

Abstract
Drug-induced neuroadaptations within the medial prefrontal cortex (mPFC) are thought to underlie the development of cocaine sensitization. Here, we report that repeated cocaine administration in vivo impaired the long-term depression (LTD) induced by bath application of group II metabotropic glutamate receptor (mGluR) agonists DCG-IV [2S, 2′R, 3′R)-2-(2′, 3′-dicarboxycyclopropyl)glycine] or LY379268 [(1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid] at excitatory synapses onto layer V pyramidal neurons of rat mPFC. In contrast, this impairment was not found in slices from rats treated with saline or a single dose of cocaine. Such effect of cocaine was selectively prevented when cocaine was coadministered with the selective D1-like receptor antagonist SCH23390 [(R)-(+)-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine]. In slices from control rats, a brief application of either protein kinase C (PKC) activator phorbol-12,13-dibutyrute or adenosine A3 receptor agonist 2-chloro-N6-(3-iodobenzyl)-adenosine-5-N-methyluronamide mimicked the effect of repeated cocaine treatment to impair the induction of LTD. Bilateral intra-mPFC infusion of PKC inhibitor bisindolylmaleimide I or adenosine A3 receptor antagonist MRS1220 (N-[9-chloro-2-(2-furanyl)[1,2,4]-triazolo[1,5-c]quinazolin-5-benzeneacetamide) before cocaine injection prevented cocaine-induced impairment of LTD induction. Furthermore, endogenous adenosine tone is greater in slices from cocaine-treated rats than from the saline-treated controls. When the metabolism of cAMP to adenosine was blocked, the extent of LTD in slices from saline and cocaine-treated rats was similar. These results suggest that cocaine-induced impairment of group II mGluR-mediated LTD is caused, at least in part, by an increase in adenosine subsequent to the rise in cAMP after D1-like receptor activation, which leads to an adenosine A3 receptor-mediated upregulation of PKC activity and thereby triggers an inhibition of group II metabotropic glutamate receptor function.
Keywords: cocaine, medial prefrontal cortex, metabotropic glutamate receptor, long-term depression, adenosine, addiction
Introduction
Repeated exposure to psychostimulants such as cocaine and amphetamine produces a progressive and enduring increase in their locomotor stimulatory effects. This enhanced behavioral response is referred to as behavioral sensitization and is postulated to contribute, in part, to drug craving and relapse to addiction (Robinson and Berridge, 1993; Wolf, 1998; Carlezon and Nestler, 2002; Hyman et al., 2006). Many studies on the neurobiological basis of behavioral sensitization have focused on the mesolimbic dopaminergic system because cocaine and amphetamine act to block the uptake or stimulate the release, respectively, of dopamine within this system (Reith et al., 1986; Seiden et al., 1993). The mesolimbic system consists of dopamine perikarya located in the ventral tegmental area (VTA) that project to numerous limbic areas including nucleus accumbens (NAc) (Oades and Halliday, 1987). Furthermore, evidence suggests that adaptive changes within the VTA appear to mediate the induction of behavioral sensitization, whereas adaptations in the NAc are involved in its long-term maintenance (Robinson and Berridge, 1993; Wolf, 1998; Carlezon and Nestler, 2002). In addition to the mesolimbic system, previous studies have implicated an important role for the mesocortical system, in particular the medial prefrontal cortex (mPFC), in the development of behavioral sensitization. For example, direct electrical stimulation of the PFC leads to rewarding effects and sensitization to cocaine, whereas lesion of the PFC or impairment of mesocorticolimbic glutamatergic transmission prevents the development of cocaine-induced behavioral sensitization (Li et al., 1997; Tzschentke, 2001). Moreover, evidence is accumulating that the alterations of multiple mPFC neurotransmitter systems, including dopamine, serotonin, glutamate, noradrenalin, acetylcholine, GABA, and peptides, and signaling by receptors activated by these neurotransmitters may also contribute to the development of cocaine sensitization and addiction (Wolf, 1998; Steketee, 2003; Williams and Steketee, 2004, 2005).
Compelling evidence indicates that drug-induced synaptic plasticity may contribute to the reorganization of neural circuitry that underlies behavioral sensitization to drugs of abuse and, thus, the development of addiction. It has been shown that a single or repeated exposure in vivo to addictive drugs induces alterations of synaptic transmission and/or long-term synaptic plasticity at excitatory synapses in in vitro slice preparations of the VTA, NAc, periaqueductal gray, hippocampus, and striatum (Ingram et al., 1998; Martin et al., 1999; Thomas et al., 2001; Ungless et al., 2001; Williams et al., 2001; Harrison et al., 2002; Robbe et al., 2002; Hoffman et al., 2003; Borgland et al., 2004; Fourgeaud et al., 2004; Liu et al., 2005; Centonze et al., 2006). Less work has been done on the impact of in vivo exposures to drugs of abuse on excitatory synaptic transmission in the mPFC. Our laboratory has demonstrated previously that repeated cocaine administration in vivo can promote the induction of long-term potentiation (LTP) in mPFC layer V pyramidal neurons (Huang et al., 2007). However, the effect of repeated cocaine exposure on the induction of long-term depression (LTD) at mPFC excitatory synapses has not yet been explored. Here, we provide the first evidence that repeated cocaine administration, which produces sensitization of locomotor activity, impairs the group II metabotropic glutamate receptor (mGluR)-mediated LTD at these synapses.
Materials and Methods
Animals and treatment.
Male Sprague Dawley rats (21–23 d old) were housed under a 12 h light/dark cycle (lights on at 6:00 A.M.) and had ad libitum access to food and water. After a 3 d acclamation period, animals were assigned randomly to two groups that received an intraperitoneal injection of saline (1 ml/kg) or cocaine HCl (15 mg/kg) once per day for 5 consecutive days followed by 3 d of withdrawal before the experiments. During this time, they were handled and weighed daily. All comparisons between saline- and cocaine-treated groups were performed by experimenters blind to group assignment. Animal care was consistent with the guidelines set by the Laboratory Animal Center of National Cheng Kung University. All experiments were approved by the National Cheng Kung University Institutional Animal Care and Use Committee governing the participating laboratories.
Cannulation.
Rats (21–23 d old) were bilaterally implanted under deep pentobarbital (50 mg/kg, i.p., supplemented as required) anesthesia with 22-gauge double guide cannulas (Plastics One, Roanoke, VA) in the mPFC. Medial PFC coordinates were +2.5 mm anterior to bregma, ±0.5 mm bilateral to midline, and 2.0 mm ventral to brain surface according to the previous studies (Huang et al., 2007) and the stereotaxic atlas of developing rat brain (Sherwood and Timiras, 1970). The cannulas were fixed to the skull with dental cement. After 3–5 d of recovery, rats received 0.5 μl microinjections in each side over a 1 min period using an infusion pump (CAM/100; CAM Microdialysis, Slona, Sweden). The infusion cannulas were kept in place for an additional 2 min to minimize backflow of the injectant. Doses of bisindolylmaleimide I (Bis-I; 0.4 nmol/side), bisindolylmaleimide V (Bis-V; 0.4 nmol/side), or N-[9-chloro-2-(2-furanyl)[1,2,4]-triazolo[1,5-c]quinazolin-5-benzeneacetamide (MRS1220; 0.5 nmol/side) were selected on the basis of published studies (Blank et al., 2003; Yang et al., 2004) and pilot experiments in our laboratory. Infusions were administered 30 min before each of the five daily cocaine injections. Rats with injection cannula placements outside the mPFC or with extensive tissue damage at the injection cannula site were excluded from analyses. We never observed any effects of cannulation itself or vehicle artificial CSF (aCSF) [10% dimethylsulfoxide (DMSO)] injection. Only slices from animals with accurate cannula placements, as verified microscopically by the cannula tips located within the mPFC, were taken for electrophysiological recordings. Although infusions spread with a radius of ∼1.0 mm (as estimated by pilot experiments using a 0.5 μl solution of 4% methylene blue in saline), we made recordings from neurons within a radius 0.3–0.5 mm from the cannula tips to better get the effects of the infused drugs.
Slice preparation and electrophysiology.
Slice preparation and whole-cell patch-clamp recordings were conducted as reported previously (Huang and Hsu, 2006; Huang et al., 2007). In brief, rats were anesthetized with halothane and decapitated with a guillotine, and coronal slices (250 μm thick) containing the prelimbic area of the mPFC (2.0–3.5 mm from the bregma) (Sherwood and Timiras, 1970) were prepared using a vibrating microtome (VT1000S; Leica, Nussloch, Germany). The slices were placed in a holding chamber of aCSF oxygenated with 95% O2-5% CO2 and kept at room temperature for at least 1 h before recording. The composition of the aCSF solution was as follows (in mm): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 glucose at pH 7.3–7.4, and equilibrated with 95% O2-5% CO2. For whole-cell patch-clamp recording, one slice was transferred to a recording chamber of standard design and fixed at the glass bottom of the chamber with a nylon grid on a platinum frame. The chamber consisted of a circular well of low volume (1–2 ml) and was perfused constantly at 32.0 ± 0.5°C with a rate of 2–3 ml/min. After a high resistance seal (>2 GΩ before breaking into whole-cell mode) was obtained, suction was applied lightly through the pipette to break through the membrane. The cell was then maintained at −70 mV for several minutes to allow diffusion of the internal solution into the cell body and dendrites. Recordings were made using a patch clamp amplifier (Axopatch 200B; Molecular Devices, Union City, CA) under and infrared-differential interference contrast microscope. Electrical signals were low-pass filtered at 2 kHz and digitized at 10 kHz using a 12 bit analog-to-digital converter (Digidata 1320; Molecular Devices). An Intel (Acer, Taipei, Taiwan) Pentium-based computer with pCLAMP software (version 8.0; Molecular Devices) was used for on-line acquisition and off-line analysis of the data. For measurement of synaptically evoked EPSCs, a bipolar stainless-steel stimulating electrode was placed in layer V, 40–60 μm away the apical dendrites of the recorded neurons to stimulate excitatory afferents at 0.05 Hz, and the superfusate routinely contained bicuculline methiodide (1 μm) to block inhibitory synaptic responses. The strength of synaptic transmission was mostly quantified by measuring the initial rising slope of EPSC (2 ms period from its onset; picoampere/millisecond), which contains only a monosynaptic component. The electrode resistance was typically 3–6 MΩ. The composition of intracellular solution was as follows (mm): 115 K-gluconate, 20 KCl, 10 HEPES, 2 MgCl2, 0.5 EGTA, 3 Na2ATP, 0.3 Na3GTP, 5 QX-314, and sucrose to bring osmolarity to 290–295 mOsM and pH to 7.3. Series resistance and input resistance were monitored on-line throughout the whole-cell recording with a 5 mV depolarizing step given after every afferent stimulus and data were discarded if resistance changed by >20%.
Biochemical measurement of surface-expressed receptors.
The procedure was similar to that described previously (Zho et al., 2002). mPFC slices were incubated with aCSF containing 1 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL) for 20 min on ice. Unreacted biotinylation reagent was washed once with ice-cold aCSF and quenched by two successive 20 min washes in aCSF containing 100 mm glycine, followed by two washes in ice-cold TBS (50 mm Tris, pH 7.5, 150 mm NaCl). The slices were then lysed in ice-cold homogenate buffer (50 mm Tris-HCl, 100 mm NaCl, 15 mm sodium pyrophosphate, 50 mm sodium fluoride, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 0.5% Triton X-100, 2 mm benzamidine, 60 μg/ml aprotinin, and 60 μg/ml leupeptin) and ground with a pellet pestle. Samples were sonicated and spun down at 14,000 × g for 15 min at 4°C. A total of 20 μg of protein was removed to measure total metabotropic glutamate receptor 2 (mGluR2). For surface protein, 150 μg of protein was incubated with 100 μl of 50% Neutravidin agarose (Pierce) for 2 h at 4°C to measure the isolated biotinylated proteins. After the Neutravidin agarose was washed five times with homogenate buffer, bound proteins were eluted with SDS sample buffer by boiling for 15 min. Total protein and isolated biotinylated proteins were analyzed by quantitative immunoblotting with monoclonal anti-mGluR2 C-terminal (1:1500; Abcam, Cambridge, UK). It was then probed with HRP-conjugated secondary antibody for 1 h and developed using the ECL immunoblotting detection system. Immunoblots were quantified by densitometric measurement.
Drug treatment.
Cocaine HCl (15 mg/kg), (R)-(+)-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH23390, 0.5 mg/kg), raclopride (0.5 mg/kg), and 8-[5-(2,4-dimethoxy-5-(4-trifluoromethylphenylsulfonamido)phenyl-5-oxopentyl)-1,3,8-triazaspiro[4.5]decane-2,4-dione (RS102221, 2 mg/kg) were dissolved in 0.9% NaCl and administered intraperitoneally. Drug doses were selected on the basis of published studies (Dong et al., 2004, 2005; Conductier et al., 2005; Huang et al., 2007). All drugs used in in vitro experiments were applied by manually switching the superfusate. Drugs were diluted from stock solutions just before application. (1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), forskolin (FSK), Bis-I, Bis-V, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), phorbol-12,13-dibutyrate (PDBu), 4α-phorbol-12,13-didecanoate (4αPDD), 2-chloro-N6-(3-lodobenzyl)-adenosine-5′-N-methyluron-amide (Cl-IB-MECA), MRS1220, and 4-(3-butoxy-4-methoxy-phenyl)methyl-2-imidazolidone (RO201724) were dissolved in DMSO stock solutions and stored at −20°C until the day of experiment. Other drugs used in this study were dissolved in distilled water. The concentration of DMSO in the perfusion medium was 0.1%, which alone had no effect on basal synaptic transmission (Huang and Hsu, 2006). LY379268, SCH23390, raclopride, RS102221, 2S, 2′R, 3′R)-2-(2′, 3′-dicarboxycyclopropyl)glycine (DCG-IV), FSK, Bis-I, IB-MECA, MRS1220, and RO201724 were purchased from Tocris Cookson (Bristol, UK); cocaine HCl, probenecid, N6-cyclopentyladenosine (CPA), and bicuculline methiodide were obtained from Sigma (St Louis, MO); adenosine-3′,5′-cyclic monophosphorothioate, 8-bromo-, Sp- isomer (Sp-8-Br-cAMPS), PDBu, 4αPDD, and Bis-V were purchased from Calbiochem (La Jolla, CA).
Statistical analysis.
The data for each experiment were normalized relative to baseline, and are presented as mean ± SEM. Numbers of experiments are indicated by n. The significance of the difference between the mean was calculated by paired or unpaired Student's t test. Probability values (p) of <0.05 were considered to represent significant differences.
Results
Repeated cocaine exposure blocks DCG-IV-LTD
In the first series of experiments, we examined whether repeated cocaine administration affects the induction of LTD at mPFC excitatory synapses. The effectiveness of the repeated cocaine treatment on rats was indicated by their heightened locomotor sensitivity to cocaine (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To induce LTD, we used a brief bath application of DCG-IV protocol that has been shown to effectively induce LTD in rat mPFC in vitro (Otani et al., 1999, 2002). As reported previously, application of DCG-IV (0.2 μm) for 10 min reliably induced a robust LTD of EPSCs of layer V pyramidal neurons termed DCG-IV-LTD. The slope of EPSCs was depressed to 76.5 ± 4.9% (n = 10; p < 0.05) measured 30 min after washout of DCG-IV in slices from rats treated with saline for 5 d (Fig. 1A). In an attempt to assign the specificity of the effect of DCG-IV on group II mGluRs at inducing LTD, a more selective group II mGluR agonist LY379268 (Monn et al., 1999) was compared. As DCG-IV did, application of LY379268 (50 nm) for 10 min also resulted in a substantial LTD of EPSCs. The slope of EPSCs was depressed to 53.5 ± 4.5% (n = 8; p < 0.05) measured 30 min after washout of LY379268 in slices from rats treated with saline for 5 d (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Both DCG-IV- and LY379268-induced LTD were completely blocked by bath application of either MCPG (0.5 mm), a nonselective antagonist of group II mGluR, or MSOPPE (100 μm), a more selective antagonist of mGluR2, suggesting that they are mainly mediated by the activation of group II mGluRs (data not shown). In contrast, the induction of LTD by DCG-IV or LY379268 was impaired in slices from rats treated with cocaine for 5 d (DCG-IV, 96.5 ± 3.6%, n = 12; LY379268, 82.7 ± 4.7%, n = 8; p < 0.05 when compared with slices from 5 d saline-treated rats) (Fig. 1B, supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). No change in the extent of DCG-IV-LTD was observed in slices from rats that were given a single injection of saline (75.3 ± 3.8%; n = 8; p < 0.05) or cocaine (79.4 ± 5.3%; n = 10; p < 0.05). These results suggest that repeated but not single cocaine administration impairs the induction of group II mGluR-mediated LTD in mPFC layer V pyramidal neurons. Because DCG-IV could effectively induce group II mGluR-mediated LTD at mPFC excitatory synapses, we chose this protocol to examine the mechanisms involved in processes by which repeated cocaine treatment impairs the induction of LTD.
Figure 1.

Repeated cocaine administration inhibits the induction of DCG-IV-LTD in mPFC layer V pyramidal neurons. A, B, Example (top) and summary (bottom) of normalized EPSC slope before and after a brief application of DCG-IV (0.2 μm) for 10 min in slices from rats treated with saline (A) or cocaine (B) for 5 d. C, D, Summary of experiments showing the effect of DCG-IV on the EPSC slope in slices from rats treated with a single dose of saline (C) or cocaine (D) for 1 d. Representative traces of EPSCs were taken at the time indicated by number. The total number of neurons examined is indicated by n. Horizontal bar denotes the period of delivery of DCG-IV. The dashed line represents 100%. Error bars indicate SEM.
To examine whether repeated cocaine exposure causes a decrease in the sensitivity of group II mGluRs and thereby leads to an apparent impairment of DCG-IV-LTD, the concentration–response relationship for DCG-IV was examined. As shown in supplemental Figure S3 (available at www.jneurosci.org as supplemental material), the concentration–response curves for DCG-IV-induced acute synaptic inhibition and LTD were shifted to the right in slices from cocaine-injected rats compared with saline-injected rats after 5 d of treatment. The maximal inhibitory effect of DCG-IV on EPSCs was also significantly reduced after 5 d of cocaine treatment. These results suggest that repeated cocaine exposure reduces group II mGluR responsiveness at mPFC excitatory synapses.
How might repeated cocaine administration reduce group II mGluR responsiveness in mPFC layer V pyramidal neurons? A straightforward hypothesis is that repeated cocaine administration stimulates postsynaptic endocytotic removal of group II mGluRs from the plasma membrane and thereby leads to an apparent abolition of DCG-IV-LTD. To test this hypothesis, we made surface biotinylation to measure levels of surface group II mGluRs in mPFC slices. However, we found no significant difference between the 5 d cocaine- and saline-treated rats in the surface and total expression of mGluR2 in the mPFC (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Thus, durable alterations of the levels of group II mGluRs after in vivo exposure to cocaine are unlikely to account for the blockade of DCG-IV-LTD observed in the mPFC of cocaine-treated rats.
Repeated cocaine exposure blocks DCG-IV-LTD via activation of D1-like receptors
Considering that cocaine blocks the dopamine reuptake transporter and, thus, acutely increases local dopamine concentrations in brain areas receiving dopaminergic inputs (Koe, 1976; Hyman, 1996), it is therefore possible that activation of dopaminergic receptors in critical brain areas is required for the aforementioned cocaine-induced impairment of DCG-IV-LTD induction. Based on gene sequence and pharmacological profile, dopaminergic receptors could be divided into two families, D1- and D2-like receptors (Civelli et al., 1993). To examine the role of these dopaminergic receptor subtypes, we administrated specific D1- or D2-like receptor antagonists before cocaine injection. As shown in Figure 2, A and C, neither the D1 receptor antagonist SCH23390 (0.5 mg/kg) nor the D2 receptor antagonist raclopride (0.5 mg/kg) had a significant effect on DCG-IV-LTD when administered with saline (SCH23390, 74.5 ± 4.8%, n = 4; raclopride, 72.5 ± 4.3%, n = 4). When coadministered with cocaine, SCH23390, but not raclopride, DCG-IV-LTD was not blocked in slices from 5 d cocaine-treated rats (SCH23390, 72.6 ± 3.9%, n = 6; raclopride, 94.5 ± 3.8%, n = 6) (Fig. 2B,D).
Figure 2.

Repeated cocaine administration impairs DCG-IV-LTD induction via the activation of D1-like dopaminergic receptors. A, B, Summary of experiments showing the induction of DCG-IV-LTD in slices from rats receiving SCH23390 (0.5 mg/kg) 15 min before saline (A) or cocaine (B) injection for 5 d. C, D, Summary of experiments showing the induction of DCG-IV-LTD in slices from rats receiving raclopride (0.5 mg/kg) before the saline (C) or cocaine (D) injection for 5 d. E, F, Summary of experiments showing the induction of DCG-IV-LTD in slices from rats receiving RS102221 (2 mg/kg) before the saline (E) or cocaine (F) injection for 5 d. The superimposed EPSCs in the inset illustrate respective recordings from example experiments taken at time indicated by number. Error bars indicate SEM.
It has been shown that administration of SCH23390 into the mPFC prevents the expression of 3,4-methylenedioxymethamphetamine-induced behavioral sensitization through the activation of 5-HT2C, but not of D1-like receptors (Ramos et al., 2005). To exclude a role for the 5-HT2C receptor activation in the development of cocaine-induced impairment of DCG-IV-LTD, we administrated specific 5-HT2C receptor antagonist RS102221 before cocaine injection. However, in contrast to SCH23390, RS102221 did not significantly affect the impairment of DCG-IV-LTD in slices from 5 d cocaine-treated rats (93.5 ± 4.1%; n = 6) (Fig. 2F). RS102221 also had no significant effect on the induction of DCG-IV-LTD when coadministered with saline (76.8 ± 3.7%; n = 4) (Fig. 2E). Together, these findings indicate that the activation of D1-like receptors is responsible for the inhibitory effect of repeated cocaine administration on DCG-IV-LTD in the mPFC.
Protein kinase C and adenosine A3 receptor activation mediate the effects of cocaine on DCG-IV-LTD
The best-characterized consequence of D1-like receptor stimulation is the activation of adenylyl cyclases, formation of cAMP, and activation of protein kinase A (PKA) (Stoof and Kebabian, 1981). We reasoned that the D1-like receptor/cAMP/PKA signaling pathway could be necessary to the cocaine-induced impairment of DCG-IV-LTD. Because the induction of DCG-IV-LTD also depends on PKA activation (Otani et al., 2002), it is not appropriate for using the PKA inhibitors to directly test this hypothesis. Therefore, we determined the effects of pharmacologically increasing cAMP levels by FSK on the induction of DCG-IV-LTD in slices from control rats. To potentiate levels of FSK-induced synaptic enhancement, the adenosine A1 receptor antagonist DPCPX was coapplied with FSK to block adenosine-induced synaptic inhibition, as reported in the NAc (Manzoni et al., 1998). As we have shown previously (Huang and Hsu, 2006), bath application of FSK (25 μm) plus DPCPX (0.2 μm) produced a rapid and sustained enhancement of EPSCs (165.5 ± 7.9%; n = 6) and also blocked the induction of DCG-IV-LTD (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). Normalizing the responses immediately before DCG-IV application to 100% reveals that the magnitude of LTD induced in the presence of FSK and DPCPX (8.2 ± 3.2%) was significantly less than control LTD (33.5 ± 4.9%). However, contrary results were obtained after treatment with the slices with Sp-8-Br-cAMPS, a membrane permeable and phosphodiesterase-resistant cAMP analog. As shown in supplemental Figure S5B (available at www.jneurosci.org as supplemental material), bath application of Sp-8-Br-cAMPS (100 μm) plus DPCPX (0.2 μm) caused the expected increase in EPSCs (143.5 ± 6.3%) that was strongly inhibited by application of DCG-IV (106.9 ± 5.7%; n = 4). The magnitude of LTD induced by DCG-IV in the presence of Sp-8-Br-cAMPs and DPCPX (36.2 ± 6.1%) was similar to control. These results suggest that the elevated cAMP may impair the induction of DCG-IV-LTD in the mPFC through a PKA-independent mechanism.
Because activation of protein kinase C (PKC) has been shown to reduce dramatically the presynaptic inhibitory function of group II mGluRs at many excitatory synapses (Swartz et al., 1993; Tyler and Lovinger, 1995; Kamiya and Yamamoto, 1997; Macek et al., 1998), we explored the possibility that cocaine-induced impairment of DCG-IV-LTD is caused by an increase in PKC activity. As an initial test of this hypothesis, we examined the effects of direct pharmacological activation of PKC with phorbol esters on the induction of DCG-IV-LTD in slices from control rats. The PKC activator PDBu (10 μm) caused a large and sustained increase in EPSCs (182.3 ± 8.7%; n = 6) and completely abolished the inhibitory effect of DCG-IV at mPFC excitatory synapses (Fig. 3A,D). To exclude the nonspecific actions of phorbol esters, we also examined the effect of DCG-IV after exposure to an inactive phorbol ester, 4αPDD. Application of 4αPDD (10 μm) for 20 min had no significant effect on EPSCs (105.6 ± 3.2%; n = 4) or the LTD induced by DCG-IV (72.5 ± 4.3%) (Fig. 3B,D). Furthermore, the response to PDBu was blocked completely by Bis-I (1 μm), a selective membrane-permeable PKC inhibitor, suggesting that this effect is indeed specific to the activation of PKC (Fig. 3C,D). If an increase in PKC activity is required for cocaine-induced impairment of DCG-IV-LTD, then preventing PKC activation should reverse the ability of DCG-IV to induce LTD in slices from rats treated with cocaine. To test this prediction, Bis-I (0.4 nmol/side) was bilateral infusion into the mPFC before each of the five daily cocaine injections. We found that Bis-I prevents the cocaine-induced impairment of DCG-IV-LTD (Fig. 3F). DCG-IV induced a robust LTD in mPFC layer V pyramidal neurons in slices from rats treated with cocaine plus Bis-I for 5 d to a similar extent as that observed for rats treated with saline plus Bis-I for 5 d (Fig. 3E,F,H). In contrast, the inactive analog Bis-V (0.4 nmol/side) did not significantly affect the effects of cocaine (Fig. 3G,H). These data are consistent with the hypothesis that the inhibitory effects of repeated cocaine treatment on group II mGluR functions in the mPFC are mediated, at least in part, through a PKC-dependent mechanism.
Figure 3.
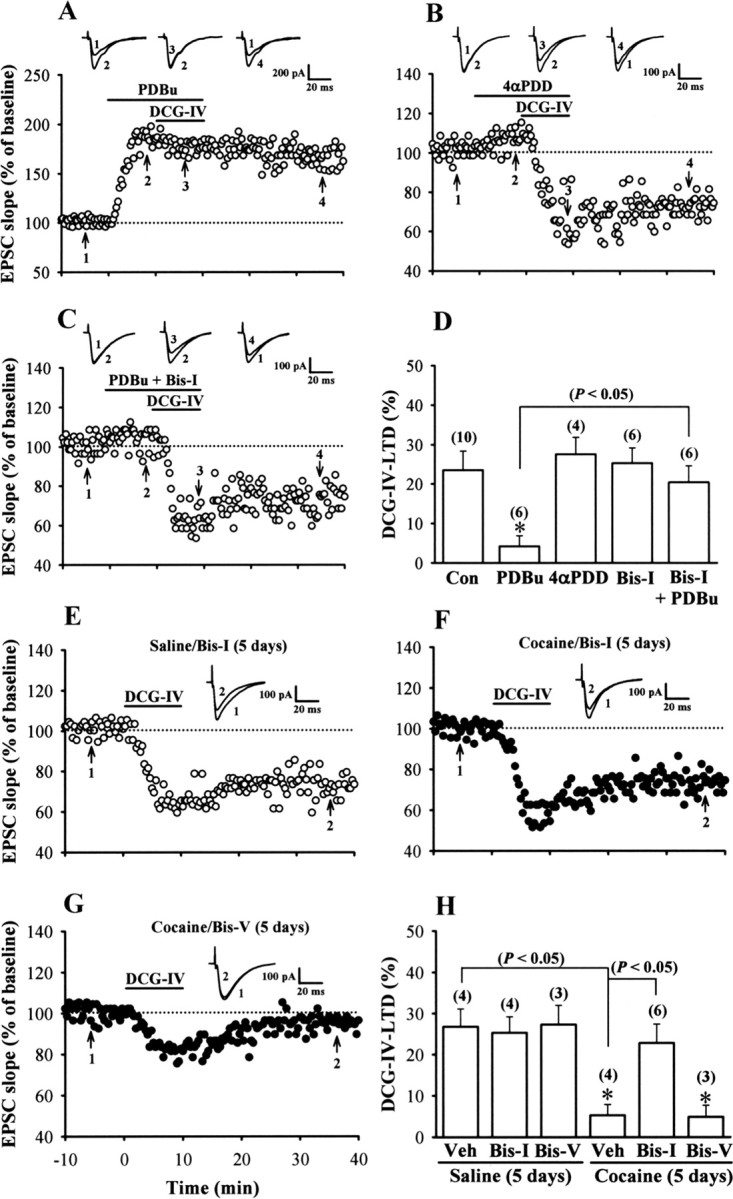
A role for PKC in cocaine-induced impairment of DCG-IV-LTD induction in the mPFC. A, Example of experiments in which PDBu (10 μm) was applied before the induction of LTD by application of DCG-IV (0.2 μm) in slices from control rats. B, Example of experiments in which 4αPDD (10 μm) was applied before application of DCG-IV. C, Example of experiments depicting that coapplication of Bis-I (1 μm) blocked the inhibitory effect of PDBu on DCG-IV-LTD. D, Summary of experiments showing the effects of phorbol esters on DCG-IV-LTD induction. The total number of neurons examined is shown in parentheses. The asterisk indicates significant difference (p < 0.05) compared with the control group. E, F, Example of normalized EPSC slope before and after a brief application of DCG-IV (0.2 μm) for 10 min in slices from rats receiving bilateral intra-mPFC infusion of Bis-I (0.4 nmol/side) before each of the five daily saline (E) or cocaine (F) injections. G, Example of normalized EPSC slope before and after application of DCG-IV (0.2 μm) for 10 min in slices from rats receiving bilateral intra-mPFC infusion of Bis-V (0.4 nmol/side) before the cocaine injection for 5 d. H, Summary of experiments showing the effects of intra-mPFC infusion of PKC inhibitor before cocaine injection on the development of cocaine-induced impairment of DCG-IV-LTD. Asterisks indicate significant difference (p < 0.05) compared with the slices from rats receiving bilateral intra-mPFC infusion of vehicle aCSF (10% DMSO) before the saline injection for 5 d. Error bars indicate SEM.
What is the cellular mechanism that couples repeated cocaine exposure to PKC activation in the mPFC? Because a role for adenosine in the long-lasting changes of synaptic transmission in response to repeated cocaine exposure has been described in the VTA and NAc (Manzoni et al., 1997, 1998) and there is evidence that activation of adenosine A3 receptor can elicit a PKC-dependent inhibition of mGluR function at the Schaffer collateral–CA1 synapses (Macek et al., 1998), the potential involvement of adenosine A3 receptor activation in the development of cocaine-induced impairment of DCG-IV-LTD was investigated. As reported previously (Macek et al., 1998), application of specific adenosine A3 receptor agonist Cl-IB-MECA (1 μm) for 20 min had no significant effect on baseline synaptic transmission at mPFC excitatory synapses but significantly blocked the inhibitory effects of DCG-IV on EPSCs (96.5 ± 3.8%; n = 6) in slices from control rats (Fig. 4A,D). The action of Cl-IB-MECA was blocked completely by either MRS1220 (10 μm), a selective antagonist of adenosine A3 receptor (Fig. 4B,D), or Bis-I (1 μm) (C,D), suggesting this effect is mediated by the activation of adenosine A3 receptors and PKC-dependent signaling pathway. To better characterize the role of adenosine A3 receptors in the development of cocaine-induced impairment of DCG-IV-LTD, MRS1220 (0.5 nmol/side) was bilateral intra-mPFC infusion before each of the five daily cocaine injections. We found that MRS1220 prevents the cocaine-induced impairment of DCG-IV-LTD induction (Fig. 4F). DCG-IV induced a stable LTD in mPFC layer V pyramidal neurons in slices from rats treated with cocaine plus MRS1220 for 5 d to a similar extent as that observed for rats treated with saline plus MRS1220 for 5 d (Fig. 4E–G). These results suggest a role of adenosine A3 receptor-mediated PKC activation in triggering the inhibitory effect of repeated cocaine treatment on the induction of DCG-IV-LTD.
Figure 4.
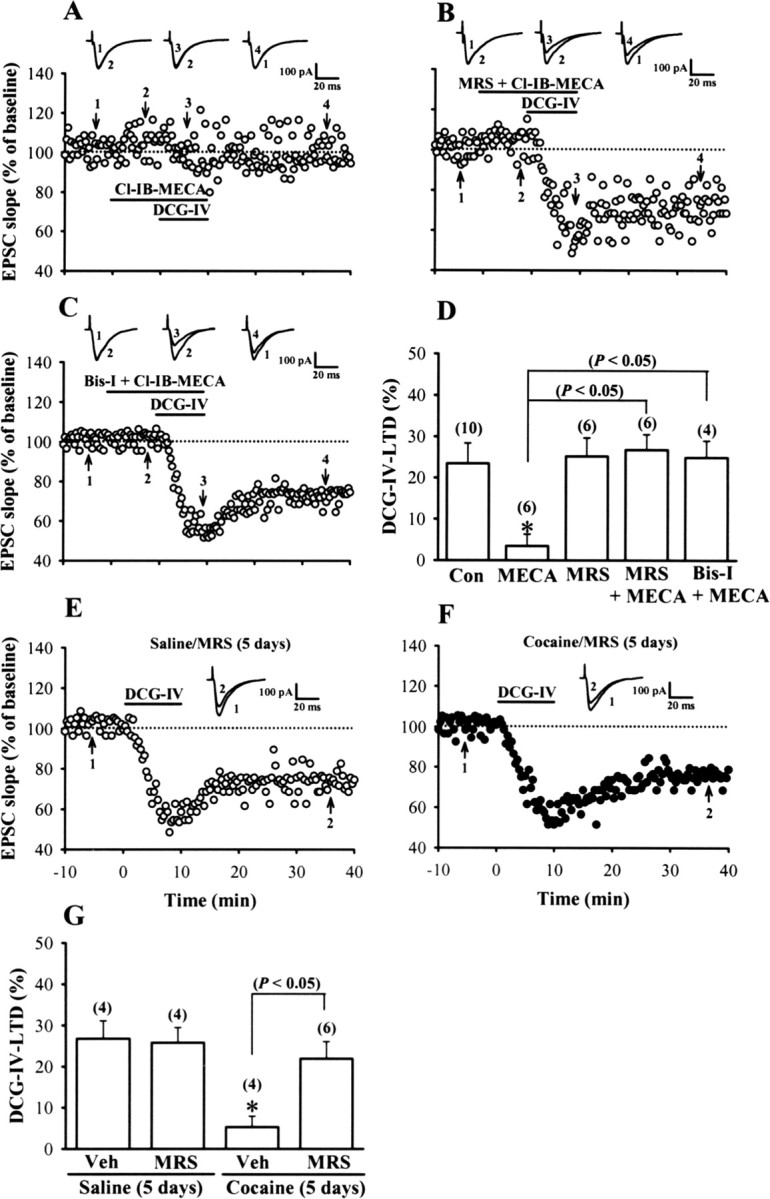
Adenosine A3 receptor activation mediates the effect of repeated cocaine exposure to impair the induction of DCG-IV-LTD. A, Example of experiments in which Cl-IB-MECA (1 μm) was applied before and together with DCG-IV (0.2 μm) in slices from control rats. B, Example of experiments showing coapplication of MRS1220 (MRS; 10 μm) blocked the effect of Cl-IB-MECA on DCG-IV-LTD. C, Example of experiments showing coapplication of Bis-I (1 μm) blocked the effect of Cl-IB-MECA on DCG-IV-LTD. D, Summary of experiments showing the effect of adenosine A3 receptor agonist and antagonist on the induction of DCG-IV-LTD. The total number of neurons examined is shown in parentheses. The asterisk indicates significant difference (p < 0.05) compared with the control (Con) group. E, F, Example of normalized EPSC slope before and after a brief application of DCG-IV (0.2 μm) for 10 min in slices from rats receiving bilateral intra-mPFC infusion of MRS1220 (0.5 nmol/side) before each of the five daily saline (E) or cocaine (F) injections. G, Summary of experiments showing the effects of intra-mPFC infusion of MRS1220 before cocaine injection on the development of cocaine-induced impairment of DCG-IV-LTD. The asterisk indicates significant difference (p < 0.05) compared with the slices from rats receiving bilateral intra-mPFC infusion of vehicle aCSF (10% DMSO) before the saline injection for 5 d. Error bars indicate SEM.
The activation of adenosine A3 receptors could be mediated via an increase in adenosine subsequent to the rise in cAMP after D1-like receptor activation. To determine whether the endogenous adenosine tone in the mPFC is upregulated after repeated cocaine administration, we examined the extent of the increased baseline EPSCs induced by the adenosine A1 receptor antagonist DPCPX (0.5 μm), a widely used protocol to measure endogenous adenosine tone in the hippocampus (Fredholm and Dunwiddie, 1988). The EPSCs were augmented by DPCPX (0.5 μm), an effect that was significantly larger in slices from cocaine-injected rats compared with those from saline-injected rats after 5 d of treatment (saline, 12.3 ± 2.1%, n = 5; cocaine, 24.8 ± 3.7%, n = 7; p < 0.05) (Fig. 5A), but not from rats treated with a single injection of cocaine (1 d) (saline, 9.8 ± 2.3%, n = 4; cocaine, 10.6 ± 2.7%, n = 5; p > 0.05) (Fig. 5B). An increase in the responsiveness of adenosine A1 receptors is not responsible for the augmented response to DPCPX because the inhibition of EPSCs induced by exogenously applied adenosine A1 receptor agonist N6-CPA was not changed (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). The EC50 for inhibition of the EPSCs was 0.17 μm in slices from cocaine-treated rats and was not significantly different in slices from saline-treated rats (0.16 μm).
Figure 5.

Endogenous adenosine tone is greater in slices from repeated cocaine-treated rats. A, Summary of normalized EPSC slope before and after application of adenosine A1 receptor antagonist DPCPX (0.5 μm) in slices from rats treated with saline or cocaine for 5 d. DPCPX increased EPSC to a significantly greater extent in slices from cocaine-treated rats than from saline-treated rats for 5 d. B, Summary of experiments showing the effect of DPCPX on the EPSC slope in slices from rats treated with a single dose of saline or cocaine for 1 d. Error bars indicate SEM.
The preceding results point to an involvement of the upregulation in the endogenous adenosine tone in mediating cocaine-induced impairment of DCG-IV-LTD. We next wanted to know further the source of endogenous adenosine. There are multiple mechanisms that can lead to an increase in the levels of endogenous adenosine, including an augmented metabolism of cAMP to adenosine or an inhibition of adenosine uptake (Rosenberg et al., 1994). To determine whether the metabolism of cAMP is one primary source of adenosine to mediate the impairment of DCG-IV-LTD after repeated cocaine administration, we examined the induction of DCG-IV-LTD in the presence of the cAMP transporter inhibitor, probenecid, or cAMP-dependent phosphodiesterase inhibitor, RO201724 (Beavo and Reifsnyder, 1990). We found that, in the presence of either probenecid (200 μm) or RO201724 (200 μm), DCG-IV normally induced LTD in mPFC pyramidal neurons in slices from rats treated with cocaine for 5 d (probenecid, 76.5 ± 3.7%, n = 6; RO201724, 81.4 ± 4.2%, n = 6), to a similar extent as that observed for rats treated with saline for 5 d (probenecid, 72.8 ± 5.1%, n = 5; RO201724, 73.6 ± 5.3%, n = 5) (Fig. 6A--D). Likewise, application of RO201726 also prevented the forskolin-induced blockade of DCG-IV-LTD (Fig. 6E). Together, these data indicate that phosphodiesterase-mediated degradation of cAMP is a major source of adenosine to account for the cocaine or forskolin-induced impairment of DCG-IV-LTD.
Figure 6.

Probenecid and RO201724 have the same effect to reverse the cocaine-induced impairment of DCG-IV-LTD. A, B, Summary of experiments in which probenecid (200 μm) was applied before the induction of LTD by application of DCG-IV (0.2 μm) in slices from rats treated with saline (A) or cocaine (B) for 5 d. C, D, Summary of experiments in which RO201724 (200 μm) applied before application of DCG-IV in slices from rats treated with saline (C) or cocaine (D) for 5 d. E, Summary of experiments showing coapplication of RO201724 (200 μm) blocked the inhibitory action of FSK (25 μm) and DPCPX (0.2 μm) on the induction of DCG-IV-LTD. Error bars indicate SEM.
Discussion
In the present study, we have demonstrated that repeated in vivo exposure to cocaine impairs the induction of group II mGluR-mediated LTD in layer V pyramidal neurons of the mPFC in vitro. The molecular mechanism mediating this change may involve an increase of endogenous adenosine tone subsequent to the rise in cAMP after D1-like receptor activation, which leads to an enhancement of adenosine A3 receptor-mediated PKC activation and thereby triggers an inhibition of group II mGluR function (Fig. 7). This is the first study, to our knowledge, to correlate the development of behavioral sensitization to cocaine with alterations in LTD of excitatory synaptic transmission within the mPFC.
Figure 7.

Schematic model illustrating the proposed mechanism for the impairment of group II mGluR-mediated LTD induction in the mPFC after repeated cocaine administration. According to this model, repeated cocaine administration increases levels of dopamine (DA) in the mPFC and then leads to increased formation of cAMP via the activation of D1 receptors. Extracellular cAMP is then metabolized to adenosine by phosphodiesterase and ecto 5′-nucleotidase and acts on the adenosine A3 receptors. The activation of adenosine A3 receptors would increase PKC activity and thereby triggers an impairment of group II mGluR function, resulting in an impairment of LTD induced by DCG-IV or LY379268.
Cocaine blocks the reuptake of dopamine, norepinephrine, and serotonin (Koe, 1976; Heikkila et al., 1979; Hyman, 1996). There is substantial evidence revealing a fundamental role for mesolimbic and mesocortical dopamine systems in the development and expression of cocaine-induced behavioral sensitization, although norepinephrine and serotonin systems have also been proposed to be involved (Cunningham et al., 1992; Drouin et al., 2002). Here, we further show that the effect of repeated cocaine administration on the induction of DCG-IV-LTD in the mPFC is likely mediated by the activation of D1-like receptors. This conclusion is mainly based on the observation that the cocaine-induced impairment of DCG-IV-LTD was prevented after pharmacological blockade of D1-like, but not of D2-like receptors. Although these experiments do not allow us to determine where in the brain D1-like receptors are required to trigger the effects of cocaine, these data are entirely consistent with the our current findings showing that the activation of D1-like receptors contributes to the facilitatory effect of repeated exposure to cocaine on LTP induction in mPFC layer V pyramidal neurons (Huang et al., 2007). A previous study showed that administration of SCH23390 into the mPFC prevents the expression of 3,4-methylenedioxymethamphetamine-induced behavioral sensitization through the activation of 5-HT2C, but not of D1-like receptors (Ramos et al., 2005). Given that SCH23390 also exhibits a high affinity to 5-HT2C receptors (Ki ≈ 0.3 nm for D1 receptor vs Ki ≈ 6.3 nm for 5-HT2C receptors) (Millan et al., 2001), it might be thought that the antagonizing effect of SCH23390 on cocaine seen in the present study is in part through its action on 5-HT2C receptors. However, this possibility appears unlikely because the selective 5-HT2C receptor antagonist RS102221 did not significantly affect the cocaine-induced impairment of DCG-IV-LTD.
Although adenosine A3 receptor is expressed in brain neurons (Lopes et al., 2003), the physiological role of this receptor is still poorly understood. The present study has demonstrated that adenosine A3 receptor is responsible for cocaine-induced impairment of DCG-IV-LTD in the mPFC. Results presented here support a link between adenosine A3 receptors and the development of cocaine effects. We found that bilateral infusion of adenosine A3 receptor antagonist MRS1220 into the mPFC before cocaine injection prevents the expression of cocaine-induced impairment of DCG-IV-LTD induction (Fig. 4F). This observation adds a novel role of this receptor in the brain. The finding that Cl-IB-MECA disrupted the normal inhibitory response to DCG-IV is consistent with a previous report showing that activation of A3 adenosine receptor induces a PKC-mediated inhibition of mGluR function at the Schaffer collateral–CA1 synapses (Macek et al., 1998). Our findings that Bis-I, the PKC inhibitor, completely prevented Cl-IB-MECA-induced suppression of DCG-IV-LTD in slices from control rats also indicate that adenosine A3 receptors mainly use PKC signaling pathway to mediate the actions of cocaine in the mPFC. In accordance with this notion, we found that bilateral infusion of Bis-I, but not Bis-V, into the mPFC before cocaine injection almost completely blocks the development of cocaine-induced impairment of DCG-IV-LTD (Fig. 3F). It is also note worthy that a similar cocaine treatment as the one used in this study produces an increase in the levels of PKCβ1 in the mPFC (Steketee et al., 1998). The mechanism by which PKC inhibits group II mGluR function remains unclear. Given that PKC activation has been reported previously to promote endocytosis of several neurotransmitter receptors including AMPA (Chung et al., 2000), somatostatin 2A (Elberg et al., 2002) and GABAA receptors (Herring et al., 2005), it is possible that PKC activation promotes group II mGluR endocytosis and decreases cell surface expression of the receptor. This seems unlikely, however, because the levels of surface mGluR2, a major receptor subtype of group II mGluR to mediate the action of DCG-IV seen in this study, remained unaffected after repeated exposure to cocaine. Macek et al. (1998) have proposed another way in which PKC can inhibit mGluR signaling. They found that activation of PKC inhibits presynaptic inhibitory functions of mGluR at several synapses in the hippocampus through a mechanism by an inhibition of coupling of receptors to G-proteins. This could occur by phosphorylation of a site on either receptors or the G-protein. Furthermore, previous work has demonstrated that elevated activator of G-protein signaling 3 (AGS3) in the PFC contributes to the reduced Giα-coupled receptor (e.g., mGluR2/3 and D2/3) signaling during withdrawal from repeated cocaine administration (Bowers et al., 2004). It has been unclear whether the increased content of AGS3 in the PFC after repeated cocaine administration requires the activation of adenosine A3 receptors and PKC-coupling signaling cascade. Additional biochemical and molecular studies are needed to delineate whether these mechanisms may account for the changes in group II mGluR function observed in the mPFC of repeated cocaine-treated rats.
The estimated affinity of rat adenosine A3 receptors for the endogenous adenosine (Ki = 1 μm) is considerably lower compared with that of A1 (Ki = 10 nm) or adenosine A2A receptors (Ki = 30 nm) (Jacobson et al., 1995). Under normal conditions, the concentrations of endogenous adenosine in the brain are estimated to be 150–200 nm (Dunwiddie and Diao, 1994), which are unlikely to activate a significant fraction of adenosine A3 receptors. Thus, the cellular processes linked to adenosine A3 receptor activation may require an elevation in endogenous adenosine levels. In support of this idea, we found that the endogenous adenosine level is augmented after repeated cocaine treatment. The observation that inhibition of cAMP transport or phosphodiesterase-mediated degradation prevented the cocaine-induced impairment of DCG-IV-LTD suggests that cAMP is the main source for the increased adenosine tone in cocaine-treated rats. The mechanisms underlying the increased level of cAMP and its subsequent conversion to adenosine remain unknown. A putative upregulation of D1-like receptor-driven adenylyl cyclase activity after repeated cocaine exposure, at least in part, could account for the observed result. Indeed, we found that coadministration of D1-like receptor antagonist SCH23390 with cocaine not only prevented the cocaine-induced impairment of DCG-IV-LTD, but also blocked the augmentation of endogenous adenosine levels observed in the drug-treated rats (C.-C. Huang and K.-S. Hsu, unpublished observations).
We found that pharmacologically increasing cAMP levels with adenylyl cyclase activator forskolin caused a large increase in the synaptic responses and also blocked the induction of DCG-IV-LTD. However, activation of PKA by a nonhydrolyzable cAMP analog Sp-8-Br-cAMPS did not affect transient synaptic depression and LTD induced by DCG-IV. Because inhibition of the conversion of cAMP to adenosine with a phosphodiesterase inhibitor RO201724 prevented the forskolin-induced blockade of DCG-IV-LTD, it seems likely that the impairment of DCG-IV-LTD by forskolin was not caused by a direct effect of cAMP, but to an elevation of extracellular concentration of adenosine.
A previous study has reported that the induction of DCG-IV-LTD in mPFC layer V pyramidal neurons requires postsynaptic activation of PKC (Otani et al., 2002). However, we found that DCG-IV still induced a stable LTD in the presence of PKC inhibitor Bis-I in slices from control rats. The basis for the difference in the present results and those of Otani et al. (2002) is uncertain but could be attributable partly to the use of different stimulation sites to elicit synaptic response (stimulating electrode was placed on layer V vs layer I-II of the prelimbic area), resulting in the activation of different synapses that may vary in their mode of synaptic plasticity. It remains to be determined whether this technical difference has a role in explaining the conflicting results. Furthermore, the lack of effect of Bis-I on DCG-IV-LTD induction is unlikely to be attributable to the invalidation of our treatment protocol to inhibit PKC activity, because the same manipulation can effectively prevent the PDBu-induced blockade of DCG-IV-LTD.
In conclusion, our data indicate that repeated cocaine exposure in vivo may impair the induction of group II mGluR-mediated LTD in the mPFC by a PKC-dependent mechanism. Our data also indicate that higher levels of adenosine subsequent to the rise in cAMP after D1-like receptor activation may activate adenosine A3 receptors, increase PKC activation, and thereby block group II mGluR function. Such a change may underlie some of the previously reported changes in glutamatergic transmission in the mPFC that occur after repeated cocaine administration (Williams and Steketee, 2004) and could also be involved in behavioral neuroadaptations associated with cocaine sensitization and craving. Targeting of this drug-induced alteration of synaptic plasticity might be a potential therapeutic strategy for the treatment of drug abuse and addiction.
Footnotes
This work was supported by National Science Council of Taiwan Research Grants NSC95-2321-B-006-001 (principal investigator) to C.C.H. and NSC95-2752-B-006-002-PAE (principal investigator) to K.S.H.
References
- Beavo JA, Reifsnyder DH. Primary sequence of cyclic nucleotide phosphodiesterase isozymes and the design of selective inhibitors. Trends Pharmacol Sci. 1990;11:150–155. doi: 10.1016/0165-6147(90)90066-H. [DOI] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Grammatopoulos DK, Randeva HS, Hillhouse EW, Spiess J. Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J Neurosci. 2003;23:700–707. doi: 10.1523/JNEUROSCI.23-02-00700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, Lanier SM, Kalivas PW. Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron. 2004;42:269–281. doi: 10.1016/s0896-6273(04)00159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Nestler EJ. Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci. 2002;25:610–615. doi: 10.1016/s0166-2236(02)02289-0. [DOI] [PubMed] [Google Scholar]
- Centonze D, Costa C, Rossi S, Prosperetti C, Pisani A, Usiello A, Bernardi G, Mercuri NB, Calabresi P. Chronic cocaine prevents depotentiation at corticostriatal synapses. Biol Psychiatry. 2006;60:436–443. doi: 10.1016/j.biopsych.2005.11.018. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL. Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci. 2000;20:7258–7267. doi: 10.1523/JNEUROSCI.20-19-07258.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O, Bunzow JR, Grandy DK. Molecular diversity of the dopamine receptors. Annu Rev Pharmacol Toxicol. 1993;33:281–307. doi: 10.1146/annurev.pa.33.040193.001433. [DOI] [PubMed] [Google Scholar]
- Conductier G, Crosson C, Hen R, Bockaert J, Compan V. 3,4-N-methlenedioxymethamphetamine-induced hypophagia is maintained in 5-HT1B receptor knockout mice, but suppressed by the 5-HT2C receptor antagonist RS102221. Neuropsychopharmacology. 2005;30:1056–1063. doi: 10.1038/sj.npp.1300662. [DOI] [PubMed] [Google Scholar]
- Cunningham KA, Paris JM, Goeders NE. Chronic cocaine enhances serotonin autoregulation and serotonin uptake binding. Synapse. 1992;11:112–123. doi: 10.1002/syn.890110204. [DOI] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc Natl Acad Sci USA. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25:936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S, Tassin JP. α1b-Adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J Neurosci. 2002;22:2873–2884. doi: 10.1523/JNEUROSCI.22-07-02873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J Pharmacol Exp Ther. 1994;268:537–545. [PubMed] [Google Scholar]
- Elberg G, Hipkin RW, Schonbrunn A. Homologous and heterologous regulation of somatostatin receptor 2. Mol Endocrinol. 2002;16:2502–2514. doi: 10.1210/me.2002-0207. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, Manzoni OJ. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–6945. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Dunwiddie TV. How does adenosine inhibit transmitter release? Trends Pharmacol Sci. 1988;9:130–134. doi: 10.1016/0165-6147(88)90194-0. [DOI] [PubMed] [Google Scholar]
- Harrison JM, Allen RG, Pellegrino MJ, Williams JT, Manzoni OJ. Chronic morphine treatment alters endogenous opioid control of hippocampal mossy fiber synaptic transmission. J Neurophysiol. 2002;87:2464–2470. doi: 10.1152/jn.2002.87.5.2464. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Cabbat FS, Manzino L, Duvoisin RC. Rotational behavior induced by cocaine analogs in rats with unilateral 6-hydroxydopamine lesions of the substantia nigra: dependence upon dopamine uptake inhibition. J Pharmacol Exp Ther. 1979;211:189–194. [PubMed] [Google Scholar]
- Herring D, Huang R, Singh M, Dillon GH, Leidenheimer NJ. PKC modulation of GABAA receptor endocytosis and function is inhibited by mutation of a dileucine motif within the receptor β2 subunit. Neuropharmacology. 2005;48:181–194. doi: 10.1016/j.neuropharm.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Caulder T, Lupica CR. Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci. 2003;23:4815–4820. doi: 10.1523/JNEUROSCI.23-12-04815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol Pharmacol. 2006;69:846–856. doi: 10.1124/mol.105.018093. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lin HJ, Hsu KS. Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex. 2007 doi: 10.1093/cercor/bhl096. in press. [DOI] [PubMed] [Google Scholar]
- Hyman SE. Addiction to cocaine and amphetamine. Neuron. 1996;16:901–904. doi: 10.1016/s0896-6273(00)80111-7. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ. Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci. 1998;18:10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Kim HO, Siddiqi SM, Olah ME, Stiles GL, von Lubitz DKJE. A3-adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future. 1995;20:689–699. doi: 10.1358/dof.1995.020.07.531583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fibre synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- Koe BK. Molecular geometry of inhibitors of the uptake of catecholamines and serotonin in synaptosomal preparations of rat brain. J Pharmacol Exp Ther. 1976;199:649–661. [PubMed] [Google Scholar]
- Li Y, Vartanian AJ, White FJ, Xue CJ, Wolf ME. Effects of the AMPA receptor antagonist NBQX on the development and expression of behavioral sensitization to cocaine and amphetamine. Psychopharmacology. 1997;134:266–276. doi: 10.1007/s002130050449. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Rebola N, Pinheiro PC, Richardson PJ, Oliverira CR, Cunha RA. Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport. 2003;14:1645–1648. doi: 10.1097/00001756-200308260-00021. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Michel JM, Bockaert J. Metabotropic glutamate receptors in the rat nucleus accumbens. Eur J Neurosci. 1997;9:1514–1523. doi: 10.1111/j.1460-9568.1997.tb01506.x. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Pujalte D, Williams J, Bockaert J. Decreased presynaptic sensitivity to adenosine after cocaine withdrawal. J Neurosci. 1998;18:7996–8002. doi: 10.1523/JNEUROSCI.18-19-07996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci. 1998;18:6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Ahmed SH, Blank T, Spiess J, Koob GF, Siggins GR. Chronic morphine treatment alters NMDA receptor-mediated synaptic transmission in the nucleus accumbens. J Neurosci. 1999;19:9081–9089. doi: 10.1523/JNEUROSCI.19-20-09081.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Newman-Tancredi A, Quentric Y, Cussac D. The “selective” dopamine D1 receptor antagonist, SCH23390, is a potent and high efficacy agonist at cloned human 5-HT2C receptors. Psychopharmacology. 2001;156:58–62. doi: 10.1007/s002130100742. [DOI] [PubMed] [Google Scholar]
- Monn JA, Valli MJ, Massey SM, Hansen MM, Kress TJ, Wepsiec JP, Harkness AR, Grutsch JL, Jr, Wright RA, Johnson BG, Andis SL, Kingston A, Tomlinson R, Lewis R, Griffey KR, Tizzano JP, Schoepp DD. Synthesis, pharmacological characterization, and molecular modeling of heterobicyclic amino acids related to (+)-2-aminobicyclo[3.1.0] hexane-2,6-dicarboxylic acid (LY354740): identification of two new potent, selective, and systemically active agonists for group II metabotropic glutamate receptors. J Med Chem. 1999;42:1027–1040. doi: 10.1021/jm980616n. [DOI] [PubMed] [Google Scholar]
- Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434:117–165. doi: 10.1016/0165-0173(87)90011-7. [DOI] [PubMed] [Google Scholar]
- Otani S, Auclair N, Desce JM, Roisin MP, Crepel F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Daniel H, Takita M, Crepel F. Long-term depression induced by postsynaptic group II metabotropic glutamate receptors linked to phospholipase C and intracellular calcium rises in rat prefrontal cortex. J Neurosci. 2002;22:3434–3444. doi: 10.1523/JNEUROSCI.22-09-03434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos M, Goni-Allo B, Aguirre N. Administration of SCH 23390 into the medial prefrontal cortex blocks the expression of MDMA-induced behavioral sensitization in rats: an effect mediated by 5-HT2C receptor stimulation and not by D1 receptor blockade. Neuropsychopharmacology. 2005;30:2180–2191. doi: 10.1038/sj.npp.1300735. [DOI] [PubMed] [Google Scholar]
- Reith ME, Meisler BE, Sershen H, Lajtha A. Structural requirements for cocaine congeners to interact with dopamine and serotonin uptake sites in mouse brain and to induce stereotyped behavior. Biochem Pharmacol. 1986;35:1123–1129. doi: 10.1016/0006-2952(86)90148-6. [DOI] [PubMed] [Google Scholar]
- Robbe D, Bockaert J, Manzoni OJ. Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci. 2002;16:2231–2235. doi: 10.1046/j.1460-9568.2002.02273.x. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Knowles R, Knowles KP, Li Y. β-adrenergic receptor-mediated regulation of extracellular adenosine in cerebral cortex in culture. J Neurosci. 1994;14:2953–2965. doi: 10.1523/JNEUROSCI.14-05-02953.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- Sherwood NM, Timiras PS. Berkeley: University of California; 1970. A stereotaxic atlas of the developing rat brain. [Google Scholar]
- Steketee JD. Neurotransmitter systems of the medial prefrontal cortex: potential role in sensitization to psychostimulants. Brain Res Brain Res Rev. 2003;41:203–228. doi: 10.1016/s0165-0173(02)00233-3. [DOI] [PubMed] [Google Scholar]
- Steketee JD, Rowe LA, Chandler LJ. The effects of acute and repeated cocaine injections on protein kinase C activity and isoform levels in dopaminergic brain regions. Neuropharmacology. 1998;37:339–347. doi: 10.1016/s0028-3908(98)00022-7. [DOI] [PubMed] [Google Scholar]
- Stoof JC, Kebabian JW. Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366–368. doi: 10.1038/294366a0. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Tyler EC, Lovinger DM. Metabotropic glutamate receptor modulation of synaptic transmission in corticostriatal co-cultures: role of calcium influx. Neuropharmacology. 1995;34:939–952. doi: 10.1016/0028-3908(95)00066-f. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. doi: 10.1016/s0301-0082(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Williams JM, Steketee JD. Cocaine increases medial prefrontal cortical glutamate overflow in cocaine-sensitized rats: a time course study. Eur J Neurosci. 2004;20:1639–1646. doi: 10.1111/j.1460-9568.2004.03618.x. [DOI] [PubMed] [Google Scholar]
- Williams JM, Steketee JD. Effects of repeated cocaine on the release and clearance of dopamine within the rat medial prefrontal cortex. Synapse. 2005;55:98–109. doi: 10.1002/syn.20093. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Yang CH, Huang CC, Hsu KS. Behavioral stress modifies hippocampal synaptic plasticity through corticosterone-induced sustained extracellular signal-regulated kinase/mitogen-activated protein kinase activation. J Neurosci. 2004;24:11029–11034. doi: 10.1523/JNEUROSCI.3968-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zho WM, You JL, Huang CC, Hsu KS. The group I metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine induces a novel form of depotentiation in the CA1 region of the hippocampus. J Neurosci. 2002;22:8838–8849. doi: 10.1523/JNEUROSCI.22-20-08838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]