

Abstract
Antipsychotics often lose efficacy in patients despite chronic continuous treatment. Why this occurs is not known. It is known, however, that withdrawal from chronic antipsychotic treatment induces behavioral dopaminergic supersensitivity in animals. How this emerging supersensitivity might interact with ongoing treatment has never been assessed. Therefore, we asked whether dopamine supersensitivity could overcome the behavioral and neurochemical effects of antipsychotics while they are still in use. Using two models of antipsychotic-like effects in rats, we show that during ongoing treatment with clinically relevant doses, haloperidol and olanzapine progressively lose their efficacy in suppressing amphetamine-induced locomotion and conditioned avoidance responding. Treatment failure occurred despite high levels of dopamine D2 receptor occupancy by the antipsychotic and was at least temporarily reversible by an additional increase in antipsychotic dose. To explore potential mechanisms, we studied presynaptic and postsynaptic elements of the dopamine system and observed that antipsychotic failure was accompanied by opposing changes across the synapse: tolerance to the ability of haloperidol to increase basal dopamine and dopamine turnover on one side, and 20–40% increases in D2 receptor number and 100–160% increases in the proportion of D2 receptors in the high-affinity state for dopamine (D2High) on the other. Thus, the loss of antipsychotic efficacy is linked to an increase in D2 receptor number and sensitivity. These results are the first to demonstrate that “breakthrough” supersensitivity during ongoing antipsychotic treatment undermines treatment efficacy. These findings provide a model and a mechanism for antipsychotic treatment failure and suggest new directions for the development of more effective antipsychotics.
Keywords: antipsychotics, dopamine, supersensitivity, dopamine D2 receptors, high-affinity receptors, striatum
Introduction
Schizophrenic patients remain on antipsychotic (i.e., antidopaminergic) treatment for years, yet remarkably little is known about what happens to dopamine (DA) function during ongoing treatment. What is known, however, is that in both humans and laboratory animals withdrawal from chronic antipsychotic treatment reveals a state of dopaminergic supersensitivity that is characterized by increased vulnerability to psychosis and to the psychomotor activating effects of dopamine agonists, respectively. In humans, this has been termed “neuroleptic-induced supersensitivity psychosis” (Chouinard et al., 1978; Chouinard and Jones, 1980), and has been observed after withdrawal from antipsychotic drugs such as quetiapine (Margolese et al., 2002), clozapine (Ekblom et al., 1984; Tollefson et al., 1999), olanzapine (Llorca et al., 2001), haloperidol (Kahne, 1989), and fluphenazine enanthate (Chouinard and Jones, 1980). In keeping with the clinical observations, animal studies show that withdrawal from antipsychotic treatment reveals an increased psychomotor response to apomorphine (Asper et al., 1973; Gianutsos et al., 1974; Sayers et al., 1975; Smith and Davis, 1975, 1976; Clow et al., 1979; Montanaro et al., 1982), amphetamine (Smith and Davis, 1975; Rebec et al., 1982; Meng et al., 1998), and dopamine injected into the caudate–putamen or nucleus accumbens (Halperin et al., 1983). Although such studies have conclusively demonstrated dopamine supersensitivity after withdrawal from an antipsychotic, much less is known about what happens during ongoing treatment.
Indeed, the concept of antipsychotic-induced dopamine supersensitivity is based almost entirely on studies examining changes in the dopamine system when drug treatment and dopamine antagonism have ceased, and a period of withdrawal has elapsed (Muller and Seeman, 1978; Rupniak et al., 1983). To our knowledge, only Clow et al. (1979, 1980) have studied antipsychotic-induced dopamine supersensitivity without an overt withdrawal period. However, antipsychotics were given in the drinking water and plasma drug levels were not monitored. Rats consume up to 90% of their daily water intake during the dark phase (Stellar and Hill, 1952; Fitzsimons, 1957) and metabolize antipsychotics much faster than humans (Cheng and Paalzow, 1992; Bezchlibnyk-Butler and Jeffries, 1999); thus, the levels of antipsychotic exposure achieved by Clow et al. (1979, 1980) were unrepresentative of the clinical condition (Farde et al., 1989; Seeman, 2002).
Given these considerations, key questions remain to be answered by preclinical models. First, can dopamine supersensitivity be observed during ongoing treatment with therapeutically relevant doses and modes of antipsychotic administration? Second, can dopamine supersensitivity overcome the effects of antipsychotics? We found that the answer to both questions is yes and we examined whether this was related to progressive changes in presynaptic dopamine levels, turnover and release, and/or to changes in the number and sensitivity of dopamine D2 receptors.
Materials and Methods
Male Sprague Dawley rats (Charles River Laboratories, Montreal, Quebec, Canada) weighing 225–250 g were housed two per cage in a climate-controlled colony room with a 12 h reverse light/dark cycle (lights off at 8:00 A.M.). Food and water were available ad libitum. All testing was conducted during the dark phase of the animals' circadian cycle and was in compliance with the institute's animal care committee.
Drugs
Haloperidol (HAL; 0.25 mg/kg/d or 0.75 mg/kg/d for all studies except experiment 3, for which 0.5 mg/kg/d was used; Sabex, Boucherville, Quebec, Canada) was dissolved in a 0.5% glacial acetic acid/H2O solution (pH adjusted to ∼5 with NaOH). Olanzapine (OLZ; 10 mg/kg/d) was dissolved in a 2% glacial acetic acid/H2O solution (pH adjusted to ∼5 with NaOH). Both drugs were given via an Alzet osmotic minipump (model 2ML2; 14 d delivery; Durect, Cupertino, CA). D-Amphetamine sulfate (AMPH; 1.5 mg/kg; US Pharmacopoeia, Rockville, MD) was dissolved in 0.9% saline and given subcutaneously in a volume of 1 ml/kg body weight.
Minipump implantation
Minipumps containing either vehicle (VEH; 0.5% glacial acetic acid/H2O solution), HAL, or OLZ were implanted under 1.5% isoflurane anesthesia. A 1.5-cm-wide incision was made in each animal's lower back and hemostats were used to loosen connective tissue between the scapulae. Minipumps were wiped with 70% ethanol and inserted to lie between the scapulae with the flow moderator away from the incision. The incision was closed using 9 mm surgical staples and cleaned with 70% ethanol.
Experiment 1: D2 occupancy with acute and chronic HAL treatment
The purpose of experiment 1 was two-fold: (1) to confirm that the HAL doses used would yield clinically relevant levels of D2 receptor blockade (Farde et al., 1988; Kapur et al., 2000b), and (2) to determine whether D2 receptor blockade by HAL would change during continued treatment.
On days 2 and 13 after minipump implantation, in vivo D2 receptor occupancy was determined in animals from the VEH (n = 4 per time point) and HAL (n = 7 per time point) groups. Individual animals from each group were placed in a Plexiglas restrainer and injected with 7.5 μCi of [3H]raclopride (PerkinElmer, Boston, MI) in 300 μl of 0.9% saline via the tail vein, which had been vasodilated in a warm water bath. Rats were killed by live decapitation 1 h after radiotracer injection and trunk blood was collected. The striata and cerebellum were rapidly dissected, weighed, and dissolved for 24 h on a shaker in 3 ml of Solvable (Canberra Packard, Montreal, Quebec, Canada) at room temperature. The next day, 6 ml of scintillation fluid (Aquasure; Canberra Packard) was added to the mixture and vials were left on a shaker for 24 h at room temperature. The next day, [3H]raclopride radioactivity was quantified in a Beckman (Fullerton, CA) LS5000 CE liquid scintillation spectrometer at 50% efficiency. Striatal and cerebellar counts were expressed as disintegrations per minute/milligram. The ratio of striatum minus cerebellum (an index of specific binding)/cerebellum (an index of nonspecific binding) was used to generate an index of the binding potential (BP) of DA D2 receptors. Percent D2 occupancy by haloperidol was calculated as follows: 100 × (BPvehicle − BPhaloperidol/BPvehicle), where BPvehicle is the pooled D2 binding potential of all the vehicle animals and BPhaloperidol is the binding potential of a haloperidol-treated rat.
Experiment 2: behavioral sensitivity to AMPH
Experiment 2a: acute versus chronic HAL
In experiment 2a, we assessed changes in the locomotor response to AMPH as a function of chronic HAL exposure.
Apparatus.
The locomotor response to AMPH was assessed in clear Plexiglas cages (27 × 48 × 20 cm) equipped with a row of 6 photocell beams placed 3 cm above the floor of the cage. Photocell beam breaks were detected and recorded by a computer.
Groups and procedures.
The influence of HAL or vehicle on AMPH-induced locomotion was assessed 2 and 12 d after minipump implantation in independent groups of animals. AMPH-induced locomotion was also examined on the fifth day of withdrawal from HAL in the animals that had been tested on day 12. On each test day, animals were brought to the locomotor activity room and left in their transport cages for 5 min. The animals were then placed in the locomotor activity cages and left undisturbed for 30 min during which baseline levels of locomotor activity were monitored. Animals were then injected with AMPH and a 60 min test period followed during which locomotor activity was recorded.
Experiment 2b: effects of further augmenting HAL levels
In experiment 2b, we assessed whether an acute injection of HAL could attenuate the locomotor response to AMPH once chronic and continuous HAL treatment via minipump was no longer able to do so.
Rats were implanted with minipumps containing VEH or 0.75 mg/kg/d HAL as described above. AMPH-induced locomotion was assessed on the 12th and 13th days after implantation. On day 12, one-half of the animals in each group received a subcutaneous injection of either saline or HAL (0.1 mg/kg) 30 min before the AMPH injection. On day 13, animals previously given saline before AMPH now received HAL, and vice versa.
Experiment 2c: acute versus chronic OLZ
Experiment 2c was conducted to assess changes in the effect of continuous OLZ exposure on AMPH-induced locomotion over time. The dose of OLZ was chosen on the basis of previous work suggesting that it would achieve clinically relevant levels of striatal D2 receptor occupancy (Kapur et al., 2003; Turrone et al., 2005). Animals were implanted with minipumps containing either VEH or OLZ and testing was conducted as described in experiment 2a.
Experiment 3: conditioned avoidance responding
In experiment 3, we monitored the ability of HAL treatment to suppress the conditioned avoidance response to an aversive conditioned stimulus over time.
Apparatus
Rats were trained and tested in six identical two-way active avoidance shuttle boxes (64 × 24 × 30 cm; Med Associates, St. Albans, VT) set in individual ventilated and sound- and light-attenuating cubicles (97 × 36 × 64 cm). Each box was divided into two equal-size compartments by a white polyvinyl chloride partition with an arch-shaped doorway (15 × 9 cm) and a 4-cm-high barrier was fixed onto the doorway. Animals had to jump over the barrier to cross from one compartment to the other. Illumination was provided by a 28 V house light mounted on the back wall of the right compartment. The shuttle boxes were equipped with a tilting grid floor and animal location was detected by microswitches. Scrambled foot shocks [0.6 mA; unconditioned stimulus (US)] lasting 20 s were delivered to the grid floor. The conditioned stimulus (CS) was a 74 dB tone lasting 10 s. Stimulus presentation was controlled by computer-run Med-Associates programs, which also recorded the behavioral measures described below.
Procedures
Each CS presentation was immediately followed by foot shock. Movement to the other compartment during the 10 s CS presentation was recorded as “avoidance.” Movement to the other compartment during presentation of the foot shock was recorded as “escape,” and failure to move to the other compartment during presentation of the foot shock was recorded as “escape failure.” Intertrial compartment crossings were also recorded. Each training/testing session consisted of 30 trials (30 CS–US presentations) with an intertrial interval of 30–60 s. Rats were first trained once a day for a total of 9 d. Only animals that reached a training criterion of ≥50% avoidance on days 4 and 5 were kept for additional training on days 6–9 (32 of 48 rats).
Finally, only animals that had ≥80% avoidance on days 8 and 9 were included in the experiment (28 of 32 rats). These animals were randomly assigned to receive vehicle or 0.5 mg/kg/d HAL via minipump. This dose of HAL was chosen because it is halfway between the 0.25 and 0.75 mg/kg/d doses used in experiment 2a. Starting on day 3 of HAL/VEH treatment, the same animals were tested for conditioned avoidance responding (CAR) once a day for five consecutive days (i.e., until day 7 of treatment), and then on days 10 and 12 of treatment.
Experiment 4: in vivo microdialysis
In experiment 4, we used in vivo microdialysis techniques in freely moving animals to examine changes in extracellular DA, dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), and norepinephrine (NE) in the nucleus accumbens during ongoing HAL treatment.
Intracranial cannulation
Animals were anesthetized with sodium pentobarbital (Nembutal, 65 mg/kg, i.p.; Sigma, Oakville, ON) and atropine sulfate (0.13 mg/kg, s.c.; Sabex) to minimize bronchial secretions. Stereotaxic procedures were used to implant a unilateral stainless-steel 20 gauge cannula (HRS Scientific, Montreal, Quebec, Canada) into the nucleus accumbens (anteroposterior, +1.7 and mediolateral, +2.6 from bregma, dorsoventral, −5.5 mm from the site) at a 10° angle to avoid damage to the lateral ventricle. The cannula was cemented in place with dental acrylic applied around the cannula and three skull screws. The hemisphere to be implanted was counterbalanced across animals. Animals were given an injection of penicillin (Pen G, i.m.; Vetoquinol, Lavaltrie, Quebec, Canada) and Ketoprofen (2 mg/kg, s.c.) after surgery. Three to 4 d after cannulation, animals were implanted with osmotic minipumps containing either VEH or HAL as described above. Animals were left to recover from intracranial surgery for 5–15 d before microdialysis testing. Two and 12 d after pump implantation, amphetamine-induced DA, DOPAC, HVA, and NE release were measured in separate groups of animals according to the following procedures.
Apparatus
Four hexagonal chambers were used for microdialysis. Each chamber (42 × 39 × 33.5 cm, home made; Concordia University, Montreal, Quebec, Canada) consisted of Plexiglas walls with wooden ceilings and stainless-steel grid floors. Chambers were individually housed in wooden cubicles and lighting was provided on a reverse cycle by overhead lights.
Microdialysis probes
Each dialysis probe consisted of a 2.5 mm length of semipermeable dialysis membrane (inner diameter, 200 μm; 13, 000 molecular weight cutoff; Spectrum Laboratories, Rancho Dominguez, CA), closed at one end with glue and inserted into a 21.5- to 22-mm-long 26 gauge piece of stainless-steel tubing. The stainless-steel shaft was inserted into the flared end of a 40–50 cm piece of PE20 tubing that connected to a single channel liquid swivel (home made, Concordia University). The swivel was in turn connected to a Harvard syringe pump (Harvard Apparatus, South Natick, MA) with PE20 tubing. Small diameter fused silica tubing (outer diameter, 108 μm; inner diameter, 40 μm; HRS Scientific) extended internally through the probe with one end resting 0.5 mm from the tip of the probe and the other end exiting the PE20 tubing 4.5 cm above the tip of the animal's guide cannula. Samples were collected at the opposite end of the silica tubing in polyethylene tubes. The external length of the PE tubing was protected from chewing by steel spring casing. The probes were inserted the day before the microdialysis testing session and were secured in place by a stainless-steel collar that was screwed onto the guide cannula. To prevent occlusion, artificial CSF [ACSF; containing (in mm)145 Na+, 2.7 K+, 1.3 Ca2+, 1.0 Mg2+, 150 Cl−, 0.2 ascorbate, and 2 Na2HPO4, pH, 7.4 ± 0.1) was perfused overnight at a rate of 0.3 μl/min. ACSF infusion rate was changed to 0.8 μl/min during dialysate sampling.
High-performance liquid chromatography
Samples were collected every 20 min and a 10 μl volume of dialysate was immediately extracted from each sample and analyzed using one of two similar high-performance liquid chromatography (HPLC) systems with electrochemical detection. The samples were loaded onto C-18 reverse-phase columns (15 cm × 5 μm, Spherisorb-ODS2; Chromatography Sciences, St. Laurent, Quebec, Canada) through manual injection ports (Reodyn 7125; 20 μl loop). Reduction and oxidation currents for DA, DOPAC, HVA, and NE were measured with dual-channel ESA (Chelmsford, MA) coulometric detectors (Coulochem III, with a model 5011 analytical cell). The currents for DA and NE (−280 mV) were measured independently of those for DOPAC and HVA (+300 mV) using separate channels of the Coulochem detectors. The mobile phases (19% acetonitrile, 0.076 m SDS, 0.1 m EDTA, 0.058 m NaPO4, 0.027 m citric acid, pH 3.35) were circulated through each closed system at a flow rate of 1.2 ml/min by Waters (Lachine, Quebec, Canada) 515 HPLC pumps. The peaks obtained for DA, DOPAC, HVA, and NE were integrated and quantified by EZChrom Chromatography Data System (Scientific Software, San Ramon, CA). The mobile phase was adjusted to allow for the separation and quantification of target analytes in a single run.
Testing procedures
Food was removed from the dialysis cages before sampling, but water was available ad libitum. Once stable baseline levels of DA and its metabolites were achieved (<10% variation in three consecutive samples), animals were injected with saline and two more samples were collected. Animals were then given AMPH (1.5 mg/kg, s.c.) and eight additional samples were collected.
Postmortem cannula placement verification
Rats were anesthetized with Somnotol (i.p.; MTC Pharmaceuticals, Cambridge, Ontario, Canada) and perfused transcardially with 0.9% saline (150 ml) and formaldehyde (150 ml, formalin 10% v/v; Anachemia, Montreal, Quebec, Canada). Brains were extracted, left in formaldehyde for 24 h, and 20 μm coronal sections were taken on a cryostat. Sections were stained with cresyl violet and probe tracts were verified under a microscope.
Experiment 5: D2 receptor binding capacity and guanine nucleotide-sensitive D2High receptors
In experiment 5, we quantified changes in striatal D2 binding capacity and D2High states as a function of chronic HAL treatment.
D2High states were measured using [3H](+)PHNO, a D2 agonist. [3H](+)PHNO (also known as naxagolide or MK 458, or (+)-4-propyl-9-hydroxynaphthoxazine) is chemically defined as (+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol. To prepare [3H](+)PHNO, the allyl precursor of (+)-PHNO was tritiated and purified by PerkinElmer Life Sciences to a specific activity of 78 Ci/mmol.
On days 2 and 12 of HAL/VEH treatment, as well as on the seventh day of withdrawal, animals in each treatment group were killed by CO2 narcosis and their striata were dissected and stored at −70°C until use. In addition, striata from naive rats (Pel-Freez Biologicals, Rogers, AZ) were used to determine whether the mere presence of HAL in the tissue could influence the density of D2High states. Thus, 0.75 and 1.5 nm haloperidol were added to these striata during processing. All striata were homogenized individually in buffer (4 mg of frozen tissue/ml of buffer), using a Teflon-glass homogenizer with the piston rotating at 500 rpm and 10 up and down strokes of the glass container. The buffer contained (in mm) 50 Tris-HC1, pH 7.4 at 20°C, 1 EDTA, 5 KCl, 1.5 CaC12, 4 MgC12, and 120 NaCl. The homogenate was not washed, centrifuged, or preincubated because previous work found that >30% of the D2 receptors were lost by these procedures (Seeman et al., 1984).
The density of [3H](+)-PHNO sites sensitive to the action of guanilylimidodiphosphate (GN), or so-called “G-sensitive [3H](+)-PHNO” sites, was determined by obtaining two saturation curves with [3H](+)-PHNO, one with and one without GN. Each incubation tube (12 × 75 mm, glass) received, in the following order, 0.5 ml of buffer (with or without a final concentration of 200 μm GN, and with or without a final concentration of 10 μm S-sulpiride to define nonspecific binding), 0.25 ml [3H](+)-PHNO (with 12 final concentrations ranging from 0.1 to 5 nm), and 0.25 ml of tissue homogenate. The tubes, containing a total volume of 1 ml, were incubated for 2 h at room temperature (20°C), after which the incubates were filtered using a 12-well cell harvester (Titertek; Skatron, Lier, Norway) and buffer-presoaked glass fiber filter mats (No. 7034; Skatron). After filtering the incubate, the filter mat was rinsed with buffer for 15 s (7.5 ml of buffer). The filters were pushed out and placed in scintillation minivials (Packard Instruments, Chicago, IL). The minivials received 4 ml each of scintillant (Ready Solve; Beckman), and were monitored 6 h later for tritium in a Packard 4660 scintillation spectrometer at 55% efficiency. Specific binding at each concentration of [3H](+)-PHNO was defined as total binding minus that in the presence of 10 μm S-sulpiride. The data for each saturation curve were graphed as a Scatchard plot, yielding the Bmax density (in picomoles per gram) and the KD (in nanomolars). The Bmax in the absence of GN minus the Bmax in the presence of GN represented the density of G-sensitive [3H](+)-PHNO-labeled D2 receptors. The Bmax in the absence of S-sulpiride minus the Bmax in the presence of S-sulpiride represented D2 receptor binding capacity, which was used as an index of D2 receptor number.
Results
Experiment 1: D2 occupancy with acute and chronic HAL treatment (data not shown)
In experiment 1, we determined whether the HAL doses tested were yielding clinically meaningful levels of D2 blockade and whether this was maintained during chronic treatment. The levels of occupancy achieved by HAL were comparable to those seen in clinical treatment: during short-term HAL treatment (day 2), average striatal D2-receptor occupancy was 84% (±SEM 2.06) for animals treated with 0.25 mg/kg/d, and 85% (±SEM 2.12) for animals treated with 0.75 mg/kg/d. With continued treatment (day 13), average D2 occupancy was similar to that seen during short-term treatment in animals given 0.75 mg/kg/d HAL (82%, ±SEM 0.64), but was reduced in animals treated with the lower dose, although still well within clinically relevant ranges (69%, ±SEM 2.7; unpaired t test on day 2 vs day 13; t = 4.12; p = 0.0021). Thus, clinical levels of D2 occupancy were achieved with both HAL doses and this was maintained during the course of treatment.
Experiment 2a: effects of acute versus chronic HAL on behavioral sensitivity to AMPH
In experiment 2a, we examined changes in the ability of HAL to block AMPH-induced locomotion with continued treatment. Early in treatment (day 2) (Fig. 1a), both doses of HAL inhibited the total locomotor response to AMPH by 60% (±SEM 9). During chronic treatment (day 12) (Fig. 1b), HAL no longer disrupted AMPH-induced locomotion. On the fifth day of withdrawal from HAL (Fig. 1c), AMPH-induced locomotion was 63% (±SEM 28) higher in animals treated previously with 0.75 mg/kg/d HAL relative to controls. No other comparisons were significant. Thus, HAL initially attenuated AMPH-induced locomotion but gradually lost this ability, and once HAL treatment was terminated, a supersensitive locomotor response to AMPH emerged.
Figure 1.
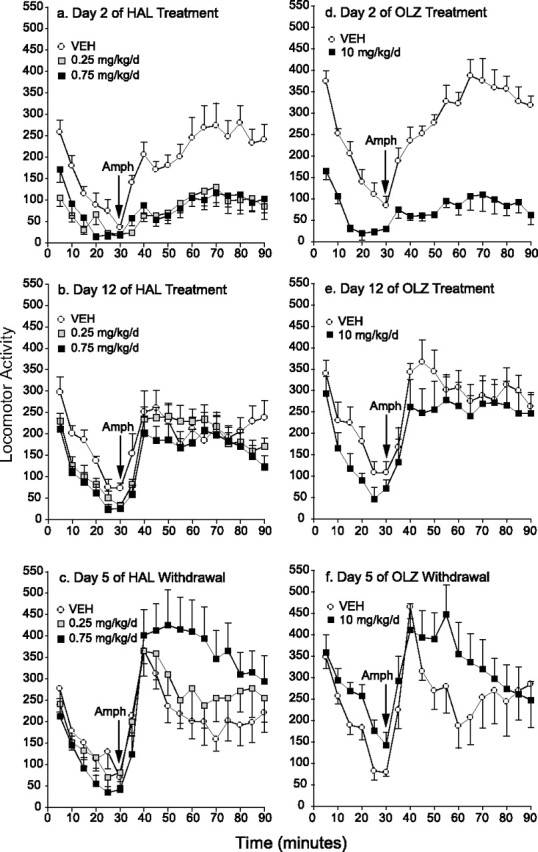
a–f, Effects of chronic haloperidol (a–c) or olanzapine (d–f) treatment on the locomotor response to AMPH over time. Initially, both HAL (a) and OLZ (d) reduced AMPH-induced locomotion relative to controls (2-way ANOVA on the 60 min after AMPH, main effects of condition, a, 0.25 mg/kg vs VEH, F = 13.13; 0.75 mg/kg vs VEH, F = 13.32; d, F = 64.36; p < 0.05). With continued treatment, HAL (b) and OLZ (e) no longer suppressed AMPH-induced locomotion (p > 0.05). In addition, amphetamine-induced locomotion was increased in all antipsychotic-treated groups from day 2 to day 12 (unpaired t tests on summed activity counts starting at 40 min; 0.25 mg/kg/d HAL, t = −3.402; 0.75 mg/kg/d HAL, t = −2.307; 10 mg/kg/d OLZ, t = −3.77; p < 0.05). Thus, both HAL and OLZ progressively lost their ability to inhibit AMPH-induced locomotion during ongoing treatment. After withdrawal, rats in the 0.75 mg/kg/d HAL group showed a potentiated response to AMPH relative to both other groups (c; condition by time, 0.75 mg/kg vs VEH, F = 4.3; 0.75 vs 0.25 mg/kg, F = 2.1; p < 0.03) and OLZ-treated rats were not different from controls (f; p = 0.15). n = 6–8 per condition per time point. Error bars indicate SEM.
Experiment 2b: effects of augmenting HAL levels (data not shown)
In experiment 2b, we determined whether augmenting chronic levels of HAL by giving an acute injection of the neuroleptic (0.1 mg/kg, s.c.) could suppress AMPH-induced locomotion when the ongoing HAL treatment (via minipump) had lost the ability to do so. AMPH-induced locomotion in rats chronically treated with HAL was similar to that of controls, indicating that HAL had lost its ability to disrupt AMPH-induced locomotion (unpaired t test, t = 0.40; p = 0.70). These results are consistent with those of experiment 2a (Fig. 1b). An acute injection of HAL suppressed AMPH-induced locomotion in both neuroleptic-naive rats and in rats maintained on HAL via minipumps (paired t tests on the locomotor response to AMPH after an acute injection of VEH or HAL, t = 2.84 and 3.96, respectively; p < 0.05). In addition, an acute HAL injection suppressed AMPH-induced locomotion to the same extent in animals maintained on HAL or VEH treatment via minipump (unpaired t test, t = −1.86; p = 0.10). Thus, during chronic treatment, the system can still respond to an additional dose of HAL, but it appears to have developed partial tolerance.
Experiment 2c: effects of acute versus chronic OLZ on behavioral sensitivity to AMPH
In experiment 2c, we determined whether these effects of HAL could be extended to an atypical antipsychotic drug, OLZ. Early in OLZ treatment (day 2) (Fig. 1d), AMPH-induced locomotion was attenuated relative to control levels. During chronic treatment (day 12) (Fig. 1e), OLZ no longer suppressed AMPH-induced locomotion. After OLZ withdrawal, AMPH-induced locomotion was not statistically different from that seen in control rats. Thus, as seen in Figure 1a–b with HAL, OLZ initially suppressed the locomotor activating effects of AMPH, but progressively lost this ability during treatment.
Experiment 3: conditioned avoidance responding
In experiment 3, we examined whether the loss of efficacy of antipsychotics could be seen in a nonpharmacological model with very strong predictive validity for antipsychotic action: the CAR test. As seen in Figure 2, HAL initially suppressed CAR relative to predrug (day 0) and control levels. The disruptive effect of HAL on CAR reached its peak on day 6, when avoidance responding was decreased by 43.3% (±SEM 8.25) relative to control levels. However, this effect diminished over time such that by day 12, HAL only suppressed CAR by 8% (±SEM 4.2), which was significantly less than on day 6 (although still different from VEH control rats).
Figure 2.

Effects of haloperidol treatment on conditioned avoidance responding over time. HAL suppressed CAR relative to controls (2-way ANOVA, main effect of condition, F = 21.44; p = 0.0001; n = 10 per condition). However, this effect diminished over time (condition by day, F = 6.03; p < 0.0001), such that by day 12, HAL only suppressed CAR by 8% (±SEM 4.2), which is lower than that seen on day 6 (paired t test; t = −4.72; p = 0.002), but still different from controls (unpaired t test; t = −2.13; p = 0.05). Error bars indicate SEM.
Experiment 4: in vivo microdialysis of DA, DOPAC, HVA, and NE
In experiment 4, we examined the possibility that behavioral supersensitivity to AMPH during chronic HAL treatment was caused by presynaptic changes in DA by assessing extracellular levels in the nucleus accumbens. Initially, HAL increased baseline levels of extracellular DA and DOPAC (Fig. 3a, inset, b), but did not significantly change HVA (Fig. 3c) or NE levels (data not shown). With continued treatment, HAL no longer enhanced levels of DA and DOPAC, and levels of all analytes were similar to those seen in controls (Fig. 3d, inset, e,f) (NE, data not shown).
Figure 3.
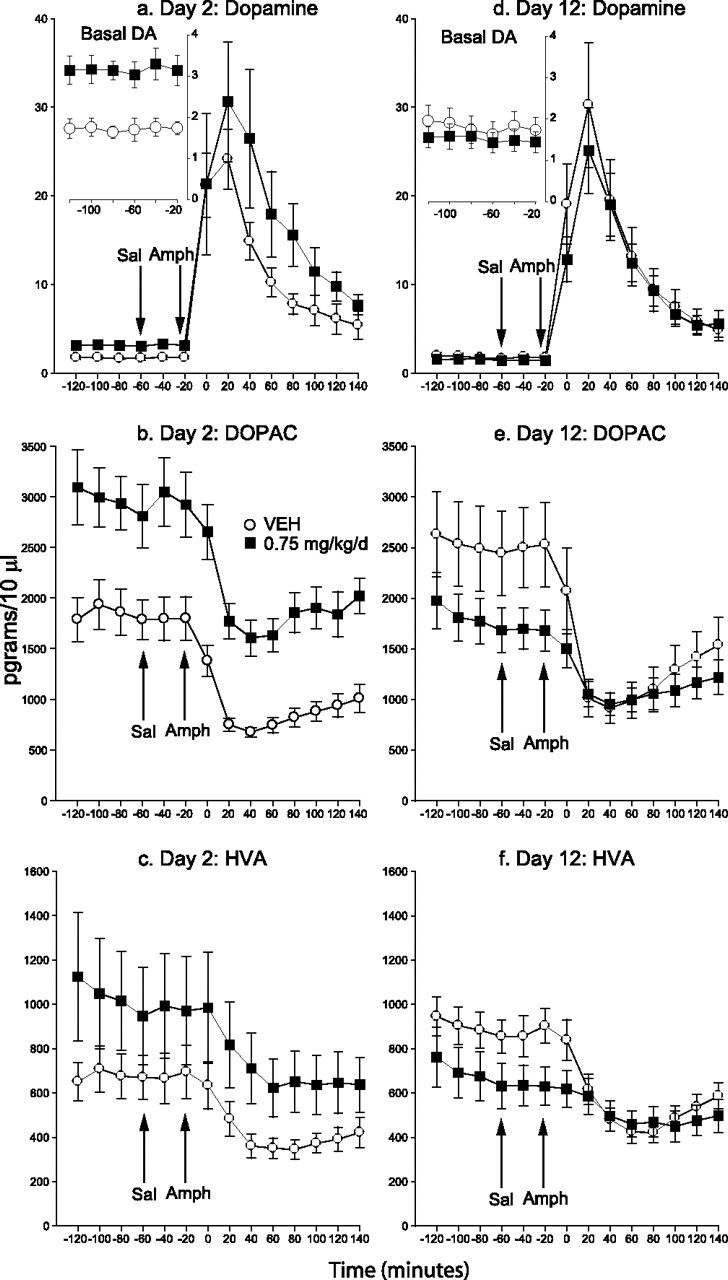
a–f, Extracellular DA, DOPAC, and HVA in the nucleus accumbens during short-term (a–c) versus longer-term (d–f) haloperidol treatment. HAL initially increased baseline levels of DA and DOPAC (a, inset, b; 2-way ANOVAs on −120 to −20 min, main effects of condition, F = 13.82 and F = 9.15, respectively; p < 0.05), but DA and DOPAC levels became similar to those of controls with chronic treatment (d, inset, e). Basal HVA levels were not significantly affected by HAL on either testing day (c, f). An injection of saline did not alter analyte levels in either group on either test day. AMPH increased DA levels above saline in both groups and on both test days (2-way ANOVAs on −40 to 140 min, main effect of time, a, F = 13.30; d, F = 37.23; p < 0.0001). HAL had no significant effect on the DA response to AMPH on either test day (a, d, 2-way ANOVAs, condition by time, p = 0.78 and 0.70, respectively). In both groups and on both test days, AMPH decreased DOPAC (main effects of time, b, F = 29.82; e, F = 56.34; p < 0.0001) and HVA (c, F = 19.28; f, F = 56.17; p < 0.0001) levels relative to saline. Initially, HAL did not alter these responses (condition by time, b, p = 0.81; c, p = 0.90), but chronic HAL treatment attenuated the AMPH-induced decrease in DOPAC and HVA (condition by time, e, F = 7.99; f, F = 11.27; p < 0.0001). n = 7–8 per condition per time point. Error bars indicate SEM.
Analyte levels were unaffected by an injection of saline in both groups and at both time points tested. AMPH increased DA (Figs. 3a,d) and NE levels (data not shown) and decreased DOPAC and HVA levels (Figs. 3b,c,e,f) at both time points tested and in both treatment groups. Early in treatment (day 2), HAL had no statistically significant effect on the neurochemical response to AMPH, but chronic HAL treatment attenuated the AMPH-induced decrease in DOPAC and HVA (Figs. 3e,f). Thus HAL-induced behavioral supersensitivity to AMPH is accompanied by a reduction in the suppressive effects of AMPH on DA turnover, but not by significant changes in extracellular levels of DA.
Experiment 5: D2 receptor binding capacity and guanine nucleotide-sensitive D2High states in the striatum
Given the results of experiment 4 showing that chronic HAL treatment did not significantly change AMPH-induced extracellular DA levels, we measured changes in D2 receptor number and sensitivity in experiment 5. Control experiments revealed that the addition of 0.75 or 1.5 nm HAL to striatal tissue did not alter D2 receptor or D2High Bmax (data not shown). Treatment with 0.25 mg/kg/d HAL did not change D2 receptor Bmax at any time point tested (Fig. 4a). Exposure to 0.75 mg/kg/d HAL increased D2 receptor Bmax relative to controls at all time points tested. Bmax in these animals was also greater after neuroleptic withdrawal (day 21) than during acute treatment (day 2). No other comparisons were significant.
Figure 4.
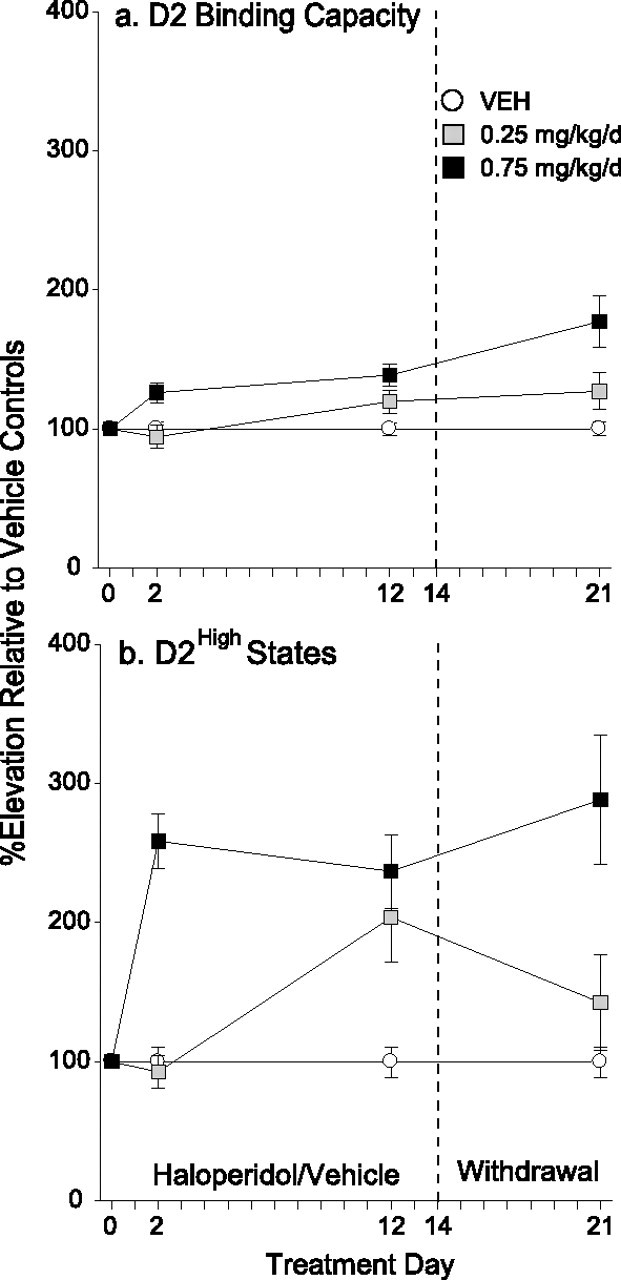
a, b, Effects of haloperidol treatment on striatal DA D2 receptor binding capacity (a) and D2High states (b) over time. a, Treatment with 0.25 mg/kg/d HAL did not change D2 receptor Bmax at any time point tested. Treatment with 0.75 mg/kg/d HAL increased D2 receptor Bmax relative to controls at all time points tested (1-way ANOVA at each time point, followed by Tukey's multiple comparison tests; day 2, F = 5.50; day 12, F = 9.91; day 21, F = 17.07; p < 0.05). In addition, D2 Bmax in the 0.75 mg/kg/d group was greater after neuroleptic withdrawal than during acute treatment (unpaired t test, day 2 vs day 21; t = −3.0; p = 0.02). b, Relative to VEH, 0.75 mg/kg/d HAL elevated striatal D2High sites from day 2 onward, and D2High levels did not change over time (day 2, F = 35.54; day 12, F = 16.08; day 21, F = 16.89; p < 0.05). In the 0.25 mg/kg/d group, D2High levels were elevated only during long-term treatment (day 12; p < 0.05). n = 8–9 per condition per time point. Error bars indicate SEM.
Antipsychotic treatment also influenced the density of D2 receptors that are in the functionally relevant high-affinity state for DA. Relative to control levels, treatment with 0.75 mg/kg/d HAL elevated striatal D2High sites by 137–188% (±SEM 20–47) from day 2 onward, and D2High levels did not change over time (Fig. 4b).Treatment with 0.25 mg/kg/d HAL elevated D2High density by 104% (±SEM 32) only during chronic treatment (day 12) and levels went back to normal after neuroleptic withdrawal. Thus, with the exception of the D2High increase seen on day 2 in the 0.75 mg/kg/d group, the changes in D2High levels predicted the behavioral response to AMPH in both HAL treated groups.
Discussion
Previous work has shown that withdrawal from chronic antipsychotic treatment leads to a supersensitive psychomotor response to dopamine agonists (Gianutsos et al., 1974; Sayers et al., 1975; Smith and Davis, 1975, 1976; Clow et al., 1979; Montanaro et al., 1982; Rebec et al., 1982; Meng et al., 1998). We show here that behavioral dopamine supersensitivity is not just evident on withdrawal, but develops early during antipsychotic exposure and significantly undermines the efficacy of ongoing treatment. The loss of efficacy was seen with typical or atypical antipsychotics in two widely used tests of antipsychotic-like effects in animals and occurred despite ongoing, clinically relevant, levels of striatal D2-receptor blockade. Thus, the effects were not likely caused by pharmacokinetic or peripheral factors, but by compensatory neurobiological changes in response to ongoing treatment.
One possible explanation for the progressive loss in the ability of antipsychotics to suppress amphetamine-induced locomotion is an increase in dopamine availability over time, which would surmount the antidopaminergic effects of the antipsychotic. However, in vivo microdialysis measurements revealed that amphetamine-induced increases in extracellular dopamine were unchanged during haloperidol treatment. This suggests that the failure of ongoing antipsychotic treatment to inhibit amphetamine-induced locomotion was not attributable to changes in dopamine availability. These data extend previous work showing that although withdrawal from haloperidol produces a supersensitive behavioral response to amphetamine (Smith and Davis, 1975; Rebec et al., 1982; Meng et al., 1998), it does not change the dopamine response to amphetamine (Compton and Johnson, 1989; Ichikawa and Meltzer, 1992; See et al., 1992). However, microdialysis measurements are a function of release and uptake and the lack of change in dopamine overflow in our study could be reflecting increased release and increased uptake or, alternatively, decreased release and decreased uptake. Thus, despite our microdialysis findings, presynaptic changes could still be involved in the development of dopamine supersensitivity and loss of antipsychotic treatment efficacy. Importantly, however, Chesi et al. (1995) demonstrated that chronic oral treatment with a dose of haloperidol very similar to the one tested here (0.7 mg/kg/d) did not alter the sensitivity of in vivo dopamine overflow to either an agonist or an antagonist at presynaptic autoreceptors (sulpiride and quinpirole, respectively). This suggests that the gradual loss of efficacy and behavioral supersensitivity that develops during antipsychotic treatment is likely caused by postsynaptic changes.
Although haloperidol did not change the dopaminergic response to amphetamine over time, it did attenuate the amphetamine-induced decrease in DOPAC and HVA. A number of explanations can be considered. First, long-term exposure to haloperidol itself reduced dopamine metabolite levels and this could have limited the extent to which amphetamine could suppress these (i.e., metabolite levels could already have been near “floor” levels). Second, amphetamine decreases dopamine metabolite levels by triggering the release of newly formed dopamine into the synapse and away from monoamine oxidase, which normally breaks down intraneuronal dopamine into DOPAC and HVA (Zetterstrom et al., 1986). However, it is unclear how haloperidol could influence this response given that amphetamine-induced dopamine overflow was not affected by the neuroleptic. Third, amphetamine also suppresses dopamine metabolite levels by inhibiting monoamine oxidase type A (Green, 1971; Zetterstrom et al., 1986). Although we did not measure monoamine oxidase levels in this study, haloperidol treatment does not appear to alter these (Van Der Krogt et al., 1982). As such, the precise mechanisms by which long-term haloperidol treatment attenuates the suppression of DOPAC and HVA by amphetamine remain to be identified.
There is evidence that postsynaptic processes contribute to antipsychotic-induced behavioral supersensitivity. For example, chronic haloperidol potentiates the locomotor activating effects of intra-accumbens and intracaudate–putamen infusions of dopamine (Halperin et al., 1983). Changes in dopamine signaling must therefore be involved. It has been proposed that withdrawal from antipsychotic treatment induces dopamine supersensitivity by increasing striatal D2 receptor number (Burt et al., 1977; Muller and Seeman, 1977; Clow et al., 1980; Seeman, 1980; Fleminger et al., 1983; Joyce, 2001). Both haloperidol doses we tested led to a supersensitive locomotor response to amphetamine, but only the higher dose increased striatal D2 receptor number. Indeed, behavioral sensitivity to dopamine is not always predicted by striatal D2 receptor density (Pierce et al., 1991; Flores et al., 1996). In contrast, animal models of dopamine supersensitivity are systematically linked to elevations in D2High receptors (Seeman et al., 2005). Our results provide additional support for this link. At the lower haloperidol dose tested, the time course of changes in D2High levels predicted behavioral supersensitivity to dopamine. Both the locomotor response to amphetamine and D2High levels were increased during antipsychotic treatment, and both returned to control levels after treatment cessation. At the higher haloperidol dose, behavioral dopamine supersensitivity was also well predicted by changes in D2High levels, with the following exception. Short-term (2 d) treatment with this dose elevated D2High sites but this did not prevent the neuroleptic from disrupting amphetamine-induced locomotion. This suggests two, nonmutually exclusive possibilities. First, different neurobiological mechanisms might underlie the dopamine supersensitivity induced by low versus higher doses of antipsychotic. Second, elevated D2High levels might be necessary although not sufficient to induce dopamine supersensitivity and loss of antipsychotic efficacy. Whatever the case might be, our results show that dopamine supersensitivity and functional tolerance to antipsychotics are due in part to changes in striatal dopamine receptor function, although the assays that we have used do not allow us to determine whether this is occurring at D2 receptors on local cells and/or on cells projecting to the striatum. In support of a role of altered dopamine receptor function was the finding that further augmenting haloperidol levels by an acute injection of the drug inhibited the locomotor response to amphetamine when ongoing haloperidol treatment had lost the ability to do so. Thus, additional haloperidol (presumably leading to additional D2 blockade) can overcome whatever mechanism is responsible for the expression of dopamine supersensitivity. It remains unknown, however, whether an increase in dose would maintain its efficacy over time, or lose to heightened breakthrough supersensitivity. This is reminiscent of evidence from the clinic, where increasing dose can, at least provisionally, reduce psychotic symptoms in patients that have developed therapeutic tolerance.
In contrast to most preclinical work where antipsychotics are given via intraperitoneal injections, we used continuous infusion via minipump. This is an important issue when one considers that the terminal half-life of haloperidol is 24 h in humans (Bezchlibnyk-Butler and Jeffries, 1999) and 1.5 h in rats (Cheng and Paalzow, 1992). Thus, 24 h after a single dose of haloperidol, D2 occupancy levels remain high in humans (Farde et al., 1989; Seeman, 2002), but fall to well below clinically relevant levels in rats (Kapur et al., 2000a). Our findings demonstrate that continuous antipsychotic treatment and D2 receptor blockade induces neuroadaptations that lead to antipsychotic failure. It is possible, therefore, that much of what is currently known about the neurobehavioural effects of antipsychotic drugs in animals might not apply if antipsychotics are given continuously rather than intermittently. Indeed, merely changing the mode of antipsychotic administration (i.e., intermittent vs continuous infusion) can have markedly different effects (Turrone et al., 2003, 2005). Animal studies that use a mode of drug administration that more closely mimics clinical antipsychotic treatment (i.e., relatively continuous treatment) might be more informative about the true effects of these drugs in humans (Kapur et al., 2003).
Although in the present study with rats haloperidol and olanzapine lost efficacy over time, not all treated patients develop therapeutic tolerance. However, a notable proportion of initially stabilized patients relapse during treatment and this cannot always be explained by nonadherence to treatment. For example, even when medication is guaranteed by depot injection, the average relapse rate at 1–2 years is still 18–55% (Carpenter et al., 1999; Schooler, 2003; De Graeve et al., 2005). Although relapse during continued treatment can be attributed to a number of reasons, our results suggest that an antipsychotic-induced increase in dopamine sensitivity might predispose certain individuals to psychotic relapse. This provides a discrete hypothesis that can be tested in patients.
Together, the present findings allow us to propose the following: (1) initially, antipsychotics block D2 receptors, and increase dopamine and dopamine turnover in a system where levels of D2 receptors and D2High receptors are normal. At this stage the balance of agonist (i.e., endogenous dopamine-related) drive and receptor blockade allows the antipsychotic to exert a net anti-dopaminergic effect. (2) With continued treatment, there is a decrease in turnover on the presynaptic side, D2 blockade by antipsychotics is maintained, but both D2 receptor numbers and D2High sites are elevated. At this later stage, endogenous dopamine drive is potentiated and can more readily oppose the anti-dopaminergic effects of the antipsychotic. This might explain why antipsychotics so often fail. The challenge now is to identify the neural processes by which dopamine supersensitivity and antipsychotic treatment tolerance develop. At the same time, the biological (i.e., increases in D2 receptors and D2High sites) and behavioral (loss of efficacy in behavioral models) markers identified here provide targets that can be used to identify strategies to overcome or prevent antipsychotic treatment failure.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR), a CIHR operating grant to J.S., a Special Initiative Grant from the Ontario Mental Health Foundation, the Stanley Medical Research Institute, the Essel Foundation, and Constance and Stephen Lieber. S.K. was supported by a Canada Research Chair. We thank Dr. H.-C. Guan, G. Reckless, and D. Rodaros for valuable technical assistance and Drs. P. Fletcher and S. Natesan for their advice during these studies.
References
- Asper H, Baggiolini M, Burki HR, Lauener H, Ruch W, Stille G. Tolerance phenomena with neuroleptics catalepsy, apomorphine stereotypies and striatal dopamine metabolism in the rat after single and repeated administration of loxapine and haloperidol. Eur J Pharmacol. 1973;22:287–294. doi: 10.1016/0014-2999(73)90028-9. [DOI] [PubMed] [Google Scholar]
- Bezchlibnyk-Butler KZ, Jeffries JJ. Toronto: Hogrefe and Huber; 1999. Clinical handbook of psychotropic drugs. [Google Scholar]
- Burt DR, Creese I, Snyder SH. Antischizophrenic drugs: chronic treatment elevates dopamine receptor binding in brain. Science. 1977;196:326–328. doi: 10.1126/science.847477. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Jr, Buchanan RW, Kirkpatrick B, Lann HD, Breier AF, Summerfelt AT. Comparative effectiveness of fluphenazine decanoate injections every 2 weeks versus every 6 weeks. Am J Psychiatry. 1999;156:412–418. doi: 10.1176/ajp.156.3.412. [DOI] [PubMed] [Google Scholar]
- Cheng YF, Paalzow LK. Linear pharmacokinetics of haloperidol in the rat. Biopharm Drug Dispos. 1992;13:69–76. doi: 10.1002/bdd.2510130106. [DOI] [PubMed] [Google Scholar]
- Chesi AJ, Feasey-Truger KJ, Alzheimer C, ten Bruggencate G. Dopamine autoreceptor sensitivity is unchanged in rat nucleus accumbens after chronic haloperidol treatment: an in vivo and in vitro voltammetric study. Eur J Neurosci. 1995;7:2450–2457. doi: 10.1111/j.1460-9568.1995.tb01043.x. [DOI] [PubMed] [Google Scholar]
- Chouinard G, Jones BD. Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics. Am J Psychiatry. 1980;137:16–21. doi: 10.1176/ajp.137.1.16. [DOI] [PubMed] [Google Scholar]
- Chouinard G, Jones BD, Annable L. Neuroleptic-induced supersensitivity psychosis. Am J Psychiatry. 1978;135:1409–1410. doi: 10.1176/ajp.135.11.1409. [DOI] [PubMed] [Google Scholar]
- Clow A, Jenner P, Marsden CD. Changes in dopamine-mediated behaviour during one year's neuroleptic administration. Eur J Pharmacol. 1979;57:365–375. doi: 10.1016/0014-2999(79)90499-0. [DOI] [PubMed] [Google Scholar]
- Clow A, Theodorou A, Jenner P, Marsden CD. Changes in rat striatal dopamine turnover and receptor activity during one years neuroleptic administration. Eur J Pharmacol. 1980;63:135–144. doi: 10.1016/0014-2999(80)90437-9. [DOI] [PubMed] [Google Scholar]
- Compton DR, Johnson KM. Effects of acute and chronic clozapine and haloperidol on in vitro release of acetylcholine and dopamine from striatum and nucleus accumbens. J Pharmacol Exp Ther. 1989;248:521–530. [PubMed] [Google Scholar]
- De Graeve D, Smet A, Mehnert A, Caleo S, Miadi-Fargier H, Mosqueda GJ, Lecompte D, Peuskens J. Long-acting risperidone compared with oral olanzapine and haloperidol depot in schizophrenia: a Belgian cost-effectiveness analysis. Pharmacoeconomics. 2005;23(Suppl 1):35–47. doi: 10.2165/00019053-200523001-00004. [DOI] [PubMed] [Google Scholar]
- Ekblom B, Eriksson K, Lindstrom LH. Supersensitivity psychosis in schizophrenic patients after sudden clozapine withdrawal. Psychopharmacology (Berl) 1984;83:293–294. doi: 10.1007/BF00464798. [DOI] [PubMed] [Google Scholar]
- Farde L, Wiesel FA, Halldin C, Sedvall G. Central D2-dopamine receptor occupancy in schizophrenic patients treated with antipsychotic drugs. Arch Gen Psychiatry. 1988;45:71–76. doi: 10.1001/archpsyc.1988.01800250087012. [DOI] [PubMed] [Google Scholar]
- Farde L, Wiesel FA, Nordstrom AL, Sedvall G. D1- and D2-dopamine receptor occupancy during treatment with conventional and atypical neuroleptics. Psychopharmacology (Berl) 1989;99(Suppl):S28–S31. doi: 10.1007/BF00442555. [DOI] [PubMed] [Google Scholar]
- Fitzsimons JT. Normal drinking in rats. J Physiology. 1957;138:39P. [Google Scholar]
- Fleminger S, Rupniak NM, Hall MD, Jenner P, Marsden CD. Changes in apomorphine-induced stereotypy as a result of subacute neuroleptic treatment correlates with increased D-2 receptors, but not with increases in D-1 receptors. Biochem Pharmacol. 1983;32:2921–2927. doi: 10.1016/0006-2952(83)90397-0. [DOI] [PubMed] [Google Scholar]
- Flores G, Barbeau D, Quirion R, Srivastava LK. Decreased binding of dopamine D3 receptors in limbic subregions after neonatal bilateral lesion of rat hippocampus. J Neurosci. 1996;16:2020–2026. doi: 10.1523/JNEUROSCI.16-06-02020.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianutsos G, Drawbaugh RB, Hynes MD, Lal H. Behavioral evidence for dopaminergic supersensitivity after chronic haloperidol. Life Sci. 1974;14:887–898. doi: 10.1016/0024-3205(74)90078-2. [DOI] [PubMed] [Google Scholar]
- Green AL. Inhibition of rat and mouse monoamine oxidase by (+)-amphetamine. Proceedings of the biochemical society; 1971. pp. 37P–38P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin R, Guerin JJ, Jr, Davis KL. Chronic administration of three neuroleptics: effects of behavioral supersensitivity mediated by two different brain regions in the rat. Life Sci. 1983;33:585–592. doi: 10.1016/0024-3205(83)90134-0. [DOI] [PubMed] [Google Scholar]
- Ichikawa J, Meltzer HY. The effect of chronic atypical antipsychotic drugs and haloperidol on amphetamine-induced dopamine release in vivo. Brain Res. 1992;574:98–104. doi: 10.1016/0006-8993(92)90805-j. [DOI] [PubMed] [Google Scholar]
- Joyce JN. D2 but not D3 receptors are elevated after 9 or 11 months chronic haloperidol treatment: influence of withdrawal period. Synapse. 2001;40:137–144. doi: 10.1002/syn.1035. [DOI] [PubMed] [Google Scholar]
- Kahne GJ. Rebound psychoses following the discontinuation of a high potency neuroleptic. Can J Psychiatry. 1989;34:227–229. doi: 10.1177/070674378903400312. [DOI] [PubMed] [Google Scholar]
- Kapur S, Wadenberg ML, Remington G. Are animal studies of antipsychotics appropriately dosed? Lessons from the bedside to the bench. Can J Psychiatry. 2000a;45:241–246. doi: 10.1177/070674370004500302. [DOI] [PubMed] [Google Scholar]
- Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000b;157:514–520. doi: 10.1176/appi.ajp.157.4.514. [DOI] [PubMed] [Google Scholar]
- Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther. 2003;305:625–631. doi: 10.1124/jpet.102.046987. [DOI] [PubMed] [Google Scholar]
- Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry. 2001;46:87–88. [PubMed] [Google Scholar]
- Margolese HC, Chouinard G, Beauclair L, Belanger MC. Therapeutic tolerance and rebound psychosis during quetiapine maintenance monotherapy in patients with schizophrenia and schizoaffective disorder. J Clin Psychopharmacol. 2002;22:347–352. doi: 10.1097/00004714-200208000-00003. [DOI] [PubMed] [Google Scholar]
- Meng ZH, Feldpaush DL, Merchant KM. Clozapine and haloperidol block the induction of behavioral sensitization to amphetamine and associated genomic responses in rats. Brain Res Mol Brain Res. 1998;61:39–50. doi: 10.1016/s0169-328x(98)00196-x. [DOI] [PubMed] [Google Scholar]
- Montanaro N, Dall'Olio R, Gandolfi O, Vaccheri A. Differential enhancement of behavioral sensitivity to apomorphine following chronic treatment of rats with (-)-sulpiride and haloperidol. Eur J Pharmacol. 1982;81:1–9. doi: 10.1016/0014-2999(82)90595-7. [DOI] [PubMed] [Google Scholar]
- Muller P, Seeman P. Brain neurotransmitter receptors after long-term haloperidol: dopamine, acetylcholine, serotonin, alpha-noradrenergic and naloxone receptors. Life Sci. 1977;21:1751–1758. doi: 10.1016/0024-3205(77)90155-2. [DOI] [PubMed] [Google Scholar]
- Muller P, Seeman P. Dopaminergic supersensitivity after neuroleptics: time-course and specificity. Psychopharmacology (Berl) 1978;60:1–11. doi: 10.1007/BF00429171. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Rowlett JK, Bardo MT, Rebec GV. Chronic ascorbate potentiates the effects of chronic haloperidol on behavioral supersensitivity but not D2 dopamine receptor binding. Neuroscience. 1991;45:373–378. doi: 10.1016/0306-4522(91)90234-f. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Peirson EE, McPherson FA, Brugge K. Differential sensitivity to amphetamine following long-term treatment with clozapine or haloperidol. Psychopharmacology (Berl) 1982;77:360–366. doi: 10.1007/BF00432771. [DOI] [PubMed] [Google Scholar]
- Rupniak MN, Jenner P, Marsden CD. The effect of chronic neuroleptic administration on cerebral dopamine receptor function. Life Sci. 1983;32:2289–2311. doi: 10.1016/0024-3205(83)90759-2. [DOI] [PubMed] [Google Scholar]
- Sayers AC, Burki HR, Ruch W, Asper H. Neuroleptic-induced hypersensitivity of striatal dopamine receptors in the rat as a model of tardive dyskinesias. Effects of clozapine, haloperidol, loxapine and chlorpromazine. Psychopharmacologia. 1975;41:97–104. doi: 10.1007/BF00421063. [DOI] [PubMed] [Google Scholar]
- Schooler NR. Relapse and rehospitalization: comparing oral and depot antipsychotics. J Clin Psychiatry. 2003;64(Suppl 16):14–17. [PubMed] [Google Scholar]
- See RE, Chapman MA, Murray CE, Aravagiri M. Regional differences in chronic neuroleptic effects on extracellular dopamine activity. Brain Res Bull. 1992;29:473–478. doi: 10.1016/0361-9230(92)90085-c. [DOI] [PubMed] [Google Scholar]
- Seeman P. Brain dopamine receptors. Pharmacol Rev. 1980;32:229–313. [PubMed] [Google Scholar]
- Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47:27–38. [PubMed] [Google Scholar]
- Seeman P, Ulpian C, Wreggett KA, Wells JW. Dopamine receptor parameters detected by [3H]spiperone depend on tissue concentration: analysis and examples. J Neurochem. 1984;43:221–235. doi: 10.1111/j.1471-4159.1984.tb06700.x. [DOI] [PubMed] [Google Scholar]
- Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, Premont RT, Sotnikova TD, Boksa P, El-Ghundi M, O'Dowd BF, George SR, Perreault ML, Mannisto PT, Robinson S, Palmiter RD, Tallerico T. Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis. Proc Natl Acad Sci USA. 2005;102:3513–3518. doi: 10.1073/pnas.0409766102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RC, Davis JM. Behavioral supersensitivity to apomorphine and amphetamine after chronic high dose haloperidol treatment. Psychopharmacol Commun. 1975;1:285–293. [PubMed] [Google Scholar]
- Smith RC, Davis JM. Behavioral evidence for supersensitivity after chronic administration of haloperidol, clozapine, and thioridazine. Life Sci. 1976;19:725–731. doi: 10.1016/0024-3205(76)90170-3. [DOI] [PubMed] [Google Scholar]
- Stellar E, Hill JH. The rats rate of drinking as a function of water deprivation. J Comp Physiol Psychol. 1952;45:96–102. doi: 10.1037/h0062150. [DOI] [PubMed] [Google Scholar]
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol. 1999;19:435–443. doi: 10.1097/00004714-199910000-00007. [DOI] [PubMed] [Google Scholar]
- Turrone P, Remington G, Kapur S, Nobrega JN. Differential effects of within-day continuous vs. transient dopamine D2 receptor occupancy in the development of vacuous chewing movements (VCMs) in rats. Neuropsychopharmacology. 2003;28:1433–1439. doi: 10.1038/sj.npp.1300233. [DOI] [PubMed] [Google Scholar]
- Turrone P, Remington G, Kapur S, Nobrega JN. Continuous but not intermittent olanzapine infusion induces vacuous chewing movements in rats. Biol Psychiatry. 2005;57:406–411. doi: 10.1016/j.biopsych.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Van Der Krogt JA, Van Valkenburg CF, Belfroid RD. Rat brain monoamine oxidase activity is not affected by repeated administration of haloperidol. J Pharm Pharmacol. 1982;34:529–531. doi: 10.1111/j.2042-7158.1982.tb04782.x. [DOI] [PubMed] [Google Scholar]
- Zetterstrom T, Sharp T, Ungerstedt U. Further evaluation of the mechanism by which amphetamine reduces striatal dopamine metabolism: a brain dialysis study. Eur J Pharmacol. 1986;132:1–9. doi: 10.1016/0014-2999(86)90002-6. [DOI] [PubMed] [Google Scholar]