

Abstract
Alzheimer's disease (AD) neuropathology is characterized by the accumulation of phosphorylated tau and amyloid-β peptides derived from the amyloid precursor protein (APP). Elevated blood levels of homocysteine are a significant risk factor for many age-related diseases, including AD. Impaired homocysteine metabolism favors the formation of S-adenosylhomocysteine, leading to inhibition of methyltransferase-dependent reactions. Here, we show that incubation of neuroblastoma cells with S-adenosylhomocysteine results in reduced methylation of protein phosphatase 2A (PP2A), a major brain Ser/Thr phosphatase, most likely by inhibiting PP2A methyltransferase (PPMT). PP2A methylation levels are also decreased after ectopic expression of PP2A methylesterase in Neuro-2a (N2a) cells. Reduced PP2A methylation promotes the downregulation of Bα-containing holoenzymes, thereby affecting PP2A substrate specificity. It is associated with the accumulation of both phosphorylated tau and APP isoforms and increased secretion of β-secretase-cleaved APP fragments and amyloid-β peptides. Conversely, incubation of N2a cells with S-adenosylmethionine and expression of PPMT enhance PP2A methylation. This leads to the accumulation of dephosphorylated tau and APP species and increased secretion of neuroprotective α-secretase-cleaved APP fragments. Remarkably, hyperhomocysteinemia induced in wild-type and cystathionine-β-synthase +/− mice by feeding a high-methionine, low-folate diet is associated with increased brain S-adenosylhomocysteine levels, PPMT downregulation, reduced PP2A methylation levels, and tau and APP phosphorylation. We reported previously that downregulation of neuronal PPMT and PP2A methylation occur in affected brain regions from AD patients. The link between homocysteine, PPMT, PP2A methylation, and key CNS proteins involved in AD pathogenesis provides new mechanistic insights into this disorder.
Keywords: Alzheimer's disease, cystathionine-β-synthase, folate, homocysteine, methylation, PP2A
Introduction
Filamentous lesions containing phosphorylated tau (Brandt et al., 2005) and amyloid-β (Aβ) plaques (Vassar, 2005) accumulate in Alzheimer's disease (AD)-affected brain regions. Alterations in tau phosphorylation affect its properties and microtubule regulatory functions (Brandt et al., 2005). In the amyloidogenic pathway, cleavage of the amyloid precursor protein (APP) by β-secretase generates an N-terminal soluble fragment (sAPPβ) and a C-terminal fragment (β-CTF) that is sequentially cleaved by γ-secretase to produce Aβ peptides (Vassar, 2005). In the alternate non-amyloidogenic pathway, APP is processed by α-secretase within the Aβ region, thereby precluding Aβ formation and favoring the release of neuroprotective sAPPα fragments (Kojro and Fahrenholz, 2005). Notably, phosphorylation of APP on Thr-668 facilitates its amyloidogenic cleavage and is elevated in AD brain (Ando et al., 2001; Lee et al., 2003; Pierrot et al., 2006). Elevated plasma levels of homocysteine (Hcy) has emerged as a significant risk factor for many age-related diseases, including AD and dementia (Seshadri et al., 2002; Morris, 2003; Irizarry et al., 2005; Ravaglia et al., 2005). Genetic polymorphisms in key enzymes regulating Hcy metabolism and dietary deficiencies in folate and vitamin B12 lead to elevated Hcy levels (Fowler, 2005). Hcy is a critical branch point metabolite that can influence cellular levels of S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH), which regulate the activity of methyltransferases important in posttranslational modification of proteins and synthesis of nucleic acids, phospholipids, and neurotransmitters (Fowler, 2005).
A candidate methylation-regulated enzyme postulated to be involved in AD pathogenesis is protein phosphatase 2A (PP2A) (Vafai and Stock, 2002), a family of heterotrimeric enzymes (Sontag, 2001). PP2A catalytic C subunit can be methylated on the conserved Leu-309 residue by a specific methyltransferase (PPMT) (De et al., 1999), and demethylated by a dedicated methylesterase (PME-1) (Ogris et al., 1999). Current studies support the hypothesis that methylation does not directly affect basal PP2A catalytic activity but alters PP2A substrate specificity by inducing changes in its subunit composition (Tolstykh et al., 2000; Yu et al., 2001). Methylation is required for efficient assembly of Bα-containing heterotrimers (Tolstykh et al., 2000; Yu et al., 2001), which are the major PP2A enzymes that bind to and dephosphorylate tau (Sontag et al., 1996). Significantly, PPMT expression and PP2A methylation become downregulated in AD (Sontag et al., 2004b), yet the cause for this deregulation and contribution of PP2A methylation/demethylation cycles in neuronal regulation are unknown. Like other methyltransferases, purified PPMT activity is dependent on SAM and inhibited by SAH (IC50 of 3 μm) (De et al., 1999), making PPMT a susceptible target for disturbances in Hcy metabolism. We show here that elevated SAH levels result in decreased PP2A methylation. Reduced PP2A methylation is associated with Bα subunit downregulation, accumulation of phosphorylated tau and APP species, reduced sAPPα secretion, and increased sAPPβ and Aβ production. Our findings establish PPMT and PP2A methylation as critical intermediates linking metabolic risk factors with the deregulation of key CNS proteins involved in AD pathogenesis.
Materials and Methods
Cell culture and transfection.
Mouse Neuro-2a (N2a) neuroblastoma cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (HyClone, Logan, UT). Cells were transfected using Metafectene following the instructions of the manufacturer (Biontex Laboratories, Munich, Germany) with the following plasmids: pcDNA3.1 (Invitrogen); pSG5 encoding human tau (Goedert et al., 2000); Rc/cytomegalovirus-small tumor antigen (small t) (Sontag et al., 1996); pcDNA 3.1-expressing the wild-type (Wt) C (Goedert et al., 2000) and the L309Δ C mutant; pBabe encoding hemagglutinin (HA)-tagged mouse PPMT; and pBabe encoding Myc-tagged mouse PME-1. The pcDNA 3.1 construct encoding the L309Δ C mutant was generated after subcloning from the corresponding pGRE5–2–L309Δ plasmid (Yu et al., 2001). The coding region of mouse PPMT was amplified by reverse transcription (RT)-PCR using RNA of NIH 3T3 cells as template. The amplified cDNA was then introduced into pBabe–hygromycin (Morgenstern and Land, 1990). The open reading frame of mouse PME-1 cDNA was cloned into the BamHI/XhoI polylinker sites of the pBabe–puromycin vector (Morgenstern and Land, 1990). All plasmids were verified by sequencing. Stable PP2A and small t-expressing clones were selected with 600 μg/ml G418 (Invitrogen). N2a–PPMT clones were generated after selection with 200 μg/ml hygromycin (Roche, Indianapolis, IN). N2a–PME-1 clones were generated after selection with 1 μg/ml puromycin (Sigma, St. Louis, MO). The expression levels of transfected proteins were constantly monitored by both immunoblotting and immunofluorescence. At least four distinct stable clones were used throughout our studies with similar results. No differences in cell viability was observed among the selected clones, and stable cell lines could be propagated for extended periods of time under normal culture conditions. Cells stably mock transfected with empty vectors were used as “controls” and behaved like nontransfected cells in our experiments.
Cell treatment and analysis.
Treatment with SAM or SAH (Sigma) was performed in exponentially growing N2a cells 24 h after plating in regular medium or transfection. Duplicate sets of dishes were treated for 16 h with the indicated concentrations of SAM or SAH, or vehicle alone. When indicated, cells were simultaneously treated with 10 nm okadaic acid (OA) (LC Laboratories, Woburn, MA). After treatment, cells were quickly washed with PBS. One set of cells was harvested in 0.4 m perchloric acid for additional SAM/SAH analyses. The other set was harvested for immunoblotting studies, by scraping on ice in buffer A [Tris 25 mm, pH 7.4, 150 mm NaCl, 2 mm EDTA, 25 μm sodium fluoride, 1 mm sodium orthovanadate, 2 mm dithiothreitol, 2 μm OA, 5 mm PMSF, and 1% NP-40, containing a mixture of protease inhibitors (Completemini; Roche)] (Sontag et al., 2004b). The samples were briefly sonicated and centrifuged for 10 min at 14,000 × g to remove insoluble material. In experiments looking at tau and APP phosphorylation, cells were serum starved for 24 h in DMEM containing 1% dialyzed fetal bovine serum (HyClone). When indicated, cells were treated for 1 h with the indicated concentrations of OA before being harvested in buffer A as described above. Protein concentrations were determined in diluted cell extracts using the Bio-Rad (Hercules, CA) protein assay kit. Equivalent amounts of proteins (∼60 μg) were resolved by SDS-PAGE on 7.5% (for APP) or 12% (for all other proteins) polyacrylamide gels (Amersham Biosciences, Piscataway, NJ).
Mouse analyses.
Mice heterozygous for disruption of the cystathionine-β-synthase (Cbs) gene were crossbred to C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) for at least nine generations to generate heterozygous (Cbs+/−) and wild-type (Cbs +/+) littermates. Genotyping was performed by PCR using a common forward primer (5′-GGTCTGGAATTCACTATGTAGC-3′) with reverse primers specific for wild-type (5-AAGAGCCCAGCAGAATGAACA-3′) or targeted (5′-GAGGTCGACGGTATCGATA-3′) Cbs alleles. At the time of weaning, mice were fed either a control diet (LM485; Harlan Teklad, Madison, WI) containing 6.7 mg/kg folic acid and 4.0 mg/kg l-methionine or a high-methionine, low-folate (HM/LF) diet (TD00205; Harlan Teklad) containing 0.2 mg/kg folic acid and 8.2 g/kg l-methionine. Mice anesthetized with sodium pentobarbital underwent thoracic dissection to expose the heart. Blood was removed from the ventricle into EDTA (5 mmol/L) and separated to obtain plasma, which was stored at −20°C. After cervical dislocation, the brain was removed and separated into two halves. The whole right hemisphere was deproteinized with 5 vol of perchloric acid (0.4 m), centrifuged to obtain a clear supernatant extract, and stored at −80°C for additional SAM/SAH analyses. The whole left hemisphere was homogenized on ice at a constant ratio of 1 g tissue/10 ml buffer A, snap frozen in liquid nitrogen, and stored at −80°C for additional Western blot analyses (Sontag et al., 2004b). During thawing, homogenates were sonicated on ice and centrifuged for 2 min at 800 × g to remove insoluble material. Equivalent aliquots (∼14 μl) of the homogenates were analyzed on 4–12% Bis-Tris gels using the Nu-PAGE system (Invitrogen). Note that all of the samples were run, immunoblotted, and exposed simultaneously to allow accurate comparison of protein expression levels across mouse groups.
Western blot analyses.
After gel electrophoresis, proteins were transferred to nitrocellulose membranes. Immunoblotting was performed with the following primary antibodies: anti-HA 16B12 (1:500; Covance Research Products, Berkeley, CA), anti-Myc 4A6 (1:500), anti-demethyl-C 4B7 (1:2000) and anti-methyl-C 2A10 (1:100; Upstate Biotechnology, Lake Placid, NY), anti-Simian virus 40 (SV40) small t (1:20) (Sontag et al., 1996), anti-B55 2G9 (1:100) (Sontag et al., 2004a), anti-Cα (1:1000; BD Biosciences, San Jose, CA), anti-tau PHF-1 (1:2000) (from Dr. Peter Davies, Albert Einstein College of Medicine, Bronx, NY) (Sontag et al., 1996), Tau-5 (1:500; Biosource International, Hopkinton, MA), rabbit anti-PPMT (1:2500) (Sontag et al., 2004b), anti-phospho-APP (Thr-668) and anti-APP (1:1000; Cell Signaling Technology, Beverly, MA); anti-N-terminal APP 22C11 and rabbit-anti-APP, Kunitz protease inhibitor (KPI) domain (1:500; Chemicon, Temecula, CA), rabbit anti-Aβ antibodies (1:250; Lab Vision, Fremont, CA) and anti-sAPPβ antibodies (1:50, from IBL-America, Minneapolis, MN; and 1:500, from Signet, Dedham, MA). The rabbit anti-phosphoepitope-specific anti-tau antibodies (Ser-199, Ser-202, Thr-212, Ser-214, Thr-231, Ser-262, Ser-404, and Ser-422) were obtained from Drs. Erik Schaefer and Lynda Zorn (1:1000; Biosource International). The anti-PME-1 rabbit antibody (1:10,000) was raised against affinity-purified His-tagged full-length mouse PME-1. Blotting with anti-tubulin and/or anti-actin antibodies (1:2000; Sigma) was systematically used to verify and normalize for protein loading. Note that extended electrophoretic conditions were used to assess the relative expression of tagged proteins (HA-tagged PPMT, Myc-tagged PME-1, and HA-tagged C) versus corresponding endogenous proteins (PPMT, PME-1, and C). Blots were developed using the SuperSignal West Pico or Femto chemiluminescence detection system (Pierce, Rockford, IL). Although representative blots are shown, several exposures of the blots were scanned by densitometry using Kodak Image software (Eastman Kodak, Rochester, NY) to ensure that the signals obtained were within the linear range (Sontag et al., 2004b). Protein expression was quantified on blots obtained from at least three separate experiments.
Analysis of PP2A methylation.
To quantify PP2A methylation, electrophoretic conditions were selected to prevent the separation of endogenous and ectopically expressed C subunit bands. Blots were probed with anti-methyl-C or anti-demethyl-C antibodies, stripped, and reprobed with anti-C antibodies to assess total C expression. The relative methylated C expression levels were determined after densitometry scanning of the blots (Sontag et al., 2004b). The specificity of the methyl-dependent antibodies was verified in parallel experiments by treating homogenates with NaOH, which induces complete PP2A demethylation; this resulted in a loss of the signal obtained with anti-methyl-C and enhanced immunoreactivity with anti-demethyl-C antibodies (Sontag et al., 2004b).
Tau analyses.
Total cell extracts were prepared 48 h after transient transfection with pSG5–tau and normalized for total tau levels (Goedert et al., 2000). To study PP2A–tau binding, cell extracts were immunoprecipitated using anti-HA-coupled affinity matrix (Goedert et al., 2000). To analyze tau phosphorylation, total cell or mouse brain extracts were boiled and centrifuged to enrich in tau (Sontag et al., 1996). Equivalent amounts of proteins (∼10 μg) were analyzed by gel electrophoresis and Western blotting for phosphorylated tau using phospho-dependent anti-tau antibodies. The blots were stripped and reprobed with Tau-5 antibody to detect total tau. The relative amounts of phosphorylated tau were determined after densitometry scanning of the blots and normalization for total tau expression levels. Note that tau isoforms migrate as several bands, the pattern of which can be affected by slight variations in electrophoretic conditions.
APP and Aβ analyses.
To assess APP phosphorylation, blots were probed with anti-phospho-APP antibodies, stripped, and reprobed with rabbit anti-APP antibodies (Cell Signaling Technology) to assess total APP expression. The relative amounts of phosphorylated APP were determined after densitometry scanning of the blots and normalization for total APP expression levels. To analyze APP secretion, exponentially growing N2a cells were plated at equivalent density (∼3 × 106 cells/60 mm dish) and grown for 24 h in regular medium. The medium was then replaced with 2 ml of serum-free Opti-MEM (Invitrogen). Conditioned media and cells were collected 1, 4, or 24 h later. Cell media were centrifuged for 5 min at 800 × g to remove debris and concentrated ∼20 times using Microcon YM-30 filter units (Millipore, Billerica, MA). The conditioned cell media were normalized to the protein concentration measured in total cell lysates prepared in buffer A. Secreted proteins were analyzed by SDS-PAGE and immunoblotting with anti-Aβ and anti-sAPPβ (Signet) antibodies. Of note, parallel immunoblotting experiments showed that both anti-sAPPβ antibodies from Signet and IBL-America recognized identical bands but not sAPPα; the anti-Aβ antibody recognized sAPPα but not sAPPβ. Corresponding cell lysates were analyzed in parallel for APP expression. Intracellular APP and total secreted APP forms were detected with anti-APP 22C11 antibodies. KPI domain-containing APP isoforms were detected with anti-KPI antibodies. Aβ40 peptide levels were determined in 10 μl of concentrated conditioned media using the commercial Mouse/rat Amyloid β(1–40)(N) Assay kit from IBL-America and following the instructions of the manufacturer.
PP2A activity assays.
PP2A activity was assayed for 5 min at 30°C in diluted cell extracts using either a synthetic phosphopeptide or phosphorylated tau as a substrate, exactly as described previously (Sontag et al., 1999, 2004a).
Metabolic assays.
Total plasma levels of Hcy (tHcy) was measured using a modified HPLC method with fluorescence detection (Ubbink et al., 1991). Methionine was measured by HPLC after precolumn derivatization with o-phthalaldehyde (Bottiglieri, 1987). SAM and SAH levels were determined by HPLC coupled to UV detection (Bottiglieri, 1990). Plasma folate was determined using a commercially available radioimmunoassay (Diagnostic Products, Los Angeles, CA).
Statistical analysis.
Data are presented as mean values ± SD and were analyzed using the Student's t test. Differences with p values <0.05 were considered statistically significant.
Results
Incubation of N2a cells with SAH is associated with a decrease in PP2A methylation
To first test the hypothesis that PPMT is a target of Hcy metabolism, we assessed SAM and SAH effects on PP2A methylation in mouse N2a neuroblastoma cells (Fig. 1). Cell exposure to exogenous SAM or SAH induced a dose-dependent increase in intracellular levels of each metabolite. Cell incubation with 100 and 200 μm SAM induced an ∼30–38% and ∼34–45% (n = 3) upregulation of PPMT expression, respectively, relative to untreated cells. PP2A methylation increased but only by ∼21–25% in cells treated with 100 μm SAM and ∼24–31% in cells incubated with 200 μm SAM (n = 4), probably because PP2A is already methylated at high levels in mammalian cells (Tolstykh et al., 2000; Sontag et al., 2004b). Treatment with increasing SAH concentrations induced a progressive decrease of the endogenous SAM/SAH ratio and reduced PP2A methylation levels without significantly affecting PPMT expression levels. Notably, methylated C subunit levels were reduced by 39–51% (n = 3) in cells incubated with 100 μm SAH.
Figure 1.

Incubation of N2a cells with SAM and SAH modulates PP2A methylation. A, Representative experiment showing a concomitant increase in endogenous SAM, PPMT expression, and methylated C levels in SAM-treated cells. B, Representative experiment showing a dose-dependent increase in endogenous SAH concentrations in SAH-treated cells. C, D, In these cells, the SAM/SAH ratio (C) and relative levels of methylated C subunits (D) are decreased, but PPMT expression levels (D) are not significantly affected by the short-term SAH treatment. For A–D, similar results were obtained in three separate experiments.
Reduced PP2A methylation is associated with enhanced tau phosphorylation at several epitopes
We next investigated how fluctuations in PP2A methylation levels can influence tau regulation. To this end, we generated and characterized N2a cells stably expressing the HA-tagged L309Δ methylation-site C subunit mutant or its wild-type counterpart (Wt C), HA-tagged PPMT, and Myc-tagged PME-1. C subunit expression levels are tightly autoregulated (Baharians and Schonthal, 1998), so that a portion of endogenous pools was replaced by expressed C subunits. Indeed, HA-tagged C represented ∼40% of total C subunit levels (Fig. 2 A), whereas total C subunit levels increased by ∼30% in selected N2a clones (n = 5) relative to controls (Fig. 2 B). Compared with controls, enhanced C expression in N2a–Wt C clones correlated with a 30–38% (n = 3) increase in PP2A activity measured in total cell extracts using either phosphorylated tau or a synthetic phosphopeptide as substrates. After expression of the methylation-competent HA-tagged Wt C, total intracellular amounts of methylated C and Bα subunits were similarly increased by ∼27–39% (n = 4). Enhanced Bα subunit expression likely results from methylation-induced formation and stabilization of Bα-containing trimeric PP2A complexes (Tolstykh et al., 2000). In contrast, there was a comparable 35–45% reduction of methylated C and Bα subunit levels in clones expressing the methylation-incompetent L309Δ C subunit (n = 5). The inability of L309Δ mutants to efficiently bind to Bα subunit and known instability of Bα monomers (Tolstykh et al., 2000; Yu et al., 2001) probably contribute to the loss of Bα in these clones.
Figure 2.

Expression of the L309Δ C subunit mutant in N2a cells decreases endogenous PP2A methylation and inhibits the ability of PP2A to bind to and dephosphorylate tau. A, Relative expression levels of endogenous and expressed C subunits in stable N2a clones expressing HA-tagged Wt C or the L309Δ C subunit mutant. B, The expression levels of methylated C and Bα subunits are enhanced in N2a–Wt C but decreased in N2a–L309Δ cells. C, D, Cells were transiently transfected with a plasmid encoding human adult tau. C, Total cell extracts and normalized HA-tag immunoprecipitates (IP) were analyzed for the presence of tau using the phosphorylation-independent Tau-5 monoclonal antibody. D, Control and stable N2a cells were analyzed by Western blotting (WB) for total tau using Tau-5 and for phosphorylated tau using a set of phosphoepitope-specific, anti-tau antibodies. A subset of control cells was treated for 1 h with 100 nm OA before harvesting. Representative blots are shown. HA-tagged C expression levels are shown for reference.
Because Bα-containing PP2A trimeric complexes are the most efficient holoenzymes in binding to and dephosphorylating tau (Sontag et al., 1996), we next investigated whether such deregulation of PP2A affects its ability to associate with and dephosphorylate tau. We reported previously that anti-HA antibodies can be successfully used to coimmunoprecipitate tau and Bα-containing PP2A heterotrimers from fibroblasts expressing HA-tagged Wt C and tau (Goedert et al., 2000). Using similar assays in our N2a clones, we found that the amounts of tau proteins that were recovered in HA-tagged L309Δ C subunit immunoprecipitates were greatly reduced compared with those present in HA-tagged Wt C immunoprecipitates (Fig. 2 C). Although expression of Wt C promoted tau dephosphorylation, steady-state tau phosphorylation was enhanced by approximately twofold to threefold in N2a–L309Δ clones at all epitopes examined (Fig. 2 D). Incubation of control cells for 1 h with 100 nm OA induced a marked increase in tau phosphorylation. OA is a potent Ser/Thr phosphatase inhibitor, the effects of which are dose and time dependent (Honkanen and Golden, 2002). Under our experimental conditions, basal PP2A activity was inhibited by ∼50–60%. A similar inhibition of PP2A activity was observed when cells were treated overnight with 10 nm OA. The OA concentrations chosen were insufficient to induce inhibition of protein phosphatase 1 (PP1) or marked changes in cell morphology or viability (data not shown). However, as reported previously (Sun et al., 2002), we observed that OA concentrations >200 nm or prolonged incubation with this toxin at 100 nm induced cell rounding and neurotoxicity.
Significantly, overexpression of PME-1, which reduced PP2A methylation and Bα expression levels by ∼47–64% (n = 5), was also associated with the accumulation of phosphorylated tau proteins (Fig. 3 A). Conversely, PPMT overexpression induced a ∼30–44% (n = 5) increase in endogenous PP2A methylation and Bα expression and enhanced the rate of tau dephosphorylation at all epitopes studied (Fig. 3 B). Moreover, SAH-induced decrease of intracellular PP2A methylation (Fig. 1 B) correlated with dose-dependent tau phosphorylation at the Ser-396/Ser-404 epitope recognized by PHF-1, a monoclonal antibody that labels phospho-tau lesions in AD (Sontag et al., 2004b) (Fig. 3 C). Conversely, steady-state tau phosphorylation was decreased in cells incubated with SAM. Notably, these effects of SAM were prevented by OA, and treatment with SAM could not overcome the phosphorylation of tau induced by expression of the L309Δ mutant in N2a cells (Fig. 3 D). Thus, PP2A functional integrity is required for effective SAM-dependent tau regulation.
Figure 3.
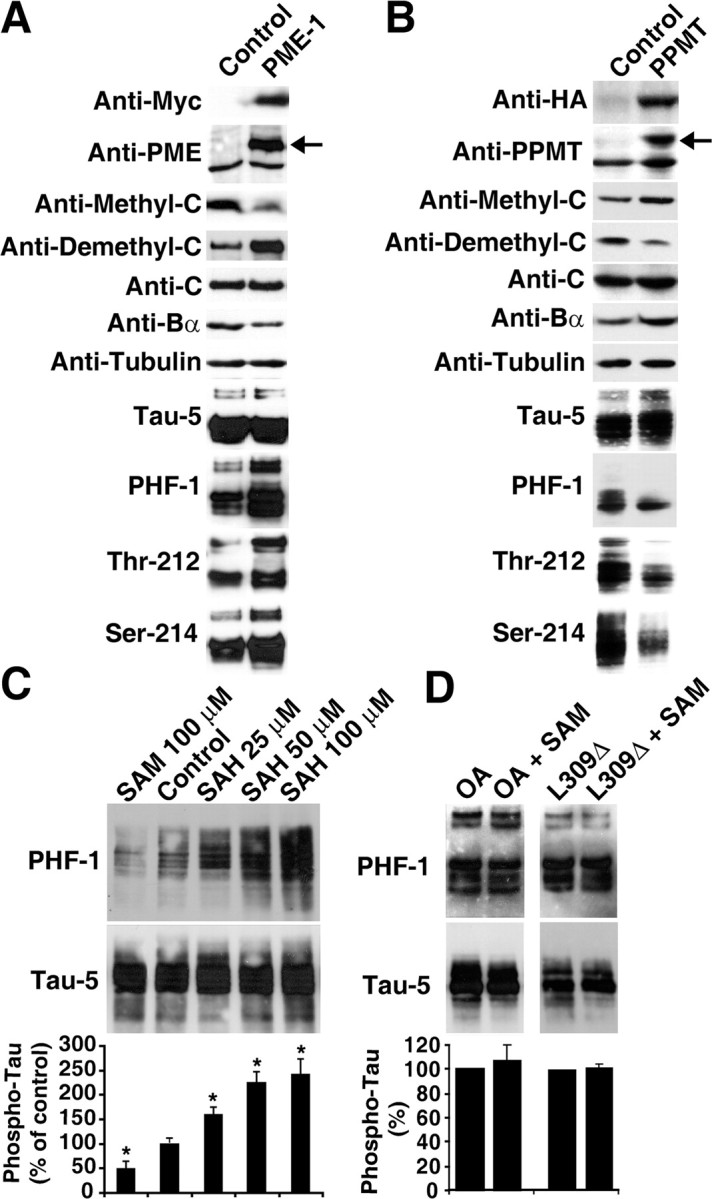
PP2A methylation-, SAM-, and SAH- dependent modulation of tau phosphorylation in N2a cells. A, Representative immunoblots showing that stable expression of Myc-tagged PME-1 is associated with PP2A demethylation, decreased Bα subunit expression, and increased tau phosphorylation. The arrow indicates Myc-tagged PME-1 recognized by the anti-PME-1 antibody. B, Representative immunoblots showing that stable expression of HA-tagged PPMT is associated with enhanced expression levels of PPMT, methylated C and Bα subunits, and tau dephosphorylation. The arrow indicates HA-tagged PPMT recognized by the anti-PPMT antibody. C, Tau-transfected N2a cells were treated with SAM or SAH and analyzed by immunoblotting for tau phosphorylated at the PHF-1 epitope and for total tau using Tau 5 antibodies. D, Concomitant incubation with 10 nm OA or expression of the L309Δ mutant in N2a cells prevent tau dephosphorylation induced by 100 μm SAM. Bottom rows in C and D show the relative amounts of phosphorylated tau (n = 3; *p < 0.001).
Reduced PP2A methylation is associated with enhanced APP phosphorylation at Thr-668
We next addressed the potential contribution of PP2A methylation in the regulation of APP in N2a cells. As reported previously (Latasa et al., 1998), endogenous APP migrated as several bands ranging from ∼95 to ∼125 kDa (Fig. 4 A). Those correspond to immature (N-glycosylated) and mature (N- and O-glycosylated, sulfated) forms of the three isoforms APP695, APP751, and APP770, which arise from alternative splicing from the APP gene. An antibody specific to the phosphorylated Thr-668 site was used to detect phosphorylated APP isoforms. The APP751 and APP770 isoforms contain a KPI domain in the extracellular domain (Menendez-Gonzalez et al., 2005) and were identified as the slowest migrating bands using anti-KPI–APP antibodies. In agreement with previous studies performed in other cells (da Cruz e Silva and da Cruz e Silva, 2003), OA induced a dose-dependent phosphorylation of endogenous APP at the Thr-668 site (Fig. 4 B). In support of a specific role for PP2A activity in modulating APP phosphorylation, enhanced Wt C expression promoted APP dephosphorylation (Fig. 4 C). Conversely, expression of SV40 small tumor antigen (small t), a viral protein that specifically interacts with and inhibits PP2A activity toward tau (Sontag et al., 1996) and other substrates (Sontag, 2001), promoted APP phosphorylation (Fig. 4 C). Phosphorylated APP also accumulated in N2a cells expressing PME-1 or the L309Δ mutant, whereas dephosphorylated APP species were prevalent in N2a–PPMT clones. We next addressed whether SAM and SAH can influence the steady-state phosphorylation of APP. As observed with tau, exposure of N2a cells to SAH led to enhanced APP phosphorylation, whereas incubation with SAM promoted its dephosphorylation (Fig. 4 D). Once again, SAM effects were abolished when cells were simultaneously incubated with OA (Fig. 4 E). SAM treatment clearly promoted APP dephosphorylation in N2a–Wt C cells but was ineffective in N2a–L309Δ cells (Fig. 4 F), suggesting that SAM-induced APP dephosphorylation is dependent on PP2A functional integrity.
Figure 4.

PP2A-, SAM-, and SAH- dependent modulation of APP phosphorylation in N2a cells. A–F, Representative immunoblots showing the relative levels of APP phosphorylated on Thr-668 and total APP in N2a cells. A, Endogenous APP migrates as several bands corresponding to mature and immature APP isoforms (Latasa et al., 1998). The fastest migrating band corresponds to immature APP695. The slowest migrating APP751 and APP770 isoforms are immunoreactive to antibodies raised against the KPI domain of APP. B, Incubation of N2a cells for 1 h with increasing OA concentrations induces dose-dependent APP phosphorylation. C, Phosphorylated APP levels are altered after deregulation of PP2A, PPMT, or PME-1. D, Incubation of N2a cells with 100 μm SAM or SAH affects APP phosphorylation. E, Concomitant incubation for 18 h with 10 nm OA prevents SAM-induced APP dephosphorylation. F, Incubation of N2a–Wt C cells with 100 μm SAM promotes APP dephosphorylation. SAM cannot induce similar effects in N2a–L309Δ cells.
Reduced PP2A methylation is associated with decreased sAPPα secretion and enhanced production of sAPPβ and Aβ
To gain some insights into the effects of deregulating PP2A activity and methylation on endogenous APP processing, we next measured the secretion of sAPPα, sAPPβ, and Aβ in our cells. As reported previously (Nakagawa et al., 2006), secreted APP (sAPP) isoforms were detected in the conditioned media of N2a cells as a triplet band (Fig. 5 A). We first measure sAPP levels 1 h after incubation of cells in conditioned media (Fig. 5 B). In agreement with previous studies performed in other cell types (Gandy et al., 1993; da Cruz e Silva et al., 1995; Holzer et al., 2000; Henriques et al., 2005), incubation of N2a cells with OA stimulated sAPPα secretion. Unexpectedly, relative to control N2a cells, expression of Wt C also increased the extracellular release of sAPPα, albeit much less efficiently than OA. sAPPα secretion was markedly enhanced in N2a–PPMT clones but inhibited in N2a cells expressing L309Δ or PME-1. Under the same experimental conditions, sAPPβ secretion was considerably increased in OA-treated N2a cells. Relative to controls, higher levels of sAPPβ were detected in N2a–PME-1 and N2a–L309Δ clones. Conversely, expression of Wt C or PPMT in N2a cells induced a decrease in released sAPPβ amounts. Immunoblotting of corresponding cell homogenates with anti-APP antibodies indicated that the promotion of sAPP release was unlikely to be the result of a significant upregulation in cellular APP holoprotein expression. Deregulation of PP2A induced similar effects on steady-state sAPPα levels measured 4 h after incubation in conditioned media (Fig. 5 C). In support of the hypothesis that PP2A methylation promotes the release of neuroprotective sAPPα fragments, incubation with SAM enhanced sAPPα secretion in N2a–Wt C cells, whereas expression of the L309Δ mutant prevented this stimulatory effect. Last, to further assess the role of PP2A in the amyloidogenic cleavage of APP, we performed ELISA assays to quantify Aβ secretion in our stable cell lines (Fig. 5 D). Relative to controls, expression of the L309Δ mutant or PME-1 was associated with increased Aβ40 secretion, whereas expression of Wt C and PPMT led to reduced levels of secreted peptides. Concomitant Western blot analysis of the conditioned media for sAPPβ secretion revealed a similar trend. Altogether, our data suggest that PP2A activity and methylation play an important role in the regulation of APP metabolism.
Figure 5.

PP2A activity and methylation regulate APP secretion and Aβ production in N2a cells. A, Secreted sAPP isoforms migrate as a triplet band recognized by the 22C11 antibody (Nakagawa et al., 2006). sAPP751 and sAPP770 isoforms are immunoreactive to the anti-KPI–APP antibody (Latasa et al., 1998). B, Steady-state levels of sAPPα and sAPPβ were measured after 1 h incubation of N2a clones in conditioned medium. A subset of control N2a cells was incubated for 1 h with 100 nm OA. Intracellular APP was detected with anti-APP 22C11 antibody. The bottom panel shows the relative amounts of sAPPα and sAPPβ determined after normalization for intracellular protein concentration (mean ± SD; n = 3; p < 0.001 in all conditions). C, The extracellular release of sAPPα was measured in the same cells analyzed for APP in Figure 4, C and F, 4 h after incubation in the conditioned media. D, Representative experiment showing the concomitant release of sAPPβ and Aβ40 peptides measured 24 h after incubation of cells in conditioned medium. Aβ40 levels (mean ± SEM) were quantified by ELISA.
PPMT and PP2A methylation become downregulated in the brain of hyperhomocysteinemic mice
Our findings in N2a cells suggest that altering SAM and SAH levels can influence tau and APP phosphorylation by affecting PP2A methylation. To test this hypothesis in vivo, we analyzed brain homogenates from Cbs+/− mice heterozygous for a targeted disruption of the gene encoding cystathionine-β-synthase (Lentz et al., 2000), a key enzyme involved in Hcy metabolism (Jhee and Kruger, 2005), and their wild-type littermates (Cbs +/+). Mice were fed an HM/LF or control diet, from weaning until 9 months of age. As expected from previous studies (Lentz et al., 2000; Devlin et al., 2005), tHcy and methionine were significantly increased, and serum folate levels were decreased in mice fed on the HM/LF diet relative to mice fed on the control diet (Fig. 6 A). Hyperhomocysteine (HHcy) was accompanied by increased brain SAH levels and lower SAM/SAH ratio. Although Hcy levels were much higher in Cbs+/− than in Cbs +/+ mice, the SAM/SAH ratio was not much different in those two groups. Brain samples from the same animals were further evaluated for PPMT expression, PP2A methylation, and tau and APP phosphorylation. PPMT expression levels were reduced by ∼25% (Fig. 6 B), and PP2A methylation was decreased by ∼27% (Fig. 6 C) in Cbs+/− mice relative to Cbs +/+ mice fed on the control diet. More extensive reduction of PPMT expression levels (>40%) and demethylation of PP2A (>75%) occurred in both Cbs+/− and Cbs+/+ mice fed on the HM/LF diet compared with mice fed on the control diet. As observed in our cellular studies, reduced PP2A methylation correlated with a substantial increase in phosphorylated APP (Fig. 6 D) and tau (Fig. 6 E). Tau and APP phosphorylation was significantly enhanced in mice fed on the HM/LF diet, being greatest in HHcy Cbs+/− mice.
Figure 6.

Effects of the high-methionine, low-folate diet on Hcy, methylation metabolites, PPMT, PP2A methylation, and tau and APP phosphorylation in Cbs+/+ and Cbs+/− mice. A–D, Groups of 1-month-old heterozygous Cbs+/− (black bars) and wild-type Cbs+/+ (white bars) mice were fed for 8 months on a control or HM/LF diet. Data are mean ± SD; n = 7 mice per group. A, Hcy and methylation metabolites were determined in plasma and brain tissue from the mice (*p < 0.01 compared with same genotype). B–D, Equivalent aliquots (∼14 μl) of total brain extracts from the same mice were simultaneously analyzed by gel electrophoresis and Western blotting. Note that the representative panels shown were assembled from the same exposed autoradiograph. B, Relative levels of PPMT in each mouse group (*p < 0.001; **p < 0.05). C, Relative levels of methylated C (*p < 0.046; **p = 0.046). D, Relative levels of Thr-668-phosphorylated APP (*p < 0.001). E, Equivalent amounts of proteins (10 μg) from boiled mouse brain homogenates were analyzed for tau phosphorylated at the PHF-1 epitope after normalization for total tau levels (*p < 0.001).
Discussion
Although a molecular explanation for the association of low folate and HHcy with degenerative diseases (Nurk et al., 2005; Ravaglia et al., 2005) is essentially unknown, it is believed that Hcy elevations can lead to a reduced cellular ratio of SAM/SAH, which results in decreased activity of methyltransferases. In support of this hypothesis are findings that SAM levels are reduced in brain tissue (Morrison et al., 1996) and CSF (Bottiglieri et al., 1991) from AD patients. Increased SAH levels in brain tissue from AD patients correlate with the inhibition of two CNS methyltransferase reactions (catechol-O-methyltransferase and phenylethanolamine-N-methyltransferase) and various markers of disease progression and cognitive impairment (Kennedy et al., 2004). However, a recent study failed to show significant differences in SAM, SAH, or folate levels in CSF from control and AD subjects (Mulder et al., 2005). Here, we provide evidence that alterations in folate and Hcy metabolism can alter PP2A methylation status, most likely by affecting PPMT activity. Long-term feeding with an HM/LF diet led to an increase in brain SAH concentration and downregulation of PPMT expression, which may account for the severe decrease of steady-state PP2A methylation in the brain of HHcy mice. By comparison, the decrease of PP2A methylation was less pronounced in cells incubated for a short-time with SAH, probably because SAH inhibited PPMT activity but did not significantly alter PPMT expression levels.
We reported that downregulation of neuronal PPMT correlates with reduced PP2A methylation and Bα subunit expression levels in AD-affected brain regions (Sontag et al., 2004a,b). Our cellular studies suggest that decreased PP2A methylation can promote Bα subunit downregulation in AD. Because ABαC is a major tau phosphatase (Sontag et al., 1996), it is not surprising that reduced PP2A methylation levels are associated with enhanced tau phosphorylation. Accordingly, downregulation of PPMT and Bα subunit inversely correlate with increased PHF-1 immunoreactivity in AD neurons (Sontag et al., 2004b). The functional implications of changes in tau phosphorylation are numerous, because epitope-specific phosphorylation differentially modulates the distribution, degradation, aggregation, binding to microtubules and other proteins such as PP2A, and microtubule-regulatory functions of tau (Brandt et al., 2005; Stoothoff and Johnson, 2005). It has been reported that exogenous Hcy and folate deficiency in cultured neurons can increase SAH and/or decrease SAM levels and promote tau phosphorylation (Ho et al., 2002, 2003). Our studies show that some of these effects are directly mediated by PP2A. They provide experimental proof for the hypothesis that PP2A methylation links Hcy metabolism with tau hyperphosphorylation in AD (Vafai and Stock, 2002).
In contrast to its well established function in tau regulation, the role of PP2A in APP regulation is poorly understood. So far, studies investigating the potential role of Ser/Thr phosphatases in APP regulation have primarily relied on the utilization of OA and have clearly implicated PP1 and/or PP2A in the regulation of APP phosphorylation and metabolism (da Cruz e Silva and da Cruz e Silva, 2003). At the same time, they have yielded conflicting or inconsistent findings. Although incubation of various cell types with OA leads to stimulation of APP secretion (Gandy et al., 1993; da Cruz e Silva et al., 1995; Holzer et al., 2000; Henriques et al., 2005), it is not clear whether these effects are preferentially mediated by PP1 (da Cruz e Silva et al., 1995) and/or PP2A (Holzer et al., 2000). Moreover, OA has been reported to either decrease (Buxbaum et al., 1993; Holzer et al., 2000) or increase (Arendt et al., 1998; Sun et al., 2002) Aβ production. All of these discrepancies could be explained by the observation that OA exerts cell-type and dose-dependent effects. Low concentrations of OA preferentially inhibit all PP2A isoforms and protein phosphatases 4, 5, and 6; higher concentrations of OA inhibit all of these enzymes as well as PP1 and can induce neurotoxicity (Honkanen and Golden, 2002). Because there are scores of OA-sensitive enzymes, many distinct phosphatase-dependent pathways with potentially overlapping or divergent regulatory effects on APP could become affected by OA. Therefore, it is quite difficult to decipher the contribution of a particular phosphatase and dissect the mechanisms involved in the regulation of APP in OA-based experiments. In the present study, incubation of N2a cells with OA, at concentrations that do not inhibit PP1, induced APP phosphorylation at Thr-668 and enhanced the secretion of both sAPPα and sAPPβ. Interestingly, overexpression of cyclin-dependent kinase 5 (cdk5) promotes similar effects (Liu et al., 2003). Because α- and β-secretases are competing for substrate in the process of APP proteolysis, it has been suggested that sAPPα elevation can be taken as indirect evidence of inhibition of amyloidogenic APP cleavage (Kojro and Fahrenholz, 2005). However, there are examples in the literature in which the production of sAPPα and Aβ are not under reciprocal regulation, probably because distinct constitutive and regulated α-secretory pathways converge to produce sAPPα (Lammich et al., 1999). Besides, APP expression, phosphorylation, protein–protein interactions, trafficking, maturation, secretase cleavage, and secretion are all regulated processes that can be differentially influenced by the complex interplay between protein kinase and phosphatase activities. Because alterations in PP2A activity can affect the activity of many protein kinases and a wide variety of cellular functions (Sontag, 2001), the regulation of APP processing by PP2A likely results from a combination of direct and indirect effects. For instance, recent findings in neurons suggest that OA-mediated inhibition of PP2A promotes the axonal accumulation of β-CTF APP fragments by inducing microtubule destabilization and deficits in APP transport (Yoon et al., 2006).
We found here that phosphorylated APP751/APP770 isoforms accumulated in response to decreased activity and reduced methylation of PP2A. Interestingly, KPI-containing APP isoform levels are elevated in AD brain and are associated with increased production and reduced clearance of Aβ (Menendez-Gonzalez et al., 2005). Moreover, high levels of phosphorylated APP and tau species colocalize in AD hippocampal neurons (Lee et al., 2003). It is of particular significance that APP can be phosphorylated on Thr-668 by cdk5 and glycogen-synthase kinase 3, two tau protein kinases that have been implicated in the neurofibrillary degeneration in AD (Stoothoff and Johnson, 2005). Notably, phosphorylation at Thr-668 has been linked to increased amyloidogenic processing of APP (Ando et al., 2001; Lee et al., 2003; Liu et al., 2003; Phiel et al., 2003; Vingtdeux et al., 2005; Chang et al., 2006; Pierrot et al., 2006). Accordingly, enhanced APP phosphorylation at the Thr-668 site correlated with increased Aβ production in our experiments. In addition, our findings suggest that enhanced PP2A activity and methylation promote the accumulation of dephosphorylated APP isoforms and favor the non-amyloidogenic cleavage of APP. Conversely, downregulation of PP2A methylation is accompanied by a concomitant decrease in the steady-state release of neuroprotective sAPPα species and increased secretion of β- and γ-secretase-cleaved APP fragments. Thus, reduced PP2A methylation levels may shift APP processing toward the amyloidogenic pathway. Like APP, the regulation of PP2A is rather intricate (Sontag, 2001). Changes in C subunit methylation modulate PP2A intracellular subunit composition, which in turn can influence PP2A compartmentalization, interactions with scaffolding and regulatory proteins, and substrate specificity. It is likely that the cumulative effects of all such alterations in PP2A regulation, and not solely changes in phosphatase activity, contribute to affect the rate of dephosphorylation and processing of APP. Additional studies will be required to elucidate the underlying mechanisms leading to the deregulation of APP metabolism by PP2A.
Interestingly, folate deficiency increases Aβ production by affecting SAM-dependent DNA methylation of APP-regulatory genes (Fuso et al., 2005). Our findings also implicate SAM in the regulation of APP phosphorylation and secretion via PPMT-dependent PP2A methylation. They suggest that SAH-mediated PP2A demethylation in response to HHcy could promote amyloidogenesis. These conclusions are further supported by recent findings showing that plasma Hcy is positively correlated with plasma Aβ40 and Aβ42 in AD patients (Irizarry et al., 2005) and that HHcy is associated with elevated brain Aβ levels in Cbs/APP/PS1 AD mice models of amyloidosis (Pacheco-Quinto et al., 2006).
In conclusion, our studies unveil a novel pathway implicating PPMT-dependent PP2A methylation in the regulation of two major CNS proteins, tau and APP, involved in neurodegenerative diseases. Our findings suggest that even relatively small changes in intracellular PP2A methylation can impact tau phosphorylation and APP processing. They support the hypothesis that impaired Hcy metabolism and deregulation of critical methylation reactions can trigger the accumulation of phosphorylated tau and APP in the brain, a process that may favor neurofibrillary tangle formation and amyloidogenesis. They provide insights into the molecular mechanisms by which dietary folate deficiency and HHcy may promote or hasten AD-like pathology in predisposed populations, as well as information that should inform the design of therapeutic trials.
Footnotes
This work was supported by National Institutes of Health Grants AG18883 (E.S.), AG12300 (E.S., J.-M.S.), AT002311 (T.B.), AG17861 (R.D.-A.), and NS24621 (S.R.L.) and Austrian Science Foundation Grant P15685 and Herzfelder Familienstiftung (E.O.). We thank Lisa Montgomery, Felicia Ware, and Dr. Leonard Craig for preliminary experiments; Claudia Juno, Ingrid Mudrak, and Patrick Piribauer for generating PPMT/PME reagents; Dr. Michael Irizarri (GlaxoSmithKline, Research Triangle Park, NC) for invaluable technical advice; Dr. Peter Davies (Albert Einstein College of Medicine, Bronx, NY) for the PHF-1 antibody; Dr. David Pallas (Emory University, Atlanta, GA) for the L309Δ cDNA; and Drs. Erik Schaefer and Lynda Zorn (Biosource International, Hopkinton, MA) for phospho-tau antibodies.
References
- Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem. 2001;276:40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- Arendt T, Holzer M, Fruth R, Bruckner MK, Gartner U. Phosphorylation of tau, Abeta-formation, and apoptosis after in vivo inhibition of PP-1 and PP-2A. Neurobiol Aging. 1998;19:3–13. doi: 10.1016/s0197-4580(98)00003-7. [DOI] [PubMed] [Google Scholar]
- Baharians Z, Schonthal AH. Autoregulation of protein phosphatase type 2A expression. J Biol Chem. 1998;273:19019–19024. doi: 10.1074/jbc.273.30.19019. [DOI] [PubMed] [Google Scholar]
- Bottiglieri T. The effect of storage on rat tissues and human plasma amino acid levels determined by HPLC. Biomed Chromatogr. 1987;2:195–196. doi: 10.1002/bmc.1130020505. [DOI] [PubMed] [Google Scholar]
- Bottiglieri T. Isocratic high performance liquid chromatographic analysis of S-adenosylmethionine and S-adenosylhomo-cysteine in animal tissues: the effect of exposure to nitrous oxide. Biomed Chromatogr. 1990;4:239–241. doi: 10.1002/bmc.1130040606. [DOI] [PubMed] [Google Scholar]
- Bottiglieri T, Reynolds EH, Toone BK, Carney MW. CSF S-adenosylmethionine in neuropsychiatric disorders. Lancet. 1991;338:121. doi: 10.1016/0140-6736(91)90114-5. [DOI] [PubMed] [Google Scholar]
- Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim Biophys Acta. 2005;1739:331–354. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Koo EH, Greengard P. Protein phosphorylation inhibits production of Alzheimer amyloid beta/A4 peptide. Proc Natl Acad Sci USA. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KA, Kim HS, Ha TY, Ha JW, Shin KY, Jeong YH, Lee JP, Park CH, Kim S, Baik TK, Suh YH. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Cell Biol. 2006;26:4327–4338. doi: 10.1128/MCB.02393-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz e Silva EF, da Cruz e Silva OA. Protein phosphorylation and APP metabolism. Neurochem Res. 2003;28:1553–1561. doi: 10.1023/a:1025630627319. [DOI] [PubMed] [Google Scholar]
- da Cruz e Silva EF, da Cruz e Silva OA, Zaia CT, Greengard P. Inhibition of protein phosphatase 1 stimulates secretion of Alzheimer amyloid precursor protein. Mol Med. 1995;1:535–541. [PMC free article] [PubMed] [Google Scholar]
- De Baere I, Derua R, Janssens V, Van Hoof C, Waelkens E, Merlevede W, Goris J. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry. 1999;38:16539–16547. doi: 10.1021/bi991646a. [DOI] [PubMed] [Google Scholar]
- Devlin AM, Bottiglieri T, Domann FE, Lentz SR. Tissue-specific changes in H19 methylation and expression in mice with hyperhomocysteinemia. J Biol Chem. 2005;280:25506–25511. doi: 10.1074/jbc.M504815200. [DOI] [PubMed] [Google Scholar]
- Fowler B. Homocysteine: overview of biochemistry, molecular biology, and role in disease processes. Semin Vasc Med. 2005;5:77–86. doi: 10.1055/s-2005-872394. [DOI] [PubMed] [Google Scholar]
- Fuso A, Seminara L, Cavallaro RA, D'Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Gandy SE, Caporaso GL, Buxbaum JD, de Cruz SO, Iverfeldt K, Nordstedt C, Suzuki T, Czernik AJ, Nairn AC, Greengard P. Protein phosphorylation regulates relative utilization of processing pathways for Alzheimer beta/A4 amyloid precursor protein. Ann NY Acad Sci. 1993;695:117–121. doi: 10.1111/j.1749-6632.1993.tb23038.x. [DOI] [PubMed] [Google Scholar]
- Goedert M, Satumtira S, Jakes R, Smith MJ, Kamibayashi C, White CL, Sontag E. Reduced binding of protein phosphatase 2A to tau protein with frontotemporal dementia and parkinsonism linked to chromosome 17 mutations. J Neurochem. 2000;75:2155–2162. doi: 10.1046/j.1471-4159.2000.0752155.x. [DOI] [PubMed] [Google Scholar]
- Henriques AG, Domingues SC, Fardilha M, da Cruz e Silva EF, da Cruz e Silva OA. Sodium azide and 2-deoxy-d-glucose-induced cellular stress affects phosphorylation-dependent AbetaPP processing. J Alzheimers Dis. 2005;7:201–212. doi: 10.3233/jad-2005-7302. [DOI] [PubMed] [Google Scholar]
- Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: glutamate excitotoxicity, kinase hyperactivation and DNA damage. J Neurosci Res. 2002;70:694–702. doi: 10.1002/jnr.10416. [DOI] [PubMed] [Google Scholar]
- Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, Rogers E. Folate deprivation induces neurodegeneration: roles of oxidative stress and increased homocysteine. Neurobiol Dis. 2003;14:32–42. doi: 10.1016/s0969-9961(03)00070-6. [DOI] [PubMed] [Google Scholar]
- Holzer M, Bruckner MK, Beck M, Bigl V, Arendt T. Modulation of APP processing and secretion by okadaic acid in primary guinea pig neurons. J Neural Transm. 2000;107:451–461. doi: 10.1007/s007020070087. [DOI] [PubMed] [Google Scholar]
- Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Curr Med Chem. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Gurol ME, Raju S, Diaz-Arrastia R, Locascio JJ, Tennis M, Hyman BT, Growdon JH, Greenberg SM, Bottiglieri T. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology. 2005;65:1402–1408. doi: 10.1212/01.wnl.0000183063.99107.5c. [DOI] [PubMed] [Google Scholar]
- Jhee KH, Kruger WD. The role of cystathionine beta-synthase in homocysteine metabolism. Antioxid Redox Signal. 2005;7:813–822. doi: 10.1089/ars.2005.7.813. [DOI] [PubMed] [Google Scholar]
- Kennedy BP, Bottiglieri T, Arning E, Ziegler MG, Hansen LA, Masliah E. Elevated S-adenosylhomocysteine in Alzheimer brain: influence on methyltransferases and cognitive function. J Neural Transm. 2004;111:547–567. doi: 10.1007/s00702-003-0096-5. [DOI] [PubMed] [Google Scholar]
- Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latasa MJ, Belandia B, Pascual A. Thyroid hormones regulate beta-amyloid gene splicing and protein secretion in neuroblastoma cells. Endocrinology. 1998;139:2692–2698. doi: 10.1210/endo.139.6.6033. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz SR, Erger RA, Dayal S, Maeda N, Malinow MR, Heistad DD, Faraci FM. Folate dependence of hyperhomocysteinemia and vascular dysfunction in cystathionine beta-synthase-deficient mice. Am J Physiol Heart Circ Physiol. 2000;279:H970–H975. doi: 10.1152/ajpheart.2000.279.3.H970. [DOI] [PubMed] [Google Scholar]
- Liu F, Su Y, Li B, Zhou Y, Ryder J, Gonzalez-DeWhitt P, May PC, Ni B. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547:193–196. doi: 10.1016/s0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- Menendez-Gonzalez M, Perez-Pinera P, Martinez-Rivera M, Calatayud MT, Blazquez MB. APP processing and the APP-KPI domain involvement in the amyloid cascade. Neurodegener Dis. 2005;2:277–283. doi: 10.1159/000092315. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MS. Homocysteine and Alzheimer's disease. Lancet Neurol. 2003;2:425–428. doi: 10.1016/s1474-4422(03)00438-1. [DOI] [PubMed] [Google Scholar]
- Morrison LD, Smith DD, Kish SJ. Brain S-adenosylmethionine levels are severely decreased in Alzheimer's disease. J Neurochem. 1996;67:1328–1331. doi: 10.1046/j.1471-4159.1996.67031328.x. [DOI] [PubMed] [Google Scholar]
- Mulder C, Schoonenboom NS, Jansen EE, Verhoeven NM, van Kamp GJ, Jakobs C, Scheltens P. The transmethylation cycle in the brain of Alzheimer patients. Neurosci Lett. 2005;386:69–71. doi: 10.1016/j.neulet.2005.03.073. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Kitazume S, Oka R, Maruyama K, Saido TC, Sato Y, Endo T, Hashimoto Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J Neurochem. 2006;96:924–933. doi: 10.1111/j.1471-4159.2005.03595.x. [DOI] [PubMed] [Google Scholar]
- Nurk E, Refsum H, Tell GS, Engedal K, Vollset SE, Ueland PM, Nygaard HA, Smith AD. Plasma total homocysteine and memory in the elderly: The Hordaland Homocysteine study. Ann Neurol. 2005;58:847–857. doi: 10.1002/ana.20645. [DOI] [PubMed] [Google Scholar]
- Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem. 1999;274:14382–14391. doi: 10.1074/jbc.274.20.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco-Quinto J, Rodriguez de Turco EB, Derosa S, Howard A, Cruz-Sanchez F, Sambamurti K, Refolo L, Petanceska S, Pappolla MA. Hyperhomocysteinemic Alzheimer's mouse model of amyloidosis shows increased brain amyloid beta peptide levels. Neurobiol Dis. 2006;22:651–656. doi: 10.1016/j.nbd.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-beta accumulation. J Biol Chem. 2006;281:39907–39914. doi: 10.1074/jbc.M606015200. [DOI] [PubMed] [Google Scholar]
- Ravaglia G, Forti P, Maioli F, Martelli M, Servadei L, Brunetti N, Porcellini E, Licastro F. Homocysteine and folate as risk factors for dementia and Alzheimer disease. Am J Clin Nutr. 2005;82:636–643. doi: 10.1093/ajcn.82.3.636. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Sontag E. Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell Signal. 2001;13:7–16. doi: 10.1016/s0898-6568(00)00123-6. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–1207. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, III, Mumby MC, Bloom GS. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem. 1999;274:25490–25498. doi: 10.1074/jbc.274.36.25490. [DOI] [PubMed] [Google Scholar]
- Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL., III Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004a;63:287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL., III Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004b;63:1080–1091. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Sun X, Cole GM, Chu T, Xia W, Galasko D, Yamaguchi H, Tanemura K, Frautschy SA, Takashima A. Intracellular Abeta is increased by okadaic acid exposure in transfected neuronal and non-neuronal cell lines. Neurobiol Aging. 2002;23:195–203. doi: 10.1016/s0197-4580(01)00265-2. [DOI] [PubMed] [Google Scholar]
- Tolstykh T, Lee J, Vafai S, Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000;19:5682–5691. doi: 10.1093/emboj/19.21.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubbink JB, Hayward Vermaak WJ, Bissbort S. Rapid high-performance liquid chromatographic assay for total homocysteine levels in human serum. J Chromatogr. 1991;565:441–446. doi: 10.1016/0378-4347(91)80407-4. [DOI] [PubMed] [Google Scholar]
- Vafai SB, Stock JB. Protein phosphatase 2A methylation: a link between elevated plasma homocysteine and Alzheimer's disease. FEBS Lett. 2002;518:1–4. doi: 10.1016/s0014-5793(02)02702-3. [DOI] [PubMed] [Google Scholar]
- Vassar R. beta-Secretase, APP and Abeta in Alzheimer's disease. Subcell Biochem. 2005;38:79–103. [PubMed] [Google Scholar]
- Vingtdeux V, Hamdane M, Gompel M, Begard S, Drobecq H, Ghestem A, Grosjean ME, Kostanjevecki V, Grognet P, Vanmechelen E, Buee L, Delacourte A, Sergeant N. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol Dis. 2005;20:625–637. doi: 10.1016/j.nbd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Yoon SY, Choi JE, Yoon JH, Huh JW, Kim DH. BACE inhibitor reduces APP-beta-C-terminal fragment accumulation in axonal swellings of okadaic acid-induced neurodegeneration. Neurobiol Dis. 2006;22:435–444. doi: 10.1016/j.nbd.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Yu XX, Du X, Moreno CS, Green RE, Ogris E, Feng Q, Chou L, McQuoid MJ, Pallas DC. Methylation of the protein phosphatase 2A catalytic subunit is Essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol Biol Cell. 2001;12:185–199. doi: 10.1091/mbc.12.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]