

Abstract
The cellular prion protein (PrPc) undergoes a physiological processing yielding the N-terminal fragment referred to as N1, the production of which can be constitutive or protein kinase C regulated. We show that activation of endogenous muscarinic receptors by carbachol and by the M1-selective agonist AF267B increases N1 recovery in an atropine-sensitive manner, in mouse embryonic primary neurons. To identify the muscarinic receptor subtype involved, we used human embryonic kidney HEK293 (HEK) cells stably overexpressing M1, M2, M3, or M4 receptor subtype. Carbachol and the selective M1 agonist AF267B dose dependently increased N1 release by HEK-M3 and HEK-M1 cells, respectively, whereas carbachol did not modify N1 production by HEK-M2 or HEK-M4 cells. We demonstrate that the increase of N1 was not attributable to modified trafficking to the membrane of either PrPc or the disintegrin metalloproteases ADAM10 or ADAM17. Furthermore, we establish that carbachol affects the overall phosphorylation of ADAM17 on its threonine and tyrosine but not serine residues, whereas levels of phosphorylated ADAM9 were not affected. Interestingly, carbachol also increases the hydrolysis of the fluorimetric substrate JMV2770, which mimicked the sequence encompassing the N1 site cleavage and was shown previously to behave as an ADAM protease substrate. Mutations of threonine 735 but not of tyrosine 702 of the ADAM17 cytoplasmic tail abolishes the carbachol-induced increase of N1, ADAM17 phosphorylation, and JMV2770-hydrolyzing activity in M1- and M3-expressing HEK293 cells. Thus, our data provide strong evidence that muscarinic receptor activation increases the physiological processing of PrPc by upregulating the phosphorylation state and activity of ADAM17 protease.
Keywords: cellular prion, regulated processing, muscarinic receptors, ADAM17, phosphorylation, HEK293, primary cultured neurons
Introduction
Prion diseases are attributable to the posttranslational conversion of a normal cellular prion protein (PrPc) into an abnormal form referred to as PrPsc or scrapie (Prusiner, 1998; Aguzzi et al., 2001). The observations that mice lacking the neuronal cell surface prion protein have a normal development and behavior (Büeler et al., 1992) and resist infection by scrapie (Büeler et al., 1993) has provided new insights into theoretical therapeutic approaches against prion pathologies. Thus, lowering endogenous levels of PrPc could constitute a potential efficient strategy to prevent or to slow down the infectious process.
In physiological conditions, PrPc undergoes both constitutive and protein kinase C (PKC)-regulated processing that generate an N-terminal fragment referred to as N1 (Chen et al., 1995; Vincent et al., 2000; Laffont-Proust et al., 2005). This cleavage occurs at the 111/112 peptidyl bond, i.e., in the middle of the 106–126 domain, which is thought to be toxic in vitro (Forloni et al., 1993; Brown et al., 1996; Jobling et al., 1999; Dupiereux et al., 2006) and in vivo (Ettaiche et al., 2000). Interestingly, in Creutzfeldt–Jakob disease-affected brains, a major cleavage occurs more N terminally, at the 90/91 site, yielding a fragment referred to as N2 (Chen et al., 1995). This shift in the PrPc cleavage site preserves the integrity of the 106–126 domain and therefore could contribute to the pathogenicity.
We previously demonstrated that ADAM10 (a disintegrin and metalloprotease) participated in the constitutive formation of the N1 fragment, whereas PKC-dependent formation of N1 appeared fully attributable to ADAM17 (Vincent et al., 2001). We also showed that ADAM9 indirectly participated in N1 formation, by modulating ADAM10 activity (Alfa Cissé et al., 2005). The mechanisms by which ADAM17 is modulated by PKC and upregulates N1 formation remained to be established.
In the present study, we explored the effect of muscarinic receptor activation on the release of N1. We show here that the nonselective agent carbachol as well as the M1-selective muscarinic agonist AF267B (Beach et al., 2001; Fisher et al., 2002) both enhance N1 production by primary cultured neurons. This effect was mainly mediated by PKC-coupled M1 and M3 but not by PKA-linked M2 and M4 muscarinic receptors. Furthermore, we establish that the M1-mediated increase of N1 was attributable to increased phosphorylation and activity of ADAM17.
Altogether, our results strongly suggest that M1 and M3 muscarinic receptors could act as upstream effectors in a signaling pathway leading to the N1-generating PrPc processing.
Materials and Methods
Antibodies and pharmacological agents.
SAF32 is a monoclonal antibody raised against the residues 79–92 of PrPc (Demart et al., 1999). Anti-phospho-serine antibody was from Zymed Laboratories (via Invitrogen, Cergy-Pontoise, France), and anti-phospho-tyrosine and phospho-threonine monoclonal antibodies were from Cell Signaling Technology (Beverly, MA). Anti-tubulin antibody was purchased from Sigma (St. Quentin-Fallavier, France). Anti-ADAM10 antibody was purchased from Euromedex (Souffelmeyersheim, France). BB3103 (hydroxamic acid-based zinc metalloprotease inhibitor) was kindly provided by Vernalis (Berkshire, UK), and GF109203X (3-[1-(3-dimethylaminopropyl)indol-3-yl]-4-(1H-indol-3-yl)pyrrole-2,5-dione hydrochloride) was from Calbiochem (La Jolla, CA). The fluorimetric substrate JMV2770 has been developed and characterized previously (Alfa Cissé et al., 2006). Phosphatase inhibitor I, phosphatase inhibitor II and carbachol were from Sigma. Atropine was from ICN Biomedicals (Aurora, OH). [(S)-2-Ethyl-8-methyl-1-thia-4,8-diazaspiro[4.5]decan-3-one] was synthesized in the laboratory of Dr. Fisher [Fisher A, Meshulam H (1996) U.S. Patent Application 5,852,029]. The batch used in the present study was >99.9% pure, as determined by chiral and achiral HPLC (data not shown).
Cell cultures.
Mock-transfected and PrPc-expressing human embryonic kidney HEK293 cells were obtained and cultured as detailed previously (Ancolio et al., 1999; Vincent et al., 2000). Previously described HEK293 M1, M2, M3, or M4 muscarinic receptor stable transfectants (Nitsch et al., 1992) were maintained at 37°C in 5% CO2 in DMEM supplemented with 10% fetal calf serum containing penicillin (100 U/ml), streptomycin (50 mg/ml), and geneticin (1 mg/ml). Initial characterization of the transfectants has been performed by radioreceptor assay (Peralta et al., 1988). Primary cultured neurons were prepared from the cerebral hemispheres of 14-d-old wild-type mouse embryos as described previously (Vincent et al., 1996). Briefly, cells were mechanically dissociated with a pipette in Ham's F-12 medium (Invitrogen) supplemented with 10% fetal calf serum and 0.6% glucose. Dissociated cells were then plated at a density of 3 × 106 cells in 35 mm dishes precoated with 10 mg/ml poly-l-lysine (Sigma) and grown in a humidified atmosphere of 5% CO2, 95% air.
Analysis of PrPc and disintegrin metalloproteases expression.
HEK293 cells grown in 35 mm dishes were washed once with PBS and resuspended in 500 μl of lysis buffer (10 mm Tris/HCl, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 0.5% deoxycholate, and 5 mm EDTA) supplemented with a protease inhibitor mixture (Sigma). Proteins (25 μg) were separated by SDS-PAGE on 12% (PrPc) or 8% (ADAM9, ADAM10, and ADAM17) Tris/glycine gels. Proteins were transferred onto nitrocellulose membranes (2 h, 100 V) and incubated overnight at 4°C with SAF32 (PrPc) (1:1000 dilution in PBS/Tween 0.05%/milk 5%), anti-ADAM9 (1:1000 dilution) (Weskamp et al., 1996), anti-ADAM10 (1:1000 dilution) (Euromedex) or anti-ADAM17 (1:1000 dilution) antibody (Black et al., 1997). Bound antibodies were detected using a goat anti-rabbit peroxidase-conjugated antibody (1:5000 dilution) (Beckman Coulter, Fullerton, CA). Chemiluminescence was recorded using a Luminescence Image Analyzer LAS-3000 (Raytest, Courbevoie, France), and quantifications were performed using NIH ImageJ analyzer software.
Immunoprecipitation and Western blot analysis of N1.
Cells cultured in 35 mm dishes were washed once with PBS and incubated for 8 h at 37°C in 1 ml of serum-depleted DMEM, in the absence or presence of various pharmacological agents. Media were collected, and cells were resuspended in 500 μl of lysis buffer. Both medium and lysates were complemented with a protease inhibitor mixture (Sigma). The medium was supplemented with radioimmunoprecipitation assay (RIPA) (0.1% SDS, 0.5% deoxycholate, and 1% NP-40, pH 8) and incubated overnight with a 500-fold dilution of monoclonal antibody SAF32 and protein A-Sepharose beads (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Beads were washed twice with 500 μl of RIPA buffer, once with 500 μl of PBS, and subjected to SDS-PAGE on a 16.5% Tris/tricine gel. Proteins were transferred onto Hybond C membrane (polyvinylidene difluoride; GE Healthcare) (45 min, 100 V; GE Healthcare) and incubated overnight at 4°C with the monoclonal antibody SAF32 (1:2000 dilution). Immunological complexes were detected with a goat anti-mouse peroxidase-conjugated antibody (1:2000 dilution) (Jackson ImmunoResearch, West Grove, PA). Chemiluminescence was recorded and quantified as above.
N-glycosidase F treatment and C1 detection.
HEK293 cells stably expressing M1 muscarinic receptors were seeded in six-well dishes and allowed to reach 80% confluency in DMEM (Invitrogen) supplemented with 10% fetal calf serum. Then, cells were cotransfected with cDNAs encoding wild-type ADAM17 and PrPc (2 μg of each) using Lipofectamine as described above. Twenty-four hours after transfection, cells were allowed to secrete for 8 h at 37°C in 1 ml of serum-depleted DMEM, in the absence or presence of various pharmacological agents. Deglycosylation was performed as described previously (Sunyach et al., 2007). Briefly, 100 μg of proteins were recovered in denaturing buffer (25 mm Tris, pH 7.5, 0.5% SDS, 1% 2- mercaptoethanol) and boiled for 5 min. One percent of NP-40 and 0.5 U of N-glycosydase F (Roche, Meylan, France) were added, and the samples were incubated for 2 h at 37°C. The reaction was stopped by addition of equal volume of loading buffer (0.5 m Tris, pH 6.8, 10% glycerol, 10% SDS, and 0.05% bromophenol blue). Samples were then loaded and resolved on a 12% SDS-PAGE gel for Western blot analysis as described above.
Construction of glutathione S-transferase-N1 fusion expressing vector and purification of N1 recombinant peptide.
A cDNA encoding the amino acids 23–110 of PrPc (N1) was generated by PCR and cloned into EcoRI and XhoI sites of pGEX-KG (Guan and Dixon, 1991). To produce and purify N1 recombinant peptide, the pGEX-KG glutathione S-transferase (GST)–N1-expressing vector was transformed into the BL21 gold strain of Escherichia coli. Cultures of E. coli were grown in Luria–Bertani broth medium (Sigma) supplemented with ampicillin (50 μg) and allowed to reach an absorbance of 0.6 at 600 nm, and then the fusion protein expression was induced with isopropyl-β-d-thiogalactopyranoside (0.5 mm; Sigma) for 4 h at 37°C. Cells were pelleted at 5000 × g for 20 min at 4°C, resuspended in PBS (50 μl/ml original culture) supplemented with complete protease inhibitor cocktail (Sigma), PMSF (Sigma), and lysozyme (150 μg/ml; Sigma), and then incubated for 30 min on ice. Protein were solubilized with 1% (v/v) Triton X-100, MgCl2 (10 mm), DNase I (5 U/ml; Promega, Madison, WI) and left on ice for 30 min. Debris were pelleted for 20 min at 10,000 × g. Glutathione-Sepharose beads (GE Healthcare) preswollen in PBS (70% slurry) were added to the crude lysate and swirled for 1 h at 4°C. The beads were pelleted and washed five times with 10 vol of PBS. Beads were then resuspended in 1 ml of PBS, and N1 peptide was released from GST with 5 U/ml thrombin (GE Healthcare). The monoisotopic mass of N1 peptide was checked by mass spectrometry and was in accordance with the theoretical mass calculated from the sequence data.
Enzymatic fluorimetric assay on intact HEK293 cells.
HEK293 cells were cultured in six-well plates at 37°C. At 80% confluence, cells were washed twice with PBS and pretreated with various inhibitors for 30 min. Cells were then incubated with 1.5 ml of PBS containing the fluorimetric substrate JMV2770 (10 μm) (Alfa Cissé et al., 2006) in the presence or absence of the disintegrin inhibitor BB3103, carbachol (100 μm), or GF109203X (2 μm) and incubated for various time periods at 37°C. At each kinetic point, 100 μl of medium were taken out, and production of hydrolysis fluorimetrically was recorded (320 and 420 nm as excitation and emission wavelengths, respectively).
Immunoprecipitation and determination of phosphorylated residues.
M1-expressing HEK293 cells grown in 100 mm dishes were incubated for 5 min in fresh serum-free media containing 100 μm carbachol, then media were discarded, and cells were collected in 1 ml of cold lysis buffer (10 mm Tris/HCl, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 0.5% deoxycholate, and 5 mm EDTA) supplemented with phosphatase inhibitor I and II cocktails (Sigma) and with a protease inhibitor mixture. Lysates were centrifuged (5 min, 13000 rpm) to remove insoluble material and then normalized for protein contents. One milligram of proteins was supplemented with RIPA (0.1% SDS, 0.5% deoxycholate, and 1% NP-40, pH 8), incubated overnight with immunoprecipitating anti-phospho antibodies and protein A-Sepharose beads (GE Healthcare) (4 μg/1 mg proteins), and then incubated overnight. Beads were washed twice with 500 μl of RIPA buffer, then once with 500 μl of PBS, and analyzed by SDS-PAGE for ADAM9 and ADAM17 phosphorylation levels, as described above.
Sucrose gradient subcellular fractionation.
Six 100 mm dishes of confluent HEK293 cells stably expressing either M1 or M3 muscarinic receptors were incubated for 4 h with fresh serum-free media without or with AF267B or carbachol. Cells were then homogenized with a Dounce homogenizer in 0.25 m sucrose and 10 mm Tris-HCl, pH 7.4, containing 1 mm MgAc2 and a protease inhibitor mixture (Sigma). Equal amounts of protein (3.5 mg) from each homogenate were loaded at the top of a step gradient and centrifuged, and then 10 fractions (1 ml) were collected in a stepwise manner from the top of each gradient. One-tenth of each fraction was precipitated overnight at 4°C with methanol (4 vol), resuspended in sample buffer, and heated for 5 min at 95°C. PrPc, ADAM10, or ADAM17 expression was analyzed by Western blot as described above. The complete characterization of fractions by organelle markers has been extensively described (Luo et al., 2003).
Site-directed mutagenesis.
Mouse ADAM17 in which tyrosine 702 was replaced by a phenylalanine (Y702F-ADAM17) or in which threonine 735 was replaced by an alanine (T735A-ADAM17) were obtained by oligonucleotide-directed mutagenesis from mouse wild-type ADAM17 cDNA by means of a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Mutagenesis was performed according to the conditions of the manufacturer using the two sets of primers 5′-CTGGACAAGCAGTTTGAATCCCTGTCTC-3′ and 5′-GAGACAGG-GATTCAAACTGCTTGTCCAG-3′ (Eurogentec, Seraing, Belgium) containing the Y702F mutation and the two primers 5′-CCTGC-ACCCCAGGCTCCAGGTCGTC-TGC-3′ and 5′-GCAGACGACCT-GGAGCCTGGGGTGC-AGG-3′ containing the T735A mutation. Final cDNA constructs were sequenced in their entirety.
Statistical analysis.
Statistical analysis was performed with Prism software (GraphPad Software, San Diego, CA) using the unpaired t test for pairwise comparisons.
Results
Endogenous muscarinic receptor activation increases N1 recovery in primary cultured neurons
We examined whether activation of endogenous muscarinic receptors could affect the N1-generating PrPc physiological processing in neuronal cells. We used primary cultured neurons derived from mice embryos that are devoid of glial cells (Vincent et al., 1996) and that express high levels of endogenous muscarinic receptors (Hamilton and Nathanson, 2001). The nonselective muscarinic agonist carbachol triggered a twofold increase in N1 recovery (Fig. 1) without altering endogenous levels of PrPc-like immunoreactivity (Fig. 1). As expected, these effects were fully blocked by the muscarinic antagonist atropine (Fig. 1), indicating that endogenous muscarinic receptors control N1 formation in neuronal cells.
Figure 1.

Activation of endogenous muscarinic receptors in mouse primary cultured cortical neurons increases N1 recovery. Primary neurons were obtained from 14-d-old mouse embryos and cultured as described in Materials and Methods. Cells were pretreated for 30 min with serum-free medium without or with atropine (Atr) (100 μm) or the vehicle DMSO. Cells were then incubated for 8 h in fresh medium containing atropine or DMSO with or without carbachol (Cch) (100 μm). The medium was then collected, and N1 was immunoprecipitated with the monoclonal antibody SAF32 and analyzed by 16.5% Tris/tricine SDS-PAGE and Western blotting using SAF32. RecN1 corresponds to the migration pattern of recombinant N1 (177 ng) analyzed under the same conditions. PrPc and tubulin were detected in cell lysates by separating proteins (25 μg) by 12% glycine SDS-PAGE as described in Materials and Methods. Bars correspond to the densitometric analysis of N1 and are expressed as the percentage of control N1 recovered under nonstimulated conditions. Values are the means ± SEM of four independent experiments. ***p < 0.0001; ns, nonstatistically significant when compared with untreated cells.
Carbachol increases N1 recovery in M1- and M3-, but not in M2- and M4-, expressing HEK293 cells
To identify the subtype of muscarinic receptors involved in N1 formation, we used HEK293 cell lines stably expressing human muscarinic receptor M1, M2, M3, or M4 subtypes (Nitsch et al., 1992). Carbachol did not affect N1 formation in mock-transfected cells (Fig. 2A); conversely, carbachol led to a dose-dependent increase in N1 recovery in M1-expressing (Fig. 2B) and M3-expressing (see Fig. 4A) cells, without significantly affecting PrPc immunoreactivity (see Figs. 2B, 4A), in agreement with the observation that only a minor percentage of membrane-associated PrPc indeed undergoes proteolytic processing (Harris et al., 1993b). Interestingly, we establish that C1 was also increased by carbachol in M1-expressing cells (Fig. 3). Both carbachol-induced increase in N1 and C1 were prevented by the nonselective muscarinic antagonist atropine (Figs. 2C, 3C–E, 4B).
Figure 2.

Influence of M1 muscarinic receptor activation on N1 secretion in HEK293 cells. HEK293 cells stably transfected with either pcDNA3 empty vector (A) or M1 muscarinic receptor cDNA (B–E) were pretreated for 30 min with serum-free media without (A, B) or with (Atr; 100 μm) either atropine (C) or BB3103 (10 μm; D, E), and then cells were incubated for 8 h in fresh media containing carbachol (Cch) at indicated concentrations (A–C) or at a final concentration of 100 μm (D, E) and monitored for their N1 content as described in Materials and Methods. Cell homogenates were prepared and analyzed for their PrPc-like immunoreactivity as described in Materials and Methods. Bars correspond to the densitometric analysis of N1 (A, B, E) and are expressed as the percentage of control N1 recovered under nonstimulated conditions. Values are the means ± SEM of four independent experiments. **p < 0.0005; ***p < 0.0001; ns, nonstatistically significant compared with control untreated cells. Cch, Carbachol.
Figure 4.

Influence of M3 muscarinic receptor activation on N1 secretion in HEK293 cells. HEK293 cells stably transfected with M3 muscarinic receptor cDNA (A, B) were pretreated for 30 min with serum-free media without (A) or with (B) the indicated concentration of atropine (Atr) and then incubated for 8 h in media containing carbachol (Cch) at the indicated concentrations and monitored as described in Materials and Methods for their N1 content. RecN1 corresponds to the migration pattern of recombinant N1 (177 ng) analyzed under the same conditions. Cell homogenates were prepared and analyzed for their PrPc-like immunoreactivity as described in Materials and Methods. Bars correspond to the densitometric analysis of N1 and are expressed as the percentage of control N1 recovered in nonstimulated conditions. Values are the means ± SEM of four independent experiments. *p < 0.005; **p < 0.0005; ***p < 0.0001; ns, nonstatistically significant.
Figure 3.

Influence of M1 muscarinic receptor activation on C1 formation in HEK293 cells. HEK293 cells overexpressing M1 muscarinic receptor cDNA were transiently cotransfected with cDNAs encoding wild-type ADAM17 and PrPc (A–E). Twenty-four hours after transfection, cells were pretreated for 30 min with serum-free media without (−) or with (+) atropine (Atr; 10 μm; C–E) and incubated for 8 h in media containing (+) or not (−) carbachol (100 μm; Cch). Media were then collected and monitored as described in Materials and Methods for their N1 content (C). Cell homogenates were prepared and analyzed for their C1 and PrPc-like immunoreactivity (D) as described in Materials and Methods. Bars in E correspond to the densitometric analysis of C1 and are expressed as the percentage of control C1 recovered in nonstimulated conditions. Values are the means ± SEM of three independent experiments. *p < 0.005; ns, nonstatistically significant. Ct, Control vehicle; Tub, tubulin; fl, full length.
Disintegrin metalloproteases are the only proteases contributing to PKC-regulated formation of N1 (Vincent et al., 2001). Interestingly, BB3103, a specific disintegrin metalloprotease inhibitor, prevented the carbachol-mediated N1 production in M1-expressing (Fig. 2D,E) and M3-expressing (data not shown) HEK293 cells, whereas other inhibitors, including 4-(2-aminoethyl)benzenesulfonyl fluoride (a serine protease inhibitor) were unable to affect N1 recovery (data not shown). Interestingly, carbachol treatment did not affect the secretion of N1 in M2-expressing (Fig. 5A) or M4-expressing (Fig. 5B) HEK293 cells. Altogether, these results suggest that N1 production is controlled by M1 or M3, but not M2 or M4, muscarinic receptors and involves BB3103-sensitive disintegrin metalloprotease activity.
Figure 5.

N1 recovery is not affected by M2 or M4 muscarinic receptor activation. HEK293 cells stably transfected with either M2 (A) or M4 (B) muscarinic receptor cDNA were incubated for 8 h in media containing carbachol (Cch) at the indicated concentrations and monitored as described in Materials and Methods for their N1 content. Cell homogenates were prepared and analyzed for their PrPc-like immunoreactivity as described in Materials and Methods. Bars correspond to the densitometric analysis of N1 and are expressed as the percentage of control N1 recovered in nonstimulated conditions. Values are the means ± SEM of three independent experiments. ns, Nonstatistically significant.
The selective M1 receptor agonist AF267B increases N1 production in HEK293 cells and primary cultured neurons
AF267B, developed as a selective M1 muscarinic agonist (Beach et al., 2001; Fisher et al., 2002), was used to confirm the selective involvement of M1 muscarinic receptors in N1 recovery in primary cultured neurons and M1-expressing HEK293 cells. AF267B increased the amount of N1 recovered in an atropine-sensitive manner in both M1-expressing HEK293 cells (Fig. 6A) and primary cultured neurons (Fig. 6B). These results further support the contribution of endogenous M1 muscarinic receptors to the physiological processing of PrPc leading to N1 formation.
Figure 6.

N1 secretion is increased by the selective muscarinic agonist AF267B in mouse primary cultured neurons and in M1-expressing HEK293 cells. HEK293 cells stably transfected with M1 muscarinic receptor cDNA (A) and mouse primary cultured cortical neurons (B) were pretreated for 30 min without (DMSO) or with atropine (Atr; 100 μm). Media were then collected, and cells were incubated for 8 h with serum-free media containing the indicated concentrations of AF267B and monitored as described in Materials and Methods for their N1 content. RecN1 corresponds to the migration pattern of recombinant N1 (177 ng) analyzed in the same conditions. Cell homogenates were prepared and analyzed for their PrPc-like immunoreactivity as described in Materials and Methods. Bars correspond to the densitometric analysis of N1 and are expressed as the percentage of control N1 recovered in nonstimulated conditions. Values are the means ± SEM of four independent experiments. ***p < 0.0001; ns, nonstatistically significant.
Activation of muscarinic receptor does not modify endogenous PrPc or disintegrin metalloprotease cellular distribution
To establish the mechanisms by which muscarinic receptor activation could control N1 production, we first examined whether muscarinic agonists could alter the normal trafficking, and therefore availability at the plasma membrane, of either ADAM proteases or PrPc. Velocity sedimentation of PrPc, ADAM10, and ADAM17 in sucrose step gradients was performed using HEK293 cells overexpressing either M1 or M3 receptor subtypes. Endogenous PrPc (Fig. 7) and ADAM10 and ADAM17 (Fig. 8) distribution profiles were similar in M1-expressing (Figs. 7A,B, 8A) or M3-expressing (Fig. 8B) cells in control, AF267B-stimulated, and carbachol-stimulated conditions, indicating that muscarinic agonists did not affect normal subcellular distribution of endogenous ADAM proteases and their substrate PrPc.
Figure 7.

Muscarinic receptors activation does not affect PrPc trafficking in M1 or M3 stably transfected HEK293 cells. A, M1-expressing HEK293 cells grown in 100 mm dishes were incubated with serum-free medium containing carbachol (100 μm; Cch), AF267B (100 μm), or vehicle (Ct) for 4 h. Cells were then homogenized, and the endogenous distribution of PrPc was analyzed by sucrose gradient fractionation as described in Materials and Methods. Bars in B represent the means ± SEM of three independent determinations. ns, Nonstatistically significant. ER/PM, endoplasmic reticulum/plasma membrane.
Figure 8.

The endogenous distribution of ADAM10 and ADAM17 is not affected by muscarinic receptor activation in HEK293 cells. M1-expressing (A) or M3-expressing (B) HEK293 cells grown in 100 mm dishes were incubated with serum-free medium containing carbachol (100 μm; Cch) (A, B), AF267B (100 μm; A), or vehicle (Ct) for 4 h. Cells was then homogenized, and the endogenous distribution of ADAM10 and ADAM17 were analyzed by sucrose gradient fractionation as described in Materials and Methods. ER/PM, Endoplasmic reticulum/plasma membrane.
Activation of M1 but not M4 muscarinic receptor increases JMV2770-hydrolyzing activity in intact HEK293 cells
We recently designed a fluorimetric substrate, JMV2770, the sequence of which overlaps the 111–112 cleavage site of PrPc, and we showed that it behaved as a disintegrin metalloprotease substrate (Alfa Cissé et al., 2006). We examined whether muscarinic activation could enhance BB3103-sensitive hydrolysis of JMV2770 on intact cells expressing membrane-bound disintegrin metalloproteases in their ectopeptidase topology. Carbachol (Fig. 9A,B) and AF267B (Fig. 9C) dose dependently increased JMV2770-hydrolyzing activity of intact HEK293 cells overexpressing the M1 receptor subtype, whereas this effect was not observed in mock-transfected or M4-expressing cells (Fig. 9A,B). These data suggest that M1 muscarinic receptors could enhance PrPc processing and N1 recovery by increasing ADAM protease activity.
Figure 9.
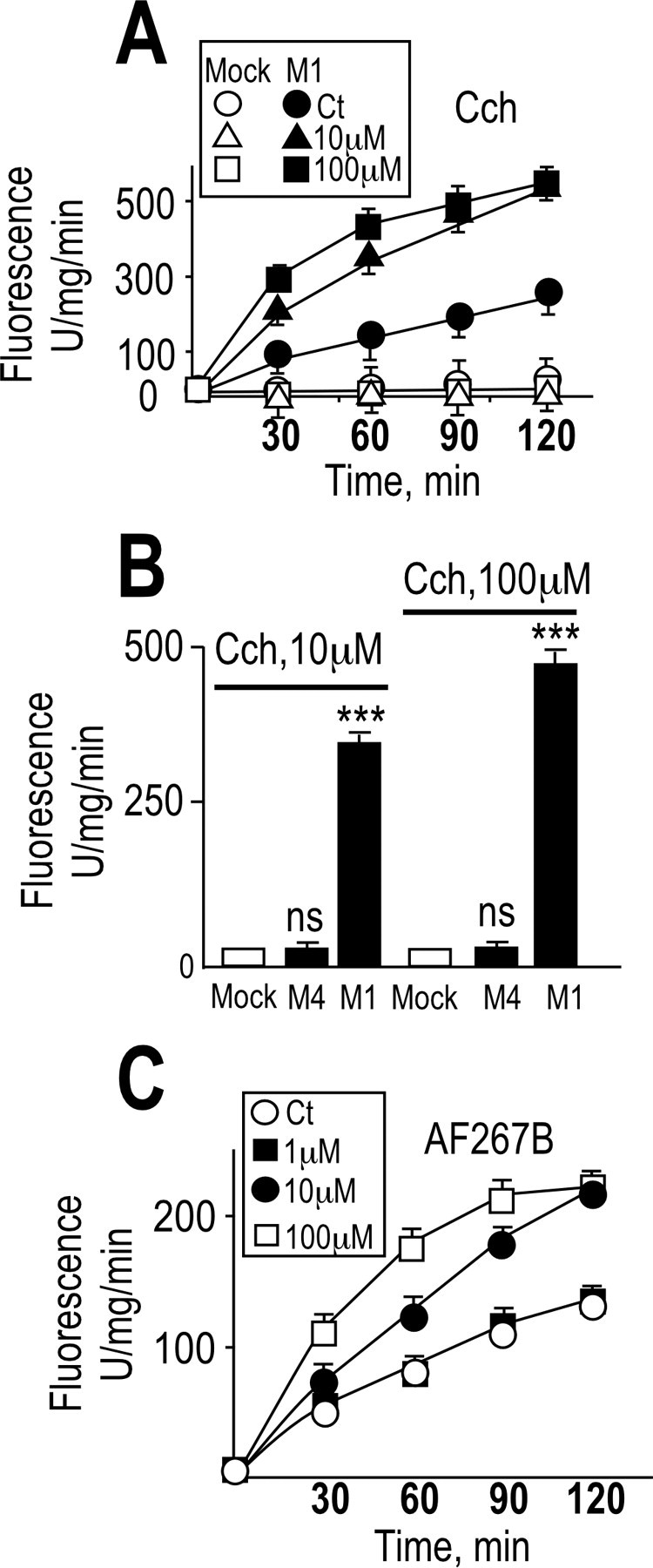
JMV2770-hydrolyzing activity is increased by muscarinic receptor activation in intact M1-expressing HEK293 cells. Plated HEK293 cells stably transfected with empty pcDNA3 (Mock) or cDNAs encoding M1 (A–C) or M4 (B) muscarinic receptors were monitored for their capacity to hydrolyze the disintegrin fluorimetric substrate JMV2770. Cells grown in six-well plates were pretreated for 30 min at 37°C with PBS in the absence or presence of the disintegrin inhibitor BB3103 (10 μm), and then cells were incubated with JMV2770 (at a final concentration of 10 μm) in the presence of the indicated concentrations of carbachol (Cch) (A, B) or AF267B (C). Aliquots of the media (100 μl) were collected at indicated time points (A, C) or after 60 min (B), and the fluorescence was recorded as described in Materials and Methods. The values represent the BB3103-sensitive activities and are means ± SEM of five independent determinations. ***p < 0.0005; ns, nonstatistically significant. Ct, Control vehicle.
Carbachol stimulation increases phosphorylation of ADAM17, but not ADAM9, in M1-expressing cells
The intracellular domains of disintegrin metalloproteases are variable in length and sequence and have been shown to regulate metalloprotease activities (Howard et al., 1999). Previous data also showed that ADAM17 can undergo phosphorylation on its cytoplasmic tail, thereby modulating its activity (Diaz-Rodriguez et al., 2002; Soond et al., 2005). The mouse ADAM17 cytoplasmic tail harbors five residues (tyrosine 702, threonine 735, and serines 723, 806, and 811) located inside putative consensus phosphorylation sites (Fig. 10A). However, a direct relationship between muscarinic receptor activation and the phosphorylation/activity status of disintegrin metalloproteases and influence on their catalytic activity remained to be established. We first monitored ADAM17 phosphorylation during carbachol stimulation of M1-overexpressing HEK293 cells, transiently expressing ADAM17. Carbachol treatment of M1-expressing cells rapidly increased ADAM17 phosphorylation on threonine and to a lesser extent on tyrosine residues (Fig. 10B), whereas carbachol treatment did not promote phosphorylation of serine residues (data not shown). Interestingly, carbachol did not trigger enhanced phosphorylation of ADAM9 (Fig. 10B) and ADAM10 (data not shown) in M1-expressing HEK293 cells. Therefore, carbachol selectively triggers ADAM17 phosphorylation.
Figure 10.
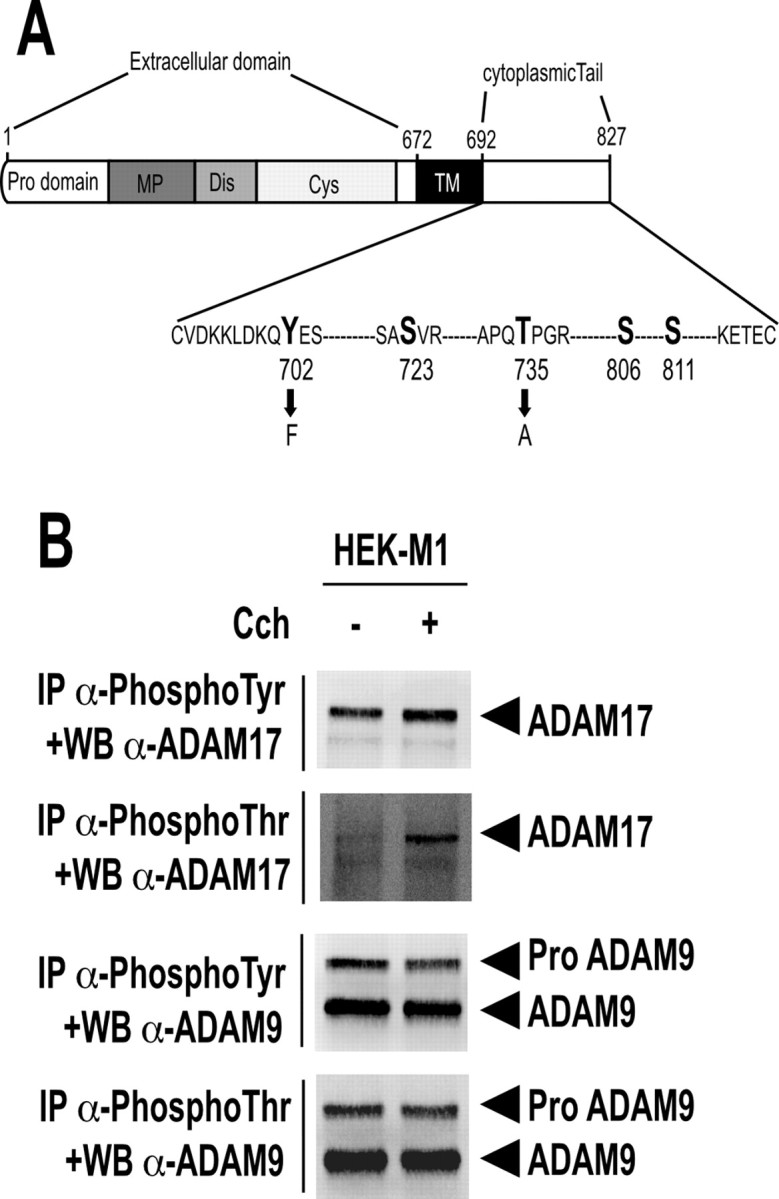
Muscarinic M1 subtype activation triggers ADAM17 phosphorylation in HEK293 cells. A, Structure of ADAM17 and its corresponding mutations. Serine, threonine, and tyrosine residues of ADAM17 cytoplasmic tail are indicated in bold. Black arrows illustrate amino acid substitutions. MP, metalloprotease domain; Dis, disintegrin domain; Cys, cystein-rich domain; TM, transmembrane domain. B, M1-expressing HEK293 cells grown at 37°C in 100 mm dishes were transiently transfected with ADAM17 or ADAM9 cDNA. Twenty-four hours after transfection, cells were treated (+) or not (−) for 5 min with carbachol (100 μm; Cch) and pelleted as described in Materials and Methods. One milligram of proteins was then submitted to immunoprecipitation (IP) using specific antibodies directed against phospho-threonine (α-phosphoThr) and phospho-tyrosine (α-phosphoTyr) residues and then analyzed by 8% SDS-PAGE and Western blotting (WB) using a specific anti-ADAM17 (α-ADAM17) or a specific anti-ADAM9 antibody. Illustrations are typical data of four independent determinations.
ADAM17 mutation of threonine 735 but not tyrosine 702 abolishes carbachol-stimulated M1/M3-mediated N1 formation and JMV2770-hydrolyzing activity
Overall, the above data clearly show that, in M1-expressing cells, carbachol triggers (1) a higher recovery of N1, (2) enhanced phosphorylation of ADAM17, and (3) increased JMV2770-hydrolyzing activity. However, these data did not directly prove that the carbachol-induced increase of N1 production in M1- and M3-expressing HEK293 cells was directly attributable to the upregulation of ADAM17 phosphorylation and activity. To address this question, we mutated ADAM17 and examined the influence on N1 production and JMV2770-hydrolyzing activity. Mutations of tyrosine 702 and threonine 735 (Fig. 10A) abrogated carbachol-stimulated phosphorylation of ADAM17 in M1-overexpressing HEK293 cells (Fig. 11A). We next examined the influence of these mutations on JMV2770-hydrolyzing activity and N1 production by M1-expressing cells. We transiently transfected the HEK293 cells with wild-type ADAM17, or Y702F or T735A ADAM17 mutant cDNAs, and we observed a carbachol-stimulated increase of N1 recovery (Fig. 11B) and JMV2770-hydrolyzing activity (Fig. 11C) in wild-type and Y702-ADAM17-expressing cells, whereas the T735A mutation abolished this phenotype (Fig. 11B,C). Importantly, wild-type, T735A-, and Y702F-ADAM17 transfectants displayed similar ectopeptidase-like JMV2770-hydrolyzing activities under basal conditions (data not shown), indicating that ADAM17 trafficking to the membrane was not impaired by the mutations. Collectively, these results demonstrate that carbachol, via M1 receptors, promotes ADAM17 phosphorylation at Thr735, modulating its proteolytic activity and thereby the physiological processing of PrPc and N1 formation in HEK293 cells.
Figure 11.

Mutation of ADAM17 on threonine 735 affects carbachol-mediated N1 release and JMV2770-hydrolyzing activity in M1-expressing HEK293 cells. A, M1-expressing HEK293 cells grown in 100 mm dishes at 37°C were transiently transfected with wild-type (Wt), Y702F-ADAM17 (Y702F), and T735A-ADAM17 (T735A) cDNAs (A–C) or empty pcDNA3 vector (Ct) (B). Twenty-four hours after transfection, cells were treated for 5 min (A) or for 8 h (B) without (−) or with (+) carbachol (100 μm; Cch). Media were collected and analyzed for their N1 content (B). One milligram of proteins was submitted to immunoprecipitation (IP) using specific antibodies directed against phospho-threonine (α-phosphoThr) or phospho-tyrosine (α-phosphoTyr) residues and analyzed by 8% SDS-PAGE and Western blotting (WB) using a specific anti-ADAM17 (α-ADAM17) antibody as described in Materials and Methods (A). Bars correspond to the densitometric analysis of N1 (B) and are expressed as the percentage of control N1 recovered in nonstimulated conditions. C, In parallel experiments, transfected plated cells described above were monitored for their BB3103-sensitive JMV2770-hydrolyzing activities as described in Materials and Methods. Bars correspond to the densitometric analysis of the BB3103-sensitive JMV2770-hydrolyzing activity recorded. Values are the means ± SE of three (B) or six (C) independent experiments. *p < 0.005; **p < 0.0005; ***p < 0.0001; ns, nonstatistically significant.
PKC inhibitors reduce both carbachol- and PDBu-induced release of N1 and JMV2770-hydrolyzing activity
It has been extensively shown that PKC plays a role in the coupling of G-protein-coupled receptors, including muscarinic M1 and M3 receptors, to mitogen-activated protein kinase/extracellular signal-regulated kinase (Desdouits-Magnen et al., 1998; Schönwasser et al., 1998; Rosenblum et al., 2000; Slack, 2000; Hamilton and Nathanson, 2001). We demonstrated previously that regulated N1 production was triggered by the PKC agonist PDBu (Vincent et al., 2000). We examined whether PKC fully accounts for the carbachol-mediated ADAM17-dependent increase of N1 recovery. We first assessed the effect of GF109203X, a nonselective PKC inhibitor (Marano et al., 1995), on PDBu-treated M1-expressing HEK293 cells. As expected, PDBu increased N1 recovery in HEK293 cells, and GFX109203X prevented this effect (Fig. 12A). Interestingly, GF109203X also drastically reduced N1 recovery in carbachol-stimulated M1-expressing (Fig. 12B) or M3-expressing (data not shown) HEK293 cells. Furthermore, M1-expressing HEK293 cells display an increased carbachol-stimulated and BB3103-sentitive JMV2770-hydrolyzing activity, which was abolished by previous treatment with GF109203X (Fig. 12C). Together, these data allow us to conclude that M1 and M3 muscarinic receptor activation, which controls the release of N1, requires the phosphorylation of ADAM17 on threonine 735 in a PKC-dependent manner.
Figure 12.

M1 receptor-mediated activation of ADAM17 activity is PKC-dependent in HEK293 cells. A, B, M1-expressing HEK293 cells grown in 35 mm dishes were pretreated for 30 min without or with the PKC inhibitor GF109203X (2 μm; GFX), then media were removed out, and cells were incubated for 8 h with fresh serum-free media without or with carbachol (100 μm; Cch; B) or PDBu (1 μm; A). Media were collected and analyzed for their N1 content, and cell homogenates were prepared and analyzed for their PrPc-like immunoreactivity as described in Materials and Methods. Bars correspond to the densitometric analysis of N1 and are expressed as the percentage of control N1 recovered in nonstimulated conditions. C, Plated M1-expressing cells were monitored for their BB3103-sensitive JMV2770-hydrolyzing activity. Cells grown in six-well plates were pretreated for 30 min at 37°C with PBS in the absence or presence of the PKC inhibitor GF109203X (2 μm; GFX), and then cells were incubated with JMV2770 (at a final concentration of 10 μm) in the presence of carbachol (10 μm; Cch) with or without GFX109203X (2 μm). Aliquots of the media (100 μl) were collected at the indicated time points, and the fluorescence was recorded as described in Materials and Methods. The values represent the BB3103-sensitive activities and are means ± SEM of three (A, B) or six (C) independent determinations. ***p < 0.0005; **p < 0.001; *p < 0.05. ns, Nonstatistically significant; Ct, control vehicle.
Discussion
PrPc is a fascinating protein that has not yet revealed its secrets. It is really striking that relatively few things are known concerning its physiology when compared with the multiplicity of studies aimed at better understanding the mechanisms underlying the pathogenicity of its scrapie counterpart (PrPsc). The major interest in the latter protein derives from the recognition that it is the agent mediating scrapie-associated diseases to human beings (Prusiner, 1998). However, it is likely that a better understanding of PrPc physiology could ultimately lead to the development of strategies to interfere with the pathological process. The first clues for such a possibility came from the seminal observation that mice devoid of PrPc resist scrapie infection and toxicity (Büeler et al., 1993). Second, it was interesting that PrPc and PrPsc are metabolized by distinct proteolytic processes in normal and pathological brains. The latter observation may be significant because we recently established that PrPc catabolites generated by these distinct cellular proteolytic pathways exhibit differing abilities to influence p53-dependent cell death (Sunyach et al., 2007).
We recently showed that PrPc undergoes both constitutive and regulated physiological cleavages within its 106–126 putative toxic domain, generating an N-terminal product referred to as N1. Regulated cleavage of PrPc appeared to be under the control of the PKC pathway because it was drastically potentiated by the PKC agonist PDBu (Vincent et al., 2000). Interestingly, although disintegrin metalloproteases are generally involved in the hydrolysis of membrane-bound proteins, we demonstrated that PrPc, which is a glycosylphosphatidylinositol-anchored protein, was also susceptible to cleavage by these proteases. We delineated the respective contribution of ADAM10 and ADAM9 in the constitutive production of N1, whereas ADAM17 [TACE (tumor necrosis α-converting enzyme)] only participated in the PDBu-stimulated PKC-regulated N1 formation (Vincent et al., 2001). The latter observation, although interesting, did not identify the physiological activator of the protein kinase C pathway that could potentiate the recovery of endogenous N1. Furthermore, the empirical observation that PKC-regulated N1 formation was fully abolished by the genetic inactivation of ADAM17 (Vincent et al., 2001) did not establish the mechanisms by which this kinase could upregulate ADAM17 activity.
We have postulated that muscarinic receptors could regulate the processing of PrPc for three main reasons. First, the study of prion processing has highlighted the striking parallel between the processing of PrPc and of the β-amyloid precursor protein (Checler and Vincent, 2002). Thus, both proteins undergo constitutive and regulated cleavages triggered by ADAM10 and ADAM17, respectively (Buxbaum et al., 1998; Lammich et al., 1999; Vincent et al., 2000, 2001). Interestingly, muscarinic receptors were shown to participate in the α-secretase ADAM-mediated cleavage of β-amyloid precursor protein (βAPP) (Nitsch et al., 1992). Second, muscarinic M1 and M3 cell-surface receptors are efficiently coupled to phosphoinositide hydrolysis, thereby generating diacylglycerol that behaves as an activator of protein kinase C (Peralta et al., 1988). Third, endogenous PrPc mRNA and proteins colocalize with acetyl choline receptors and cholinergic neurons in brain and periphery (Harris et al., 1993a).
Our data show that atropine-sensitive carbachol stimulation enhances N1 formation by primary cultured neurons. Carbachol did not affect N1 production in mock-transfected HEK293 cells. This agrees with a previous study showing that carbachol did not affect the secretory form of APPα secretion in these cells that, therefore, do not express muscarinic receptors (Nitsch et al., 1992). This cell system was particularly useful for examining the putative contribution of each of the muscarinic receptor to N1 production. M1 and M3 expression increased carbachol-stimulated recovery of N1, whereas M2 and M4 expression did not modify N1 formation. The involvement of M1 receptors was further supported by the similar effect triggered by the selective M1 agonist AF267B (Beach et al., 2001). The lack of involvement of M2/M4 muscarinic receptors, which modulate the adenylyl cyclase-mediated pathway (Ashkenazi et al., 1987), fits perfectly with our previous observation that, unlike PKC, cAMP-dependent protein kinase A was unable to affect N1 recovery (Vincent et al., 2000). It should be noted here that, although displaying common features, βAPP and PrPc processing also exhibit differences because we established that α-secretase-mediated βAPP cleavage could be modulated by the cAMP–PKA-dependent pathway (Marambaud et al., 1998a,b).
M1- or M3-mediated enhancement of the recovery of N1 release is not attributable to the cellular redistribution of either PrPc or ADAMs by carbachol. We show that this effect is likely attributable to enhanced phosphorylation of ADAM17, as demonstrated by three independent types of experiments: (1) mutation of ADAM17 at the threonine 735 residue completely abolished M1-mediated phosphorylation of ADAM17; (2) T735A-ADAM17 did not cleave the fluorimetric ADAM17 substrate JMV2770 (Alfa Cissé et al., 2006); and (3) mutation of threonine 735 abolished the carbachol-stimulated M1-mediated increase of N1. The demonstration that ADAM17 phosphorylation modulated its activity and thereby N1 production agrees very well with a series of present and previous observations. Thus, several reports showed that ADAM17 undergoes phosphorylation on residues located in its cytoplasmic tail, thereby regulating its activity (Diaz-Rodriguez et al., 2002; Fan et al., 2003; Soond et al., 2005), although none of these studies identified muscarinic agonists as potentiators of ADAM17 activity. Second, it is striking that our study shows that, unlike ADAM17, carbachol does not affect ADAM9 phosphorylation, in line with our previous demonstration that only ADAM17 contributed to the regulated cleavage of PrPc (Vincent et al., 2001). Although the mutation at residue 735 was reported to alter the trafficking of ADAM17 to the plasma membrane (Soond et al., 2005), our data showing that wild-type and mutated ADAM17 display similar activities, on intact cells, in basal conditions strongly suggests that the abolishment of carbachol-stimulated N1 production by the ADAM17 mutation was not attributable to an impairment of the enzyme trafficking to the membrane.
Does muscarinic transmission act via PKC to increase the recovery of N1? Although carbachol mimicked the effect triggered by PDBu, the final proof that both agonists act through a unique signaling pathway came from our demonstration that carbachol-induced stimulation of N1 was fully prevented by GFX109203X, a broad-spectrum inhibitor of multiple PKC isoforms (Marano et al., 1995). One cannot rule out the possibility that PKC could have targeted a downstream kinase such as the extracellular signal-regulated protein kinase, which has been shown to trigger ADAM17 phosphorylation at its threonine 735 residue in Cos-7 and HEK293 cells (Diaz-Rodriguez et al., 2002; Soond et al., 2005). However, it seems reasonable to propose that M1/M3 muscarinic transmission controls the physiological processing of PrPc through PKC-dependent phosphorylation of ADAM17.
PrPc depletion protects mice from scrapie-mediated infection (Büeler et al., 1993). We were first to propose (Checler and Vincent, 2002) that enhanced physiological cleavage of PrPc, which inactivates the toxic core of PrPc and, therefore, diminishes the ability of this protein to act as a substratum for disease propagation, could be seen as a means to slow down infection. It is interesting to note that a study indicated that cellular prion interferes with the release of acetylcholine (Re et al., 2006). The alteration of various effectors of the cholinergic pathway could also occur in the pathological situation. Thus, an altered glycosylated form of acetylcholinesterase with lowered activity has been documented in Creutzfeldt–Jakob CSF (Silveyra et al., 2006a,b). Furthermore, the levels of choline acetyltransferase, the enzyme that is involved in acetylcholine synthesis, are modified after central administration of the scrapie agent (Durand-Gorde et al., 1985). The alteration of activities involved in both genesis and degradation of acetylcholine strongly suggests that acetylcholine levels are indeed altered in pathological brains. Along with this hypothesis, a previous study indicated that, in PC12 cells, scrapie infection led to decreased activity of the cholinergic-associated pathway (Rubenstein et al., 1991). Whether the putative alteration of cholinergic transmission could be causal for, or consequential of, the pathogenic process remains to be established.
It is interesting to note that M1 stimulation by AF267B was recently used to attenuate the major histopathological hallmarks associated with Alzheimer's disease and to rescue the cognitive deficits in a triple transgenic mice that recapitulates the major hallmarks of Alzheimer's disease (Caccamo et al., 2006). It is therefore reasonable to consider the use of M1/M3 muscarinic agonists as a putative means to potentiate PrPc processing, thereby pharmacologically downregulating endogenous PrPc and slowing down or arresting scrapie propagation and transmission.
Footnotes
This work was supported by the Fondation pour la Recherche Médicale, the Fondation pour la Recherche sur le Cerveau, and APOPIS (Abnormal proteins in the pathogenesis of neurodegenerative disorders) integrated project Grant PL 503330. We thank Drs. R. Black, C. Lunn, and C. Blobel for providing ADAM17, ADAM10, and ADAM9 cDNAs, respectively, and Dr. B. Mari for providing BB3103. We thank Dr. J. Grassi for providing SAF32 and Drs. J. F. Hernandez and J. Martinez for providing JMV2770.
References
- Aguzzi A, Montrasio F, Kaeser PS. Prions: health scare and biological challenge. Nat Rev Mol Cell Biol. 2001;2:118–126. doi: 10.1038/35052063. [DOI] [PubMed] [Google Scholar]
- Alfa Cissé M, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem. 2005;281:40624–40631. doi: 10.1074/jbc.M506069200. [DOI] [PubMed] [Google Scholar]
- Alfa Cissé M, Gandreuil C, Hernandez J, Martinez J, Checler F, Vincent B. Design and characterization of a novel cellular prion-derived quenched fluorimetric substrate of alpha-secretase. Biochem Biophys Res Commun. 2006;347:254–260. doi: 10.1016/j.bbrc.2006.06.065. [DOI] [PubMed] [Google Scholar]
- Ancolio K, Dumanchin C, Barelli H, Warter JM, Brice A, Campion D, Frébourg T, Checler F. Unusual phenotypic alteration of β amyloid precursor protein (βAPP) maturation by a new Val→Met βAPP-770 mutation responsible for probable early-onset Alzheimer's disease. Proc Natl Acad Sci USA. 1999;96:4119–4124. doi: 10.1073/pnas.96.7.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A, Winslow J, Peralta E, Peterson G, Schimerlik M, Capon D, Ramachandran J. An M2 muscarinic receptor subtype coupled to both adenylyl cyclase and phosphoinositide turnover. Science. 1987;238:672–675. doi: 10.1126/science.2823384. [DOI] [PubMed] [Google Scholar]
- Beach TG, Walker DG, Potter PE, Sue LI, Fisher A. Reduction of cerebrospinal fluid amyloid beta after systemic administration of M1 muscarinic agonists. Brain Res. 2001;905:220–223. doi: 10.1016/s0006-8993(01)02484-2. [DOI] [PubMed] [Google Scholar]
- Black RA, Durie CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, Kretzschmar HA. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature. 1996;380:345–347. doi: 10.1038/380345a0. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp H, DeArmond S, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Büeler H, Aguzzi A, Sailer A, Greiner R, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Liu K-N, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Checler F, Vincent B. Alzheimer and prion diseases: distinct pathologies, common proteolytic denominators. Trends Neurosci. 2002;25:616–620. doi: 10.1016/s0166-2236(02)02263-4. [DOI] [PubMed] [Google Scholar]
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion disease. J Biol Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- Demart S, Fournier JG, Cremignon C, Frobert Y, Lamoury F, Marce D, Lasmézas C, Dormont D, Grassi J, Deslys J-P. New insight into abnormal prion protein using monoclonal antibodies. Biochem Biophys Res Commun. 1999;265:652–657. doi: 10.1006/bbrc.1999.1730. [DOI] [PubMed] [Google Scholar]
- Desdouits-Magnen J, Desdouits F, Takeda S, Syu L-J, Saltiel AR, Buxbaum JD, Czernik AJ, Nairn AC, Greengard P. Regulation of secretion of Alzheimer amyloid precursor protein by the mitogen-activated protein kinase cascade. J Neurochem. 1998;70:524–530. doi: 10.1046/j.1471-4159.1998.70020524.x. [DOI] [PubMed] [Google Scholar]
- Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell. 2002;13:2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupiereux I, Zorzi W, Rachidi W, Zorzi D, Pierard O, Lheureux B, Heinen E, Elmoualij B. Study on the toxic mechanism of prion protein peptide 106–126 in neuronal and non neuronal cells. J Neurosci Res. 2006;84:637–646. doi: 10.1002/jnr.20965. [DOI] [PubMed] [Google Scholar]
- Durand-Gorde JM, Bert J, Nieoullon A. Changes in tyrosine hydroxylase, glutamic acid decarboxylase and choline acetyltransferase after local microinoculation of scrapie agent into the nigrostriatal system of the golden hamster. Brain Res. 1985;341:243–251. doi: 10.1016/0006-8993(85)91063-7. [DOI] [PubMed] [Google Scholar]
- Ettaiche M, Pichot R, Vincent JP, Chabry J. In vivo cytotoxicity of the prion protein fragment 106–126. J Biol Chem. 2000;275:36487–36490. doi: 10.1074/jbc.C000579200. [DOI] [PubMed] [Google Scholar]
- Fan H, Turck CW, Derynck R. Characterization of growth factor-induced serine phosphorylation of tumor-necrosis factor-α converting enzyme and on alternative translated polypeptide. J Biol Chem. 2003;278:18617–18627. doi: 10.1074/jbc.M300331200. [DOI] [PubMed] [Google Scholar]
- Fisher A, Brandeis R, Bar-Ner RH, Kliger-Spatz M, Natan N, Sonego H, Marcovitch I, Pittel Z. AF150(S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer's disease. J Mol Neurosci. 2002;19:145–153. doi: 10.1007/s12031-002-0025-3. [DOI] [PubMed] [Google Scholar]
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Hamilton SE, Nathanson NM. The M1 receptor is required for muscarinic activation of mitogen-activated protein (MAP) kinase in murine cerebral cortical neurons. J Biol Chem. 2001;276:15850–15853. doi: 10.1074/jbc.M011563200. [DOI] [PubMed] [Google Scholar]
- Harris DA, Lele P, Snider WD. Localization of the mRNA for a chicken prion protein by in situ hybridisation. Proc Natl Acad Sci USA. 1993a;90:4309–4313. doi: 10.1073/pnas.90.9.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA, Huber MT, van Dijken P, Shyng S-L, Chait BT, Wang R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry. 1993b;32:1009–1016. doi: 10.1021/bi00055a003. [DOI] [PubMed] [Google Scholar]
- Howard L, Nelson KK, Maciewicz RA, Blobel C. Interaction of the metalloprotease disintegrin MDC 9 and MDC 15 with two SH3-domain-containing proteins endophilin-1 and SH3PX1. J Biol Chem. 1999;274:31693–31699. doi: 10.1074/jbc.274.44.31693. [DOI] [PubMed] [Google Scholar]
- Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, B. K., Masters CL, Barrow CJ, Collins SJ, Cappai R. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106–126. J Neurochem. 1999;73:1557–1565. doi: 10.1046/j.1471-4159.1999.0731557.x. [DOI] [PubMed] [Google Scholar]
- Laffont-Proust I, Faucheux BA, Hässig R, Sazdovitch V, Simon S, Grassi J, Hauw JJ, Moya KL, Haïk S. The N-terminal cleavage of cellular prion protein in the human brain. FEBS Lett. 2005;579:6333–6337. doi: 10.1016/j.febslet.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee H-J, Thinakaran G, Kim T-W, Yu G, Xu H. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem. 2003;278:7850–7854. doi: 10.1074/jbc.C200648200. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Ancolio K, Checler F. Protein kinase A inhibitors drastically reduce constitutive production of Aβ40 and Aβ42 by human cells expressing normal and familial Alzheimer's disease-linked mutated βAPP and presenilin1. Br J Pharmacol. 1998a;126:1186–1190. doi: 10.1038/sj.bjp.0702406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Chevallier N, Ancolio K, Checler F. Post-transcriptional contribution of a cAMP-dependent pathway to the formation of α-and β/γ-secretases-derived products of βAPP maturation in human cells expressing wild type and Swedish mutated βAPP. Mol Med. 1998b;4:715–723. [PMC free article] [PubMed] [Google Scholar]
- Marano C, Laughlin K, Russo L, Mullin J. The protein kinase C inhibitor, bisindolylmaleimide, inhibits the TPA-induced but not the TNF-induced increase in LLC-PK1 transepithelial permeability. Biochem Biophys Res Commun. 1995;209:669–676. doi: 10.1006/bbrc.1995.1551. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Peralta E, Ashkenazi A, Winslow J, Ramachandran J, Capon D. Differential regulation of PI hydrolysis and adenylyl cyclase by muscarinic receptor subtypes. Nature. 1988;334:434–437. doi: 10.1038/334434a0. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re L, Rossini F, Re F, Bordicchia M, Mercanti A, Fernandez OS, Barocci S. Prion protein potentiates acetylcholine release at the neuromuscular junction. Pharmacol Res. 2006;53:62–68. doi: 10.1016/j.phrs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Rosenblum K, Futter M, Jones M, Hulme EC, Bliss TVP. ERKI/II regulation by the muscarinic acetylcholine receptors in neurons. J Neurosci. 2000;20:977–985. doi: 10.1523/JNEUROSCI.20-03-00977.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein R, Deng H, Scalici C, Papini M. Alterations in neurotransmitter-related enzyme activity in scrapie-infected PC12 cells. J Gen Virol. 1991;72:1279–1285. doi: 10.1099/0022-1317-72-6-1279. [DOI] [PubMed] [Google Scholar]
- Schönwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveyra MX, Garcia-Ayllon MS, Calero M, Saez-Valero J. Altered glycosylation of acetylcholinesterase in the Creutzfeldt-Jakob cerebrospinal fluid. J Mol Neurosci. 2006a;30:65–66. doi: 10.1385/JMN:30:1:65. [DOI] [PubMed] [Google Scholar]
- Silveyra MX, Cuadrado-Corrales N, Marcos A, Barquero MS, Rabano A, Calero M, Saez-Valero J. Altered glycosylation of acetylcholinesterase in Creutzfeldt-Jakob disease. J Neurochem. 2006b;96:97–104. doi: 10.1111/j.1471-4159.2005.03514.x. [DOI] [PubMed] [Google Scholar]
- Slack BE. The m3 muscarinic acetylcholine receptor is coupled to mitogen-activated protein kinase via protein kinase C and epidermal growth factor receptor kinase. Biochem J. 2000;348:381–387. [PMC free article] [PubMed] [Google Scholar]
- Soond S, Everson B, Riches DWH, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- Sunyach C, Alfa Cissé M, Alves da Costa C, Vincent B, Checler F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J Biol Chem. 2007;282:1956–1963. doi: 10.1074/jbc.M609663200. [DOI] [PubMed] [Google Scholar]
- Vincent B, Beaudet A, Dauch P, Vincent JP, Checler F. Distinct properties of neuronal and astrocytic endopeptidase 3.4.24.16: a study on differentiation, subcellular distribution and secretion processes. J Neurosci. 1996;16:5049–5059. doi: 10.1523/JNEUROSCI.16-16-05049.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent B, Paitel E, Frobert Y, Lehmann S, Grassi J, Checler F. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J Biol Chem. 2000;275:35612–35616. doi: 10.1074/jbc.M004628200. [DOI] [PubMed] [Google Scholar]
- Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, de Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol-esters-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–37746. doi: 10.1074/jbc.M105677200. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Kratzschmar J, Reid M, Blobel C. MDC9, a widely expressed cellular disintegrin containing cytoplasmic SH3 ligand domains. J Cell Biol. 1996;132:717–726. doi: 10.1083/jcb.132.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]