and X11α/β.
Figure 3.
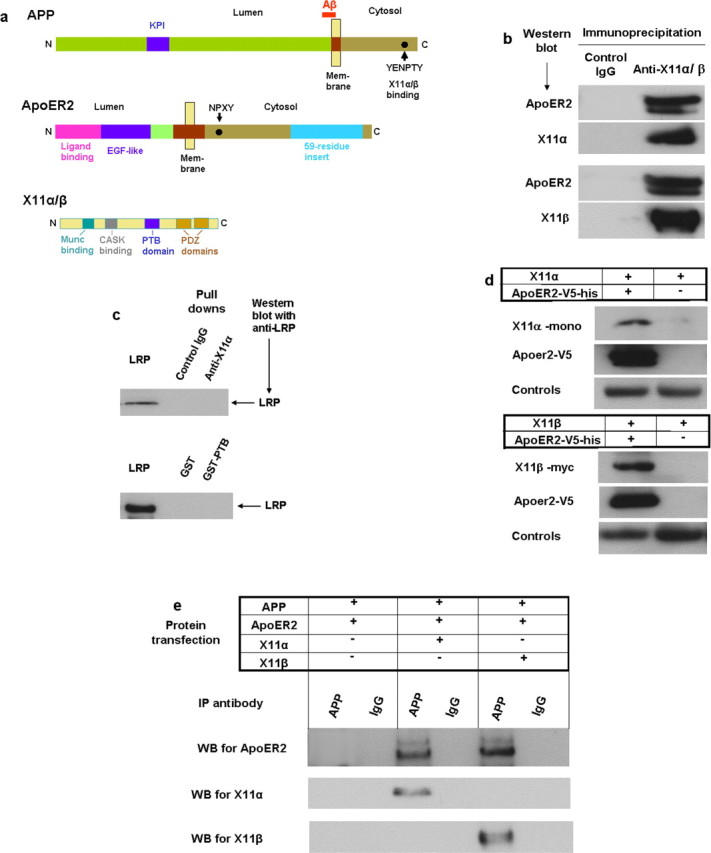
Binding of X11α/β with ApoER2. a, Diagrams of some functional domains of APP, ApoER2, and X11. In APP, a dot denotes the location of the YENPTY motif, which involves in binding of several adaptor proteins including X11α/β. KPI is a Kunitz protease inhibitor domain. In ApoER2, the alternative spliced 59-residue insertion is shown in blue. N, N terminus; C, C terminus. b, ApoER2 is recovered in immunoprecipitation of X11α/β. Lysates of HEK293 cells transiently expressing ApoER2 (with V5 tag) and X11α or X11β (with myc tag) were immunoprecipitated separately with anti-X11α, anti-myc (for X11β), or control IgG and visualized in Western blots with anti-V5, anti-X11α, or anti-myc. ApoER2 band was present in both immunoprecipitation for X11α and X11β. c, LRP did not coimmunoprecipitate with X11α. Experimental conditions are the same as in a. d, Presence of X11α/β in ApoER2 pull-down experiments. Lysates of cells expressing ApoER2 (with both V5 and hexahistidine tags) and X11α/β were subjected to binding by Ni-affinity gel to retrieve ApoER2. Western blots showed that both X11α (top) and X11β (bottom) were recovered. e, ApoER2 and X11α/β are immunoprecipitated (IP) with APP. Lysates of HEK293 cells transiently expressing APP, ApoER2, and X11α or X11β were immunoprecipitated with anti-APP antibody 5228 and visualized by Western blots (WB). ApoER2 was present when X11α/β was expressed (lanes 3 and 5) but was absent when X11 was not expressed (lane 1).
Domain interaction between ApoER2 and X11α/β
PTB domain of X11α but not the PDZ domain was shown to pull down ApoER2 from the cell lysate (Fig. 4a), indicating that it is responsible for ApoER2 binding. To identify the binding motif in ApoER2, various GST fusions from different regions of ApoER2 cytosolic domain were tested for X11α/β pull down. Constructs containing the 59-residue insertion were able to bind X11 (Fig. 4b), indicating that this region contains an X11-binding motif. Various deletions or mutations were made for potential motif sites in the cytosolic domain of ApoER2. Deletions of a NPXY motif (Fig. 4c, mutants 1 and 2) or mutations of two PXXP motifs (Fig. 4c, mutants 3 and 4) did not affect the binding of X11–PTB domain to ApoER2. The deletion of residues YDRPLW (residues 899–904) within the 59-residue insertion abolished X11-PTB pull-down (Fig. 4c, mutant 5). This same deletion also abolished the pull-down of a separately expressed PTB domain of X11α (Fig. 4d). These results indicate that the motif YDRPLW mediates the binding of ApoER2 to the PTB domain of X11α/β.
Figure 4.

Domains and motif involved in ApoER2/X11α/β interaction. a, PTB domain of X11α binds ApoER2. Lysates from cells expressing ApoER2 and GST (glutathione-S-transferase) fusions of PTB or PDZ domains of X11α were subjected to GST pull down using a glutathione affinity column. Western blots showed that ApoER2 was associated with PTB domain (third lane) but not PDZ domains (second lane). b, X11α pull-down by GST (white bar) fusion constructs containing different regions of ApoER2 intracellular regions (gray bar). GST fusion to APP cytosolic domain (slash bar) was the positive control. Constructs containing the 59-residue insertion (check bar) in GST–ApoC1 and GST–ApoC3 were able to pull down X11α from cell lysate, as shown in the Western blot. c, GST–PTB (X11α) pull-down of ApoER2 mutants as shown by Western blot (top). The bottom shows a diagram of mutation positions. Deletion of the NPTY motif (mutants 1 and 2) or mutation of a PXXP motif (mutants 3 and 4) did not affect the pull-down. Only the deletion of the YDRPLW motif abolished the pull-down. GST was the negative control in pull-down and Western blot. c, Reversed pull-down from d, in which wild-type ApoER2 pulled down X11α (lane 2), but mutant 5 (ApoER2 M5) did not (lane 4). IP, Immunoprecipitation; WB, Western blot.
X11α/β involves in ApoE stimulated Aβ production
Because X11α/β binds to both ApoER2 and APP and is required for the formation of a complex containing these three proteins (Figs. 3, 4), we hypothesize that it is involved in facilitating endocytosis of these proteins leading to ApoE-induced Aβ production. This hypothesis is supported by the intracellular colocalization of APP with ApoER2 (Fig. 5A, a–c) and ApoER2 with X11α/β (Fig. 5A, d–i), especially in subcellular compartments consistent with endosomes (Fig. 5A, arrows). However, it is also known (Borg et al., 1998; Sastre et al., 1998) and we have confirmed (results not shown) that the expression of X11α/β inhibits Aβ production. Such a response seems to contradict with the hypothesis. Therefore, we seek evidence on the involvement of endogenous X11α/β in the ApoE-induced Aβ increase from N2a cells. First, transfected PTB domain from X11α was used as dominant-negative inhibitor for the function of endogenous X11α/β. We observed that ApoE4-induced Aβ increase (Fig. 5B, compare columns 1 and 3) was largely abolished by PTB expression (Fig. 5B, column 4). Second, the expression of mutant ApoER2 with a deletion of the X11α/β-binding motif (Fig. 4c, mutant 5) also greatly reduced Aβ increase induced by ApoE4 (Fig. 5C). These observations suggest that X11α/β binding at least to ApoER2 plays a role in ApoE-induced endocytosis leading to the production of Aβ.
Figure 5.
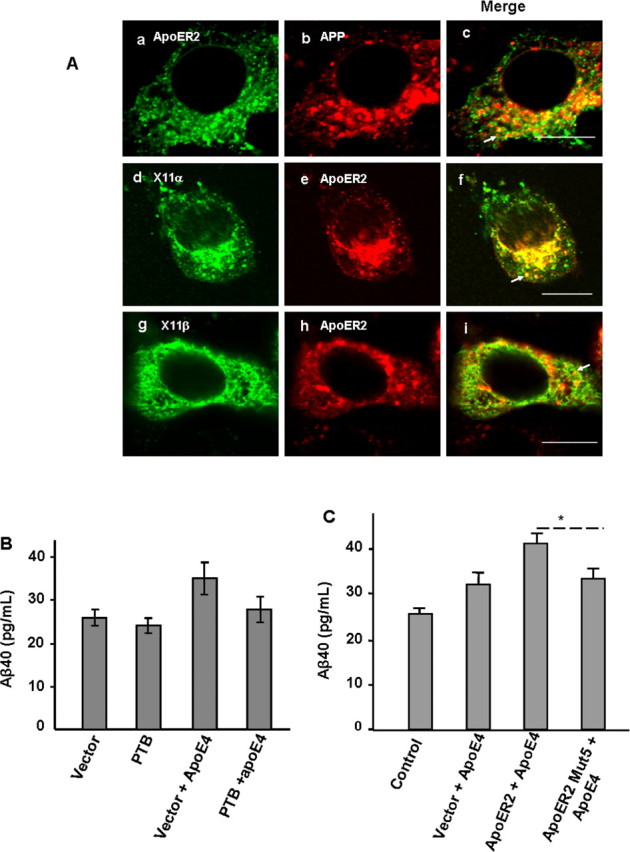
Involvement of X11α/β in APP and ApoER2 internalization and ApoE-induced Aβ production. A, Intercellular localization of APP with ApoER2 (a–c) and ApoER2 with X11α/β (d–i). Arrows in the merged images point to subcellular compartments consistent with endosomes. Scale bar, 10 μm. B, Expression of PTB domain of X11α/β abolished ApoE-induced Aβ production. C, Deletion of X11α/β-binding motif YDRPLW in ApoER2 (Mut5; mutant 5 in Fig. 4c) abolished ApoE-induced Aβ production (*p < 0.05).
ApoER2-mediated Aβ increase by ApoE is independent of Reelin
ApoER2 is also a receptor for the neuronal signaling protein Reelin (D'Arcangelo et al., 1999; Hiesberger et al., 1999), which is known to reduce cellular Aβ production (Hoe et al., 2006). It is of interest then to determine whether ApoER2-mediated Aβ increase by ApoE is related to Reelin–ApoER2 interaction. Western blots of N2a-APPsw culture medium or cell lysate concentrated from cell culture in T25 flasks did not detect the presence of Reelin (Fig. 6a, two left lanes), whereas the Reelin bands were clearly visible in cells transfected with a Reelin expression vector (Fig. 6a, two right lanes). Moreover, the addition of anti-Reelin antibody to the culture medium did not significantly alter the ApoE-stimulated Aβ increase in N2a-APPsw cells (Fig. 6b). These observations indicate that ApoER2-mediated Aβ increase by ApoE is independent of Reelin–ApoER2 interaction.
Figure 6.

Absence of Reelin and a Reelin effect in ApoE triggered Aβ increase in N2a-APPsw cells. a, Western blots for Reelin in cell lysate and culture medium of N2a-APPsw cells (left two lanes) and N2a-APPsw cells transfected with a Reelin expression vector. b, Aβ40 production in N2a-APPsw cells in the presence (right column) and absence of anti-Reelin antibody (left and center columns). Values were averaged from three determinations.
Discussion
Data presented above suggest that ApoE induces the production of Aβ through a mechanism as follows (Fig. 7). At cell surface, ApoER2 and APP are part of a complex mediated by X11α/β. After the binding of ApoE to ApoER2 in the complex, the entire complex, along with β-secretase, is endocytosed. The hydrolysis of APP by β-secretase and the subsequent generation of Aβ by this process take place in acidic intracellular compartments, such as endocytic vesicles and endosomes, as is consistent with the observation of ApoE-induced increase of intracellular C99 (Fig. 2d).
Figure 7.

A schematic presentation of ApoE-triggered Aβ production. At the cell surface, ApoER2 (white) and APP (gray) each bind a molecule of X11α/β (green) to form a complex with the possible involvement of other cytosolic proteins (black and ?) during the recruitment and packaging of endocytic vesicles. β-Secretase (β-Sec) is associated with this complex by virtue of its recognition of APP, in which the protease is inactive at pH 7. In the presence of ApoE-containing lipid particles, ApoE binds ApoER2 to trigger endocytosis of the complex with β-secretase to intracellular compartments in which, at pH 4.5, APP is cleaved by β-secretase and γ-secretase to generate Aβ. In this mechanism, ApoE triggers the production of Aβ, in which ApoE4 generates more Aβ than do ApoE2 and ApoE3, possibly as a result of a stronger association with ApoER2. This mechanism also links Aβ production to lipid uptake, which may be associated with neuronal activities.
The observation that ApoE4 generates more Aβ than does ApoE2 or ApoE3 in an ApoER2-mediated system (Fig. 1) suggests that the ApoE4 may bind ApoER2 more intensely than ApoE2 or ApoE3. Three isomorphic forms of ApoEs differ not only in two residues but also in conformations (for review, see Y. Huang et al., 2004); thus, their binding to receptors could be significantly different. Although we used plasma VLDL and purified ApoE to reach the above conclusions, these observations are likely applicable to brain lipoproteins despite the difference of its density from that of plasma VLDL. Although the density is determined by the lipid content in the interior of the lipoprotein particles, ApoE at the surface of the particles should interact in the same manner to ApoER2, thus triggering the same downstream events as shown in Figure 7. This is supported by the fact that we observed the same responses with isomorphic ApoEs in VLDL and in lipid-free form. Therefore, the current observations suggest that the ApoER2-mediated Aβ production may play a role in linking ApoE polymorphism with the risk of the sporadic form of AD. Other mechanisms that link ApoE4 to AD risk have been reported. For example, the association of isomorphic forms of ApoE with LRP, another member of the LDL receptor family, has been shown to affect the efflux of Aβ from the brain (Tanzi et al., 2004). ApoE-stimulated increase of Aβ mediated by LPR has also been reported recently (Ye et al., 2005). Although the underlying mechanism of this association is unknown, it seems possible that a mechanism similar to the one discussed here is also operative for LPR. However, ApoER2 knockdown completely abolished Aβ response to ApoE4, suggesting the absence of a parallel system in N2a cells. The fact that Aβ40 and Aβ42 respond to ApoE in a similar manner (Fig. 1c) suggests that the effect of an ApoE-triggered mechanism does not significantly influence the hydrolysis of C99 by γ-secretase. The proposed mechanism is also indirectly supported by the reduction of sα-APP by ApoE species (Fig. 2e) because the increased APP endocytosis leads to a reduction of APP available at the cell surface for the activity of α-secretase. We observed that ApoE4 was more effective than ApoE in the increase of APP endocytosis and decrease of sα-APP. A previous report described ApoE3-increased and ApoE4-decreased sα-APP in PC12 cells (Wolozin et al., 1996). Although PC12 is a neuronal cell line, the different ApoE4 effects could be derived from a difference in ApoER2 or X11α/β level from that in N2a-APP cells. Other possibilities include the presence of other ApoE-driven mechanisms in PC12.
How X11α/β bridges ApoER2 to APP is unclear. X11α/β has only one PTB domain, which binds ApoER2 or APP. Although an X11 homolog in Caenorhabditis elegans (lin-10) has been shown to form a dimmer (Walhout et al., 2000), it is uncertain that this can be extended to X11α/β. It seems plausible that the PTB domain of different X11α/β molecules binds to APP and ApoER2 separately (Fig. 7). The complex is likely formed in the lipid raft at cell surface as a prelude to endocytosis. Considering the complexity of the endocytic assembly, it seems probable that other cytosolic proteins are involved in the formation of this complex. As an adaptor protein with multiple binding domains, it is reasonable to assume that the bound X11α/β may interact with other partners (Fig. 7, question mark) to facilitate the recruitment and packaging of the endocytic vesicles. The clarification of the detailed process will require additional studies. The binding of ApoE-containing lipoprotein particles to ApoER2 triggers endocytosis of not only ApoER2 and APP but also β-secretase (Fig. 2). The coordinated internalization of APP can be explained by the X11α/β-mediated complex formation. The coordinated endocytosis of β-secretase is particularly interesting, because it did not immunoprecipitate with the ApoER2 complex (results not shown). We previously reported that APP mediates the endocytosis of ectodomain of β-secretase (X. P. Huang et al., 2004), suggesting that β-secretase is recognized by the network of the APP-containing endocytic assembly at cell surface. This association does not involve the catalytic site of the protease (X. P. Huang et al., 2004) and at the pH of the lumen, the protease has little activity, thus the complex should be stable before reaching the acid intracellular compartments. Such association is consistent with the current observations and could play a role in the coordinated internalization, although we have not provided evidence of its specific involvement. The results in Figure 2 may be interpreted as the amount of internalization is in the order of APP, ApoER2, and β-secretase. However, these data were collected at a single time point, and a firm conclusion on relative internalization rates would require more detailed kinetic studies. The scheme in Figure 7 can also explain the inhibition of Aβ production by the expression of X11α/β (Borg et al., 1998; Sastre et al., 1998). In the absence of ApoE, extra amounts of X11α/β would keep a larger fraction of cell-surface APP in a complex involving ApoER2; thus, less APP is available for internalization and Aβ generation by alternative mechanisms.
The scheme in Figure 7 can be thought of as a triggering mechanism for the production of Aβ. In the absence of the triggering of ApoE, X11α/β actually inhibits Aβ production. In the presence of ApoE, ApoER2, and X11α/β facilitate Aβ production. This mechanism provides a link between the neuronal uptake of lipid particles and Aβ production. The relationship of lipid, especially cholesterol, level to Aβ production and AD has been well documented in cellular and animal models and in clinical trials (for review, see Puglielli et al., 2003; Wolozin, 2004). The mechanism described here may conceivably play a role in this pathologenic relationship. The physiological function of such a link may also be important. ApoE is the major lipoprotein carrier in the brain for the delivery of lipids, predominantly cholesterol, to neurons (Huang, 2006). Such a process is initiated on cell surface by the binding of ApoE to LDL receptor family, including ApoER2, followed by endocytosis. Cholesterol and lipids supplied in this process are essential in remodeling neuronal membranes and developing new growth terminals. These processes are important to neuronal activities and synaptic plasticity. Thus, it is logical to expect that high neuronal activity is associated with the uptake of ApoE-coated lipid particles, the predominant supplier of lipids in the brain. Kamenetz et al. described a mechanism for the physiological function of Aβ in which high neuronal activity leads to an enhanced β-secretase activity and Aβ production. Aβ then feedback inhibits synaptic activity as a downregulation mechanism of neuronal functions (Kamenetz et al., 2003). The scheme in Figure 7 is consistent with the mechanism proposed by Kamenetz et al. In linking these two mechanisms, high neuronal activity leads to ApoE-triggered endocytosis of APP and β-secretase; then, the increased Aβ production affects the feedback inhibition. The possible connection of these two mechanisms warrants additional investigation.
Our data from N2a cells show that the mechanism in Figure 7 is independent of Reelin. The increase of Aβ production by ApoE can be accounted for from the increased endocytosis shown in Figure 2 and does not involved in a mechanism of reversing Aβ inhibition of Reelin by ApoE. The absence of Reelin in N2a cells (Fig. 6a) is not surprising because Reelin is produced specifically by Cajal-Retzius neurons. As discussed above, the current mechanism in Figure 7 is likely to be a general one that links neuronal functions to Aβ production, whereas Reelin signaling appears to have more regulatory functions. Our results do not rule out, however, that Reelin and ApoE do compete for ApoER2 under Reelin signaling conditions as suggested by previous studies (D'Arcangelo et al., 1999; Hiesberger et al., 1999).
ApoER2 motif (mouse, YDRPLW; human, FDRPLW) recognized for X11α/β binding is only moderately related to that in APP (YENPTY). Another similar motif (FDNPVY) in the cytosolic domain of ApoER2 is not recognized by X11α/β, perhaps because of the presence of a valine. These observations suggest a consensus X11α/β-binding sequence with F/Y at residue 1, E/D at residue 2, P at residue 4, and a hydrophobic residue at residue 6 of the motif. This motif is located in the 59-residue insertion (Fig. 3a) and resulted from a brain-specific alternative splicing of exon 19, a region known to modulate synaptic plasticity and learning (Beffert et al., 2005). Our observation that the motif in this region binds X11α/β, leading to the production of Aβ, which in turn regulates the synaptic activities, is consistent with such a function.
Footnotes
This work was supported by National Institutes of Health Grant AG-18933 and an Alzheimer Association Pioneer Award (to J. T.). J.T. is holder of the J.G. Puterbaugh Chair in Biomedical Research at the Oklahoma Medical Research Foundation. We thank Drs. Riqiang Yan, Chistopher Miller, Ben Margolis, Guojun Bu, Johannes Nimpf, Tom Currant, and Falk Fahrenholz for their generous gifts of cell line and plasmids and Dr. J. Donald Capra for helpful suggestions.
References
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Borg JP, Yang Y, De Taddeo-Borg M, Margolis B, Turner RS. The X11alpha protein slows cellular amyloid precursor protein processing and reduces Abeta40 and Abeta42 secretion. J Biol Chem. 1998;273:14761–14766. doi: 10.1074/jbc.273.24.14761. [DOI] [PubMed] [Google Scholar]
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer's A beta(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- He X, Chang WP, Koelsch G, Tang J. Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 2002;524:183–187. doi: 10.1016/s0014-5793(02)03052-1. [DOI] [PubMed] [Google Scholar]
- He X, Zhu G, Koelsch G, Rodgers KK, Zhang XC, Tang J. Biochemical and structural characterization of the interaction of memapsin 2 (beta-secretase) cytosolic domain with the VHS domain of GGA proteins. Biochemistry. 2003;42:12174–12180. doi: 10.1021/bi035199h. [DOI] [PubMed] [Google Scholar]
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE) J Biol Chem. 2005;280:11696–11703. doi: 10.1074/jbc.M411296200. [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem. 2006;281:35176–35185. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- Huang XP, Chang WP, Koelsch G, Turner RT, III, Lupu F, Tang J. Internalization of exogenously added memapsin 2 (beta-secretase) ectodomain by cells is mediated by amyloid precursor protein. J Biol Chem. 2004;279:37886–37894. doi: 10.1074/jbc.M402130200. [DOI] [PubMed] [Google Scholar]
- Huang Y. Apolipoprotein E and Alzheimer disease. Neurology. 2006;66:S79–S85. doi: 10.1212/01.wnl.0000192102.41141.9e. [DOI] [PubMed] [Google Scholar]
- Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci. 2004;23:189–204. doi: 10.1385/JMN:23:3:189. [DOI] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000;275:33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- King GD, Scott Turner R. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci USA. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchekar M, Richardson PE, Forte TM, Datta G, Segrest JP, Dashti N. Apolipoprotein B-containing lipoprotein particle assembly: lipid capacity of the nascent lipoprotein particle. J Biol Chem. 2004;279:39757–39766. doi: 10.1074/jbc.M406302200. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Ikin AF, Nairn AC, Pursnani A, Buxbaum JD. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total A(beta) Mol Cell Neurosci. 2002;19:175–185. doi: 10.1006/mcne.2001.1065. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- Sastre M, Turner RS, Levy E. X11 interaction with beta-amyloid precursor protein modulates its cellular stabilization and reduces amyloid beta-protein secretion. J Biol Chem. 1998;273:22351–22357. doi: 10.1074/jbc.273.35.22351. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wahle T, Prager K, Raffler N, Haass C, Famulok M, Walter J. GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol Cell Neurosci. 2005;29:453–461. doi: 10.1016/j.mcn.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, Lammich S, Multhaup G, Haass C. Phosphorylation regulates intracellular trafficking of beta-secretase. J Biol Chem. 2001;276:14634–14641. doi: 10.1074/jbc.M011116200. [DOI] [PubMed] [Google Scholar]
- Wolozin B. Cholesterol and the biology of Alzheimer's disease. Neuron. 2004;41:7–10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- Wolozin BL, Basaric-Keys J, Canter R, Li Y, Vanderputten D, Sunderland T. Differential regulation of APP secretion by apolipoprotein E3 and E4. Ann NY Acad Sci. 1996;777:322–326. doi: 10.1111/j.1749-6632.1996.tb34440.x. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH, Mahley RW. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]