

Abstract
Well-documented experimental evidence from both in vitro and in vivo models of stroke strongly supports the critical involvement of NMDA receptor-mediated excitotoxicity in neuronal damage after stroke. Despite this, the results of clinical trials testing NMDA receptor antagonists as neuroprotectants after stroke and brain trauma have been discouraging. Here, we report that in mature cortical cultures, activation of either synaptic or extrasynaptic NR2B-containing NMDA receptors results in excitotoxicity, increasing neuronal apoptosis. In contrast, activation of either synaptic or extrasynaptic NR2A-containing NMDA receptors promotes neuronal survival and exerts a neuroprotective action against both NMDA receptor-mediated and non-NMDA receptor-mediated neuronal damage. A similar opposing action of NR2B and NR2A in mediating cell death and cell survival was also observed in an in vivo rat model of focal ischemic stroke. Moreover, we found that blocking NR2B-mediated cell death was effective in reducing infarct volume only when the receptor antagonist was given before the onset of stroke and not 4.5 h after stroke. In great contrast, activation of NR2A-mediated cell survival signaling with administration of either glycine alone or in the presence of NR2B antagonist significantly attenuated ischemic brain damage even when delivered 4.5 h after stroke onset. Together, the present work provides a molecular basis for the dual roles of NMDA receptors in promoting neuronal survival and mediating neuronal damage and suggests that selective enhancement of NR2A-containing NMDA receptor activation with glycine may constitute a promising therapy for stroke.
Keywords: stroke, focal ischemia, NMDA receptor, glycine, neuroprotection, rat
Introduction
Neurodegeneration after stroke is becoming a major cause of morbidity and mortality in today's society. Understanding the molecular mechanisms mediating stroke-related brain damage, and the use of this knowledge in the development of effective therapeutics, are urgently required to address this problem. Although molecular and experimental animal studies have consistently demonstrated that overactivation of the NMDA subtype of glutamate receptors is the primary step leading to neuronal injury after insults of stroke and brain trauma (Lipton and Rosenberg, 1994; Mattson, 1997; Lee et al., 1999; Arundine and Tymianski, 2004), several large-scale clinical trials have failed to find the expected efficacy of NMDA receptor antagonists in reducing brain injuries (Lee et al., 1999; Ikonomidou and Turski, 2002; Kemp and McKernan, 2002). The reasons underlying the apparent contradiction between basic research results and clinical trials remain unknown but are likely multifactorial (Albensi et al., 2004). NMDA receptors are believed to be tetrameric protein complexes comprised of NR1 subunits with at least one type of NR2 subunit. Different NR2 subunits confer distinct electrophysiological and pharmacological properties on the receptors and couple them with different signaling machineries. For instance, NR2A- and NR2B-containing NMDA receptor subtypes have opposing roles in influencing the direction of synaptic plasticity (Liu et al., 2004; Massey et al., 2004). Because NR2A and NR2B are the predominant NR2 subunits in the adult forebrain, where stroke most frequently occurs, we hypothesize that NR2A- and NR2B-containing NMDA receptors may have differential roles in supporting neuronal survival and mediating neuronal death and hence have opposing impacts on excitotoxic brain damage after acute brain insults such as stroke and brain trauma. Such opposing actions may explain, at least in part, the failure of NMDA receptor antagonism-based clinical trials of stroke and provide a scientific basis for developing novel and effective NMDA receptor-based stroke therapies.
Materials and Methods
Primary culture of cortical neurons.
Dissociated cultures of rat cortical neurons were prepared from 18-d-old Sprague Dawley rat embryos as described previously (Mielke and Wang, 2005). To obtain mixed cortical cultures enriched with neurons, uridine (10 μm) and 5-Fluor-2′-deoxyuridine (10 μm) were added to the culture medium at 3 d in vitro (DIV) and maintained for 48 h to inhibit non-neuronal cell proliferation before the cultures were returned to the normal culture medium. Mature neurons (11–14 DIV) were used for experiments. Mouse cortical cultures were prepared using embryos at 18 d postcoitum from litters resulting from heterozygote NR2A+/− or NR2B+/− matings (Sakimura et al., 1995; Kutsuwada et al., 1996). To obtain homozygous and wild-type (WT) littermate control neuronal cultures, cortical cells from individual embryos were plated separately. Genotyping was performed as described previously (Tovar et al., 2000; Thomas et al., 2006) using tail samples collected from each embryo. To induce neuronal apoptosis, cortical cultures were stimulated with NMDA (50 μm) and glycine (10 μm) for 20 min, or STS (100 nm) for 1 h in Mg2+-free extracellular solution (ECS) containing the following (in mm): 25 HEPES acid, 140 NaCl, 33 glucose, 5.4 KCl, and 1.3 CaCl2, with pH 7.35 and osmolarity 320–330 mOsm. Specific blockade of synaptic NMDA receptors was achieved by treatment with (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (10 μm) in the presence of bicuculline (50 μm) for 10–15 min in Mg2+-free ECS, followed by thorough wash with ECS containing 1 mm MgCl2 (normal ECS) to remove any trace of MK-801. NR2A-specific antagonist NVP-AAM077 (0.4 μm; generous gift from YP Auberson, Novartis Pharma AG, Basel, Switzerland) or NR2B-specific antagonist Ro 25–6981 (0.5 μm) was added to the bath medium 10 min before and throughout the treatments.
Assessment of neuronal death.
Necrotic neuronal death was quantified by measuring lactate dehydrogenase (LDH) release 20 h after treatments using a Cyto Tox 96 assay kit (Promega, Madison, WI). Apoptotic neuronal death was determined either by visualizing neurons stained with Hoechst-33342 or using a cell ELISA assay. For visualizing apoptotic neurons, Hoechest-33342 (10 μg/ml) was added to the culture medium 20 h after treatments and incubated for 45 min at 37°C. Images were taken with a Leica DMIRE2 fluorescence microscope. Cells with condensed or fragmented chromatin were considered apoptotic. These observations were quantitated by double-blind counting of apoptotic and total neurons in each visual field and expressed as percentage apoptosis. Cell ELISA quantitative assessment of neuronal apoptosis was performed 20 h after treatments using a Cell Death Detection ELISAPLUS Kit (Roche Products, Welwyn Garden City, UK). Absorbance readings were measured using a spectrophotometric microplate reader. Data analyses were performed according to the instructions of the manufacturer. Data are expressed as the difference in apoptosis relative to control and are expressed as a percentage.
Recording of miniature EPSCs and whole-cell NMDA currents.
Neurons on coverslips (11 DIV) were transferred to a recording chamber that was continuously perfused with normal ECS. Bicuculline (10 μm) and tetrodotoxin (0.5 μm) were added to isolate action potential-independent miniature EPSCs (mEPSCs). Patch pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) and filled with an intracellular solution (pH 7.2; 300–310 mOsm) composed of the following (in mm): 140 CsCl gluconate, 0.1 CaCl2, 10 HEPES, 2 MgCl2, 10 BAPTA, and 4 ATP. A MultiClamp 700A amplifier (Molecular Devices, Palo Alto, CA) was used for the recording. The series resistance was monitored throughout each recording and if it varied by >10%, the recording was rejected. No electronic compensation for series resistance was used. Whole-cell patch-clamp recordings were performed under voltage-clamp mode at a holding membrane potential of −60 mV. Recordings were low-pass filtered at 2 kHz, sampled at 10 kHz, and stored as data files using Clampex 8.0 (Molecular Devices). Synaptic events were analyzed off-line using the Mini Analysis Program 6.0 (Synaptosoft, Decatur, GA). During recording, Mg2+-free ECS was used so that mEPSCs comprising both AMPA and NMDA receptor-mediated components could be measured. NMDA receptor antagonists (APV, NVP-AAM077, or Ro 25–6981) were bath applied for at least 10 min to obtain sufficient recording data for analysis after achieving a stable level of NMDA receptor blockade. Synaptic events before and after application of NMDA receptor antagonists were automatically detected from computer-stored recordings using the same detection parameters in Mini Analysis Program. Subtraction of averaged traces was done in Microsoft Excel (Microsoft, Seattle, WA). Whole-cell NMDA currents were recorded at a holding membrane potential of −60 mV under voltage-clamped configuration, and the currents were evoked by NMDA at concentrations specified in the results in Mg2+-free ECS using a fast perfusion system (Warner Instruments, Hamden, CT).
Western blotting.
Twelve hours after treatments, proteins were extracted from neurons using a lysis buffer composed of 150 mm NaCl, 50 mm Tris, pH 7.4, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 1 mm EDTA, 1 mm Na3VO4, 10 μg/ml each of leupeptin and aprotinin, and 1 mm phenylmethylsulfonyl fluoride. To determine the state of Akt phosphorylation, the samples were separated on 10% SDS-PAGE gels, transferred to polyvinylidene difluoride (PVDF) membrane, and immunoblotted with anti-phosphoSer473-Akt antibody (Cell Signaling Technology, Beverly, MA). The same membrane was stripped and reprobed with anti-Akt antibody (Cell Signaling Technology). To determine the activity of caspase-3, the samples were separated on 15% SDS-PAGE gels and transferred to PVDF membranes, which were then sequentially probed with antibodies against cleaved caspase-3 (Asp175; Cell Signaling Technology) and β-tubulin (Sigma, St. Louis, MO).
Experimental stroke in vitro and in vivo.
Oxygen-glucose deprivation (OGD) was achieved by transferring cortical cultures to an anaerobic chamber (Thermo EC) containing a 5% CO2, 10% H2, and 85% N2 (<0.01% O2) atmosphere (Goldberg and Choi, 1993; Aarts et al., 2002; Mielke and Wang, 2005). Cultured cells were then washed three times with glucose-free bicarbonate-buffered solution (deoxygenated in the anaerobic chamber for 30 min before use) and maintained anoxic for 1 h at 37°C. OGD was terminated by removing the cultures from the chamber, washing them twice with normal ECS, and returning them to the original growth conditions until additional assay.
Transient cerebral focal ischemia was produced by middle cerebral artery occlusion (MCAo) as described previously (Bederson et al., 1986; Longa et al., 1989; Aarts et al., 2002). Briefly, male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) weighing ∼300 g were anesthetized, and MCAo was achieved by introducing a 3–0 monofilament suture into the MCA via the internal carotid artery. Body temperature was maintained at 37.0 ± 0.5°C, and blood pressure and blood gases were monitored during the experiments. Animals were killed 24 h after MCAo onset. Cerebral infarction was analyzed using brain sections stained with hematoxylin and eosin (H&E) or 2,3,5-triphenyltetrazolium chloride. Ten minutes before the animals were killed, two tests [the postural reflex test to examine upper body posture (Bederson et al., 1986) and the forelimb placing test to examine sensorimotor integration in forelimb placing responses to visual, tactile, and proprioceptive stimuli (De Ryck et al., 1989)] were performed to grade neurological function on a scale of 0–12 (0, normal; 12, worst). In the pretreatment study, a single bolus of drugs was infused intravenously 30 min before a 1 h MCAo. For posttreatment experiments, animals were subjected to a 1.5 h MCAo, and drug treatments were then given via intraperitoneal injection 3 h after reperfusion (4.5 h after the onset of MCAo).
Data analysis.
Data are expressed as mean ± SEM. ANOVA was used for comparison among multiple groups, followed by the Holm–Sidak test for comparison between two groups. Statistical significance was defined as p < 0.05.
Results
We first tested our hypothesis that NR2A- and NR2B-containing NMDA receptors exert differential roles in mediating NMDA-induced neuronal death in rat cortical cultures of 11–14 DIV using subunit-specific NMDA receptor antagonists: NVP-AAM077, which preferentially inhibits NR2A-containing receptors at concentrations of 0.4–1 μm (Liu et al., 2004; Massey et al., 2004; Tigaret et al., 2006), and Ro 25–6981, which specifically blocks NR2B-containing receptors (Fischer et al., 1997; Mutel et al., 1998). Because the subunit specificity of these antagonists, NVP-AAM077 in particular, is debated (Berberich et al., 2005; Weitlauf et al., 2005), and because the expression of NR2A and NR2B varies depending on the developmental stage of the neurons (Monyer et al., 1994; Zhou and Baudry, 2006), it is critically important for us to determine whether both subtypes of NMDA receptors exist in these neurons and, if so, whether NVP-AAM077 and Ro 25–6981 function as respective subunit-selective antagonists. To do this, we examined the ability of these antagonists to inhibit whole-cell currents evoked with a rapid and brief application of NMDA (50 μm NMDA, 10 μm glycine, 5 μm strychnine). As shown in Figure 1A, bath application of either NVP-AAM077 (0.4 μm) or Ro 25–6981 (0.5 μm) alone produced a partial, but significant, blockade of the NMDA-induced currents. To determine the potentially nonselective, overlapping blockade of these antagonists, we applied the two antagonists sequentially and compared the degree of blockade produced by each antagonist alone and when applied after the other antagonist (Fig. 1A,B). NVP-AAM077 produced a similar blockade when applied alone (42.9 ± 5.9%) and after Ro 25–6981 blockade (43.4 ± 12.4%), confirming that Ro 25–6981, at the concentration used here, is a very specific NR2B subunit antagonist with little effect on NR2A-containing receptors (Fischer et al., 1997; Mutel et al., 1998), and that NVP-AAM077 effectively blocked NR2A-containing receptor-mediated currents (Liu et al., 2004; Massey et al., 2004; Tigaret et al., 2006). After NVP-AAM077 blockade, NMDA current inhibition by Ro 25–6981 was reduced by ∼5.6% (34.6 ± 1.8% when applied alone vs 29.0 ± 3.3% after NVP-AAM077; p = 0.19). Although the reduction did not reach statistical significance, as suggested by several recent studies (Berberich et al., 2005; Weitlauf et al., 2005; Tigaret et al., 2006), it may reflect a small degree of cross-inhibition of NR2B receptors by NVP-AAM077 in these neurons under our experimental conditions. However, several recent studies have demonstrated that such a marginal effect may not significantly alter the utility of NVP-AAM077 as an NR2A-subunit preferential antagonist under experimental conditions such as in the present study (Liu et al., 2004; Massey et al., 2004; Tigaret et al., 2006). Previous studies have indicated that Ro 25–6981 specifically blocks NR1/NR2B diheteromeric NMDA receptors with little effect on NR1/NR2A/NR2B triheteromeric NMDA receptors (Hatton and Paoletti, 2005). The relative potency of NVP-AAM077 for NR1/NR2A/NR2B versus NR1/NR2A receptors remains to be determined; however, considering that NVP-AAM077 is a competitive antagonist at glutamate binding sites, and that both glutamate binding sites must be occupied to open the channel (Clements and Westbrook, 1991, 1994), it is likely able to block all NR2A-containing NMDA receptors. Together, our results suggest that both NR2A-containing and NR1/NR2B receptor subtypes are expressed in these neurons and, under our experimental conditions, the two antagonists selectively block respective receptor subtypes with insignificant cross-receptor subtype antagonism at the concentrations used in the present study.
Figure 1.

Functional NR2A- and NR2B-containing NMDA receptors are present in cultured neurons and are preferentially blocked by their respective antagonists. Whole-cell recording was performed at a holding membrane potential of −60 mV in an extracellular solution supplemented with 10 μm CNQX, 0.5 μm TTX, and 10 μm bicuculline. A, Example traces of whole-cell currents evoked by a brief perfusion of 50 μm NMDA (plus 10 μm glycine and 5 μm strychnine) from a multibarrel fast perfusion system in the absence or presence of specific NR2A (NVP-AAM077, 0.4 μm), NR2B antagonists (Ro 25–6981, 0.5 μm), or both. The percentage blockade of the NMDA-induced currents by sequential application of these two antagonists is summarized in the histogram in B. Preapplication of Ro 25–6981 did not alter the percentage blockade produced by NVP-AAM077 (p = 0.97; in the absence vs the presence of Ro 25–6981), whereas preapplication of NVP-AAM077 produced a small, albeit statistically insignificant, reduction in the percentage blockade of the currents produced by Ro 25–6981 (5.6%; p = 0.19).
Antagonist effects on NMDA receptor-mediated neuronal death were investigated next. Because NMDA receptor stimulation can induce both necrotic and apoptotic neuronal death (Bonfoco et al., 1995), we first investigated subunit roles in necrotic neuronal death. Neuronal death was induced by incubating cultures with 50 μm NMDA plus 10 μm glycine for 20 min (NMDA-mediated excitotoxicity) and 20 h after treatments, LDH release from cells was measured. LDH release is an accepted marker of loss of plasma membrane integrity, a major indicator of necrotic neuronal death. As shown in Figure 2A, NMDA treatment produced a significant increase in the release of LDH. The NMDA-induced neuronal damage was a result of specific activation of NMDA receptors, because it was fully blocked by the NMDA receptor antagonist APV (50 μm; data not shown). To determine the individual roles of NR2A-containing and NR1/NR2B NMDA receptor subtypes in NMDA-induced neuronal necrosis, we compared the effects of a blockade of these receptors with subunit-specific antagonists. Bath application of NR2B antagonist Ro 25–6981 (0.5 μm), but not NR2A-containing receptor antagonist NVP-AAM077 (0.4 μm), prevented NMDA-induced neuronal necrosis, indicating the critical involvement of NR1/NR2B, but not NR2A-containing, NMDA receptor subtypes in mediating the necrotic component of NMDA receptor-mediated neuronal death.
Figure 2.

NR2A- and NR2B-containing NMDA receptors exert opposing effects on NMDA-induced excitotoxic neuronal damage. Cortical neuronal cultures were treated without NMDA (control) or with NMDA (50 μm plus 10 μm glycine) for 20 min and examined for neuronal cell death after 20 h. A, NMDA-induced necrotic neuronal injury was mediated by NR1/NR2B- but not NR2A-containing NMDA receptors. Necrotic neuronal injury was quantitatively determined by measuring LDH release. NR2B preferential antagonist Ro 25–6981 (Ro; 0.5 μm), but not NR2A preferential antagonist NVP-AAM077 (NVP; 0.4 μm), prevented NMDA-induced necrosis. Data are presented as the difference in LDH levels as a percentage of control. **p < 0.01 compared with nontreated control; n = 18 tissue culture wells from three separate experiments for each group. B, C, NR2A-containing and NR1/NR2B receptors exert opposing effects on NMDA-induced neuronal apoptosis. Representative images of Hoechst-33342-stained neurons in B illustrate the differential effects of coapplication of NMDA with NR2A antagonist NVP-AAM077 (NVP; 0.4 μm) or NR2B antagonist Ro 25–6981 (Ro; 0.5 μm) on NMDA-induced neuronal apoptosis. NMDA stimulation produced neuronal damage such as chromatin condensation and/or fragmentation, which were aggravated in the presence of NVP-AAM077 but eliminated in the presence of Ro 25–6981. The cell death ELISA assay in C quantifies the differential effects of NVP-AAM077 and Ro 25–6981 on NMDA-induced neuronal apoptosis. Data are presented as the difference in apoptosis levels as a percentage of control. **p < 0.01 compared with nontreated control; #p < 0.05 and ##p < 0.01, compared with NMDA treatment alone; n = 18 tissue culture wells from three separate experiments for each group. D, NR2A-containing and NR1/NR2B receptors have opposing actions on cell survival (Akt activation) and death (caspase-3 activation) signaling pathways. Top, Cell lysates from cultured neurons treated as indicated were sequentially immunoblotted with antibodies specific to Akt phosphorylated on serine 473, the active form of the enzyme (P-Akt), and total Akt (Akt). Bottom, Cell lysates were sequentially immunoblotted with antibodies that specifically recognize cleaved caspase-3 (activated caspase-3; Casp3) and β-tubulin as a load control (Tubulin).
We then turned our attention to apoptosis. Neuronal apoptosis was determined 20 h after treatment by microscopic visualization of neurons after nuclear staining with Hoechst-33342 and quantifying DNA damage using a Cell ELISA assay. Consistent with previous reports (Budd and Lipton, 1999; Hardingham et al., 2002; Wang et al., 2004;), NMDA treatment induced characteristic neuronal apoptosis as indicated by an increase in the proportion of neurons displaying nuclear condensation and/or fragmentation (Fig. 2B). Neuronal apoptosis was confirmed using a quantitative biochemical measurement of intranucleosomal fragmentation (Fig. 2C). Bath application of Ro 25–6981 (0.5 μm) prevented NMDA-induced neuronal apoptosis, indicating the apoptotic component of neuronal injury is also primarily mediated by the NR1/NR2B NMDA receptor subtype. In striking contrast, we found that application of NVP-AAM077 (0.4 μm) failed to block, and in fact significantly enhanced, NMDA-induced apoptosis (Fig. 2B,C) (p < 0.05 compared with NMDA alone). These unexpected results strongly suggest that activation of NR2A-containing NMDA receptors may activate cell survival-promoting signals, counteracting the apoptotic action produced by NR1/NR2B receptors.
One well-characterized neuronal survival signaling pathway activated by NMDA receptors is the serine/threonine kinase Akt/protein kinase B-dependent cell survival signaling pathway (Dudek et al., 1997), and inhibition of this kinase activity contributes to NMDA receptor-mediated apoptosis (Chalecka-Franaszek and Chuang, 1999). As shown in Figure 2D, treatment of neurons with NMDA resulted in a significant reduction in Akt kinase activity as gauged by the dephosphorylation of Serine473 (Coffer et al., 1998; Wang et al., 2004). Blocking NR2A receptors with NVP-AAM077 not only failed to prevent, but slightly increased, the NMDA-induced reduction in Akt activity. In contrast, after NR2B blockade, the NMDA-induced Akt inhibition was virtually eliminated (Fig. 2D). Activation of certain caspases, such as caspase-3 and -7 (Okamoto et al., 2002; Wang et al., 2004), has been suggested to be a critical step in NMDA-induced neuronal apoptosis. In the present study, we found that NMDA treatment dramatically increased the level of the activated form of caspase-3, as shown by Western blots using an antibody that specifically recognizes activated/cleaved caspase-3 (Fig. 2D). The activation of the caspase-mediated death signal was inhibited by blocking NR2B receptors but slightly enhanced after NR2A blockade. These unexpected results strongly suggest that activation of NR2A-containing NMDA receptors exerts a cell survival-promoting effect that counteracts the apoptotic action produced by NR2B-containing receptors.
To corroborate results obtained with the pharmacological blockade, we investigated the differential roles of NMDA receptor subtypes in mediating NMDA receptor-induced apoptosis in mature cortical cultures prepared from NR2A (ε1) or NR2B (ε2) knock-out mice (Sakimura et al., 1995; Kutsuwada et al., 1996). NMDA receptor-mediated neuronal apoptosis was produced by bath application of NMDA as described previously and quantified by analysis of Hoechst-33342-stained neurons (Fig. 3A,B). We found that mouse neurons lacking the NR2A subunit showed a degree of neuronal apoptosis similar to wild-type neurons after NMDA stimulation (Fig. 3A), consistent with little contribution of NR2A-containing receptor to the NMDA receptor-mediated apoptosis. The lack of enhancement of NMDA-induced apoptosis may be attributable to a compensation in the NR2A-dependent neuronal survival signaling pathways during neuronal development in the absence of NR2A subunits. In contrast, NMDA-induced neuronal apoptosis was significantly reduced in NR2B-lacking neurons relative to wild-type cortical neurons (Fig. 3B) (p < 0.05 compared with NMDA-treated WT). These results demonstrate that the NR2B subunit has a primary role in mediating NMDA receptor-induced neuronal apoptosis in wild-type animals and hence are in reasonable agreement with the results obtained by pharmacological NR1/NR2B blockade.
Figure 3.

Genetic deletions of NR2A and NR2B subunits have differential effects on NMDA-induced neuronal apoptosis. Cultured mouse cortical neurons from NR2A−/− embryos or NR2B−/− embryos and WT littermate control mice were treated with NMDA (50 μm plus 10 μm glycine) for 20 min. Neurons were stained with Hoechst-33342 20 h after NMDA treatments and examined by microscopy. Apoptosis was quantified as the percentage of cells demonstrating cromatin condensation/fragmentation. NMDA-induced apoptosis was significantly reduced by genetic deletion of NR2B (B), but not NR2A (A), subunit (n = 4–6 tissue culture wells with >150 cells counted in each group). *p < 0.05 and **p < 0.01, compared with nontreated WT control; #p < 0.05, compared with NMDA-treated WT group. KO, Knock-out.
In a recent study, Hardingham et al. (2002) reported that selective activation of synaptic or extrasynaptic NMDA receptors mediates neuronal survival or death, respectively. Furthermore, available immunochemical and electrophysiological evidence suggests that, at least in the mature neurons of rat hippocampus and cortex, NR2A- and NR2B-containing receptors are differentially expressed at synaptic and extrasynaptic sites (Tovar and Westbrook, 1999). These data raise the possibility that it is the anatomical location (synaptic vs extrasynaptic), rather than subunit composition, of NMDA receptors that mediates the differential actions observed in the present study. To differentiate the effects of NMDA receptor subunit compositions from their anatomical localizations, we functionally mapped the expression of NR2A- and NR2B-containing NMDA receptors at synaptic and extrasynaptic sites and investigated their roles in promoting cell survival or death in cultured cortical neurons after pharmacological isolation. In CA1 neurons in hippocampal slices prepared from rats 3–4 weeks of age, although the vast majority of synaptic NMDA receptors are NR2A-containing, clear electrophysiological evidence indicates that a small proportion of functional NR1/NR2B (Ro 25–6981 sensitive) receptors do express at synapses (Liu et al., 2004; Wong et al., 2005). We therefore examined whether functional NR1/NR2B receptors are also expressed at the synaptic sites of the cultured cortical neurons used in the present study using whole-cell recording of spontaneous mEPSCs. As shown in Figure 4A, under these recording conditions, mEPSCs are comprised of both a fast AMPA subtype glutamate receptor-mediated component, which was completely blocked by the non-NMDA receptor antagonist DNQX (data not shown), and a slow NMDA receptor-mediated component, which was fully blocked by the NMDA receptor antagonist APV (Fig. 4Aa,Ab,Ad). Consistent with the presence of a proportion of functional synaptic NR1/NR2B receptors, the NMDA component was significantly reduced by bath application of NR2B antagonist Ro 25–6981 (0.5 μm) (Fig. 4Ac,Ad). Because mEPSCs are primarily mediated by synaptically localized receptors activated by glutamate spontaneously released from presynaptic terminals, the sensitivity to NR2B antagonist demonstrates that functional NR1/NR2B NMDA receptors are present within the glutamatergic synapses of the neurons under study. On average, the NR1/NR2B receptor-mediated component accounted for 32.4 ± 3.6% of the synaptic NMDA currents (n = 5) (Fig. 4Ac,Ad), and the remainder was primarily mediated by NR2A-containing receptors as it was essentially eliminated in the presence of the NR2A-specific antagonist NVP-AAM077 (0.4 μm; n = 5). Thus, similar to hippocampal CA1 neurons in brain slices (Liu et al., 2004; Wong et al., 2005), functional subpopulations of both NR2A-containing and NR1/NR2B NMDA receptors, although the former is predominant, are expressed at the synapses of the cultured neurons used in the present study.
Figure 4.

Activation of synaptic NR1/NR2B NMDA receptors produces a proapoptotic action, which is masked by a predominant synaptic NR2A-containing receptor-mediated cell survival-promoting effect. A, Functional synaptic NR2B-containing receptors are present in cortical neurons in culture. Spontaneous mEPSCs were recorded in whole-cell voltage-clamp mode at a holding membrane potential of −60 mV in the presence of TTX (0.5 μm) and bicuculline (10 μm) with 0 added Mg2+. Aa, Examples of mEPSC traces (averaged from 100 individual events) obtained in the absence (Control) and presence of Ro 25–6981 (Ro; 0.5 μm) or the broad spectrum NMDA receptor antagonist APV (APV; 50 μm). Ab, Total NMDA receptor-mediated component of mEPSCs was obtained by subtracting the averaged mEPSC recorded in the presence of APV from the averaged control mEPSC (Control-APV; shaded area). Ac, The NR1/NR2B receptor component was obtained by subtracting the averaged mEPSC recorded in the presence of Ro 25–6981 (Ro) from the averaged control mEPSC (Control-Ro; shaded area). Ad, Bar graph summarizes data obtained from five individual neurons. Charge transfer is equivalent to the area of the shaded regions. B, Enhanced activation of synaptic NR2A-containing and NR1/NR2B receptors exerts opposing actions on neuronal survival and death. Potentiation of synaptic NMDA receptor activation was achieved by increasing the presynaptic release of glutamate by incubating cultured neurons with bicuculline (Bic; 50 μm) for 4 h in the absence or presence of NR2-containing receptor-specific antagonists. Blockade of NR2A-containing (Bic+NVP), but not NR1/NR2B (Bic+Ro), NMDA receptors increased neuronal apoptosis. The NR2A blockade-induced apoptosis was prevented by an additional blockade of NR1/NR2B receptors (Bic+NVP+Ro). C, Spontaneously activated synaptic NR2A- and NR2B-containing receptors also have opposing roles in promoting neuronal survival and death. Incubation of neurons with NVP-AAM077 (NVP), but not Ro 25–6981 (Ro), for an extended duration (48 h) in the absence of bicuculline stimulation was sufficient to produce an increase in apoptosis. The NVP-AAM077-induced apoptosis was prevented by addition of Ro 25–6981 (NVP+Ro). Thus, both synaptic NR2A-containing and NR1/NR2B subpopulations of NMDA receptors are spontaneously activated by presynaptically released glutamate, exerting counteracting effects on cell survival and death, but synaptic NR2A-containing receptor activation is predominant and required for maintaining normal neuronal survival. **p < 0.01 compared with control; n = 16 (B) and 10–12 (C) for each group from three separate experiments.
The function of the two synaptic receptor subpopulations in mediating neuronal survival or death was examined next. We reasoned that if the location of the receptors is the determining factor, activation of either receptor population at the synapse should promote neuronal survival. However, if the subunit composition is the determinant, one would expect that the two populations, despite both being synaptically localized, should have different actions. To increase activation of synaptic NMDA receptors by synaptically released glutamate, neurons were incubated with the GABAA receptor antagonist bicuculline (50 μm; 4 h). Bicuculline increases neuronal excitation by blocking the GABAA receptor-mediated synaptic inhibition and thereby enhances action potential-dependent synchronized release of glutamate from presynaptic terminals. Neuronal apoptosis was quantified 20 h after the treatments. We found that stimulation of synaptic NMDA receptors by application of bicuculline alone, or in the presence of NR2B antagonist Ro 25–6981, did not cause apoptotic cell death (Fig. 4B). In contrast, blocking synaptic NR2A-containing receptors by coapplication of NVP-AAM077 with bicuculline significantly increased neuronal apoptosis (p < 0.01) (Fig. 4B). The NR2A blockade-induced neuronal apoptosis was mediated by synaptic NR1/NR2B, because it was prevented in the presence of Ro 25–6981 (Fig. 3B). Because, under bicuculline incubation, the increased action potential-dependent synaptic release of glutamate may lead to activation of extrasynaptic NMDA receptors by glutamate spillover, we next examined the impact of a blockade of synaptic NMDA receptor activation by glutamate spontaneously released from terminals under basal, nonstimulated conditions. Incubation of neurons with NVP-AAM077 for 4 h failed to increase neuronal apoptosis (data not shown). However, when the incubation time was increased to 48 h, a significant increase in neuronal apoptosis was observed (Fig. 4C) (p < 0.01). The synaptic NR2A antagonist-induced apoptosis was also prevented by the blockade of synaptic NR2B receptors with Ro 25–6981. In contrast, blockade of synaptic NR2B alone for up to 48 h did not increase neuronal apoptosis (Fig. 4C). Together, these results strongly suggest that both synaptic NR2A-containing and NR1/NR2B receptors are activated by spontaneously released glutamate from the presynaptic terminal and hence tonically exert opposing influences with respect to promoting cell survival or death. Under physiological conditions, the NR2A-mediated cell survival-promoting effect counteracts the tonic apoptotic action of NR2B, thereby maintaining normal neuronal survival. As such, synaptic NR2B-mediated neuronal death can only be unmasked after pharmacological blockade of the NR2A-mediated cell survival signaling pathway.
In contrast to the predominant expression of NR2A-containing receptors at synapses, NR2B-containing receptors are thought to be the predominant NMDA receptor expressed at extrasynaptic sites in mature neurons (Tovar and Westbrook, 2002; Massey et al., 2004). To determine if some, albeit small, proportion of extrasynaptic NMDA receptors contain NR2A in the neurons under study, we first pharmacologically blocked all NMDA receptors expressed at synapses and then examined whether currents gated through extrasynaptic NMDA receptors are sensitive to NR2A subunit-specific antagonism. The selective blockade of synaptic NMDA receptors was achieved by coapplication of bicuculline (50 μm) and MK-801 (10 μm) for 10 min. Bicuculline enhances synaptic release of glutamate and thereby selectively activates synaptic NMDA receptors (Hardingham et al., 2002). MK-801, as an irreversible blocker of open NMDA receptor channels (Huettner and Bean, 1988; Tovar and Westbrook, 2002), can only block the bicuculline-activated synaptic NMDA receptors and cannot block extrasynaptic channels that are not activated during bicuculline application. The complete blockade of synaptic NMDA receptors could be achieved within 10 min of bicuculline and MK-801 coapplication as indicated by the virtual elimination of the slow, APV-sensitive component of mEPSCs (Fig. 5Aa,Ab). Little recovery was observed 1 h after wash-out of the drugs. The currents gated through extrasynaptic NMDA receptors were then induced by application of NMDA (200 μm) via a fast perfusion system after washing out bicuculline and MK-801. The extrasynaptic NMDA receptor-mediated currents were mostly reduced by the NR2B antagonist Ro 25–6981 (Fig. 5Ac,Ad), consistent with the idea that extrasynaptic NMDA receptors are predominantly NR1/NR2B receptor subtypes (Stocca and Vicini, 1998; Tovar and Westbrook, 1999; Tovar and Westbrook, 2002). The residual, NR2B antagonist-resistant current was virtually blocked by the NR2A antagonist NVP-AAM077 (Fig. 5Ac,Ad), indicating that the non-NR2B-containing extrasynaptic NMDA receptors were primarily NR2A-containing receptors. On average, ∼26.6 ± 2.3% (n = 5) of total currents gated by extrasynaptic NMDA receptors were mediated by NR2A-containing receptors (Fig. 5Ad). These results provide additional support for the existence of a substantial number of functional extrasynaptic NR2A-containing NMDA receptors in mature cultured cortical neurons (Thomas et al., 2006).
Figure 5.

Activation of extrasynaptic NR2A-containing NMDA receptors promotes cell survival, protecting against extrasynaptic NR1/NR2B receptor-mediated and non-NMDA receptor-dependent neuronal death. A, Functional NR2A-containing NMDA receptors are present at extrasynaptic sites. Whole-cell recordings were performed at a holding membrane potential of −60 mV. Aa, Averaged traces of mEPSCs showing an APV-sensitive (50 μm) NMDA receptor-mediated component (Control-APV; shaded area). Ab, Averaged traces of mEPSCs showing the blockade of synaptic NMDA receptors by the open channel blocker MK-801 (10 μm plus 50 μm bicuculline; 10 min), as demonstrated by the elimination of the NMDA receptor-mediated component of the mEPSCs (Control-APV; shaded area). Ac, Example traces of whole-cell currents evoked by NMDA (200 μm) after the blockade of synaptic NMDA receptors with MK-801 in the absence (A; control) or presence (B; 0.5 μm) of Ro 25–6981 or Ro 25–6981 plus NVP-AAM077 (C; 0.4 μm). NMDA receptor-mediated currents were evoked by fast application of NMDA within 10 min of washing out MK-801 and bicuculline. Currents remaining after the blockade of extrasynaptic NR1/NR2B receptors were virtually abolished by the addition of NVP-AAM077, suggesting the presence of functional extrasynaptic NR2A-containing NMDA receptors in these neurons. Ad, Histogram summarizes data from five individual neurons. B, Activation of extrasynaptic NR2A-containing NMDA receptors protects against neuronal death mediated by extrasynaptic NR1/NR2B NMDA receptors. Excitotoxic neuronal death was induced in cortical neurons by bath application of NMDA (50 μm; 20 min) after the blockade of synaptic NMDA receptors with MK-801 plus bicuculline, and cell death was assayed 20 h later. NMDA elicited neuronal apoptosis, which was exacerbated when the NR1/NR2B component was selectively stimulated (NVP+NMDA) but eradicated when the NR2A-containing component was specifically activated (Ro+NMDA). **p < 0.01 compared with control. #p < 0.05, ##p < 0.01 compared with NMDA treatment. n = 11–12 from two separate experiments for each group.
We next addressed the role of extrasynaptic NR2A-containing and NR1/NR2B receptors in mediating NMDA-induced cell survival and death. After a specific blockade of synaptic NMDA receptors and wash-out of bicuculline and MK-801, the neurons were treated with NMDA (50 μm plus 10 μm glycine) for 20 min in the absence or presence of NVP-AAM077 (0.4 μm) or Ro 25–6981 (0.5 μm). Quantitative neuronal apoptosis assays performed 20 h after the treatments showed that NMDA application alone (nonselective activation of extrasynaptic NMDA receptors) elicited significant apoptosis (p < 0.01) (Fig. 5B), which was prevented by a selective blockade of NR1/NR2B extrasynaptic NMDA receptors with Ro 25–6981. In sharp contrast, blockade of the NR2A-containing receptors with NVP-AAM077 (i.e., leaving NR1/NR2B NMDA receptors intact) did not prevent but instead potentiated NMDA-mediated apoptosis (p < 0.05 compared with NMDA treatment). Thus, as with synaptic NMDA receptors, activation of extrasynaptic NR2A-containing receptors has a role in promoting cell survival, counteracting NR1/NR2B receptor-mediated neuronal apoptosis. Together, the data illustrated in Figures 4 and 5 strongly indicate that, regardless of their anatomical locations (synaptic vs extrasynaptic), NR2A-containing and NR1/NR2B receptors have opposing roles in mediating NMDA-elicited neuronal survival and apoptosis.
These results strongly suggest that selective activation of NR2A-containing receptors may be an effective therapeutic means of protecting neurons from apoptotic injuries. Because of the lack of an NR2A-specific agonist, we attempted to achieve specific activation of NR2A using two different strategies. First, we examined the effect of selective activation of extrasynaptic NR2A-containing receptor activation on neuronal apoptosis induced by staurosporine (STS), a potent apoptosis inducer that does not act through the NMDA receptor (Budd et al., 2000). In these experiments, all synaptic NMDA receptors were irreversibly blocked by pretreatment of the neurons with coapplication of bicuculline and MK-801, and extrasynaptic NR1/NR2B NMDA receptors were blocked by the addition of Ro 25–6981 (0.5 μm) in the medium throughout the experiments. As shown in Figure 6A, STS treatment (100 nm; 1 h) triggered tremendous neuronal apoptosis. Bath application of NMDA (200 μm; 5 min) did not increase neuronal apoptosis on its own, confirming the effective blockade of NR1/NR2B receptor-mediated apoptotic actions by Ro 25–6981. However, the application of NMDA, which would primarily activate extrasynaptic NR2A-containing receptors under these conditions, significantly reduced STS-induced apoptosis (p < 0.01 compared with STS alone) (Fig. 6A). The NMDA-induced neuronal protection was indeed mediated by NR2A-containing receptors as it was prevented by coapplication of NVP-AAM077. Second, we examined the effect of enhancement of synaptic NR2A activation on reducing NMDA-induced excitotoxicity. We accomplished the selective enhancement of synaptic NMDA receptor activation by a brief bath application of a suprasaturating concentration of glycine (Lu et al., 2001; Man et al., 2003). As an NMDA receptor coagonist (McBain and Mayer, 1994), glycine applied through bath can enhance the function of synaptic NMDA receptors that are activated by glutamate spontaneously released from presynaptic terminals under nonstimulated conditions but not extrasynaptic NMDA receptors, which are not activated under the nonstimulated condition because of the low ambient level of extracellular glutamate (Lu et al., 2001; Man et al., 2003). Because, as shown in Figure 4, synaptic NMDA receptors in these neurons are predominantly NR2A-containing, and their activation produces a dominant cell survival-promoting action (Fig. 4), we reasoned that selective enhancement of activation of synaptic NMDA receptors with a suprasaturating concentration of glycine would lead to an increase in synaptic NR2A-dependent neuronal survival action. Consistent with our prediction, a 10 min pretreatment of neurons with glycine (300 μm; plus 10 μm strychnine to block glycine Cl− channels) significantly reduced the neuronal apoptosis produced by a subsequent application of NMDA (p < 0.05, compared with NMDA treatment alone) (Fig. 6B). The survival-promoting effect of glycine was mediated by activation of the synaptic NR2A-containing NMDA receptor subpopulation because the neuroprotective effects of glycine pretreatment were prevented by NR2A antagonist NVP-AAM077 but not affected by NR2B antagonist Ro 25–6981 (Fig. 6B). Together, these results strongly suggest that preferential activation of NR2A receptors can stimulate a prosurvival pathway that is able to guard against both NMDA receptor-mediated and non-NMDA receptor (such as STS)-mediated neuronal damage.
Figure 6.

Selective activation of NR2A-containing NMDA receptors protects neurons from NMDA receptor-mediated or non-NMDA receptor-mediated neuronal apoptosis. A, Activation of extrasynaptic NR2A-containing NMDA receptors can counteract NMDA receptor-independent apoptosis. Bath application of STS (100 nm; 1 h) after blockade of synaptic NMDA receptors by pretreatment with MK-801 plus bicuculline and of extrasynaptic NR1/NR2B receptors by Ro 25–6981 (Control) induced a significant increase in neuronal apoptosis (STS). Brief application of NMDA (200 μm; 5 min) did not produce neuronal apoptosis on its own (NMDA) but significantly reduced the STS-induced neuronal apoptosis (NMDA+STS), and the NMDA-induced neuroprotective action was abrogated by coapplication of NVP-AAM077 (0.4 μm; NVP+NMDA+STS). **p < 0.01 compared with Ro 25–6981 treatment (Control). ##p < 0.01 compared with STS treatment. n = 8–12 for each group from three separate experiments. B, Pretreatment of neuronal cultures with glycine (300 μm plus 10 μm strychnine) for 10 min significantly reduced the neuronal apoptosis produced by NMDA applied thereafter (Gly+NMDA). This neuroprotective effect was abolished by coapplication of NR2A antagonist NVP-AAM077 (0.4 μm) with glycine (NVP+Gly+NMDA) but not by coapplication of NR2B-specific antagonist Ro 25–6981 (0.5 μm) with glycine (Ro+Gly+NMDA), indicating that the neuroprotective effect of glycine is primarily mediated through enhancing the activation of NR2A-containing NMDA receptors. *p < 0.05, **p < 0.01 compared with control. #p < 0.05, ##p < 0.01 compared with NMDA. n = 17–18 for each group from three separate experiments.
In view of the opposing roles of NR2A- and NR2B-containing NMDA receptors in mediating survival and death, we predicted that these two subpopulations may also play different roles in mediating neuronal injury after acute brain insults such as stroke and brain trauma. To examine this prediction, we used a well-characterized in vitro stroke model, OGD (Goldberg and Choi, 1993; Aarts et al., 2002). Cortical cultures of 11–14 DIV were exposed to an anaerobic atmosphere for 1 h in a glucose-free solution in the absence or presence of either NVP-AAM077 (0.4 μm) or Ro 25–6981 (0.5 μm). Neuronal apoptosis was quantitatively determined 20 h after OGD. As shown in Figure 7A, 1 h of OGD produced a pronounced increase in neuronal apoptosis. As we expected, selective inhibition of the NR2A-containing NMDA receptors with NVP-AAM077 significantly enhanced OGD-induced neuronal apoptosis (p < 0.05 compared with OGD), and in contrast, a specific blockade of the NR1/NR2B NMDA receptors by Ro 25–6981 drastically reduced the ODG-induced apoptosis (p < 0.01 compared with OGD) (Fig. 6A).
Figure 7.

Pretreatments with NR2A- and NR2B-specific antagonists promote neuronal survival and death, respectively, in both in vitro and in vivo models of ischemia. A, NR2A-containing and NR1/NR2B receptors exert opposing effects in ischemic neuronal injuries in vitro. Cortical cultures were challenged with a 1 h OGD, and apoptosis was assayed 23 h after the challenge. OGD resulted in a significant increase in neuronal apoptosis compared with nonchallenged controls (Control), and the OGD-induced apoptosis was respectively potentiated by the NR2A preferential antagonist NVP-AAM077 (NVP+OGD; 0.4 μm) and inhibited by the NR2B antagonist Ro 25–6981 (Ro+OGD; 0.5 μm) when bath applied 30 min before, and during, the OGD challenge. **p < 0.01 compared with control. #p < 0.05, ##p < 0.01 compared with OGD. n = 17–18 for each group from three separate experiments. B, C, NR2A- and NR2B-containing receptors exert opposing effects in ischemic neuronal injuries in vivo. Adult rats were subjected to a 1 h focal cerebral ischemia produced by MCAo, and cerebral infarction was assessed 24 h after MCAo onset. Intravenous infusion 30 min before MCAo onset of NVP-AAM077 (NVP+MCAo; 2.4 mg/kg; n = 5) and Ro 25–6981 (Ro+MCAo; 6 mg/kg; n = 6) increased and decreased, respectively, both infarct area (B) and total infarct volume (C). *p < 0.05, **p < 0.01 compared with MCAo. D, Neurological scores assessed 24 h after stroke onset in the same groups of animals shown in B and C indicate that blockade of the NR2A-containing NMDA receptors resulted in a trend toward worsening neurological function, whereas blockade of NR1/NR2B NMDA receptors markedly improved neurological behavior. **p < 0.01 compared with MCAo.
We next determined whether these findings observed in vitro could be reproduced in vivo using a rat focal ischemic stroke model, MCAo (Bederson et al., 1986; Aarts et al., 2002). We first infused NVP-AAM077 [2.4 mg/kg, a dose selected based on two times the anticonvulsant ED50 in vivo (Y. Auberson, personal communication)], Ro 25–6981 (6 mg/kg) (Loschmann et al., 2004), or vehicle (saline) intravenously in the rats 30 min before stroke onset. The animals were then subjected to a 1 h transient ischemic stroke induced by MCAo. This relatively short duration of ischemia was chosen to unmask the potential neuroprotective effects mediated by NR2A-containing receptors activated during the stroke challenge. Neurological score and cerebral infarction were examined 24 h after the MCAo onset. Similar to the results observed with OGD in vitro, we found that blockade of NR2A-containing NMDA receptors significantly increased the infarct areas and the total infarct volume, whereas, in sharp contrast, the stroke-induced brain injuries were remarkably reduced by NR2B antagonism (Fig. 7B,C). Specifically, when compared with saline-treated animals, NVP-AAM077 pretreatment gave rise to a 67.0 ± 17.9% increase in total infarct volume (n = 5; p < 0.05), whereas Ro 25–6981 treatment decreased the total infarct volume by 67.8 ± 4.3% (n = 6; p < 0.01). Neurological behavioral tests showed that the NVP-AAM077-treated animals exhibited a trend toward poorer neurological function, whereas Ro 25–6981 treatment produced a significant protective effect (Fig. 6D). Together, these observations indicate that both NR2A-containing and NR1/NR2B NMDA receptor subtypes are activated during stroke, exerting opposing effects on ischemic brain damage.
In a clinical setting, a practical therapy for stroke would be one that could be implemented after the onset of stroke. Thus, we determined whether postischemic blockade of NR2B or potentiation of NR2A would be effective in reducing ischemic brain injury. We predicted that, as the extracellular glutamate concentration may rapidly recover to prestroke levels [as quickly as 30 min after the stroke onset (Benveniste et al., 1984)] and extrasynaptic NR1/NR2B receptors may not be activated thereafter, the NR2B antagonist may have a narrow window of efficacy. In contrast, selective activation of NR2A-containing receptors would initiate cell survival-promoting signals, protecting neurons against ischemic damage regardless of the time in relation to the stroke event and may therefore be expected to have a much broader therapeutic window. As expected, we found that treatment with either nonsubunit-specific NMDA receptor antagonist MK-801 (1 mg/kg) (Fig. 8A) (Margaill et al., 1996) or NR2B-specific antagonist Ro 25–6981 (6 mg/kg) (Fig. 8B) (Loschmann et al., 2004) 3 h after a 1.5 h MCAo challenge (4.5 h after stroke onset) did not provide any noticeable neuroprotection when compared with MCAo alone (Fig. 8A), which was in line with previous reports (Xue et al., 1994; Margaill et al., 1996). In contrast, selective activation of synaptic NMDA receptors, which are predominantly NR2A-containing receptors, with the application of NMDA receptor coagonist glycine [800 mg/kg (De et al., 2000)] resulted in a remarkable reduction in total infarct volume (Fig. 8A). The glycine effect is mediated through enhancement of synaptic NMDA receptors and not its action on glycine receptor-mediated Cl− channels, as it was virtually abolished by coadministration of MK-801 (Fig. 8A). Moreover, the glycine effect was resistant to NR2B antagonist Ro 25–6981 but prevented by NR2A antagonist NVP-AAM077, demonstrating that the efficacy of glycine was a result of specific enhancement of NR2A-containing NMDA receptor-mediated cell survival-promoting action (Fig. 8B,C). These results suggest that postischemic potentiation of the prosurvival action of NR2A-containing NMDA receptors, rather than NMDA receptor antagonism, is an effective neuroprotection strategy for the reduction of ischemic brain damage.
Figure 8.
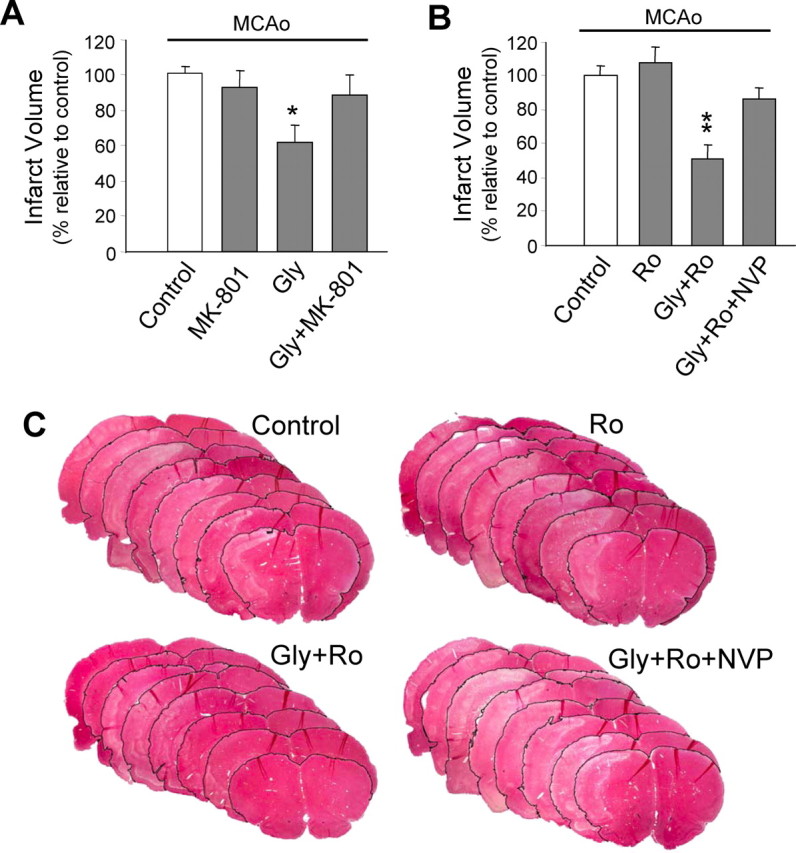
Postischemic potentiation of NR2A-containing NMDA receptors through administration of glycine reduces ischemic brain damage in an in vivo focal ischemic stroke model. Adult rats received either drug or saline treatment 3 h after a 1.5 h MCAo challenge (4.5 h after MCAo onset). A, General blockade of NMDA receptors with nonsubunit-specific antagonist MK-801 (1 mg/kg; n = 8) after stroke was no longer neuroprotective, whereas a poststroke treatment with NMDA receptor coagonist glycine (Gly; 800 mg/kg; n = 8) significantly reduced total infarct volume. The glycine effect was fully blocked by coapplication of MK-801 (Gly+MK-801; n = 7), indicating mediation by NMDA receptors. *p < 0.05 compared with Control (MCAo alone). B, Postischemic treatment with NR2B-selective antagonist Ro 25–6981 (Ro; 6 mg/kg; n = 10) was ineffective, whereas the glycine effect persisted in the presence of Ro 25–6981 (6 mg/kg) (Gly+Ro; n = 9). The addition of NR2A antagonist NVP-AAM077 (2.4 mg/kg; Gly+Ro+NVP; n = 10) abolished the neuroprotection offered by glycine, indicating glycine acts via selective potentiation of NR2A-containing NMDA receptors. **p < 0.01 compared with control (MCAo alone). C, Representative rat brain sections stained with H&E from each treatment group in B. Pale staining indicates infarct.
Discussion
Our results demonstrate that NMDA receptor activation can produce either neuronal survival or death-promoting actions and that this dual action is mediated by receptor subunit composition. The cell survival action can be fully blocked by the NR2A preferential antagonist NVP-AAM077. Although NVP-AAM077 may have a small contaminant inhibition of NR2B-containing receptors, the lack of blockade of the NMDA receptor-mediated cell survival action by the NR2B antagonist Ro 25–6981 essentially rules out the contribution of this subunit. Moreover, the lack of effect on NMDA-induced necrotic neuronal injury suggests that NR2A-containing receptor-mediated cell survival actions preferentially protect against apoptosis, and this appears to be mediated at least in part by the activation of an Akt-dependent anti-apoptotic signal. In contrast, the efficient blockade of NMDA receptor-dependent cell death by Ro 25–6981, but not by NVP-AAM077, strongly suggests that it is the NR1/NR2B, not the NR2A-containing, NMDA receptor subpopulation that plays a primary role in triggering intracellular cascades that lead to NMDA- or ischemia-induced neuronal apoptosis. The inability of NVP-AAM077 to block cell death is an additional indication that any small fraction of NR2B inhibition provided by this antagonist is not sufficient to block the NR2B-dependent cell death.
In addition to NR1/NR2A and NR1/NR2B diheteromers, cortical neurons are also believed to express an NR1/NR2A/NR2B triheteromeric NMDA receptor subpopulation. Because of the lack of a selective antagonist for these receptors, their contributions to cell survival and/or death remain to be determined. Given that Ro 25–6981 has a much higher potency toward the NR1/NR2B diheteromeric receptors than NR1/NR2A/NR2B triheteromeric receptors (Hatton and Paoletti, 2005), it is reasonable to speculate that most of the NMDA-induced cell death actions observed in the present study are primarily a result of activation of NR1/NR2B receptors. In contrast, although the sensitivity of NVP-AAM077 toward the NR1/NR2A/NR2B triheteromeric receptors has not been investigated previously, because NVP-AAM077 is a competitive antagonist at the agonist binding sites of NR2A subunits and because both binding sites need to be occupied to open the channel, we speculate that NVP-AAM077 may inhibit all NR2A-containing receptors. Thus, without an antagonist that blocks only NR1/NR2A/NR2B triheteromeric receptors, we tentatively attribute the NVP-AAM077- and Ro 20–6891-sensitive actions observed here to NR2A-containing (both NR1/NR2A and NR1/NR2A/NR2B) and NR1/NR2B receptor subpopulations, respectively. Therefore, together, our study suggests that activation of NR1/NR2B NMDA receptors, synaptic or extrasynaptic, initiates apoptotic signaling cascades and promotes neuronal death, whereas selective activation of NR2A-containing NMDA receptors stimulates prosurvival signaling, thereby exerting a neuroprotective action against both NMDA receptor-dependent and non-NMDA receptor-dependent apoptotic neuronal injuries. The net impact of NMDA receptor activation on neuronal survival and death is dependent on the balance between the activation of these two receptor subpopulations. The differential actions of NMDA receptor subunits in mediating cell survival and death revealed by the subunit-preferential antagonists are also primarily corroborated by results obtained with genetic deletions of NR2A or NR2B subunits (Fig. 3). Some differences between these approaches, particularly in the complete blockade of NMDA-induced toxicity by Ro 20–6891 versus partial loss of toxicity by genetic deletion of NR2B and the lack of significant enhancement in excitotoxicity by the deletion of NR2A, suggest the occurrence of alterations in expression, localization, or signaling downstream of other NMDA receptor subunits in neurons developing without the respective NR2 subunits.
The observed involvement of NR2B receptors in NMDA-mediated excitotoxicity is in good agreement with a recent study using brain slices (Zhou and Baudry, 2006), and the NR2A-mediated neuronal survival has also been demonstrated in a traumatic mechanical injury model in vitro (DeRidder et al., 2006). It is interesting, however, to note that NR2A was also recently reported to be involved in human immunodeficiency virus-mediated neurotoxicity (O'Donnell et al., 2006). As the reasons underlying the conflict between these results remain to be determined, we should be cautious of generalizations concerning the roles of NR2A and NR2B in mediating NMDA receptor-mediated neuronal survival and death. Roles may vary depending on developmental stage, brain area, and the model of disease.
Results from this work strongly suggest that the receptor subcellular localization has little influence on the differential roles of NR2A-containing and NR1/NR2B receptors in promoting cell survival and death. However, because of the preferential expression of the two subunits at synaptic versus extrasynaptic sites, selective activation of synaptic and extrasynaptic NMDA receptors could be expected to result in the activation of predominantly NR2A-containing receptor-dependent cell survival and NR1/NR2B receptor-mediated apoptotic pathways, respectively. Thus, our results may not be in conflict with recently reported differential actions on cell survival and death after selective activation of synaptic and extrasynaptic NMDA receptors (Hardingham et al., 2002). Our work may also provide a molecular basis to explain the paradoxical roles of NMDA receptor antagonism in producing extensive neuronal apoptosis in developing animals (Ikonomidou et al., 1999) and neuroprotection against ischemic brain damage in animal studies (Lee et al., 1999; Arundine and Tymianski, 2004). For instance, under normal conditions, spontaneously released glutamate preferentially stimulates synaptic NMDA receptors and hence activates predominantly the NR2A-containing receptor-dependent cell survival signaling, thereby playing an essential role in maintaining the physiological survival of the neurons in some areas of the brain under certain developmental stages. As demonstrated in the present study, NR2A-containing receptor activation, in addition to counteracting NR2B-containing receptor-mediated cell death, has the ability to guard against non-NMDA receptor-mediated apoptotic processes, which may be particularly active as part of the process of eliminating excess numbers of neurons in the developing animal. Thus, blocking these synaptically activated NMDA receptors may lead to extensive neuronal apoptosis. However, under some pathological conditions, such as stroke and brain trauma, there is usually a transient and rapid increase in extracellular glutamate concentrations and consequently extrasynaptic receptors, which are predominantly NR1/NR2B receptors and not usually activated by synaptically released glutamate under normal synaptic transmission, become activated, resulting in a predominant activation of the NR1/NR2B receptor-dependent cell death pathway. Thus, blocking these receptors can be neuroprotective under these pathological conditions.
Despite a large body of well-documented evidence for a critical role of NMDA receptor-mediated excitotoxicity in brain damage after stroke and brain trauma, clinical trials of NMDA receptor antagonists as neuroprotectants in stroke and traumatic brain injuries have not been successful (Lee et al., 1999; Ikonomidou and Turski, 2002; Kemp and McKernan, 2002). The reasons underlying the apparent contradiction between basic research results and clinical trials remain unknown, but several reasons have been proposed. These include, but are not limited to, the inability to use the antagonists at the doses required for neuroprotection resulting from side effects, the inability to administer the drugs within their neuroprotective windows, poor experimental designs, and heterogeneity in the patient population (Corbett and Nurse, 1998; Gladstone et al., 2002; Ikonomidou and Turski, 2002; Albensi et al., 2004). In the present study, we demonstrate that the NMDA receptor-mediated excitotoxic neuronal injuries after stroke in the rat MCAo model of focal ischemia are primarily mediated by NR1/NR2B receptors, because NR2B preferential antagonist applied before the stroke onset significantly reduced the brain damage. However, the NR2B antagonist appears to have a relatively narrow therapeutic window, because it offers little protection when administered 4.5 h after the stroke onset. This may be attributable to, at least in part, the rapid recovery of extracellular glutamate concentrations to prestroke levels (Benveniste et al., 1984; Ikonomidou and Turski, 2002), which are not sufficient to activate extrasynaptic NMDA receptors. Thus, NR2B-specific antagonists would be expected to have no effect after this point. Administration of nonsubunit-specific NMDA receptor antagonists such as MK-801 and amantadine at this point may even be harmful because of their blockade of NR2A-containing receptor-dependent prosurvival signaling. Unfortunately, in most clinical settings, because of the time required to transport a patient to the hospital and obtain a definitive diagnosis, treatment is not usually possible until several hours after the stroke onset, which is most likely outside the window of efficacy for NMDA receptor blockers. Thus, consistent with previous suspicions (Ikonomidou and Turski, 2002), our results suggest that the failure to administer NMDA receptor antagonists within their efficacy window might be one of the major reasons for their failure in the clinical trials of stroke and neurotrauma conducted thus far.
In contrast, activation of NR2A-containing NMDA receptors may have several obvious advantages over previously proposed NMDA receptor antagonism-based stroke therapies: first, as demonstrated in the present work, it possesses a broader therapeutic window than NR1/NR2B receptor blockade. In fact, it should theoretically have no therapeutic window limitation as it protects neurons against brain damage by promoting neuronal survival, rather than blocking the activation of a death signal initiated by the stroke insult; second, it is significant to point out that NR2A-containing receptor activation is not only effective against NMDA receptor-mediated cell death (primary neuronal injuries), but can also guard against non-NMDA receptor-mediated cell death (secondary neuronal injuries). Increasing evidence supports the fact that some of the non-NMDA receptor-mediated mechanisms, while secondary to NMDA receptor activation, may contribute significantly to brain damage, particularly after severe stroke insults (Aarts et al., 2003; Xiong et al., 2004). Thus, activation of NR2A-containing receptors, rather than blockade of all NMDA receptors, appears to be a more effective after stroke neuroprotective therapy. In addition to the neuronal injuries caused by acute brain insults such as stroke and brain trauma, activation of NR2A-containing receptor-dependent prosurvival signaling may also prove to be a potential neuroprotective therapy for a number of chronic neurodegenerative disorders, such as Parkinson's disease, Huntington's disease, amyotrophic lateral sclerosis, and Alzheimer's disease, where a “slow” NMDA receptor-mediated excitotoxicity has been implicated (Lipton and Rosenberg, 1994; Zoghbi et al., 2000; Ikonomidou and Turski, 2002).
The mechanisms by which NR2A-containing and NR1/NR2B receptors exert their opposing roles remain to be determined. The two receptor subpopulations mediate calcium influx with distinct kinetics (Cull-Candy and Leszkiewicz, 2004); therefore, selective activation of one or the other may provide differentially required levels and kinetics of rising [Ca2+]i in the postsynaptic neurons (Choi, 1994; Lipton and Nicotera, 1998). In addition, mounting evidence suggests that different NR2 subunits serve distinct functions by coupling to different postsynaptic signaling pathways, most likely via differential protein interactions of their cytoplasmic C-tail regions (Sheng and Pak, 2000; Kohr et al., 2003). As such, the activation of NR2A-containing NMDA receptors could trigger different signaling events than the activation of their NR1/NR2B counterparts, resulting in subunit-specific signaling outcomes (Bliss and Schoepfer, 2004; Liu et al., 2004). Consistent with this hypothesis, a phosphatidyl inositol 3-kinase-dependent proneuronal survival signaling has been linked with NR2A-containing NMDA receptor activation (Lee et al., 2002), whereas an interference peptide derived from part of the sequence in the NR2B tail has been shown to be able to protect against excitotoxic neuronal death by disrupting NR2B C-tail interaction with its binding partner(s) (Aarts et al., 2002). Several other NR2A- and/or NR2B-specific signaling pathways have been characterized recently (Kim et al., 2005; Zhu et al., 2005; Li et al., 2006), and it would be interesting to determine whether some of these subunit-specific signaling pathways play a major determinant role in mediating NMDA receptor subtype-specific neuronal survival and death-promoting actions. In addition to potential signals differentially coupled to these receptor subpopulations, evidence accumulated from several recent studies also suggests that there may be some functional interactions between the receptor subtypes and/or downstream signaling pathways coupled to these receptors (Mallon et al., 2004; Waxman and Lynch, 2005). Future studies are needed to clarify how these interactions contribute to the differential actions for promoting neuronal survival and death after activation of these NMDA receptor subpopulations.
Whatever the precise mechanism, our study establishes that NR2A- and NR2B-containing subpopulations of NMDA receptors have differential roles in mediating neuronal survival and death and hence provides a molecular basis for the paradoxical actions of NMDA receptor antagonism in producing both proapoptotic and antiapoptotic effects under different conditions. Our work also provides an explanation for the failure of NMDA receptor antagonism-based clinical trials of stroke treatment and leads us to propose a novel NR2A-containing receptor activation-based neuroprotective therapy for acute brain injuries after stroke, neurotrauma, and chronic brain damage associated with a large number of neurodegenerative diseases. Therefore, this work calls for the urgent development of highly specific agonists for the NR2A-containing NMDA receptor subtype. In the absence of such agonists, a specific enhancement of NR2A-containing NMDA receptor function, as demonstrated in the present work, may be achieved by the combination of a nonsubunit-specific NMDA receptor enhancer, such as glycine, and an NR2B-specific antagonist. It is particularly interesting to note, in this regard, that both NMDA receptor glycine site agonists, such as d-cycloserine (Posey et al., 2004), and NR2B-specific antagonists (Chazot, 2000) are now available for clinical trials. Therefore, our study may have an immediate impact on the treatment of stroke.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and the Heart and Stroke Foundation of British Columbia and Yukon (HSFBC&Y) (to Y.T.W.) and from the Canadian Stroke Network (CSN) (to Y.T.W., M.T.). Y.T.W. is a Howard Hughes Medical Institute International Scholar, CIHR Investigator, Michael Smith Foundation for Health Research (MSFHR) Senior Scholar, and HSFBC&Y Chair in Stroke Research. A.M.C. is a Canada Research Chair and MSFHR Senior Scholar. T.P.W. is supported by fellowships from CIHR and MSFHR, and Y.L. is supported by doctoral awards from CIHR/Heart and Stroke Foundation of Canada/CSN and MSFHR. We thank Dr. Y. P. Auberson (Novartis Pharma AG, Basel, Switzerland) for the generous gift of NVP-AAM077.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Albensi BC, Igoechi C, Janigro D, Ilkanich E. Why do many NMDA antagonists fail, while others are safe and effective at blocking excitotoxicity associated with dementia and acute injury? Am J Alzheimers Dis Other Demen. 2004;19:269–274. doi: 10.1177/153331750401900502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss T, Schoepfer R. Neuroscience. Controlling the ups and downs of synaptic strength. Science. 2004;304:973–974. doi: 10.1126/science.1098805. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Lipton SA. Signaling events in NMDA receptor-induced apoptosis in cerebrocortical cultures. Ann NY Acad Sci. 1999;893:261–264. doi: 10.1111/j.1749-6632.1999.tb07832.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA. 2000;97:6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazot PL. CP-101606 Pfizer Inc. Curr Opin Investig Drugs. 2000;1:370–374. [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-d-aspartate receptor. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- Clements JD, Westbrook GL. Kinetics of AP5 dissociation from NMDA receptors: evidence for two identical cooperative binding sites. J Neurophysiol. 1994;71:2566–2569. doi: 10.1152/jn.1994.71.6.2566. [DOI] [PubMed] [Google Scholar]
- Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett D, Nurse S. The problem of assessing effective neuroprotection in experimental cerebral ischemia. Prog Neurobiol. 1998;54:531–548. doi: 10.1016/s0301-0082(97)00078-6. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- DeRidder MN, Simon MJ, Siman R, Auberson YP, Raghupathi R, Meaney DF. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol Dis. 2006;22:165–176. doi: 10.1016/j.nbd.2005.10.011. [DOI] [PubMed] [Google Scholar]
- De Ryck M, Van RJ, Borgers M, Wauquier A, Janssen PA. Photochemical stroke model: flunarizine prevents sensorimotor deficits after neocortical infarcts in rats. Stroke. 1989;20:1383–1390. doi: 10.1161/01.str.20.10.1383. [DOI] [PubMed] [Google Scholar]
- De Sarro G, Siniscalchi A, Ferreri G, Gallelli L, De SA. NMDA and AMPA/kainate receptors are involved in the anticonvulsant activity of riluzole in DBA/2 mice. Eur J Pharmacol. 2000;408:25–34. doi: 10.1016/s0014-2999(00)00709-3. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Gladstone DJ, Black SE, Hakim AM. Toward wisdom from failure: lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33:2123–2136. doi: 10.1161/01.str.0000025518.34157.51. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-d-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci USA. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5(Suppl):1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kohr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby O. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Li S, Tian X, Hartley DM, Feig LA. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26:1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Nicotera P. Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium. 1998;23:165–171. doi: 10.1016/s0143-4160(98)90115-4. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Mechanisms of disease: excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Loschmann PA, De GC, Smith L, Wullner U, Fischer G, Kemp JA, Jenner P, Klockgether T. Antiparkinsonian activity of Ro 25–6981, a NR2B subunit specific NMDA receptor antagonist, in animal models of Parkinson's disease. Exp Neurol. 2004;187:86–93. doi: 10.1016/j.expneurol.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Mallon AP, Auberson YP, Stone TW. Selective subunit antagonists suggest an inhibitory relationship between NR2B and NR2A-subunit containing N-methyl-d-aspartate receptors in hippocampal slices. Exp Brain Res. 2004;162:374–383. doi: 10.1007/s00221-004-2193-6. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Margaill I, Parmentier S, Callebert J, Allix M, Boulu RG, Plotkine M. Short therapeutic window for MK-801 in transient focal cerebral ischemia in normotensive rats. J Cereb Blood Flow Metab. 1996;16:107–113. doi: 10.1097/00004647-199601000-00013. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Neuroprotective signal transduction: relevance to stroke. Neurosci Biobehav Rev. 1997;21:193–206. doi: 10.1016/s0149-7634(96)00010-3. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- Mielke JG, Wang YT. Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABA receptors following oxygen-glucose deprivation in cultured cortical neurons. J Neurochem. 2005;92:103–113. doi: 10.1111/j.1471-4159.2004.02841.x. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mutel V, Buchy D, Klingelschmidt A, Messer J, Bleuel Z, Kemp JA, Richards JG. In vitro binding properties in rat brain of [3H]Ro 25–6981, a potent and selective antagonist of NMDA receptors containing NR2B subunits. J Neurochem. 1998;70:2147–2155. doi: 10.1046/j.1471-4159.1998.70052147.x. [DOI] [PubMed] [Google Scholar]
- O'Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson DL. Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. J Neurosci. 2006;26:981–990. doi: 10.1523/JNEUROSCI.4617-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto SS, Li Z, Ju C, Scholzke MN, Mathews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2002;99:3974–3979. doi: 10.1073/pnas.022036399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey DJ, Kem DL, Swiezy NB, Sweeten TL, Wiegand RE, McDougle CJ. A pilot study of D-cycloserine in subjects with autistic disorder. Am J Psychiatry. 2004;161:2115–2117. doi: 10.1176/appi.ajp.161.11.2115. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Sheng M, Pak DT. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000;6:755–778. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol (Lond) 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tigaret CM, Thalhammer A, Rast GF, Specht CG, Auberson YP, Stewart MG, Schoepfer R. Subunit dependencies of NMDA receptor-induced AMPA receptor internalization. Mol Pharmacol. 2006;69:1251–1259. doi: 10.1124/mol.105.018580. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Sprouffske K, Westbrook GL. Fast NMDA receptor-mediated synaptic currents in neurons from mice lacking the epsilon2 (NR2B) subunit. J Neurophysiol. 2000;83:616–620. doi: 10.1152/jn.2000.83.1.616. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ju W, Liu L, Fam S, D'Souza S, Taghibiglou C, Salter M, Wang YT. alpha-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptor (AMPAR) endocytosis is essential for N-methyl-d-aspartate-induced neuronal apoptosis. J Biol Chem. 2004;279:41267–41270. doi: 10.1074/jbc.C400199200. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-d-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. J Biol Chem. 2005;280:29322–29333. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci. 2005;25:8386–8390. doi: 10.1523/JNEUROSCI.2388-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TP, Liu L, Sheng M, Wang YT. Response to comment on “role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity.”. Science. 2005;305:1912b. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Xue D, Huang Z-G, Barnes K, Lesiuk HJ, Smith KE, Buchan AM. Delayed treatment with AMPA, but not NMDA, antagonists reduces neocortical infarction. J Cereb Blood Flow Metab. 1994;14:251–261. doi: 10.1038/jcbfm.1994.32. [DOI] [PubMed] [Google Scholar]
- Zhou M, Baudry M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J Neurosci. 2006;26:2956–2963. doi: 10.1523/JNEUROSCI.4299-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, van AL, Sheng M, Zhu JJ. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Gage FH, Choi DW. Neurobiology of disease. Curr Opin Neurobiol. 2000;10:655–660. doi: 10.1016/s0959-4388(00)00135-5. [DOI] [PubMed] [Google Scholar]