

Abstract
The perirhinal cortex plays a critical role in memory formation, in part because it forms reciprocal connections with the neocortex and entorhinal cortex and is thus in a position to integrate and transfer higher-order information to and from the hippocampus. However, for reasons that remain unclear, perirhinal transfer of neocortical inputs to the entorhinal cortex occurs with a low probability. Using patch recordings in vitro and tract-tracing combined with GAD-67 immunohistochemistry, we show that the perirhinal cortex contains GABAergic neurons with long-range projections to superficial entorhinal cells. This finding challenges the traditional model of cortical inhibition in which all trans-areal inhibition is thought to be disynaptic because the axons of GABAergic interneurons are assumed to be confined within the area in which their somata are located. Moreover, consistent with recent studies indicating that the formation of perirhinal-dependent memories requires activation of muscarinic receptors, long-range IPSPs were presynaptically inhibited by M2 receptor activation. Overall, these results suggest that long-range feedforward inhibition regulates perirhinal transfer of neocortical inputs to the entorhinal cortex, but that cholinergic inputs can presynaptically adjust the impact of this control mechanism as a function of environmental contingencies.
Keywords: perirhinal, entorhinal, hippocampus, inhibition, interneurons, learning, memory
Introduction
The perirhinal cortex (areas 36–35) plays a key role in memory. For instance, many perirhinal neurons display reduced responses to previously viewed stimuli (Fahy et al., 1993; Li et al., 1993; Sobotka and Ringo, 1993), a property thought to reflect their ability to discriminate familiar objects. Consistent with this, perirhinal lesions produce impairments in recognition memory (Zola-Morgan et al., 1989; Gaffan and Murray, 1992; Meunier et al., 1993, 1996; Suzuki et al., 1993) that are comparable with, or more severe than, those caused by hippocampal and entorhinal lesions (Aggleton et al., 1986; Murray and Mishkin, 1986; Meunier et al., 1993; Leonard et al., 1995; Murray et al., 2005). Moreover, perirhinal lesions disrupt hippocampal-dependent processes such as the stability of place fields (Muir and Bilkey, 2001) and spatial memory (Wiig and Bilkey, 1994; Nagahara et al., 1995; Murray et al., 1998).
Another line of research indicates that rhinal contributions to memory require cholinergic activation (Hasselmo, 2006). Indeed, perirhinal infusions of muscarinic receptor antagonists impair recognition memory and learned taste aversion (Tang et al., 1997; Warburton et al., 2003; Abe et al., 2004; Gutierrez et al., 2004). Similarly, cholinergic deafferentation of the rhinal cortices impairs recognition memory (McGaughy et al., 2005; Turchi et al., 2005; Winters and Bussey, 2005). Together, these results indicate that the perirhinal cortex, under cholinergic control, contributes to various types of memory-related functions that involve the integration and transfer of neocortical information to the hippocampus.
What is the anatomical substrate of perirhinal contributions to hippocampal-dependent memories? Tract-tracing studies suggests that information transfer between the neocortex and hippocampus occurs via the sequential activation of the perirhinal and entorhinal cortices (Burwell and Witter, 2002). Axons arising from associative neocortical areas project to area 36, which in turn, sends axons toward area 35 of the perirhinal cortex (Van Hoesen and Pandya, 1975; Witter and Groenewegen, 1986; Insausti et al., 1987). In turn, areas 35 and 36 project to superficial entorhinal neurons (Shi and Cassell, 1999), the main source of inputs to the hippocampus (Burwell and Witter, 2002). However, in vitro and in vivo physiological studies indicate that perirhinal transmission of neocortical activity to the entorhinal cortex occurs with a low probability (de Curtis and Paré, 2004), suggesting that strong inhibitory pressures regulate rhinal excitability.
Consistent with this, a recent study reported that Phaseolus vulgaris leucoagglutinin (PHA-L) injections in the perirhinal cortex anterogradely labeled a population of GABA-immunoreactive axon terminals that form symmetric synapses in the entorhinal cortex (Pinto et al., 2006), suggesting that the perirhinal cortex is endowed with long-range GABAergic neurons. However, the fact that PHA-L is not only transported anterogradely but also along the axon collaterals of retrogradely labeled cells (Smith et al., 1998) raises the possibility that these GABAergic terminals did not arise from the perirhinal cortex but from corticopetal GABAergic neurons, such as those described previously in the basal forebrain (Fisher et al., 1988), with branching axons to the perirhinal and entorhinal cortices. Thus, the present experiments were undertaken to shed light on the organization of the rhinal inhibitory circuit and its modulation by cholinergic inputs, using direct microstimulation of the perirhinal cortex coupled to pharmacological manipulations and retrograde tracing techniques.
Materials and Methods
Preparation of rhinal slices.
Experiments were conducted on Hartley guinea pigs (200–250 g, 3–5 weeks old) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with the approval of the Institutional Animal Care and Use committee of Rutgers University. Animals were deeply anesthetized with pentobarbital (40 mg/kg, i.p.) and ketamine (100 mg/kg, i.p.). The brain was removed and placed in an artificial CSF (aCSF) (4°C) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose. Using a vibrating microtome, the brain was cut in 400 μm coronal sections. Slices were stored for 1 h in an oxygenated chamber at room temperature and then transferred to a recording chamber perfused with an oxygenated aCSF solution at a rate of 2 ml/min. The temperature of the chamber was gradually increased to 32°C before the recordings began.
Recording methods.
Patch recordings of rhinal neurons were obtained under visual control with differential interference contrast and infrared video microscopy. Recording pipettes (4–6 MΩ) were pulled from borosilicate glass capillaries and filled with a solution containing the following (in mm): 130 K-gluconate, 10 HEPES, 10 KCl, 2 MgCl2, 2 ATP-Mg, and 0.2 GTP-Tris. The pH was adjusted to 7.2 and osmolarity to 280–290 mOsm. The liquid junction potential was measured, and membrane potential values were corrected accordingly. Bridge balance was monitored regularly. Unstable recordings or with access resistance higher than 15 MΩ were discarded. Current-clamp recordings were performed with an Axon-2B amplifier driven by Clampex acquisition software using a DigiData 1200 interface (Molecular Devices, Palo Alto, CA).
Electrical and chemical stimulation.
An array of seven tungsten electrodes (tip diameter, 25 μm; intertip spacing, 160 μm) was positioned in either area 35 or area 36 to activate afferents to the ventrolateral entorhinal cortex (vlEC) or area 35 cells, respectively. On average, the distance between the area 35 stimulating electrodes and the recorded vlEC cells was 0.95 ± 0.02 mm compared with 0.75 ± 0.02 for the distance between the area 36 stimulating electrodes and the recorded area 35 cells. Bipolar electrical stimuli consisted of 50–200 μs current pulses (0.1–1.2 mA). To chemically activate neurons projecting to the recorded cells, extracellular pressure applications of glutamate (500 μm in aCSF) were performed. In this case, brief air pressure pulses (10–100 ms; 18 psi) were applied to micropipettes (tip outer diameter, 1 μm). In these experiments, slices were oriented so that the aCSF flow would facilitate glutamate diffusion away from the recorded cells, toward the chamber outlet. To determine the reversal potential of evoked responses, all tests were performed at various membrane potentials, as determined by intracellular current injection.
Morphological identification of recorded neurons.
In some experiments, Neurobiotin (0.2%) was added to the pipette solution to reveal the morphology of recorded cells. Only one cell was labeled per slice. After the recording, the slice was placed in a fixative containing 2% paraformaldehyde and 1% glutaraldehyde in 0.1 PBS, pH 7.4, for 1–3 d, then embedded in agar (4%), and sectioned on a vibrating microtome (60 μm). Cells were visualized by incubating the sections in the avidin–biotin–peroxidase solution (ABC Elite kit; Vector Laboratories, Burlingame, CA). Sections were placed in a sodium borohydride solution [1% in 0.1 m phosphate buffer (PB) for 30 min], rinsed in PB, and incubated overnight in a solution containing 1% bovine serum albumin (BSA), 0.3% Triton X-100, and 1% solutions A and B of ABC kit in PBS. The next day, the sections were washed in PBS followed by multiple washes in 0.1 m TBS. Neurobiotin was visualized by incubating the sections in a Tris buffer containing 700 μm DAB and 0.3% H2O2 for 8–10 min. Then, sections were washed in PBS (six times for 5 min), mounted onto slides, dehydrated, and coverslipped.
Retrograde tracing and immunofluorescence protocol used to reveal GAD-67 immunoreactivity.
The animals were anesthetized with a mixture of ketamine, acepromazine, and xylazine (50, 2.5, and 5 mg/kg, i.p., respectively) and placed in a stereotaxic apparatus. Perirhinal cells projecting to the vlEC were retrogradely labeled by performing iontophoretic injections (6 μA, 7 s ON/OFF for 15 min/site) of hydroxystilbamidine [Fluorogold (FG) equivalent] (peak absorption, 361 nm, peak emission, 536 nm) in the vlEC through a glass micropipette (tip diameter of 25–30 μm). After a 4 d survival period, animals were deeply anesthetized as above and perfused with saline (0.9%), followed by a fixative containing 4% paraformaldehyde, 1.4% l-lysine, and 0.2% sodium metaperiodate in 0.1 m PB, pH 7.4. After extraction from the skull, the brains were postfixed overnight in the same fixative and then sectioned at 60 μm in PB. The sections were then incubated in sodium borohydride (1% in 0.1 m PB for 30 min), washed repeatedly in PB followed by rinses in TBS, and placed in a blocking solution containing 3% normal goat serum, 1% BSA, and 0.3% Triton X-100 in 0.1 m TBS for 30 min. Sections were then incubated for 48 h at 4°C with monoclonal mouse anti-GAD-67 (Chemicon, Temecula, CA) diluted (1:1000) in the same blocking solution. After several washes in TBS, sections were incubated with a cyanine 3 (Cy3)-conjugated donkey anti-mouse secondary antibody (peak absorption, 552 nm, peak emission, 570 nm; Jackson ImmunoResearch, West Grove, PA) diluted (1:800) in the same blocking solution for 1 h. After several washes in TBS and PBS, the sections were mounted on glass slides, dehydrated, and coverslipped with Cytoseal.
Approach used for cell counts.
For each of the three cases in which FG injections were confined to the entorhinal cortex, we analyzed three perirhinal sections located at the same rostrocaudal level as the center of the injection site. Areas 36 and 35 of the perirhinal cortex were defined before counting using the criteria defined by Uva et al. (2004) for the guinea pig. FG-labeled and GAD-67-immunoreactive neurons were visualized with a Nikon (Tokyo, Japan) microscope (Eclipse E800) equipped with a fluorescence illumination system (X-cite 120). Images were captured with a digital camera (Nikon DXM 1200F) controlled by the software ACT-1. Cells labeled with FG or GAD-67 were differentially visualized with an ultraviolet or Cy3 filter (528–553 nm excitation and 590–650 nm emission), respectively. Only cells that were clearly labeled were considered. The luminosity threshold we used to identify labeled neurons was more than three times background, as measured with Photoshop (Adobe Systems, San Jose, CA). Because we were not interested in estimating the absolute number of long-range GABA cells but only in obtaining an estimate of the proportion of FG or GAD-67 neurons that were double labeled, we did not use unbiased stereological counting methods. This is line with the guidelines used by the Journal of Comparative Neurology, as defined in the editorial of Saper (1996).
For perirhinal area 36, layers III–IV were defined as the region located between the densely packed cells of layer II and the larger tightly packed layer V neurons. In a study on the cytoarchitecture of the guinea pig perirhinal cortex (Uva et al., 2004), layer II cells of area 35 were described as being more dispersed making the border between layers II and III rather vague. After Uva et al. (2004), we delineated layer III by extrapolating from the layer II–III border of area 36. Layer IV of the perirhinal cortex gradually thins from lateral to medial almost to the point of disappearing in area 35. For this reason, we have chosen not to try to make a distinction between the two and have grouped the region between layers II and V of area 35 into layers III–IV.
Results
Electrophysiological experiments
To shed light on the perirhinal inhibitory pressures that limit information transfer from the neocortex to the entorhinal region, coronal slices preserving the connections between these cortical fields were prepared, and patch recordings of principal neurons were obtained under visual guidance in the vlEC (n = 128) and perirhinal area 35 (n = 74). All recorded cells displayed physiological properties characteristic of principal neurons (e.g., spiny pyramidal or stellate cells), including spike frequency accommodation (Fig. 1A) and, in a subset of cells, various degrees of time-dependent inward rectification in response to hyperpolarizing current pulses (Bilkey and Heinemann, 1999; Faulkner et al., 1999; Martina et al., 2001; Alonso, 2002). Consistent with this, all morphologically identified cells (vlEC, n = 7; area 35, n = 6) had a pyramidal (Fig. 1B1) or stellate morphology, and their dendrites were covered with spines (Fig. 1B2).
Figure 1.

Identification of recorded cells and experimental setup. Physiological (A) and morphological (B) identification of principal rhinal neurons. Whole-cell patch recordings of perirhinal or entorhinal neurons were obtained under visual guidance, and their electroresponsive properties were studied by injecting a series of rectangular current pulses of gradually increasing amplitude. We only considered rhinal neurons that displayed a regular spiking pattern, as shown in the representative example of layer II entorhinal neurons depicted in A. Intracellular Neurobiotin injection was performed to reveal the morphology of recorded cells. As shown in B, all recovered neurons had multipolar dendritic trees (B1) that were covered with spines (B2). C, Experimental setup. A stimulating electrode array (stim.; red dots) was positioned in area 35 (C1) or area 36 (C2), and evoked responses were monitored in cells of the vlEC (C1) or area 35 (C2), respectively. The cross indicates orientation of the scheme in which D, V, L, and M stand for dorsal, ventral, lateral, and medial, respectively. NC, Neocortex.
Two sets of experiments were performed. The first focused on the projection from perirhinal area 35 to the vlEC (Fig. 1C1). The second examined transmission from perirhinal area 36 to area 35 (Fig. 1C2). However, because identical results were obtained, these experiments will be described in parallel.
We first tested the effects of electrical stimuli delivered in area 35 on superficial (layer II–III) vlEC neurons. During these tests, their membrane potential was set to −60 mV by intracellular current injection to allow distinction of EPSPs and IPSPs on the basis of polarity. At each stimulation site (Fig. 1C1, red dots), the stimulus intensity was gradually increased in steps of 50 μA in search of a site in which threshold stimuli would first evoke inhibitory responses. In 25% of superficial vlEC neurons (or 7 of 28 tested cells), one or more stimulation sites in area 35 evoked apparently pure inhibitory responses (average amplitude, 2.7 ± 0.4 mV at −60 mV) at threshold intensities.
An example of this phenomenon is shown in Figure 2, for a layer II vlEC cell. Note that, as the stimulation intensity was increased (Fig. 2A1, numbers on the right), the first response to be evoked was an apparently pure IPSP. At the threshold intensity (Fig. 2A1, 150 μA), its latency was variable but it stabilized with a slight increase in stimulation intensity (Fig. 2A1, 200 μA). Presumably, these slight latency variations are caused by intertrial differences in the membrane potential of the GABAergic cells in the face of a gradually increasing afferent volley. With additional increases in stimulation intensity, EPSPs appeared in all tested cells (arrows in Fig. 2A1, 300 μA), the response eventually evolving into a canonical EPSP–IPSPa-b response (Fig. 2A1, 500 μA).
Figure 2.

Threshold electrical stimulation of area 35 often elicits CNQX- and AP-5-resistant IPSPs in vlEC neurons. A1, Response of a superficial vlEC neuron to area 35 stimuli of gradually increasing intensity (numbers on the right). A2, Response seen in A1 with 200 μA stimulus is not abolished by addition of CNQX and AP-5 to the aCSF. B, In many vlEC cells, IPSPs preceded EPSPs after threshold stimulation of area 35. B1, Example of IPSP–EPSP sequence evoked from area 35 at three different membrane potentials (numbers on the right), as determined by intracellular current injection. B2, The initial IPSP persisted after addition of CNQX and AP-5 to the aCSF. B3, CNQX- and AP-5-resistant IPSPs evoked in vlEC cells by threshold stimulation of area 35 reverse just below −70 mV (top) and are abolished by picrotoxin (bottom).
In the other superficial vlEC neurons (n = 21), EPSPs and IPSPs appeared at the same threshold stimulation intensity. However, in 33% of these cells (or 7 of 21), the IPSP onset occurred before that of the EPSP. An example of this is shown in Figure 2B1 (trace labeled, −60 mV). That these early negative potentials truly are IPSPs, as opposed to field responses seen through the membrane of the recorded cells, was indicated by the fact that they reversed in polarity during membrane hyperpolarization (Fig. 2B1).
The above findings are difficult to reconcile with the traditional model of cortical inhibition. Indeed, this model is based on the premise that the axons of GABAergic interneurons are confined within the area in which their somata are located. It is thus typically assumed that all trans-areal inhibition is disynaptic. That is, cortical afferents to a second cortical region would inhibit target cells indirectly, via the glutamatergic activation of local-circuit neurons located in the target region. As a result, electrically evoked IPSPs should always follow EPSPs. Moreover, stimulation of area 35 should not trigger pure IPSPs unless the glutamatergic afferents recruited by our stimuli happened to avoid the recorded vlEC cells but not the feedforward interneurons contacting them. A more parsimonious explanation, however, would be that area 35 stimuli directly activated GABAergic neurons of the perirhinal cortex with axons contacting superficial vlEC cells.
To test this possibility, we examined how the addition of the glutamate receptor antagonists CNQX and AP-5 (respectively, 20 and 100 μm in the extracellular perfusate, aCSF) affected the responses evoked in vlEC neurons by threshold stimulation of area 35. Our hypothesis predicts that IPSPs should persist despite glutamate receptor blockade, whereas the traditional model predicts their abolition. In support of our hypothesis, the IPSPs evoked by juxtathreshold stimuli persisted in the presence of CNQX and AP-5 in five of six vlEC cells (Fig. 2A2, 200 μA), although they were reduced in amplitude (42 ± 7% reduction). Moreover, in all vlEC cells in which threshold stimuli elicited IPSPs that preceded EPSPs (n = 7), the IPSPs persisted in the presence of the glutamate receptor antagonists (Fig. 2B2). The CNQX- and AP-5-resistant IPSPs seen at threshold stimulation intensities had a peak amplitude of −2.3 ± 0.3mV, reversed in polarity at −74.5 ± 1.3 mV, and were abolished by picrotoxin (n = 5) (Fig. 2B3), suggesting that they were mediated by GABAA receptors. Overall, whether threshold area 35 stimuli evoked responses consisting of pure IPSPs, IPSP–EPSP, or EPSP–IPSP sequences, evoked IPSPs persisted in the presence of glutamate antagonists in 67% of superficial vlEC neurons (or 18 of 27).
Similar results were obtained when we tested the effect of area 36 stimulation on area 35 cells. That is, in 32% of tested layer II–III cells (or 9 of 28), area 36 stimuli just above threshold evoked apparently pure IPSPs that persisted in the presence of CNQX and AP-5 (Fig. 3A). In the rest of our sample, EPSPs and IPSPs appeared at the same threshold stimulus intensity, although the IPSP onset preceded that of the EPSPs in 26% (or 5 of 19) of cases. These early IPSPs persisted in the presence of CNQX and AP-5 (40 ± 6% amplitude reduction), whereas the EPSPs were abolished (Fig. 3B). The CNQX- and AP-5-resistant IPSPs (−1.9 ± 0.2 mV) reversed at −71.2 ± 1.1 mV (Fig. 3C, top) and were abolished by picrotoxin (n = 5) (Fig. 3C, bottom). In total, 75% of superficial area 35 neurons (or 21 of 28) showed CNQX/AP-5-resistant IPSPs after threshold stimulation of area 36.
Figure 3.

Threshold electrical stimulation of area 36 often elicits CNQX- and AP-5-resistant IPSPs in area 35 neurons. A, Response of a superficial area 35 neuron to area 36 stimuli of gradually increasing intensity (numbers on the right). B, Response seen in A with 200 μA stimulus is not abolished by addition of CNQX and AP-5 to the aCSF. C, CNQX- and AP-5-resistant IPSPs evoked in area 35 cells by threshold stimulation of area 36 reverse just below −70 mV (top) and are abolished by picrotoxin (bottom).
In the above experiments, it is unclear whether the electrically evoked IPSPs were attributable to the activation of GABAergic neurons located within the perirhinal cortex or to axons coursing through the perirhinal cortex but arising from elsewhere. In addition, although we used low-intensity electrical stimuli, it could be argued that current diffused enough to activate GABAergic neurons located outside the stimulated region. To address these concerns, we attempted to evoke long-range IPSPs in vlEC or area 35 neurons by performing local pressure applications of glutamate (500 μm) via a patch pipette (1 μm tip) in areas 35 or 36, respectively. To ensure spatial specificity of the glutamate microinjection method, the following precautions were taken. First, the slices were oriented such that the ejected glutamate would be carried by the aCSF flow, away from the recording site, toward the chamber outlet. Second, we only considered responses that could be elicited by glutamate puff durations of ≤60 ms or less because we estimated that ≤5 nl of glutamate is ejected with such short-duration stimuli. Third, we performed these experiments in the presence of CNQX and AP-5 to ensure that the ejected glutamate would only exert significant effects where it is most concentrated, immediately adjacent to the glutamate ejection pipette.
Figure 4 illustrates control experiments designed to test the feasibility of this approach. Patch recordings of perirhinal cells (n = 16) were obtained (Fig. 4A) and glutamate puffs performed at various distances with respect to the recorded cell (Fig. 4A). In addition, an array of stimulating electrodes (Fig. 4A, dots) was positioned in area 36 to test whether CNQX and AP-5 would exert differential effects on synaptically released versus exogenous glutamate. Figure 4B shows the response of a representative cell to local pressure applications of glutamate (60 ms pulse) (Fig. 4B, left) and electrical stimulation of area 36 (Fig. 4B, right) in control media (Fig. 4B1) and in the presence of CNQX and AP-5 (Fig. 4B2). Note that these glutamate receptor antagonists abolished electrically evoked EPSPs. In contrast, CNQX and AP-5 only delayed the response to the glutamate puff, a phenomenon that presumably reflects the time required for glutamate to compete with CNQX and AP-5 for NMDA and non-NMDA receptors. Similar pressure applications of aCSF had no effect (n = 4).
Figure 4.

Contrasting effects of CNQX and AP-5 on synaptically released versus pressure-applied glutamate. Control experiments designed to test the feasibility and spatial selectivity of local pressure glutamate applications in the presence of glutamate receptor antagonists. A, Scheme showing the experimental approach used in these control experiments. An array of stimulating electrodes (dots) was positioned in area 36 while monitoring evoked responses with the patch method in superficial area 35 neurons. A second pipette containing 0.5 mm glutamate (dissolved in aCSF) was positioned at proximity of the recorded neuron. B, Responses to pressure-applied (left) or synaptically released (right) glutamate in control aCSF (B1) versus after addition of CNQX and AP-5 to the aCSF (B2). C, Spatial selectivity of responses to pressure-applied glutamate. Pressure applications of glutamate were performed at various locations (100 μm grid pattern; red squares) around a perirhinal cell recorded in whole-cell mode. Two stimulus durations (60 and 100 ms) were tested (bottom and top traces, respectively). Only two of the ejection sites (C1, C3), both located in the immediate vicinity of the soma, evoked suprathreshold responses in this perirhinal neuron. D, Dorsal; V, ventral; L, lateral; M, medial; NC, neocortex.
Having established that CNQX and AP-5 could block the effects of synaptically released but not exogenous glutamate, we next tested the spatial selectivity of our pressure application method. Figure 4C shows a representative control experiment in which glutamate was applied at various positions with respect to the recorded cell (see scale bar in the top right corner of Fig. 4C). At each ejection site, two stimulus durations were used (60 and 100 ms; bottom and top traces, respectively). As shown in Figure 4C, even with the longest stimulus durations, only two closely spaced ejection sites immediately adjacent to the perisomatic region could fire this cell. Similar experiments were performed in 13 perirhinal neurons, and identical results were obtained. Thus, we are confident that, with the short pulse durations used here, this glutamate ejection method is spatially selective, only firing neurons that are immediately adjacent to the tip of the ejection pipette.
To search for long-range GABAergic neurons, we systematically scanned the stimulation region in a grid of 100 μm, applying 100 ms glutamate puffs at each site in the presence of CNQX and AP-5. When a site effective in evoking a response was found, the glutamate pulse duration was reduced and the pipette position optimized to elicit the largest inhibitory response with the shortest pulse durations possible. Then, as a control, we moved the ejection pipette 50 μm toward the recording site. Figure 5A illustrates examples of IPSPs evoked in a vlEC neuron by applying glutamate (60 ms puff) in area 35 in presence of CNQX and AP-5, at a concentration that was sufficient to block the effects of synaptically released glutamate (Fig. 5B). A second example is shown in Figure 5C in which glutamate puffs of various durations were used, as indicated by the numbers on the left. Note that, in this example, IPSPs were evoked using a stimulus duration of 20 ms (≤2 nl). When the glutamate ejection pipette was moved 50 μm toward the recording site, the response was abolished (data not shown), indicating that the long-range IPSP was not mediated by glutamate diffusion to GABAergic cells located in the vlEC. Similar results were obtained in a total of five vlEC with stimulus durations ≤60 ms. Long-range IPSPs could also be seen in area 35 neurons in response to brief (≤60 ms) pressure application of glutamate in area 36 (n = 5).
Figure 5.

Local pressure application of glutamate in area 35 elicits CNQX- and AP-5-resistant IPSPs in superficial vlEC neurons. A, Response of a layer II vlEC neuron to local pressure application of glutamate in area 35. The same test was performed at three different membrane potentials (numbers on the right), as determined by intracellular current injection. B, In the same neuron, CNQX and AP-5 block the EPSP but not the IPSP elicited by threshold electrical stimulation of area 35. C, A second example of long-range IPSPs evoked in a vlEC neurons by pressure application of glutamate in area 35 in the presence of CNQX and AP-5. Various pulses durations (numbers on the left) were tested.
Tract-tracing combined with GAD-67 immunohistochemistry
To further test whether the perirhinal cortex contains GABAergic neurons with long-range projections to the vlEC, we combined injections of the retrograde tracer FG with immunohistochemistry for GAD-67. Under stereotaxic guidance, FG injections were aimed to the vlEC in five animals. The FG injection site was confined to the vlEC in three of these cases (see examples in Fig. 6A1,A2). After these FG injections, numerous retrogradely labeled neurons could be seen within areas 36 and 35 (Fig. 6A3). FG-positive (FG+) perirhinal neurons were most numerous in superficial layers (II–III), but some were also present in deeper layers (Fig. 6A3). Although most FG+ perirhinal neurons were immunonegative for GAD-67 (Fig. 6B), some of them were immunoreactive for this enzyme (Fig. 6C,D).
Figure 6.
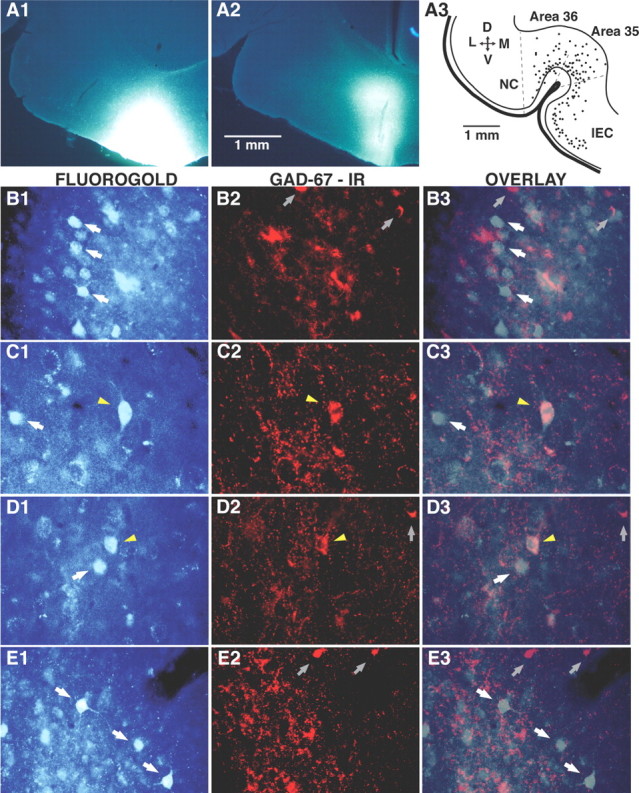
Iontophoretic injection of Fluorogold in the vlEC retrogradely labels perirhinal neurons, a proportion of which are immunoreactive for GAD-67. A1, A2, Representative examples of Fluorogold injection sites in the vlEC. A3, Mapping of retrogradely labeled neurons (dots) in the perirhinal cortex on a representative 60-μm-thick coronal section. B–E are all organized in the same manner. The left column shows neurons retrogradely labeled with Fluorogold. The center column shows GAD-67 immunoreactivity (IR) in the same field. The right column overlays the Fluorogold and GAD-67 labeling. The photomicrographs shown in B and C were obtained in area 35, those in D were obtained in area 36, and those in E were obtained in the lateral amygdala. White arrows indicate FG-positive neurons that were immunonegative for GAD-67. Yellow arrowheads mark FG-positive neurons that were immunoreactive for GAD-67. Gray arrows point to neurons immunoreactive for GAD-67 only. D, Dorsal; V, ventral; L, lateral; M, medial; NC, neocortex.
To quantify the relative importance of these two populations of perirhinal cells, we counted FG+, GAD-67+, and double-labeled neurons in three sections per animal and averaged the results (see Materials and Methods). On average, FG injections retrogradely labeled 53.1 ± 5.4 area 35 neurons and 46.9 ± 3.6 area 36 neurons per section. Although many GAD-67+ neurons were observed in these cortical fields (105.3 ± 7.4 and 72.4 ± 3.7 per section, respectively), only 2.1 ± 0.3 and 1.5 ± 0.4% of them were also FG+ in area 35 (Fig. 6C) and area 36 (Fig. 6D), respectively. These doubled-labeled neurons, respectively, accounted for 4.0 ± 0.6 and 2.4 ± 0.5% of FG+ area 35 and 36 neurons and were mostly confined to layers III–IV of the perirhinal cortex. Indeed, restricting the analysis to these layers revealed that 3.6 ± 0.6% of area 36 and 4.5 ± 0.6% area 35 neurons that were GAD-67+ were FG+. As a control, we performed similar analyses in the basolateral amygdala (BLA), a structure thought to send exclusively glutamatergic projections to the vlEC (Smith and Paré, 1994; Paré et al., 1995). Consistent with this view, none of the FG+ BLA neurons (13.4 ± 2.8 per section) (Fig. 6E1) displayed immunoreactivity for GAD-67 (Fig. 6E2,E3).
Overall, these tract-tracing experiments provide independent support for the notion that the perirhinal cortex contains long-range GABAergic cells projecting to the vlEC. It should be noted that, because we performed small FG injections medial to the vlEC sector studied in our physiological experiments, the approach we used likely underestimated the numerical importance of long-range GABAergic cells. Also, although long-range GABA cells account for a small proportion of GABAergic perirhinal neurons, this does not imply that they have a negligible impact on entorhinal excitability as their axons may branch profusely. Consistent with this, it was reported that, after PHA-L injections in the perirhinal cortex, as many as 12% of anterogradely labeled axon terminals seen in the entorhinal cortex were GABA immunoreactive (Pinto et al., 2006).
Regulation of long-range GABAergic inhibition by acetylcholine
Having established that the perirhinal cortex contains GABAergic neurons projecting to the vlEC, we turned our attention to the modulation of this long-range inhibition by acetylcholine because previous work revealed that the memory functions of the perirhinal cortex require muscarinic receptor activation (Warburton et al., 2003; Abe et al., 2004; Gutierrez et al., 2004). Acetylcholine is known to exert various effects on inhibition, the most common of which is to presynaptically inhibit GABA release via muscarinic receptors (Ben-Ari et al., 1981; Behrends and ten Bruggencate, 1993) (for review, see Miller, 1998). Consistent with this, M2 receptors are commonly expressed by GABAergic terminals (Hersch et al., 1994; Hajos et al., 1998; Erisir et al., 2001), including those originating from long-range GABAergic hippocampal neurons projecting to the septum (Hajos et al., 1998). These observations led us to test the impact of M2 receptor activation on the long-range IPSPs in the perirhinal to entorhinal pathway. However, M1 receptor activation is known to depolarize principal layer II entorhinal neurons via the activation of a Ca2+-dependent nonspecific cationic current (Klink and Alonso, 1997; Shalinsky et al., 2002; Magistretti et al., 2004). Thus, to isolate the effects of muscarine on long-range inhibition from these postsynaptic actions, the M1 receptor antagonist (pirenzepine, 800 nm) was added to the control aCSF. These tests were performed in current-clamp (n = 20) or voltage-clamp (n = 34) modes in different subsets of cells, and qualitatively identical results were obtained.
As in the first set of experiments (Fig. 2B), long-range IPSPs or IPSCs were evoked in vlEC cells with threshold stimulation of the perirhinal cortex in the presence of CNQX and AP-5. Addition of the M1 receptor antagonist pirenzepine to the aCSF had no effect on the input resistance of the cells (change of 3.7 ± 2.9%; paired t test, p > 0.05; n = 16) and on the amplitude of long-range IPSPs or IPSCs (change of 4.1 ± 3.8%; paired t test, p > 0.05; n = 36). In contrast, application of muscarine chloride (5 μm) in the presence of pirenzepine significantly reduced IPSP and IPSC amplitudes (to 59.5% ± 7.0% of control; p < 0.05, n = 36) (Fig. 7A1) without altering the GABAA reversal potential (change of 1.4 ± 0.86 mV; paired t test, p > 0.05), the input resistance of the cells (change of 3.3 ± 2.4%; paired t test, p > 0.05), or the holding current (change of −3.4 ± 5.3 pA; p > 0.05). However, when the M2 receptor antagonist methoctramine (0.8 μm) was applied before muscarine in the presence of pirenzepine, the reduction in IPSP (n = 4) and IPSC (n = 5) amplitude was not observed (800 nm) (92.0 ± 6.0% of control; p > 0.05) (Fig. 7A2). The M2-mediated inhibition of long-range IPSPs was also seen when the experiments were performed with a Cs+-based intracellular solution (64.5 ± 8.6% of control; p < 0.05; n = 11) (Fig. 7A3), further supporting the view that these effects are not mediated by the inhibition of the hyperpolarization-activated mixed cationic current Ih (Klink and Alonso, 1993; Dickson et al., 2000) or of voltage-dependent K+ conductances (Klink and Alonso, 1997; Shalinsky et al., 2002; Magistretti et al., 2004) expressed by the recorded cells.
Figure 7.

Inhibition of long-range IPSCs by M2 receptor activation. CNQX, AP-5, and the M1 receptor antagonist pirenzepine are present in the aCSF throughout. A, Voltage-clamp recordings of long-range IPSCs evoked in three different layer II vlEC cells by threshold stimulation of area 35. A1, Effect of muscarine (5 μm) on long-range IPSCs. Left, Control response. Right, After addition of muscarine to the aCSF (A2), application of the M2 antagonist methoctramine (0.8 μm) before muscarine completely blocks the inhibition of long-range IPSCs. Left, Control response in the presence of methoctramine. Right, Response elicited in the presence of methoctramine and muscarine. A3, The muscarinic inhibition of long-range IPSCs is also observed when vlEC cells are recorded with pipettes containing a CS+-based internal solution. Left, Control response. Right, Response elicited in the presence of muscarine. B, Muscarine produces an increase in paired-pulse ratio. B1, Top, Control conditions. Bottom, After addition of muscarine. B2, Same IPSCs as in B1 after scaling of the first IPSC seen in the presence of muscarine to that observed in control conditions. C, Graph plotting the ratio of experimental to control 1/CV2 (y-axis) against the ratio of experimental to control response amplitudes (x-axis). The solid line is a linear fit to the data.
To test whether the M2-mediated inhibition of long-range inhibition resulted from a presynaptic inhibition of GABA release, we tested the effects of muscarine on paired-pulse facilitation (PPF) of long-range IPSCs. In this analysis (Katz and Miledi, 1968), two stimuli of equal intensity are applied in brief succession. When the interval between the two shocks is sufficiently brief, the second one typically elicits a larger response that is believed to result from residual Ca2+ in the presynaptic terminals (for review, see Zucker and Regeher, 2002). The amount of PPF is thought to be inversely proportional to transmitter release probability because manipulations that increase release probability decrease PPF and conversely (Creager et al., 1980; Manabe et al., 1993).
Thus, we used the same protocol as above with the exception that control and post-muscarine stimuli were applied in pairs with an interstimulus interval of 50 ms. Below, the term PPF will be used when referring to the ratio of the second to the first IPSC. In control conditions, the paired-pulse ratio was 1.11 ± 0.07 (n = 8). In the presence of pirenzepine, muscarine application increased the PPF ratio to 1.44 ± 0.13 (n = 8) (Fig. 7B1,B2). Also consistent with a presynaptic site of action was the observation that muscarine reduced the input resistance drop associated with the long-range inhibition (by 28.9 ± 2.1%; paired t test, p < 0.05; n = 23).
To obtain independent evidence for the notion that M2 effects on long-range inhibition depend on a reduction of GABA release, we analyzed the variability of long-range IPSCs before versus after the application of muscarine. This variability is known to reflect the probabilistic process underlying transmitter release and can be estimated by computing the coefficient of variation (CV) (SD/mean) of long-range IPSCs. The parameter 1/CV2 is primarily determined by the presynaptic factors controlling transmitter release. By plotting the ratio of experimental to control 1/CV2 against the ratio of experimental to control response amplitudes, the dependence of presynaptic versus postsynaptic function can be determined (Bekkers and Stevens, 1990; Manabe et al., 1993; Kimura and Baughman, 1997). A positive correlation between the two was shown to reflect a presynaptic mechanism, whereas a horizontal regression is indicative of a purely postsynaptic action. Consistent with a presynaptic locus of action for the M2 effect, a strong correlation was found between the experimental to control 1/CV2 and IPSC amplitude ratios (r = 0.62; p < 0.05) (Fig. 7C).
Discussion
The present study was undertaken to shed light on the inhibitory mechanisms dampening perirhinal transfer of neocortical inputs to the entorhinal cortex. The interest of this issue stems from previous findings indicating that, although the rhinal cortices constitutes the main access route for neocortical inputs into the hippocampus (Burwell and Witter, 2002), perirhinal transmission of neocortical impulses occurs with a low probability (for review, see de Curtis and Paré, 2004). However, the underlying causes were unknown. A previous anterograde tracer study had reported evidence consistent with the existence of long-range GABA cells in the perirhinal cortex (Pinto et al., 2006). However, the fact that anterograde tracers are not only transported anterogradely but also along the axon collaterals of retrogradely labeled cells (Smith et al., 1998) raised the possibility that this GABAergic projection arose from subcortical corticopetal GABAergic neurons, such as those described previously in the basal forebrain (Fisher et al., 1988). The present study sheds light on this issue by establishing that the perirhinal cortex does contain a population of long-range GABAergic neurons with axons extending to neighboring cortical regions, including the entorhinal cortex, the main source of inputs to the hippocampus. Moreover, in keeping with previous findings indicating that perirhinal contributions to memory require muscarinic receptor activation, the impact of these long-range GABAergic neurons could be presynaptically inhibited by M2 receptor activation. In the following account, we discuss the significance of these findings for cortical physiology and perirhinal–entorhinal interactions in memory.
Glutamatergic neurons do not constitute the sole substrate of interactions between different rhinal areas
In the traditional view of cortical physiology, communication between different cortical areas is thought to depend entirely on glutamatergic neurons. According to this model, GABAergic cells act locally, within the area in which their somata are located. Thus, all IPSPs generated in the course of interactions between different cortical areas are believed to be generated disynaptically, after the glutamatergic activation of local-circuit GABAergic neurons.
In contrast with this view, we obtained evidence that some GABAergic neurons of the perirhinal cortex participate in communication between different rhinal areas. The evidence supporting this claim can be summarized as follows. First, threshold electrical stimuli delivered in areas 36 and 35 could evoke apparently pure IPSPs in principal neurons of area 35 or of the vlEC, respectively. Second, when threshold stimuli evoked mixed excitatory and inhibitory responses, the IPSP onset often preceded that of the EPSP. Third, these IPSPs persisted in the presence of glutamate receptor antagonists. Fourth, local pressure application of glutamate could evoke IPSPs in neighboring regions in the presence of glutamate receptor antagonists. Finally, entorhinal injections of the tracer FG retrogradely labeled a population of GAD-67-immunopositive neurons in the perirhinal cortex.
Although the suggestion that the rhinal cortices contain long-range GABAergic neurons might seem unorthodox, there are numerous precedents for this claim in the cortical literature. For instance, in the primary visual and somatosensory cortices, some GABAergic neurons project to the homonymous cortical field in the contralateral hemisphere (Hughes and Peters, 1992; Gonchar et al., 1995). Similarly, it was shown that a small proportion of cortical inhibitory neurons have axons that span millimeters within (Wang et al., 2004) (for review, see Markram et al., 2004) and across (McDonald and Burkhalter, 1993; Fabri and Manzoni, 1996; Tomioka et al., 2005; Pinto et al., 2006) cortical regions. Moreover, long-range GABA cells are not only present in the neocortex but also in the hippocampal formation. For instance, one class of GABAergic neuron located in the dentate gyrus projects to the subiculum (Ceranik et al., 1997), yet other types of CA1 interneurons have long-range projections to field CA3 (Sik et al., 1994) or the entorhinal cortex (Ino et al., 1990; Tamamaki and Nojyo, 1995). It was further reported that there are GABAergic projections from the presubiculum to the entorhinal cortex (van Haeften et al., 1997).
Impact of long-range GABAergic inhibition on rhinal transfer of neocortical impulses to the hippocampus
Although the rhinal cortices make unique contributions to memory (Murray and Richmond, 2001), they also play an essential role in hippocampal-dependent learning because they constitute the main access route for neocortical impulses into the hippocampus. However, previous work has revealed that perirhinal transfer of neocortical impulses occurs with a low probability (for review, see de Curtis and Paré, 2004). This conclusion was based on experiments in which both electrically evoked and spontaneous activity was analyzed (Biella et al., 2002; Kajiwara et al., 2003; Pelletier et al., 2004). In part, this reflects the fact that the rhinal pathway linking the neocortex to the hippocampus is multisynaptic and that feedforward inhibition regulates neuronal excitability at every stage of this pathway. Moreover, physiological and anatomical studies suggest that an important part of the connectivity in this multisynaptic pathway involves excitatory synapses onto local-circuit GABAergic neurons (Finch et al., 1988; Jones, 1994; Funahashi and Stewart, 1998; Martina et al., 2001; Garden et al., 2002; Pinto et al., 2006).
The findings of the present study suggest that an additional mechanism contributes to reduce impulse transmission in this pathway: the participation of long-range GABAergic neurons in the projections from perirhinal areas 36–35 as well as from the perirhinal to the entorhinal cortex. Although our retrograde tracing results suggest that long-range GABA cells account for a low proportion (1.5–2%) of the total population of perirhinal GABAergic neurons, that proportion doubles when expressed as a function of the number of retrogradely labeled neurons. However, the property that truly matters when assessing the functional importance of long-range inhibition in this circuit is the number of synapses this small subset of GABAergic cells forms compared with glutamatergic neurons. Insights in this question come from our previous study (Pinto et al., 2006) in which we used anterograde tracing with PHA-L. This approach revealed that as many as 12% of synapses found in the entorhinal cortex after PHA-L injections in the perirhinal cortex were immunopositive for GABA. A last point to consider when assessing the relative importance of long-range versus local feedforward inhibition in regulating rhinal transfer of neocortical inputs to the hippocampus is the timing of these two forms of inhibition. In the case of the conventional feedforward inhibition, IPSPs are generated disynaptically, thus, after EPSPs have impinged on principal cells. In contrast, long-range inhibition, although it probably involves fewer synapses, does not occur after the fact but at the same time as the EPSPs. Thus, long-range inhibition has a strategic temporal advantage.
For these various reasons, we propose that long-range inhibition likely has a significant impact on rhinal transfer of neocortical inputs to the hippocampus. Combined with the intrinsic properties of rhinal neurons (Faulkner and Brown, 1999; McGann et al., 2001), the cumulative effects of local and long-range GABAergic inhibition at successive stages of this multisynaptic pathway probably account for the low probability of impulse transfer between the neocortex and entorhinal cortex.
Cholinergic control of long-range inhibition in the rhinal cortices
Previous studies have established that rhinal contributions to memory require muscarinic receptor activation. Indeed, local infusion of scopolamine in the perirhinal cortex or cholinergic deafferentation of the rhinal cortices produce deficits in tasks probing various types of memory (Warburton et al., 2003; Abe et al., 2004; Gutierrez et al., 2004; McGaughy et al., 2005; Turchi et al., 2005; Winters and Bussey, 2005). Although it was proposed that muscarinic receptor activation regulates rhinal contributions to memory by modulating synaptic plasticity in the rhinal cortices (Warburton et al., 2003), the results of the present study suggest that an additional mechanism might be at play. Indeed, we observed that M2 receptor activation produces a presynaptic inhibition of GABA release by long-range inhibitory neurons of the perirhinal cortex projecting to the entorhinal cortex.
To understand the significance of these observations, we must consider the origin of this cholinergic input and how activity in this pathway relates to behavior. Cholinergic afferents to the rhinal cortices arise from several components of the basal forebrain including nucleus basalis and the diagonal band of Broca (Wainer and Mesulam, 1990). The firing rate of cholinergic corticopetal neurons is known to fluctuate in a state-dependent manner, with higher discharge rates seen in EEG-desynchronized states (Zaborszky and Duque, 2000; Jones, 2004). Consistent with this, acetylcholine release in cortex is lowest in slow-wave sleep and increases in paradoxical sleep and waking (Celesia and Jasper, 1966; Jasper and Tessier, 1971), particularly in behaviorally arousing conditions (Pepeu and Giovannini, 2004).
Combined with our findings, these observations suggest that, via a presynaptic reduction of long-range inhibition by cholinergic afferents, rhinal transfer of neocortical inputs to the hippocampus can be modulated as a function of environmental contingencies. In particular, behaviorally salient and arousing events, because they produce the largest increases in acetylcholine release (Pepeu and Giovannini, 2004), would result in a more pronounced M2-mediated reduction of long-range inhibition in the rhinal cortices. Because the hippocampus is required for the formation of long-term declarative memories, such context-dependent gating of neocortical–hippocampal interactions at the level of the rhinal cortices could serve a highly adaptive function: allow organisms to select from their experiences those that should be committed to long-term memory.
Footnotes
This material is based on work supported by the National Science Foundation under Grant 0208712 and by National Institute of Mental Health Grant RO1 MH073610-01. We thank members of the Paré laboratory for comments on a previous version of this paper.
References
- Abe H, Ishida Y, Iwasaki T. Perirhinal N-methyl-d-aspartate and muscarinic systems participate in object recognition in rats. Neurosci Lett. 2004;356:191–194. doi: 10.1016/j.neulet.2003.11.049. [DOI] [PubMed] [Google Scholar]
- Aggleton JP, Hunt PR, Rawlins JN. The effects of hippocampal lesions upon spatial and non-spatial tests of working memory. Behav Brain Res. 1986;19:133–146. doi: 10.1016/0166-4328(86)90011-2. [DOI] [PubMed] [Google Scholar]
- Alonso A. Electrophysiology of neurones in the perirhinal and entorhinal cortices and neuromodulatory changes in firing patterns. In: Witter MP, Wouterlood F, editors. The parahippocampal region. Oxford: Oxford UP; 2002. pp. 89–105. [Google Scholar]
- Behrends JC, ten Bruggencate G. Cholinergic modulation of synaptic inhibition in the guinea pig hippocampus in vitro: excitation of GABAergic interneurons and inhibition of GABA-release. J Neurophysiol. 1993;69:626–629. doi: 10.1152/jn.1993.69.2.626. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Krnjevic K, Reinhardt W, Ropert N. Intracellular observations on the disinhibitory action of acetylcholine in the hippocampus. Neuroscience. 1981;6:2475–2484. doi: 10.1016/0306-4522(81)90093-2. [DOI] [PubMed] [Google Scholar]
- Biella G, Uva L, de Curtis M. Propagation of neuronal activity along the neocortical–perirhinal–entorhinal pathway in the guinea pig. J Neurosci. 2002;22:9972–9979. doi: 10.1523/JNEUROSCI.22-22-09972.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilkey DK, Heinemann U. Intrinsic theta-frequency membrane potential oscillations in layer III/V perirhinal cortex neurons of the rat. Hippocampus. 1999;9:510–518. doi: 10.1002/(SICI)1098-1063(1999)9:5<510::AID-HIPO4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Burwell RD, Witter MP. Basic anatomy of the parahippocampal region in monkeys and rats. In: Witter MP, Wouterlood F, editors. The parahippocampal region. Oxford: Oxford UP; 2002. pp. 89–105. [Google Scholar]
- Celesia GG, Jasper HH. Acetylcholine released from cerebral cortex in relation to state of activation. Neurology. 1966;16:1053–1063. doi: 10.1212/wnl.16.11.1053. [DOI] [PubMed] [Google Scholar]
- Ceranik K, Bender R, Geiger JR, Monyer H, Jonas P, Frotscher M, Lubke J. A novel type of GABAergic interneuron connecting the input and the output regions of the hippocampus. J Neurosci. 1997;17:5380–5394. doi: 10.1523/JNEUROSCI.17-14-05380.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creager R, Dunwiddie T, Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis M, Paré D. The rhinal cortices: a wall of inhibition between the neocortex and the hippocampus. Prog Neurobiol. 2004;74:101–110. doi: 10.1016/j.pneurobio.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Dickson CT, Magistretti J, Shalinsky MH, Fransen E, Hasselmo ME, Alonso A. Properties and role of I(h) in the pacing of subthreshold oscillations in entorhinal cortex layer II neurons. J Neurophysiol. 2000;83:2562–2579. doi: 10.1152/jn.2000.83.5.2562. [DOI] [PubMed] [Google Scholar]
- Erisir A, Levey AI, Aoki C. Muscarinic receptor M(2) in cat visual cortex: laminar distribution, relationship to gamma-aminobutyric acidergic neurons, and effect of cingulate lesions. J Comp Neurol. 2001;441:168–185. doi: 10.1002/cne.1405. [DOI] [PubMed] [Google Scholar]
- Fabri M, Manzoni T. Glutamate decarboxylase immunoreactivity in corticocortical projecting neurons of rat somatic sensory cortex. Neuroscience. 1996;72:435–448. doi: 10.1016/0306-4522(95)00568-4. [DOI] [PubMed] [Google Scholar]
- Fahy FL, Riches IP, Brown MW. Neuronal activity related to visual recognition memory: long-term memory and the encoding of recency and familiarity information in the primate anterior and medial inferior temporal and rhinal cortex. Exp Brain Res. 1993;96:457–472. doi: 10.1007/BF00234113. [DOI] [PubMed] [Google Scholar]
- Faulkner B, Brown TH. Morphology and physiology of neurons in the rat perirhinal-lateral amygdala area. J Comp Neurol. 1999;411:613–642. [PubMed] [Google Scholar]
- Finch DM, Tan AM, Isokawa-Akesson M. Feedforward inhibition of the rat entorhinal cortex and subicular complex. J Neurosci. 1988;8:2213–2226. doi: 10.1523/JNEUROSCI.08-07-02213.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, Buchwald NA, Hull CD, Levine MS. GABAergic basal forebrain neurons project to the neocortex: the localization of glutamic acid decarboxylase and choline acetyltransferase in feline corticopetal neurons. J Comp Neurol. 1988;272:489–502. doi: 10.1002/cne.902720404. [DOI] [PubMed] [Google Scholar]
- Funahashi M, Stewart M. GABA receptor-mediated post-synaptic potentials in the retrohippocampal cortices: regional, laminar and cellular comparisons. Brain Res. 1998;787:19–33. doi: 10.1016/s0006-8993(97)01384-x. [DOI] [PubMed] [Google Scholar]
- Gaffan D, Murray EA. Monkeys (Macaca fascicularis) with rhinal cortex ablations succeed in object discrimination learning despite 24-hr intertrial intervals and fail at matching to sample despite double sample presentations. Behav Neurosci. 1992;106:30–38. doi: 10.1037//0735-7044.106.1.30. [DOI] [PubMed] [Google Scholar]
- Garden DL, Kemp N, Bashir ZI. Differences in GABAergic transmission between two inputs into the perirhinal cortex. Eur J Neurosci. 2002;16:437–444. doi: 10.1046/j.1460-9568.2002.02096.x. [DOI] [PubMed] [Google Scholar]
- Gonchar YA, Johnson PB, Weinberg RJ. GABA-immunopositive neurons in rat neocortex with contralateral projections to S-I. Brain Res. 1995;697:27–34. doi: 10.1016/0006-8993(95)00746-d. [DOI] [PubMed] [Google Scholar]
- Gutierrez R, De la Cruz V, Rodriguez-Ortiz CJ, Bermudez-Rattoni F. Perirhinal cortex muscarinic receptor blockade impairs taste recognition memory formation. Learn Mem. 2004;11:95–101. doi: 10.1101/lm.69704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Papp EC, Acsady L, Levey AI, Freund TF. Distinct interneuron types express m2 muscarinic receptor immunoreactivity on their dendrites or axon terminals in the hippocampus. Neuroscience. 1998;82:355–376. doi: 10.1016/s0306-4522(97)00300-x. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:710–715. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Peters A. Symmetric synapses formed by callosal afferents in rat visual cortex. Brain Res. 1992;583:271–278. doi: 10.1016/s0006-8993(10)80033-2. [DOI] [PubMed] [Google Scholar]
- Ino T, Matsuzaki S, Shinonaga Y, Ohishi H, Ogawa-Meguro R, Mizuno N. Direct projections of non-pyramidal neurons of Ammon's horn to the amygdala and the entorhinal cortex. Neurosci Lett. 1990;115:161–166. doi: 10.1016/0304-3940(90)90448-i. [DOI] [PubMed] [Google Scholar]
- Insausti R, Amaral DG, Cowan WM. The entorhinal cortex of the monkey. II. Cortical afferents. J Comp Neurol. 1987;264:356–395. doi: 10.1002/cne.902640306. [DOI] [PubMed] [Google Scholar]
- Jasper HH, Tessier J. Acetylcholine liberation from cerebral cortex during paradoxical (REM) sleep. Science. 1971;172:601–602. doi: 10.1126/science.172.3983.601. [DOI] [PubMed] [Google Scholar]
- Jones BE. Activity, modulation and role of basal forebrain cholinergic neurons innervating the cerebral cortex. Prog Brain Res. 2004;145:157–169. doi: 10.1016/S0079-6123(03)45011-5. [DOI] [PubMed] [Google Scholar]
- Jones RS. Synaptic and intrinsic properties of neurons of origin of the perforant path in layer II of the rat entorhinal cortex in vitro. Hippocampus. 1994;4:335–353. doi: 10.1002/hipo.450040317. [DOI] [PubMed] [Google Scholar]
- Kajiwara R, Takashima I, Mimura Y, Witter MP, Iijima T. Amygdala input promotes spread of excitatory neural activity from perirhinal cortex to the entorhinal-hippocampal circuit. J Neurophysiol. 2003;89:2176–2184. doi: 10.1152/jn.01033.2002. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura F, Baughman RW. Distinct muscarinic receptor subtypes suppress excitatory and inhibitory synaptic responses in cortical neurons. J Neurophysiol. 1997;77:709–716. doi: 10.1152/jn.1997.77.2.709. [DOI] [PubMed] [Google Scholar]
- Klink R, Alonso A. Ionic mechanisms for the subthreshold oscillations and differential electroresponsiveness of medial entorhinal cortex layer II neurons. J Neurophysiol. 1993;70:144–157. doi: 10.1152/jn.1993.70.1.144. [DOI] [PubMed] [Google Scholar]
- Klink R, Alonso A. Ionic mechanisms of muscarinic depolarization in entorhinal cortex layer II neurons. J Neurophysiol. 1997;77:1829–1843. doi: 10.1152/jn.1997.77.4.1829. [DOI] [PubMed] [Google Scholar]
- Leonard BW, Amaral DG, Squire LR, Zola-Morgan S. Transient memory impairment in monkeys with bilateral lesions of the entorhinal cortex. J Neurosci. 1995;15:5637–5659. doi: 10.1523/JNEUROSCI.15-08-05637.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Miller EK, Desimone R. The representation of stimulus familiarity in anterior inferior temporal cortex. J Neurophysiol. 1993;69:1918–1929. doi: 10.1152/jn.1993.69.6.1918. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Ma L, Shalinsky MH, Lin W, Klink R, Alonso A. Spike patterning by Ca2+-dependent regulation of a muscarinic cation current in entorhinal cortex layer II neurons. J Neurophysiol. 2004;92:1644–1657. doi: 10.1152/jn.00036.2004. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Martina M, Royer S, Paré D. Propagation of neocortical inputs in the perirhinal cortex. J Neurosci. 2001;21:2878–2888. doi: 10.1523/JNEUROSCI.21-08-02878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald CT, Burkhalter A. Organization of long-range inhibitory connections with rat visual cortex. J Neurosci. 1993;13:768–781. doi: 10.1523/JNEUROSCI.13-02-00768.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann JP, Moyer JR, Jr, Brown TH. Predominance of late-spiking neurons in layer VI of rat perirhinal cortex. J Neurosci. 2001;21:4969–4976. doi: 10.1523/JNEUROSCI.21-14-04969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughy J, Koene RA, Eichenbaum H, Hasselmo ME. Cholinergic deafferentation of the entorhinal cortex in rats impairs encoding of novel but not familiar stimuli in a delayed nonmatch-to-sample task. J Neurosci. 2005;25:10273–10281. doi: 10.1523/JNEUROSCI.2386-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier M, Bachevalier J, Mishkin M, Murray EA. Effects on visual recognition of combined and separate ablations of the entorhinal and perirhinal cortex in rhesus monkeys. J Neurosci. 1993;13:5418–5432. doi: 10.1523/JNEUROSCI.13-12-05418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier M, Hadfield W, Bachevalier J, Murray EA. Effects of rhinal cortex lesions combined with hippocampectomy on visual recognition memory in rhesus monkeys. J Neurophysiol. 1996;75:1190–1205. doi: 10.1152/jn.1996.75.3.1190. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annu Rev Pharmacol Toxicol. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Muir GM, Bilkey DK. Instability in the place field location of hippocampal place cells after lesions centered on the perirhinal cortex. J Neurosci. 2001;21:4016–4025. doi: 10.1523/JNEUROSCI.21-11-04016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray EA, Mishkin M. Visual recognition in monkeys following rhinal cortical ablations combined with either amygdalectomy or hippocampectomy. J Neurosci. 1986;6:1991–2003. doi: 10.1523/JNEUROSCI.06-07-01991.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray EA, Richmond BJ. Role of perirhinal cortex in object perception, memory, and associations. Curr Opin Neurobiol. 2001;11:188–193. doi: 10.1016/s0959-4388(00)00195-1. [DOI] [PubMed] [Google Scholar]
- Murray EA, Baxter MG, Gaffan D. Monkeys with rhinal cortex damage or neurotoxic hippocampal lesions are impaired on spatial scene learning and object reversals. Behav Neurosci. 1998;112:1291–1303. doi: 10.1037//0735-7044.112.6.1291. [DOI] [PubMed] [Google Scholar]
- Murray EA, Graham KS, Gaffan D. Perirhinal cortex and its neighbours in the medial temporal lobe: contributions to memory and perception. Q J Exp Psychol B. 2005;58:378–396. doi: 10.1080/02724990544000077. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Otto T, Gallagher M. Entorhinal-perirhinal lesions impair performance of rats on two versions of place learning in the Morris water maze. Behav Neurosci. 1995;109:3–9. doi: 10.1037//0735-7044.109.1.3. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y, Pare JF. Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat: Phaseolus vulgaris-leucoagglutinin anterograde tracing at the light and electron microscopic level. Neuroscience. 1995;69:567–583. doi: 10.1016/0306-4522(95)00272-k. [DOI] [PubMed] [Google Scholar]
- Pelletier JG, Apergis J, Paré D. Low-probability transmission of neocortical and entorhinal impulses through the perirhinal cortex. J Neurophysiol. 2004;91:2079–2089. doi: 10.1152/jn.01197.2003. [DOI] [PubMed] [Google Scholar]
- Pepeu G, Giovannini MG. Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem. 2004;11:21–27. doi: 10.1101/lm.68104. [DOI] [PubMed] [Google Scholar]
- Pinto A, Fuentes C, Paré D. Feedforward inhibition regulates perirhinal transmission of neocortical inputs to the entorhinal cortex: ultrastructural study in guinea pigs. J Comp Neurol. 2006;495:722–734. doi: 10.1002/cne.20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB. Any way you cut it: a new journal policy for the use of unbiased counting methods. J Comp Neurol. 1996;364:5. doi: 10.1002/(SICI)1096-9861(19960101)364:1<5::AID-CNE1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Shalinsky MH, Magistretti J, Ma L, Alonso AA. Muscarinic activation of a cation current and associated current noise in entorhinal-cortex layer-II neurons. J Neurophysiol. 2002;88:1197–1211. doi: 10.1152/jn.2002.88.3.1197. [DOI] [PubMed] [Google Scholar]
- Shi CJ, Cassell MD. Perirhinal cortex projections to the amygdaloid complex and hippocampal formation in the rat. J Comp Neurol. 1999;406:299–328. doi: 10.1002/(sici)1096-9861(19990412)406:3<299::aid-cne2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Sik A, Ylinen A, Penttonen M, Buzsaki G. Inhibitory CA1-CA3-hilar region feedback in the hippocampus. Science. 1994;265:1722–1724. doi: 10.1126/science.8085161. [DOI] [PubMed] [Google Scholar]
- Smith Y, Paré D. Intra-amygdaloid projections of the lateral nucleus in the cat: PHA-L anterograde labeling combined with postembedding GABA and glutamate immunocytochemistry. J Comp Neurol. 1994;342:232–248. doi: 10.1002/cne.903420207. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, Bolam JP. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience. 1998;86:353–387. doi: 10.1016/s0306-4522(98)00004-9. [DOI] [PubMed] [Google Scholar]
- Sobotka S, Ringo JL. Investigation of long-term recognition and association memory in unit responses from inferotemporal cortex. Exp Brain Res. 1993;9:28–38. doi: 10.1007/BF00230436. [DOI] [PubMed] [Google Scholar]
- Suzuki WA, Zola-Morgan S, Squire LR, Amaral DG. Lesions of the perirhinal and parahippocampal cortices in the monkey produce long-lasting memory impairment in the visual and tactual modalities. J Neurosci. 1993;13:2430–2451. doi: 10.1523/JNEUROSCI.13-06-02430.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Nojyo Y. Preservation of topography in the connections between the subiculum, field CA1, and the entorhinal cortex in rats. J Comp Neurol. 1995;353:379–390. doi: 10.1002/cne.903530306. [DOI] [PubMed] [Google Scholar]
- Tang Y, Mishkin M, Aigner TG. Effects of muscarinic blockade in perirhinal cortex during visual recognition. Proc Natl Acad Sci USA. 1997;94:12667–12669. doi: 10.1073/pnas.94.23.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomioka R, Okamoto K, Furuta T, Fujiyama F, Iwasato T, Yanagawa Y, Obata K, Kaneko T, Tamamaki N. Demonstration of long-range GABAergic connections distributed throughout the mouse neocortex. Eur J Neurosci. 2005;21:1587–1600. doi: 10.1111/j.1460-9568.2005.03989.x. [DOI] [PubMed] [Google Scholar]
- Turchi J, Saunders RC, Mishkin M. Effects of cholinergic deafferentation of the rhinal cortex on visual recognition memory in monkeys. Proc Natl Acad Sci USA. 2005;102:2158–2161. doi: 10.1073/pnas.0409708102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uva L, Gruschke S, Biella G, De Curtis M, Witter MP. Cytoarchitectonic characterization of the parahippocampal region of the guinea pig. J Comp Neurol. 2004;474:289–303. doi: 10.1002/cne.20121. [DOI] [PubMed] [Google Scholar]
- van Haeften T, Wouterlood FG, Jorritsma-Byham B, Witter MP. GABAergic presubicular projections to the medial entorhinal cortex of the rat. J Neurosci. 1997;17:862–874. doi: 10.1523/JNEUROSCI.17-02-00862.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoesen G, Pandya DN. Some connections of the entorhinal (area 28) and perirhinal (area 35) cortices of the rhesus monkey. I. Temporal lobe afferents. Brain Res. 1975;95:1–24. doi: 10.1016/0006-8993(75)90204-8. [DOI] [PubMed] [Google Scholar]
- Wainer BH, Mesulam MM. Ascending cholinergic pathways in the rat brain. In: Steriade M, Biesold D, editors. Brain cholinergic systems. Oxford: Oxford UP; 1990. pp. 65–119. [Google Scholar]
- Wang Y, Toledo-Rodriguez M, Gupta A, Wu C, Silberberg G, Luo J, Markram H. Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J Physiol (Lond) 2004;561:65–90. doi: 10.1113/jphysiol.2004.073353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton EC, Koder T, Cho K, Massey PV, Duguid G, Barker GR, Aggleton JP, Bashir ZI, Brown MW. Cholinergic neurotransmission is essential for perirhinal cortical plasticity and recognition memory. Neuron. 2003;38:987–996. doi: 10.1016/s0896-6273(03)00358-1. [DOI] [PubMed] [Google Scholar]
- Wiig KA, Bilkey DK. The effects of perirhinal cortical lesions on spatial reference memory in the rat. Behav Brain Res. 1994;63:101–109. doi: 10.1016/0166-4328(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Winters BD, Bussey TJ. Removal of cholinergic input to perirhinal cortex disrupts object recognition but not spatial working memory in the rat. Eur J Neurosci. 2005;21:2263–2270. doi: 10.1111/j.1460-9568.2005.04055.x. [DOI] [PubMed] [Google Scholar]
- Witter MP, Groenewegen HJ. Connections of the parahippocampal cortex in the cat. III. Cortical and thalamic efferents. J Comp Neurol. 1986;252:1–31. doi: 10.1002/cne.902520102. [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Duque A. Local synaptic connections of basal forebrain neurons. Behav Brain Res. 2000;115:143–158. doi: 10.1016/s0166-4328(00)00255-2. [DOI] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG, Suzuki WA. Lesions of perirhinal and parahippocampal cortex that spare the amygdala and hippocampal formation produce severe memory impairment. J Neurosci. 1989;9:4355–4370. doi: 10.1523/JNEUROSCI.09-12-04355.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]