

Abstract
Research on the detrimental effects of stress in the brain has mainly focused on the hippocampus. Because prefrontal cortex (PFC) dysfunction characterizes many stress-related disorders, we here analyzed the impact of chronic stress in rats on the integrity of the hippocampal–PFC pathway, monitored by behavioral and electrophysiological function and morphological assessment. We show that chronic stress impairs synaptic plasticity by reducing LTP induction in the hippocampal–PFC connection; in addition, it induces selective atrophy within the PFC and severely disrupts working memory and behavioral flexibility, two functions that depend on PFC integrity. We also demonstrate that short periods of stress exposure induce spatial reference memory deficits before affecting PFC-dependent tasks, thus suggesting that the impairment of synaptic plasticity within the hippocampus-to-PFC connection is of relevance to the stress-induced PFC dysfunction. These findings evidence a fundamental role of the PFC in maladaptive responses to stress and identify this area as a target for intervention in stress-related disorders.
Keywords: plasticity, stereology, working memory, LTP, chronic stress, rat
Introduction
Stressful events predispose to a variety of mental disorders (McEwen, 2004). Normally, after exposure to a stressor, adrenoglucocorticoids act in the brain to restore physiological and behavioral homeostasis (de Kloet et al., 2005); however, when the intensity or duration of stressors exceeds a certain individual-specific threshold, activation of the stress response can be deleterious (McEwen, 2005). Chronic stress induces profound behavioral changes in humans and rodents, manifested as depressive-like symptoms, a hyperanxious state, and learning/memory deficits (Mizoguchi et al., 2000; McEwen, 2004), paralleled by structural damage (Sousa and Almeida, 2002) and impaired synaptic plasticity (Sousa et al., 1998; Kim and Diamond, 2002), mainly in the hippocampus. However, not all stress-induced dysfunctions can be explained by hippocampal injury, the deficits in executive function that characterize stress-related disorders being a prototypic example.
Working memory, which involves transient storage and manipulation of information to guide subsequent behavior, represents one key function of the prefrontal cortex (PFC) (Goldman-Rakic, 1995) that is affected by stress. In addition, the PFC is implicated in processing of emotional stimuli (Davidson, 2002) as well as in complex cognitive tasks, including temporal organization of behavior, decision-making, rule learning, and behavioral flexibility (Clark et al., 2004). Previously, independent studies (Wellman, 2001; Radley et al., 2004; Brown et al., 2005; Cerqueira et al., 2007) described that chronic stressors or glucocorticoids produce profound rearrangements of the apical dendrites of layer II–III PFC pyramidal neurons, resulting in atrophy of the distal dendritic branches and loss of dendritic spines.
The hippocampus is connected with the PFC by axons originating in the subiculum and ventral CA1 subfields, which terminate in the pyramidal cells and interneurons of the medial PFC (Jay and Witter, 1995; Tierney et al., 2004). Modulation of synaptic activity in the connection between these two areas contributes to a synergistic regulation of learning/memory processes (Wall and Messier, 2001) and of the stress response (de Kloet et al., 2005). In fact, training in associative learning task increases synaptic transmission in the hippocampal-PFC pathway (Doyère et al., 1993) and both regions are strongly involved in the regulation of the stress response by inhibiting corticotrophin-releasing hormone (CRH) in the paraventricular hypothalamic nuclei (Sullivan and Gratton, 2002; de Kloet et al., 2005).
Therefore, we hypothesized that the constellation of memory deficits observed after stress might result from damage to the neuronal network linking the hippocampus to the PFC. This hypothesis was tested in a series of complementary experiments. In the first, chronically stressed and control rats were subjected to behavioral tests of PFC- and hippocampus-dependent cognitive function (behavioral flexibility and spatial working and reference memory, respectively). In the second, we induced long-term potentiation (LTP) to examine the effects of chronic unpredictable stress (CUS) on synaptic plasticity in the hippocampal-PFC pathway. In the third, we used stereological measurements to assess the impact of cell and volumetric changes in the PFC and subiculum. Finally, we reported the behavioral tests and stereological analysis in animals submitted to shorter periods (3 and 6 d) of stress to determine the sequential pattern of stress-induced changes.
Materials and Methods
Animals and treatments
Experiments were conducted in accordance with local regulations (European Union Directive 86/609/EEC) and National Institutes of Health guidelines on animal care and experimentation.
Adult (2 months old at the beginning of the experiment) male Wistar rats (Charles River Laboratories, Barcelona, Spain) were housed in groups of 3–4 under standard laboratory conditions (lights on from 8:00 A.M. to 8:00 P.M.; room temperature 22°C; ad libitum access to food and drink). A group of 10 rats were submitted to 4 weeks of CUS. Briefly, animals were exposed once daily to a stressor (1 h/d) of one of several aversive stimuli [cold water (18°C), vibration, restraint, overcrowding, exposure to a hot air stream]; the stressors were presented in random order for the duration of the experiment. This stress paradigm was shown previously to result in persistently elevated plasma levels of corticosterone, the primary glucocorticoid of the rat (for details, see Sousa et al., 1998). Another group of 10 rats were handled daily and served as controls (CONs).
Body weights were recorded on a weekly basis throughout the study as an indication of treatment efficacy; postmortem thymus weights also provided information on treatment efficacy. Corticosterone levels were measured in blood serum sampled between 8:00 and 9:00 A.M. (3 h before the electrophysiological recordings and >12 h after the last exposure to stress) using a commercially available ELISA kit (R & D Systems, Minneapolis, MN).
To determine the sequential pattern of chronic stress-induced disturbances in the hippocampus–PFC system, we submitted a different set of rats to shorter periods of the same stress paradigm: 10 animals were exposed to unpredictable stress for 3 d and another 10 rats were stressed for 6 d. Two groups of 10 animals, handled daily for the same period, served as controls.
Behavioral testing
Behavioral tests were conducted in a circular black tank (170 cm diameter) filled to a depth of 31 cm with water at 22°C, colored with a black nontoxic dye (Jazz Gloss Tempera black ink; Van Aken International, Rancho Cucamonga, CA) and placed in a dimly lit room with extrinsic clues. The tank was divided in imaginary quadrants and had a black platform (12 cm diameter, 30 cm high) placed in one of them. Data were collected using a video camera fixed to the ceiling and connected to a video-tracking system (Viewpoint, Champagne au mont d'or, France).
Working memory task.
The test used was described by Kesner (2000) as a test of PFC function: its goal is to assess the ability of rats to learn the position of the hidden platform and to keep this information online during four consecutive trials. This working memory task, a modification of the spatial reference memory test (Morris, 1984), consists of 4 d of acquisition. The position of the platform is kept constant during the four trials of each day, but varies on each successive day such that all four quadrants are used. Rats are placed, facing the wall of the maze, at a different starting point (north, east, south, or west) at the beginning of each of the four daily trials. A trial is considered complete when the rat escapes onto the platform; when this escape fails to occur within 120 s, the animal is gently guided to the platform and an escape latency of 120 s is recorded for that trial. Rats are allowed to spend 30 s on the escape platform before being positioned at a new starting point. Length of the path described (distance swam) and time spent to reach the platform (escape latency) are recorded in the consecutive trials.
Reference memory task.
After the working memory procedure (days 1–4), the platform remained in the same quadrant as on day 4 and animals were tested for an additional 3 d (days 5–7) to ensure that they had correctly learnt the position of the platform before assessment of reversal learning (de Bruin et al., 1994). All of the remaining procedures were similar to the ones described for the working memory task. Data on average escape latencies and distances swam on days 4–7 were analyzed as a reference memory test, which is a hippocampal-dependent task (Morris, 1984).
Reversal learning task.
The reversal learning task is a PFC-dependent function (de Bruin et al., 1994). On day 8, the escape platform was positioned in a new (opposite) quadrant and rats were tested in a 4-trial paradigm, similar to that described above. For this reverse-learning task, distance and time spent swimming in each quadrant were recorded and analyzed.
Electrophysiology
Chronically stressed (n = 5) and control (n = 5) rats (350–400 g) were anesthetized with sodium pentobarbital (60 mg/kg, i.p., supplemented when necessary throughout the experiment) and placed in a stereotaxic frame. Rectal temperature was maintained at 37°C by a homeothermic warming blanket. Experimental procedures for implantation and recording extracellular field potentials in the prelimbic area (PL) of the PFC have been described previously (Rocher et al., 2004). Briefly, recording electrodes were placed in the PL (coordinates, 3.3 mm anterior to bregma, 0.8 mm lateral to the midline, 3.0–3.8 mm below cortical surface) and a concentric bipolar stainless-steel stimulating electrode was positioned into the ipsilateral CA1/subicular region of the ventral hippocampus (coordinates, 6.3–6.5 mm posterior to bregma, 5.5 mm lateral to the midline, 4.9–6.0 mm below cortical surface), according to the atlas of Paxinos and Watson (1998). Stimulation of the CA1/subicular region induces a characteristic monosynaptic negative going field postsynaptic potential (PSP) in the PFC with a peak latency of 18–22 ms (Rocher et al., 2004). Test pulses (100 μs) were delivered every 30 s at an intensity that evokes a response of 70% of its maximum (range, 250–500 μA). At this intensity, the field potential is most likely to reflect summated PSPs. High-frequency stimulation (HFS) to induce LTP consisted of two series of 10 trains (250 Hz, 200 ms) at 0.1 Hz, 6 min apart, delivered at test intensity. PSP amplitudes were analyzed using A/Dvance software (Fine Science Instruments, Vancouver, Canada), expressed as a percentage change of the mean response over a 30 min baseline period, and presented in figures as the mean ± SEM for 2 min epochs.
Histological procedures
Five controls and five chronically stressed rats were perfused transcardially with fixative (4% paraformaldehyde) under deep pentobarbital anesthesia. Brains were removed and placed in fixative, whereas excised thymi were maintained on saline-soaked filter papers until being weighed. After ∼4 weeks in fixative, brains were split into two hemispheres by a midsagittal section and processed for stereology, according to the procedure described previously by Keuker et al. (2001). Briefly, brain hemispheres were embedded in glycolmethacrylate (Tecnovit 7100; Heraeus Kulzer, Werheim, Germany) and every other microtome-cut section (30 μm) was then collected on a noncoated glass slide, stained with Giemsa, and mounted with Entellan New (Merck, Darmstadt, Germany). The shrinkage factor was calculated, as described previously (Cerqueira et al., 2005), as 1.07 for both controls and stressed rats.
Region and layer boundaries.
We analyzed the three areas of the medial PFC (mPFC) [cingulate (Cg), PL, and infralimbic (IL) cortices] and the subiculum. These regions were outlined according to the atlas of Paxinos and Watson (1998), based on clear cytoarchitectural differences (Vogt et al., 2004; Witter and Amaral, 2004). Each mPFC region was further divided, parallel to the surface, into three easily recognizable levels (layer I, layer II, and layers III–VI) based on cell packing. The third level was considered as a whole because a clear boundary between its layers could not be easily discerned in the mPFC, especially in the more ventral regions. The nomenclature of Zilles and Wree (1995) of mPFC areas was adopted for easier reference to the most widely used atlases and previously reported data.
Stereological procedures.
Volume and neuronal number estimations were performed using StereoInvestigator software (Microbrightfield, Williston, VT) and a camera (DXC-390; Sony, Tokyo, Japan) attached to a motorized microscope (Axioplan 2; Carl Zeiss, Oberkochen, Germany). Cavalieri's principle (Gundersen et al., 1988) was used to assess the volume of each region. Briefly, every eighth (for IL and PL), 12th (for Cg), and 20th (for subiculum) section was used and its cross-sectional area was estimated by point counting at a final magnification ×112. For this we randomly superimposed onto each area a test point grid in which the interpoint distance, at tissue level, was (1) 75 μm for IL levels 1–2, (2) 100 μm for IL level 3 and PL levels 1–2, (3) 150 μm for PL level 3 and levels 1–2 of Cg, and (4) 250 μm for level 3 of Cg and the two layers of the subiculum. The volume of the region of interest was calculated from the number of points that fell within its boundaries and the distance between the systematically sampled sections.
Average cell numbers were estimated using the optical fractionator method, described previously (West et al., 1991). Briefly, the following sampling scheme was used: (1) every eighth, 12th, or 20th section, depending on the region being analyzed (see previous paragraph), was measured; (2) beginning at a random starting position, a grid of virtual three-dimensional boxes (30 × 30 × 15 μm) that were equally spaced (same grid as for the volume estimations) was superimposed within the predefined borders; and (3) neurons were counted whenever their nucleus came into focus within the counting box. Neurons were differentiated from other cells on the basis of nuclear size (larger in neurons than in glia cells), a prominent nucleolus, and the shape of their perikarya caused by dendritic emergence (Peinado et al., 1997).
Coefficients of error were automatically computed according to the formulas of Gundersen et al. (1999) and were all within the optimal range (<0.10).
Statistical analysis
All results are expressed as group means ± SE. Body weight gain, thymus to body weight ratio, and corticosterone levels were compared between groups using Student's t test. To account for multiple comparisons, working memory and reference memory task performances were analyzed using repeated-measures ANOVA on the average results of each trial or day, respectively, and reversal learning task results were analyzed using multivariate ANOVA. Electrophysiological data were averaged in consecutive 30 min periods (t0–30 min; t30–60 min; t60–90 min; t90–120 min) after LTP induction and analyzed using repeated-measures ANOVA. Student's t test was then used to discriminate the effects of treatment for each 30 min period. Stereological measurements were analyzed using Mann–Whitney nonparametric test. In all cases, differences between groups were considered to be significant if p < 0.05.
Results
Biometric parameters and hormonal determinations
The CUS protocol decreased body-weight gain (CON, 96.1 ± 3.1 g; CUS, 79.3 ± 2.4 g; Student's t test, t = 4.264, p < 0.001) and reduced the thymus/body-weight ratio (CON, 1.03 ± 0.090 × 10−3; CUS, 0.773 ± 0.082 × 10−3; t = 2.106; p = 0.049). Exposure to chronic stress resulted also in persistently raised plasma corticosterone levels, which were still significantly higher than in controls >12 h after the last stress exposure (CON, 31 ± 6.0 ng/ml; CUS, 117 ± 11 ng/ml; t = 6.801; p < 0.001).
Behavioral data
Analysis of the learning curves revealed that chronic stress impaired the acquisition of both the working and reference memory tasks, inducing higher escape latencies (working memory, F(1,18) = 20.8, p < 0.001; reference memory, F(1,18) = 21.2, p < 0.001) and distances swam (working memory, F(1,18) = 18.7, p < 0.001; reference memory, F(1,18) = 7.2, p = 0.015) (Fig. 1A,B). However, performance between the groups did not differ on the last day of the reference memory task (p > 0.05) (Fig. 1B). Stress was also deleterious to behavioral flexibility, because it reduced the distance swam and time spent on the quadrant of the platform (new quadrant) at the cost of distance swam and time spent on the previous location of the platform (old quadrant) in the reversal learning test (distance, F(3,16) = 4.6, p = 0.016; time, F(3,16) = 3.5, p = 0.03) (Fig. 1C).
Figure 1.
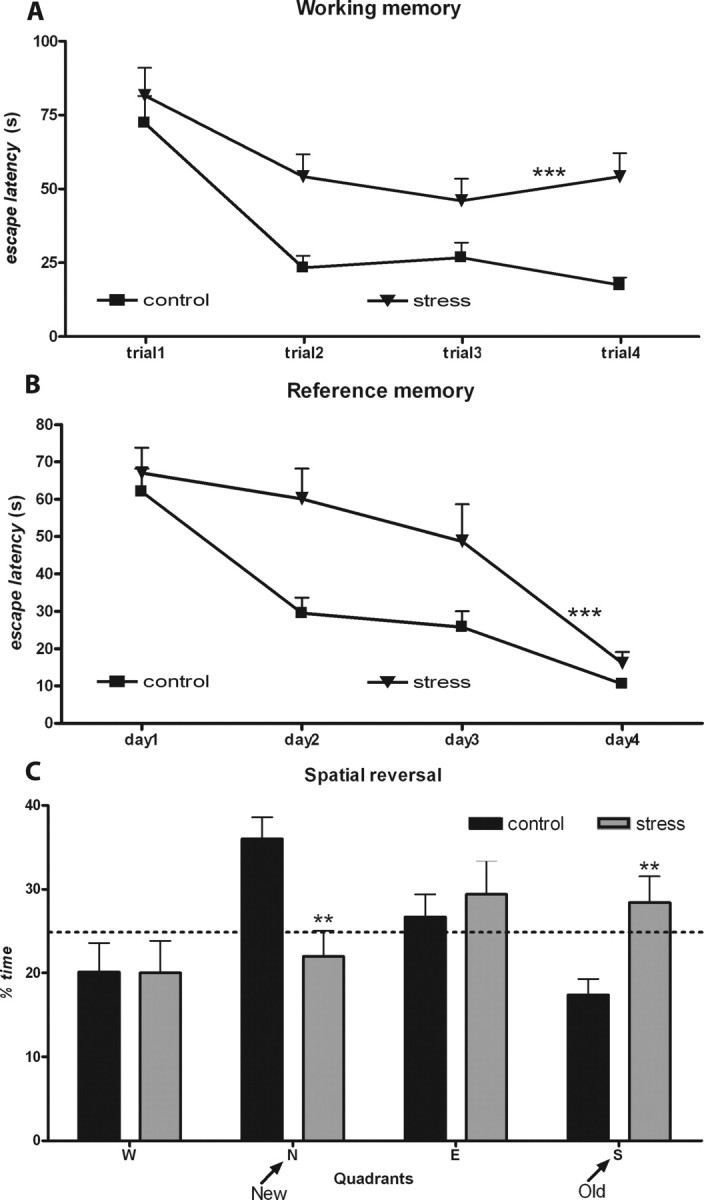
Behavioral impairments induced by chronic stress. A, B, Learning curves in the working (A) and reference memory task (B) of control (n = 10) and chronically stressed (n = 10) rats. The higher escape latencies of the stressed animals are easily appreciated. C, Results from the reverse task experiment. Average time spent on the four trials in each imaginary quadrant is given as a percentage of the total escape latency. Dotted line represents performance at the chance level (25%). Results of distance swam are not presented. **p < 0.01 and ***p < 0.001 compared with controls. Error bars represent SEM.
Exposure to 3 d of stress was deleterious to spatial reference memory, resulting in higher escape latencies (F(1,18) = 5.78; p = 0.027) and distances (F(1,18) = 7.91; p = 0.012), but induced a milder impairment on the spatial working memory task that was statistically significant only in the analysis of swam distances (latencies, F(1,18) = 4.06, p = 0.059; distances, F(1,18) = 7.31, p = 0.014) (Fig. 2A,C), without affecting behavioral flexibility (latencies, F(3,16) = 0.083, p = 0.968; distances, F(4,15) = 0.54, p = 0.711) (Fig. 2E). On the contrary, unpredictable stress for 6 d was deleterious to all behavioral functions tested. On the spatial reference and working memory tasks, stressed rats displayed higher escape latencies (reference memory, F(1,18) = 10.6, p = 0.004; working memory, F(1,18) = 13.1, p = 0.002) and distances swam (reference memory, F(1,18) = 15.96, p < 0.001; working memory, F(1,18) = 8.26, p = 0.01) (Fig. 2B,D). On the behavioral flexibility task, stressed animals spent less time and swam less in the quadrant of the platform (new quadrant) at the cost of distance swam and time spent on the previous location of the platform (old quadrant) (distance, F(4,15) = 3.43, p = 0.035; time: F(4,15) = 3.72, p = 0.027) (Fig. 2F).
Figure 2.

Behavioral tests after 3 and 6 d of unpredictable stress. A–D, Learning curves in the working (A, B) and reference memory task (C, D) of control (n = 10) and stressed (n = 10) rats. E, F, Results from the reverse task experiment. Average time spent on the four trials in each imaginary quadrant is given as a percentage of the total escape latency. Dotted line represents performance at the chance level (25%). Graphs on the left refer to animals stressed for 3 d, and those on the right refer to 6 d of stress. Results of distance swam are not presented. *p < 0.05 and **p < 0.01 compared with controls. Error bars represent SEM.
Electrophysiology
In CON animals, HFS of the CA1/subiculum region induced a lasting increase in the amplitude of the PSP in the ipsilateral PFC (for change in PSP amplitude recorded between baseline and post-HFS period, see Fig. 3, top; for time course of normalized PSP, see Fig. 3, middle). Exposure to CUS significantly and robustly impaired LTP in the PFC (F(1,8) = 18.9; p < 0.01) for the whole 120 min of recording after HFS (Fig. 3, bottom) (CUS-treated rats vs control rats, t0–30 min, 126 ± 7% vs 159 ± 8%, p = 0.015; t30–60 min, 121 ± 3% vs 158 ± 10%, p = 0.007; t60–90 min, 115 ± 5% vs 150 ± 9%, p = 0.011; t90–120 min, 117 ± 6% vs 150 ± 9%, p = 0.015).
Figure 3.
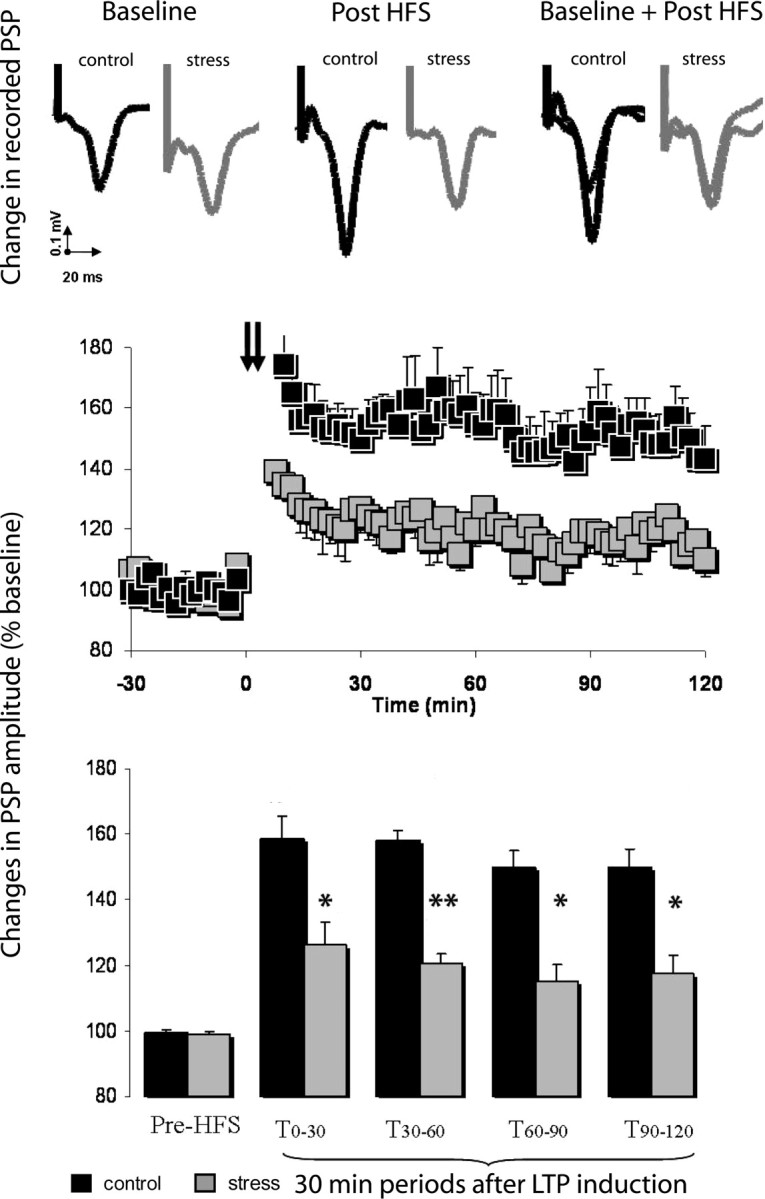
Effects of chronic stress on hippocampal-prefrontal cortex LTP. Top, Representative PSP recorded from the prefrontal cortex of chronic-stressed (n = 5) and control (n = 5) animals before and after hippocampal HFS. Middle, Chronically stressed rats displayed a deficit in HFS-induced LTP when compared with controls. Squares are mean ± SEM of the normalized PSP amplitude for 2 min periods. Hippocampal HFS is indicated by arrows. Bottom, LTP in chronically stressed rats and controls represented at different time periods. The first and following pairs of columns represent mean ± SEM of the average normalized PSP amplitude in consecutive 30 min periods before and after HFS, respectively. *p < 0.05 and **p < 0.01 compared with controls. Error bars represent SEM.
Volumes and neuronal numbers
Layers I and II of all regions of the medial PFC were significantly smaller in CUS-treated rats (cingulate cortex, p = 0.009 and p = 0.040; prelimbic cortex, p = 0.015 and p = 0.002; infralimbic cortex, p = 0.010 and p = 0.028, respectively), although layer III–VI volumes were unaffected (Fig. 4A); the reduced volumes were not accompanied by changes in the number of neurons (Fig. 4B). Volumes of the pyramidal and molecular layer of the subiculum as well as the number of neurons in the pyramidal layer did not differ between groups (Fig. 4B).
Figure 4.

Effects of chronic stress on the volumes and number of neurons of the different mPFC areas and subiculum. A, Average volumes of layers I, II, and III–VI of the IL, PL, and Cg regions of the mPFC and the molecular (Mol) and pyramidal (Pyr) layers of the subiculum. B, Estimated number of neurons in layers II and III–VI of the IL, PL, and Cg regions and the Pyr layer of the subiculum. Note the different scales used and that with the exception of the numerical values for layers I and II, which should be read off against the left axis, all other values should be read off against the right one. Control animals, n = 5; stress rats, n = 5. *p < 0.05 and **p < 0.01 compared with CON. Error bars represent SEM.
Exposure to 3 or 6 d of unpredictable stress did not influence the volume or the number of neurons in any of the analyzed areas (Fig. 5).
Figure 5.

Volumes and number of neurons of the different mPFC areas and subiculum after 3 and 6 d of unpredictable stress. A, B, Average volumes of layers I, II, and III–VI of the IL, PL, and Cg regions of the mPFC and the molecular (Mol) and pyramidal (Pyr) layers of the subiculum. C, D, Estimated number of neurons in layers II and III–VI of the IL, PL, and Cg regions and the Pyr layer of the subiculum. Graphs on the left refer to animals stressed for 3 d, and those on the right refer to 6 d of stress. Control animals, n = 5; stressed rats, n = 5. Error bars represent SEM.
Discussion
This study shows that chronic stress severely disrupts two key processes attributed to the PFC: working memory and behavioral flexibility; these behavioral deficits closely correlated with impaired plasticity at hippocampus-to-PFC synapses, which was accompanied by a selective reduction in the volume of the upper prefrontal layers (I and II). Chronic stress is known to influence several cognitive processes, including spatial reference (Gouirand and Matuszewich, 2005) and working memory (Mizoguchi et al., 2000). Using a water-maze task, we here confirmed previous findings that chronic stress impairs working memory and further, showed damages in behavioral flexibility, another PFC-dependent function. Behavioral flexibility refers to the ability to adapt behavior to a change in environmental circumstances (de Bruin et al., 1994), thereby facilitating survival. Although first described as a PFC-specific task in humans (Verin et al., 1993), impairments in behavioral flexibility have been demonstrated in primates and rodents bearing specific lesions of the PFC (de Bruin et al., 1994). In humans, the prototypic test for behavioral flexibility is the Wisconsin-card-sorting test, in which the subject has to sort a deck of cards according to rules that change unpredictably throughout the test (Goldberg and Bougakov, 2005). Here, behavioral flexibility was assessed in rats by monitoring performance in a reversal learning task in which the animal has to adapt previously learned behavior to a new set of “rules” (de Bruin et al., 1994). Because previous studies showed that chronic high-dose corticosterone administration impairs performance in reverse learning without affecting working memory (Cerqueira et al., 2005), stress-induced deficits in working memory can obviously not be attributed solely to the increased corticosteroid secretion that accompanies stress. It is therefore likely that other mediators of the stress response, including among others CRH and vasopressin (AVP), are involved in the herein observed stress induced impairments. CRH and AVP are peptides released as part of the stress response, that have receptors in several regions of the brain, including the hippocampus and the PFC (Szot et al., 1994; Herman et al., 2003); importantly, their release influences brain function and structure (Bayatti and Behl, 2005; Landgraf, 2006).
The mPFC receives innervation from the hippocampus. Hippocampal afferents, originating in the pyramidal cells of the subiculum and ventral CA1 regions, travel through the fimbria-fornix system and terminate in the mPFC, where they establish glutamatergic contacts with both pyramidal cells and interneurons (Jay et al., 1992; Jay and Witter, 1995; Tierney et al., 2004). Therefore, the occurrence and strength of LTP in the hippocampus-to-PFC pathway reflects synaptic plasticity in the PFC (Jay et al., 1992). Although it can be argued that deficits in the hippocampus-to-PFC projections may be solely the consequence of damage to the projecting areas, there are several reasons why this is an unlikely explanation for the present results. First, although chronic stress can impose structural damage to particular hippocampal subfields (Sousa et al., 2000; McEwen, 2001), it does not affect the volume and number of neurons in the subiculum (present study) and CA1 (Sousa et al., 1998). Second, identical stimulation intensities produced similar baseline PSPs (waveform and amplitude) in stressed and control animals, which indicates the intactness of both the projecting area and connecting pathway. Finally, LTP induction in the hippocampus-to-PFC pathway has also been shown to be impaired after a single episode of acute stress (Rocher et al., 2004), a condition which impairs working memory while facilitating hippocampus-dependent reference memory storage (Shors, 2004). However, although no gross structural changes were observed in the CA1 and subiculum, chronic stress imposes functional and structural damage to other hippocampal subfields (Sousa et al., 2000; McEwen, 2001) that may result in functional alterations of the main projecting areas. As an example, an altered glutamate release in the PFC was observed after stress exposure (Bagley and Moghaddam, 1997); taking into consideration that the hippocampal-to-PFC projection is glutamatergic, it is admissible that these findings are associated.
In the present study, we also determined the sequential pattern of stress-induced behavioral changes. Whereas reference memory and, to a lesser extent, working memory were impaired after 3 d of stress, behavioral flexibility was still unaffected. Interestingly, after 6 d of stress exposure, the full spectrum of behavioral deficits was already present. This sequential pattern of deficits demonstrates that stress-induced functional deficits propagate from a hippocampus-dependent task to a PFC-dependent task. This observation, together with the fact that the volumetric reductions observed in the mPFC were specifically confined to the upper layers, where most hippocampal projections terminate (Jay and Witter, 1995), strongly implicates the connection between both structures in the genesis of stress-induced PFC damage.
Interestingly, the observed PFC structural changes correlated closely with both the behavioral and the synaptic plasticity impairments discussed above. Because the volumetric reductions were not accountable for by neuronal loss, it is highly plausible that they reflect dendritic atrophy/retraction. Indeed, previous work has shown that chronic stress induces profound atrophy and remodeling of the apical, but not the basal, dendrites of pyramidal neurons of layers II/III of the PFC, changes which result in reduction of average spine density of the more superficial part of the dendritic tree (Wellman, 2001; Radley et al., 2004; Brown et al., 2005; Cerqueira et al., 2007). Moreover, a previous study (Radley et al., 2005) demonstrated that this stress-induced atrophy of mPFC apical dendrites is reversible.
In summary, the present study identifies the structural and electrophysiological changes that underpin stress-induced impairments in PFC function. These results reinforce the view that synaptic adaptations, at both the structural and physiological levels, are pivotal in mediating stress effects in the brain. Importantly, our results highlight the fact that stress can influence the integrity of the hippocampus-to-PFC pathway, thereby helping to explain some of the neurological deficits triggered by stress that cannot be attributed to hippocampal lesions. These observations may help the design of experimental approaches aimed at exploiting synaptic reinforcement as a means for ameliorating the impact of stress on brain dysfunction.
Footnotes
This work was supported in part by an Acções Integradas Luso-Alemãs grant from the German Academic Exchange Service and the Portuguese Rectors' Conference, and Mobility Grant 183/2006 from the Institut National de la Santé et de la Recherche Médicale and the Cabinet for International Relations of the Portuguese Ministry of Science and Higher Education.
References
- Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience. 1997;77:65–73. doi: 10.1016/s0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- Bayatti N, Behl C. The neuroprotective actions of corticotropin releasing hormone. Ageing Res Rev. 2005;4:258–270. doi: 10.1016/j.arr.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Brown SM, Henning S, Wellman CL. Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex. 2005;15:1714–1722. doi: 10.1093/cercor/bhi048. [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Pêgo JM, Taipa R, Bessa-Peixoto JM, Almeida OFX, Sousa N. Morphological correlates of corticosteroid-induced changes in prefrontal cortex-dependent behaviours. J Neurosci. 2005;25:7792–7800. doi: 10.1523/JNEUROSCI.1598-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira JJ, Taipa R, Uylings HBM, Almeida OFX, Sousa N. Specific configuration of dendritic degeneration in pyramidal neurons of the medial prefrontal cortex induced by differing corticosteroid regimens. Cereb Cortex. 2007 doi: 10.1093/cercor/bhl108. in press. [DOI] [PubMed] [Google Scholar]
- Clark L, Cools R, Robbins TW. The neuropsychology of ventral prefrontal cortex: decision-making and reversal learning. Brain Cogn. 2004;55:41–53. doi: 10.1016/S0278-2626(03)00284-7. [DOI] [PubMed] [Google Scholar]
- Davidson RJ. Anxiety and affective style: role of prefrontal cortex and amygdala. Biol Psychiatry. 2002;51:68–80. doi: 10.1016/s0006-3223(01)01328-2. [DOI] [PubMed] [Google Scholar]
- de Bruin JP, Sànchez-Santed F, Heinsbroek RP, Donker A, Postmes P. A behavioral analysis of rats with damage to the medial prefrontal cortex using the Morris water maze: evidence for behavioral flexibility, but not for impaired spatial navigation. Brain Res. 1994;652:323–333. doi: 10.1016/0006-8993(94)90243-7. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Doyère V, Burette F, Negro CR, Laroche S. Long-term potentiation of hippocampal afferents and efferents to prefrontal cortex: implications for associative learning. Neuropsychologia. 1993;31:1031–1053. doi: 10.1016/0028-3932(93)90031-t. [DOI] [PubMed] [Google Scholar]
- Goldberg E, Bougakov D. Neuropsychologic assessment of frontal lobe dysfunction. Psychiatr Clin North Am. 2005;28:567–580. doi: 10.1016/j.psc.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Architecture of the prefrontal cortex and the central executive. Ann NY Acad Sci. 1995;769:71–83. doi: 10.1111/j.1749-6632.1995.tb38132.x. [DOI] [PubMed] [Google Scholar]
- Gouirand AM, Matuszewich L. The effects of chronic unpredictable stress on male rats in the water maze. Physiol Behav. 2005;86:21–31. doi: 10.1016/j.physbeh.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology-reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo–pituitary–adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1995;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Jay TM, Thierry AM, Wiklund L, Glowinski J. Excitatory amino acid pathway from the hippocampus to the prefrontal cortex. Contribution of AMPA receptors in hippocampo-prefrontal cortex transmission. Eur J Neurosci. 1992;4:1285–1295. doi: 10.1111/j.1460-9568.1992.tb00154.x. [DOI] [PubMed] [Google Scholar]
- Kesner RP. Subregional analysis of mnemonic functions of the prefrontal cortex in the rat. Psychobiology. 2000;28:219–228. [Google Scholar]
- Keuker JI, Vollmann-Honsdorf GK, Fuchs E. How to use the optical fractionator: an example based on the estimation of neurons in the hippocampal CA1 and CA3 regions of tree shrews. Brain Res Brain Res Protoc. 2001;7:211–221. doi: 10.1016/s1385-299x(01)00064-2. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Landgraf R. The involvement of the vasopressin system in stress-related disorders. CNS Neurol Disord Drug Targets. 2006;5:167–179. doi: 10.2174/187152706776359664. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann NY Acad Sci. 2001;933:265–277. doi: 10.1111/j.1749-6632.2001.tb05830.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann NY Acad Sci. 2004;1032:1–7. doi: 10.1196/annals.1314.001. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stressed or stressed out: what is the difference? J Psychiatry Neurosci. 2005;30:315–318. [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui DH, Tabira T. Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J Neurosci. 2000;20:1568–1574. doi: 10.1523/JNEUROSCI.20-04-01568.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Peinado MA, Quesada A, Pedrosa JA, Martinez M, Esteban FJ, Del Moral ML, Peinado JM. Light microscopic quantification of morphological changes during aging in neurons and glia of the rat parietal cortex. Anat Rec. 1997;247:420–425. doi: 10.1002/(SICI)1097-0185(199703)247:3<420::AID-AR14>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Sisti HM, Hao J, Rocher AB, McCall T, Hof PR, McEwen BS, Morrison JH. Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex. Neuroscience. 2004;125:1–6. doi: 10.1016/j.neuroscience.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Janssen WG, Hof PR, McEwen BS, Morrison JH. Reversibility of apical dendritic retraction in the rat medial prefrontal cortex following repeated stress. Exp Neurol. 2005;196:199–203. doi: 10.1016/j.expneurol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Rocher C, Spedding M, Munoz C, Jay TM. Acute stress-induced changes in hippocampal/prefrontal circuits in rats: effects of antidepressants. Cereb Cortex. 2004;14:224–229. doi: 10.1093/cercor/bhg122. [DOI] [PubMed] [Google Scholar]
- Shors TJ. Learning during stressful times. Learn Mem. 2004;11:137–144. doi: 10.1101/lm.66604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa N, Almeida OF. Corticosteroids: sculptors of the hippocampal formation. Rev Neurosci. 2002;13:59–84. doi: 10.1515/revneuro.2002.13.1.59. [DOI] [PubMed] [Google Scholar]
- Sousa N, Almeida OF, Holsboer F, Paula-Barbosa MM, Madeira MD. Maintenance of hippocampal cell numbers in young and aged rats submitted to chronic unpredictable stress. Comparison with the effects of corticosterone treatment. Stress. 1998;2:237–249. doi: 10.3109/10253899809167288. [DOI] [PubMed] [Google Scholar]
- Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Gratton A. Prefrontal cortical regulation of hypothalamic-pituitary-adrenal function in the rat and implications for psychopathology: side matters. Psychoneuroendocrinology. 2002;27:99–114. doi: 10.1016/s0306-4530(01)00038-5. [DOI] [PubMed] [Google Scholar]
- Szot P, Bale TL, Dorsa DM. Distribution of messenger RNA for the vasopressin V1a receptor in the CNS of male and female rats. Brain Res Mol Brain Res. 1994;24:1–10. doi: 10.1016/0169-328x(94)90111-2. [DOI] [PubMed] [Google Scholar]
- Tierney PL, Degenetais E, Thierry AM, Glowinski J, Gioanni Y. Influence of the hippocampus on interneurons of the rat prefrontal cortex. Eur J Neurosci. 2004;20:514–524. doi: 10.1111/j.1460-9568.2004.03501.x. [DOI] [PubMed] [Google Scholar]
- Verin M, Partiot A, Pillon B, Malapani C, Agid Y, Dubois B. Delayed response tasks and prefrontal lesions in man: evidence for self generated patterns of behaviour with poor environmental modulation. Neuropsychologia. 1993;31:1379–1396. doi: 10.1016/0028-3932(93)90105-9. [DOI] [PubMed] [Google Scholar]
- Vogt BA, Vogt L, Farber N. Cingulate cortex and disease models. In: Paxinos G, editor. The rat nervous system. Ed 3. San Diego: Academic; 2004. pp. 705–727. [Google Scholar]
- Wall PM, Messier C. The hippocampal formation: orbitomedial prefrontal cortex circuit in the attentional control of active memory. Behav Brain Res. 2001;127:99–117. doi: 10.1016/s0166-4328(01)00355-2. [DOI] [PubMed] [Google Scholar]
- Wellman CL. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol. 2001;49:245–253. doi: 10.1002/neu.1079. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Witter MP, Amaral DG. Hippocampal formation. In: Paxinos G, editor. The rat nervous system. Ed 3. San Diego: Academic; 2004. pp. 729–757. [Google Scholar]
- Zilles K, Wree A. Cortex: areal and laminar structure. In: Paxinos G, editor. The rat nervous system. Ed 2. San Diego: Academic; 1995. pp. 649–685. [Google Scholar]