

Abstract
The neuropeptide transmitter cocaine- and amphetamine-regulated transcript (CART) inhibits food intake and is expressed by both vagal afferent and hypothalamic neurons. Here we report that cholecystokinin (CCK) regulates CART expression in rat vagal afferent neurons. Thus, CART was virtually undetectable after energy restriction for 24 h, but administration of CCK to fasted rats increased CART immunoreactivity, and refeeding of fasted animals promptly increased CART by a mechanism sensitive to a CCK-1 receptor antagonist. In vagal afferent neurons incubated in serum-free medium, CART was virtually undetectable, whereas the orexigenic peptide melanin-concentrating hormone (MCH) was readily detected. The addition of CCK rapidly induced CART expression and downregulated MCH. Using a CART promoter–luciferase reporter vector transfected into cultured vagal afferent neurons, we showed that CCK stimulation of CART transcription was mediated by activation of protein kinase C and cAMP response element-binding protein (CREB). The action of CCK on CART expression was inhibited by the orexigenic peptide ghrelin, through a mechanism that involved exclusion of phosphorylated CREB from the nucleus. Thus, CCK reciprocally regulates expression of CART and MCH within the same vagal afferent neuron; ghrelin inhibits the effect of CCK at least in part through control of the nuclear localization of phosphoCREB, revealing previously unsuspected modulation of gut–brain signals implicated in control of food intake.
Keywords: satiety, CREB, melanin-concentrating hormone, ghrelin, vagus, food
Introduction
The mechanisms controlling food intake are attracting considerable attention, not the least because of their significance in understanding the recent dramatic increase in obesity (Schwartz et al., 2000; Woods, 2004). Termination of feeding involves gastrointestinal signals (Dockray, 2004; Murphy and Bloom, 2004; Woods, 2004) that include non-nutrient distension of the stomach and nutrient-dependent release of gastrointestinal satiety hormones, of which cholecystokinin (CCK) is one of the best-studied examples (Liddle, 1997; Dockray, 2004). Both gastric distension and gut hormones such as CCK inhibit food intake via vagal afferent neurons (Gonzalez and Deutsch, 1981; Smith et al., 1985; Moran and Kinzig, 2004). Transmission at the central terminals of these neurons in the nucleus tractus solitarius activates ascending pathways to the hypothalamus where there is integration of a variety of different peripheral signals including those from adipose tissue (e.g., leptin) and the endocrine pancreas (e.g., insulin) (Schwartz et al., 2000; Luckman and Lawrence, 2003).
The neuropeptide transmitter systems implicated in the control of feeding behavior within the hypothalamus include those associated with the stimulation of food intake [e.g., neuropeptide Y, melanin-concentrating hormone (MCH), agouti-related peptide] or with inhibition of food intake [e.g., cocaine- and amphetamine-regulated transcript (CART)] (Kristensen et al., 1998; Lambert et al., 1998; Schwartz et al., 2000; Ellacott and Cone, 2004; Hunter et al., 2004). The latter was discovered by differential mRNA display as a transcript exhibiting increased expression in rats treated with amphetamine or cocaine (Douglass et al., 1995). Interestingly, CART is expressed in vagal afferent neurons that also express CCK-1 receptors (Broberger et al., 1999), and there is evidence that it can inhibit ingestive behavior from both hindbrain and hypothalamic sites (Aja et al., 2001; Zheng et al., 2001; Smedh and Moran, 2003).
Recent studies suggest that in addition to expression of CCK-1 receptors, vagal afferent neurons also express the leptin receptor (Ob-R) and receptors associated with stimulation of food intake including the ghrelin (GHS-1), cannabinoid (CB1), orexin (OX-R1), and MCH-1 receptors (Kirkup et al., 2001; Burdyga et al., 2002, 2003, 2004, 2006b; Dockray, 2003). Orexin A and ghrelin inhibit the discharge of vagal afferent neurons in response to CCK compatible with integration of both orexigenic and anorexigenic stimuli outside the CNS (Burdyga et al., 2003; Date et al., 2005). In addition, a growing body of evidence now indicates regulation by CCK of gene expression in these neurons. Thus, energy restriction of rats for 12–18 h or longer sharply increases expression of CB1 and MCH-1 receptors, and of MCH, and refeeding downregulates expression via CCK (Burdyga et al., 2004, 2006b). This is not a general response of all receptors associated with orexigenic function because OX-R1 and GHS-1 expression appears unchanged in these circumstances (Burdyga et al., 2003, 2006a). The phenotype of vagal afferent neurons therefore reflects energy ingestion over the previous 24 h. Because these neurons express CART, which has been linked to satiety effects (Kristensen et al., 1998; Lambert et al., 1998; Broberger et al., 1999), we hypothesized that CCK regulates CART expression in vagal afferent neurons, and we report here evidence consistent with the hypothesis.
Materials and Methods
Materials.
Unless stated otherwise, chemicals were obtained from Sigma (Poole, Dorset, UK); CCK8 and ghrelin were obtained from Bachem (St. Helens, UK), lorglumide was a gift from Rotta Pharmaceuticals (Milan, Italy), and Ro-32-0432 was obtained from Calbiochem (La Jolla, CA). Dr. M. Kuhar (Yerkes National Primate Research Center, Emory University, Atlanta, GA) and Dr. P. Barrett (Rowett Research Institute, Aberdeen Center for Energy Regulation and Obesity, Bucksburn, UK) provided CART promoter–luciferase reporter constructs of 120, 620, 3451, and 3100 bp upstream of the transcriptional start site (i.e., 120 CART-Luc, 620 CART-Luc, 3451 CART-Luc, and 3100 CART-Luc). Dr. J. Nevins (Duke University Medical Center, Durham, NC) kindly provided plasmids for the adenovirus product E1A and the inactive mutant Δ2-36 E1A. Dr. D. Ginty (The Johns Hopkins University School of Medicine, Baltimore, MD) provided the cAMP response element-binding protein (CREB) dominant-negative mutant, ACREB plasmid.
Animals.
Studies were performed using adult male Wistar rats (250–400 g) housed at 22°C under a 12 h light/dark cycle with ad libitum access to food and water, unless stated otherwise.
Fasting–feeding experiments.
Rats were fasted for 24 h (water ad libitum) and subsequently refed for up to 2 h. Some fasted animals received CCK8 (10 nmol, i.p.) or saline and were killed 2 h later. In addition, some rats that were fasted for 24 h received the CCK-1 receptor antagonist lorglumide (10 mg/kg, i.p.) 15 min before refeeding and were killed 1 h after refeeding. Rats were killed by CO2 inhalation, and nodose ganglia were rapidly immersed in 4% paraformaldehyde for 1 h at 22°C, followed by 25% sucrose in 1 mol/L PBS overnight at 4°C.
Cell culture.
Nodose ganglia were dissected under aseptic conditions and digested for 90 min at 37°C in 3 ml of Ca2+- and Mg2+-free HBSS containing 50 μl of 100 mm DTT and 1 mg/ml collagenase type Ia (Roche Diagnostics, Indianapolis, IN). Cells were dispersed by gentle trituration through siliconized Pasteur pipettes, washed twice with HEPES-buffered DMEM (HDMEM) containing 10% fetal calf serum (FCS) (Hyclone/Perbio Science, Cramlington, UK) supplemented with 1% penicillin/streptomycin solution and 2% antibiotic/antimycotic solution, plated onto four-well chamber slides, and maintained in HDMEM with 10% FCS at 37°C in 5% CO2, and the medium was changed every 48 h. For many experiments, cells were transferred to serum-free medium for 2–10 h before experimental treatments.
Immunohistochemistry.
Cryostat sections of fixed nodose ganglia (10–15 μm) were mounted on polysine-coated slides (Polysine; Menzel-Gläser, Braunschweig, Germany) and processed for immunohistochemistry. Cultured neurons were fixed in 4% paraformaldehyde in PBS. The following antibodies were used: affinity-purified rabbit polyclonal antibody to CART (Phoenix Laboratories, San Antonio, TX); affinity-purified rabbit polyclonal antibody to phosphoCREB (Cell Signaling Technology, Beverly, MA); affinity-purified goat polyclonal antibody to MCH; rabbit polyclonal antibody to the CCK-1 receptor; rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP; Santa Cruz Biotechnology, Calne, Wiltshire, UK); affinity-purified mouse monoclonal antibody to neuron-specific enolase (NSE; Abcam, Cambridge, UK); and affinity-purified mouse anti-Flag monoclonal antibody (Upstate Biotechnology, Hampshire, UK). Secondary antibodies were used as appropriate and included donkey anti-rabbit Ig conjugated to fluorescein isothiocyanate and donkey anti-mouse Ig labeled with Texas Red (Jackson ImmunoResearch, West Grove, PA). Specificity of immunostaining was determined by omitting the primary antibody and by preincubation with an excess of appropriate peptide where available. Samples were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Peterborough, UK) for nuclear localization and examined using an Axiopan Universal microscope (Zeiss, Oberkochen, Germany), and images were processed using the Axio Vision 3.0 Imaging system (Zeiss).
Luciferase assay.
Vagal afferent neurons were cultured in full medium for 2 d in 12-well plates. The medium was removed, and cells were transfected with firefly-luciferase constructs and a constitutively active Renilla luciferase plasmid as an internal control (Promega, Madison, WI); cells were transfected with 1 μg of DNA per well, using CombiMag (OzBiosciences, Marseille, France) together with Transfast reagent (Promega), on a magnetic plate according to the manufacturer's instructions. Cells were further incubated for 24 h before stimulation. Luciferase activity was measured by dual luciferase assay (Promega) according to the manufacturer's instructions in a Lumat LB9507 luminometer (Berthold, Redbourne, Herts, UK). Results are presented as fold increase over unstimulated control, so 1.0 signifies no change in luciferase activity.
Mutation of cAMP response element.
To investigate the importance of the cAMP response element site for CCK-mediated CART transcription, this site was mutated from tgacgtca to gtcagtca in the 34510 CART-Luc construct using a QuikChange II XL site-directed mutagenesis kit (Stratagene Europe, Amsterdam, The Netherlands).
Results
Energy restriction depresses CART in vagal afferent neurons in vivo
In initial studies, we examined the localization of CART in vagal afferent neurons of rats fed ad libitum or fasted for 24 h. In the middle and caudal regions of the nodose ganglion from rats fed ad libitum, 45 ± 2% of all neurons expressed CART, but after fasting for 24 h, CART immunoreactivity was detected in <10% of neurons. Administration of CCK8 (10 nmol, i.p.) to fasted rats increased CART-expressing cells within 1 h. In addition, 1 h after refeeding fasted rats, CART expression was restored, and this increase was inhibited by administration of the CCK-1 receptor antagonist lorglumide (Fig. 1).
Figure 1.
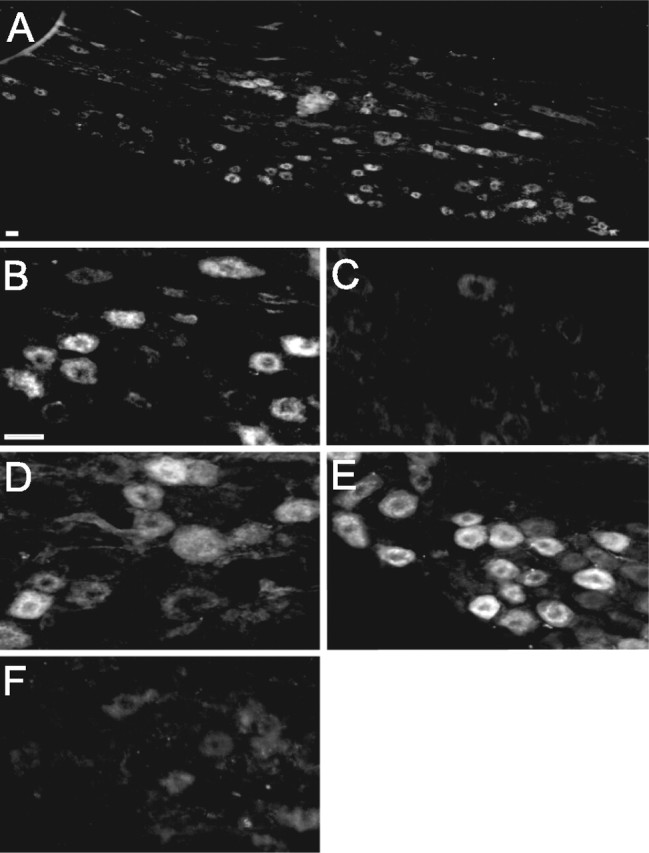
CCK-dependent expression of CART in vagal afferent neurons. Nodose ganglia from rats are shown. A, B, Rats fed ad libitum exhibiting CART-immunoreactive neurons. C, Reduced CART immunoreactivity after fasting for 24 h. D, Stimulation of CART immunoreactivity after fasting for 24 h and 1 h after an intraperitoneal injection of 10 nmol of CCK. E, F, Stimulation of CART after fasting for 24 h and refeeding for 1 h (E), which was inhibited by administration of the CCK-1 receptor antagonist lorglumide (F; 10 mg · kg−1, i.p.) 15 min before refeeding. Representative images from 10 independent experiments are shown. Magnification: A, 20×; B–F, 40×.
CART expression in vagal afferent neurons in culture
The mechanisms controlling CART abundance in response to CCK were then examined in cultured vagal afferent neurons. After culture of dissociated rat nodose ganglia for 96 h, 79.1 ± 1.8% of cells were identified as neurons on the basis of expression of NSE, and most of the remainder (12.7 ± 1.3%) were identified as glial cells on the basis of GFAP staining. Approximately 75% of neurons expressed CART (61.0 ± 1.2% of all cells), and as expected, CART was expressed by <1% of GFAP-positive cells. Serum withdrawal for 2 h reduced the population of cells expressing CART immunoreactivity to 6.2 ± 0.8% of the total (Fig. 2). However, the addition of CCK (10 nm) to serum-deprived vagal afferent neurons restored the proportion of neurons expressing CART to that of control cultures (Fig. 2A,C). The addition of 12-myristate-13-acetate phorbol ester (PMA) to neurons in serum-free conditions also increased CART immunoreactivity (Fig. 2E); moreover, the PKC inhibitor Ro-32-0432 blocked the effect of both CCK and PMA (data not shown), compatible with the idea that CCK induces CART via PKC (Fig. 2G). The orexigenic hormone ghrelin alone had no effect on CART expression in vagal afferent neurons in serum-free medium (Fig. 2I). However, the addition of ghrelin inhibited the action of CCK, indicating an interplay between orexigenic and anorexic peptides in regulating CART expression (Fig. 2K).
Figure 2.

CART expression in cultured vagal afferent neurons. Vagal afferent neurons cultured for >48 h showing CART immunoreactivity (A, C, E, G, I, K) or nuclei stained with DAPI in the respective fields (B, D, F, H, J, L). A, B, CART is undetectable after transfer of neurons to serum-free medium for 2 h. C, D, CART immunoreactivity is readily detected in cells in serum-free medium 2 h after the addition of 10 nm CCK. E, F, CART is also induced in cells in serum-free medium 2 h after the addition of 100 nm PMA. G, H, The effect of CCK is reduced by the PKC inhibitor Ro-32-0432 (1 μm). I–L, In serum-free medium, ghrelin (10 nm) has no effect on CART abundance (I, J) but inhibits the effect of CCK (K, L). Arrows identify a representative neuron in each pair of images; filled arrows show CART-positive neurons, and open arrows show CART-negative neurons. Representative images from six independent experiments are shown. Scale bar, 50 μm.
CCK stimulation of CREB phosphorylation in vagal afferent neurons
Because it is known that CART expression is regulated by CREB (Lakatos et al., 2002; Dominguez and Kuhar, 2004; Jones and Kuhar, 2006), we asked whether CCK-1 receptor activation led to CREB phosphorylation in vagal afferent neurons. In vagal afferent neurons transferred to serum free-medium for 2 h, phosphoCREB was undetectable, but in the presence of 10 nm CCK for 30 min, there was abundant nuclear phosphoCREB (Fig. 3). The PKC inhibitor Ro-32-0432 blocked the action of CCK in stimulating phosphoCREB in cultured vagal afferent neurons, and PMA stimulated it (Fig. 3E,G), compatible with CCK phosphorylation of CREB via PKC. On its own, ghrelin did not increase nuclear phosphoCREB (Fig. 3I). However, when cells were treated with ghrelin in the presence of CCK, there was a striking exclusion of phosphoCREB from the nucleus that was associated with localization to the cytosol (Fig. 3K). In response to CCK alone, antibody to total (i.e., phosphorylated and unphosphorylated) CREB revealed a predominantly nuclear localization, but when CCK was combined with ghrelin, there was strong cytosolic localization similar to that of phosphoCREB (Fig. 3M,O).
Figure 3.

CCK induces CREB phosphorylation through activation of PKC in vagal afferent neurons. Vagal afferent neurons cultured for >48 h and stained with antibody to phosphoCREB (A, C, E, G, I, K) or CREB (M, O) and DAPI (B, D, F, H, J, L, N, P), in the respective fields, are shown. A, B, Nuclear phosphoCREB staining was undetectable after transfer to serum-free medium for 2 h. C–F, Nuclear phosphoCREB was readily detectable after treatment with 10 nm CCK (C, D) and 100 nm PMA (E, F) for 30 min. G, H, The effect of CCK was inhibited by pretreatment with 1 μm Ro-32-0432. I, J, Nuclear phosphoCREB was undetectable after treatment with ghrelin (10 nm). K, L, Importantly, ghrelin (10 nm) in the presence of 10 nm CCK led to exclusion of phosphoCREB from the nucleus. M–P, Total CREB (M, N) was unchanged by CCK and showed a cytoplasmic localization in the presence of ghrelin plus CCK (O, P). A–L, Arrows identify a representative neuron in each pair of images; filled arrows show phosphoCREB-positive neurons, and open arrows show phosphoCREB-negative neurons. M–O, Arrows indicate a representative neuron exhibiting nuclear localization of total CREB (M, N) and a representative neuron exhibiting cytosolic localization of total CREB (M, O). Representative images from six independent experiments are shown. Scale bar, 50 μm.
CCK stimulation of CART promoter–luciferase reporter expression
To explore the mechanisms by which CCK regulates CART abundance in more detail, we then examined the expression of CART promoter–luciferase reporter constructs after transfection into vagal afferent neurons (Fig. 4). In response to CCK, there was a 2.3 ± 0.1-fold increase in expression of a 3.45 kb sequence of the CART promoter region coupled to luciferase. Truncation of the sequence to 620 bp reduced the response to CCK, and further truncation to 120 bp virtually abolished it (Fig. 4A). The increase in luciferase activity in response to CCK was significantly reduced by the PKC inhibitor Ro-32-0432 (Fig. 4B) and by ghrelin in combination with CCK (Fig. 4C). Compatible with the idea that CCK regulates CART expression via CREB, cotransfection with a vector encoding a dominant-negative CREB protein, ACREB, significantly inhibited the response to CCK. Moreover, cotransfection with a plasmid encoding E1A (which inhibits the CREB coactivator CBP) also significantly reduced the response to CCK, whereas an inactive E1A mutant (Δ 2-36 E1A) had no effect (Fig. 5). Finally, mutation of the Cre site in the 3.45 kb sequence virtually abolished responses to CCK. Together, the data are therefore compatible with a role for PKC, CREB, CBP, and the Cre element in mediating the effects of CCK.
Figure 4.
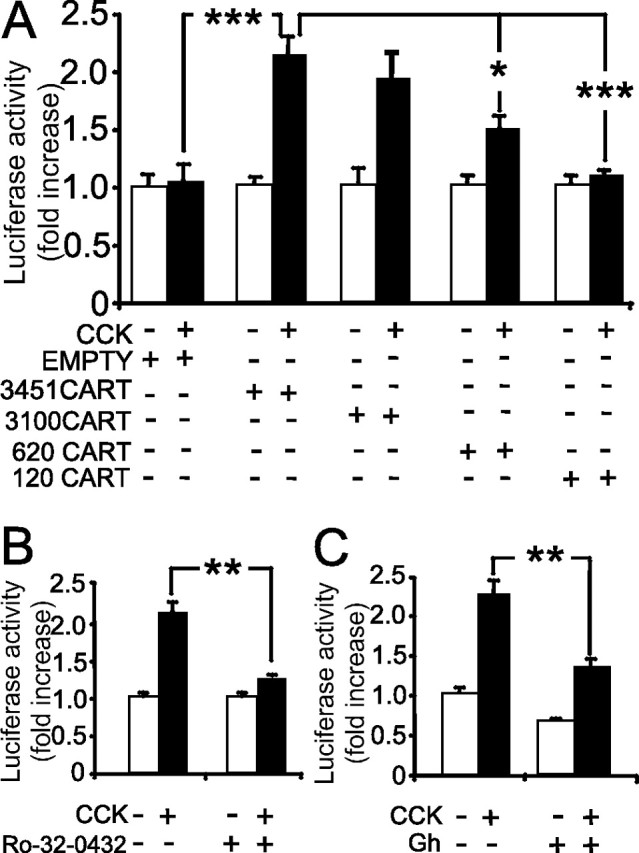
CCK regulates CART transcription in luciferase promoter–reporter assays. A, Cultured vagal afferent neurons were transfected with empty vector (empty) and deletional mutants of the CART promoter of 3451, 3100, 620, and 120 bp upstream of the transcriptional start site in luciferase reporter vectors (empty, empty vector). Studies were performed 16 h after transfection when cells were transferred to serum-free medium and stimulated (filled bars) or not (open bars) with 10 nm CCK for 6 h. B, In vagal afferent neurons transfected with 3451CART-Luc, the response to 10 nm CCK was inhibited by 1 μm Ro-32-0432. C, In vagal afferent neurons transfected with 3451 CART-Luc, the effect of 10 nm CCK was inhibited by preincubation for 30 min with 10 nm ghrelin (Gh). Responses to CCK are expressed relative to the appropriate control (1.00). *p < 0.05, **p < 0.01,*** p < 0.001 (t test; n = 6–15). Error bars indicate SEM.
Figure 5.

CCK-stimulated CART transcription is mediated by Cre, CREB, and CBP in vagal afferent neurons. A, In cultured vagal afferent neurons cotransfected with 3451CART-Luc and ACREB, stimulation of luciferase activity by CCK was inhibited compared with a control vector (EMPTY). B, An inhibitor of the CREB transcriptional coactivator CBP, E1A, inhibited the CART luciferase vector response to CCK compared with an appropriate E1A control vector (E1AΔ2-36). WT, Wild type. C, Mutation of the Cre site in the 3451CART-Luc vector (ΔCRE) abolished the response to CCK. ***p < 0.001 (n = 6). Error bars indicate SEM.
CREB-mediated CART–MCH switch
Previous studies have shown that expression of the orexigenic peptide MCH in vagal afferent neurons is increased by fasting and decreased by CCK (Burdyga et al., 2006b). We therefore asked whether the loss of CART in serum-starved vagal afferent neurons was associated with increased MCH (Fig. 6). Serum withdrawal for 8 h was found to be associated with MCH expression in a substantial proportion of cells (53.0 ± 0.9% of all cells); in parallel with the increased expression of CART on exposure to CCK, there was rapid loss of MCH expression so that after 120 min MCH was detectable in only 2.8 ± 1.2% of cells (Fig. 6). At ∼85 min after the addition of CCK, it was possible to identify separate populations of vesicles containing CART and MCH within the same neuron, indicating that these neurons switched between expression of CART and MCH depending on exposure to CCK (Fig. 6); in particular, at this time point, <10% of cells expressed either CART or MCH alone, but 48.9 ± 2.7% expressed both.
Figure 6.

CCK switches between expression of CART and MCH in vagal afferent neurons. A–D, Immunocytochemical localization of CART and MCH in vagal afferent neurons exposed to serum-free medium for 8 h (A, DAPI; B, MCH; C, CART; D, overlay of B and C). Note the expression of MCH but not CART. E–H, In neurons incubated in serum-free medium with the addition of CCK (10 nm) for 85 min, coexpression of CART and MCH in the same neuron can be demonstrated (E, DAPI; F, MCH in neurite terminals; G, CART in the cell soma and neurite terminals; H, overlay of F and G). I–L, In neurons incubated in serum-free medium (6 h) with the addition of CCK (10 nm) for 2 h, there is loss of MCH and stimulation of CART (I, DAPI; J, MCH; K, CART; L, overlay of J and K). M–P, In neurons transfected with ACREB, incubated in serum-free medium (6 h) with the addition of CCK (10 nm) for 2 h, the loss of MCH is inhibited (M, DAPI; N, ACREB revealed by immunostaining for FLAG epitope; O, MCH; P, overlay of N and O). Filled arrows indicate the position of nuclei for reference, and open arrows indicate the position of a terminal neurite containing both CART- and MCH-immunoreactive vesicles. Representative images from six independent experiments are shown. Scale bar, 50 μm.
To determine whether CREB regulated expression of wild-type CART and MCH in vagal afferent neurons, we then examined the consequences of transfecting cultured vagal afferent neurons with the dominant-negative mutant ACREB, identified by expression of the Flag epitope. Cells expressing ACREB made up 10.9% of the total population; in the same cultures, CCK-1 receptor expression was identified in 59.1% of all cells, and cells positive for both ACREB and CCK-1 receptor made up 10.2% of the total population. Thus, 93.1% of ACREB-transfected cells were CCK-1 receptor positive, reflecting a transfection efficiency for this population of 18%. Of the cells that expressed ACREB, only 15 ± 2% expressed CART in response to CCK (data not shown), whereas untransfected cells in the same cultures exhibited CART expression in response to CCK similar to that described above. In control experiments in which cells were transfected with a vector encoding green fluorescent protein (GFP), the proportions exhibiting CART expression in response to CCK were again similar to controls (data not shown). Importantly, however, 96.6 ± 1% of cells expressing ACREB also exhibited MCH after treatment with CCK, indicating that ACREB prevented CCK-induced downregulation of MCH.
Discussion
The present study shows that CCK regulates CART expression in vagal afferent neurons and that this depends on activation of PKC and CREB. The idea that CART might be a peptide mediator of the satiety effects of CCK has been recognized for some time (Broberger et al., 1999; Hokfelt et al., 2001). However, the regulation of CART expression by CCK in vagal afferent neurons adds an additional dimension to our understanding of this system. Although other examples of CCK-regulated expression of signaling molecules associated with feeding behavior have been described previously (Burdyga et al., 2004, 2006b), in each case CCK was found to depress expression of genes associated with orexigenic function (e.g., CB1 and MCH-1 receptors and MCH). The present findings suggest that activation of CCK-1 receptors on vagal afferent neurons is also associated with increased expression of a gene associated with satiety signaling. Two features of this system are noteworthy. First, within the same neuron, CCK acts as a switch that relatively rapidly depresses production of the orexigenic peptide MCH and stimulates expression of the satiety peptide CART. Second, orexigenic peptides such as ghrelin inhibit the action of CCK and appear to do so by restricting the nuclear pool of phosphoCREB.
Stimulation of CCK-1 receptors on vagal afferent neurons is well known to lead to inhibition of food intake, inhibition of gastric emptying, and stimulation of pancreatic secretion (Dockray, 2003; Moran and Kinzig, 2004), thereby limiting delivery of nutrient to the small intestine after a meal and ensuring optimal digestion of food that has already progressed beyond the pyloric sphincter. Recent evidence indicates that the effect of CCK on vagal afferent neurons is potentiated by leptin and by gastric distension (Schwartz et al., 1993; Wang et al., 1997) and inhibited by ghrelin and orexin A (Burdyga et al., 2003; Date et al., 2005). Together, with the observation that CCK regulates the expression of some receptors (CB1, OX-R1) and peptides (MCH, CART) controlling feeding behavior, the data indicate considerably more sophisticated patterns of integration of information at the level of vagal afferent neurons than previously supposed.
Broberger et al. (1999) previously noted that CART expression in vagal afferent neurons was not changed by feeding different diets. The present data indicate that both food withdrawal in vivo and serum starvation in vitro lead to loss of CART immunoreactivity that is reversed by CCK. The loss of CART in serum-deprived cultured neurons is not attributable to a generalized depression of protein synthesis because MCH exhibits increased expression. Instead, it seems that CCK is able to switch the neurochemistry of vagal afferent neurons by decreasing the capacity for orexigenic signaling while enhancing the capacity for satiety signaling. The CART–MCH switch within these neurons is presumably part of a mechanism that allows control of meal size to be influenced by previous energy intake so that after energy restriction there is a transient and compensatory increase in food intake and a corresponding acceleration of gastric emptying to ensure accelerated delivery of food to the intestine.
The observation that CCK regulates CART expression via CREB in vagal afferent neurons, and is blocked by ghrelin, is consistent with previous studies showing that CREB regulates CART expression (Lakatos et al., 2002; Dominguez and Kuhar, 2004; Jones and Kuhar, 2006), that CCK stimulates CREB phosphorylation (Burdyga et al., 2004; Sutton et al., 2004), and that ghrelin inhibits the effect of CCK. However, the observation that ghrelin inhibits the nuclear localization of phosphoCREB in the presence of CCK was unanticipated. Recent work in other systems has identified nuclear exclusion of CREB [e.g., in 6-hydroxydopamine-treated neurons (Chalovich et al., 2006)] as a novel regulatory event. The precise mechanism by which this is achieved is presently uncertain, and clearly additional work is required in this area.
Until quite recently, the gastrointestinal mechanisms regulating feeding behavior were considered to depend on a relatively simple relay function on the part of vagal afferent neurons serving the gut with major sites of integration at the level of the brainstem and hypothalamus. It is now clear that this view should be revised. In particular, there appears to be two distinct neurochemical phenotypes adopted by vagal afferent neurons. In response to energy restriction, there is increased expression of CB1, MCH-1, and MCH and decreased expression of CART, whereas the converse pattern is characteristic of states of unrestricted energy supply. CCK is able, on its own, to switch neurons between the two states, and although ghrelin alone appears not to determine the phenotype, it inhibits the action of CCK, at least in part by nuclear exclusion of phosphoCREB. A more detailed understanding of this system may well be useful in developing novel strategies for the therapeutic modulation of food intake.
Footnotes
We thank the Wellcome Trust and the Medical Research Council for financial support. We are grateful to Drs. Michael Kuhar, Perry Barrett, Joseph Nevins, and David Ginty for the supply of plasmids and to Rotta Pharmaceuticals for the gift of lorglumide.
References
- Aja S, Sahandy S, Ladenheim EE, Schwartz GJ, Moran TH. Intracerebroventricular CART peptide reduces food intake and alters motor behavior at a hindbrain site. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1862–R1867. doi: 10.1152/ajpregu.2001.281.6.R1862. [DOI] [PubMed] [Google Scholar]
- Broberger C, Holmberg K, Kuhar MJ, Hokfelt T. Cocaine- and amphetamine-regulated transcript in the rat vagus nerve: a putative mediator of cholecystokinin-induced satiety. Proc Natl Acad Sci USA. 1999;96:13506–13511. doi: 10.1073/pnas.96.23.13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Spiller D, Morris R, Lal S, Thompson DG, Saeed S, Dimaline R, Varro A, Dockray GJ. Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience. 2002;109:339–347. doi: 10.1016/s0306-4522(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Lal S, Spiller D, Jiang W, Thompson DG, Attwood S, Saeed S, Grundy D, Varro A, Dimaline R, Dockray GJ. Localization of orexin-1 receptors to vagal afferent neurons in the rat and humans. Gastroenterology. 2003;124:129–139. doi: 10.1053/gast.2003.50020. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Lal S, Varro A, Dimaline R, Thompson DG, Dockray GJ. Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J Neurosci. 2004;24:2708–2715. doi: 10.1523/JNEUROSCI.5404-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Ghrelin receptors in rat and human nodose ganglia: putative role in regulating CB-1 and MCH receptor abundance. Am J Physiol Gastrointest Liver Physiol. 2006a;290:G1289–G1297. doi: 10.1152/ajpgi.00543.2005. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Varro A, Dimaline R, Thompson DG, Dockray GJ. Feeding-dependent depression of melanin-concentrating hormone and melanin-concentrating hormone receptor-1 expression in vagal afferent neurones. Neuroscience. 2006b;137:1405–1415. doi: 10.1016/j.neuroscience.2005.10.057. [DOI] [PubMed] [Google Scholar]
- Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem. 2006;281:17870–17881. doi: 10.1074/jbc.M602632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date Y, Toshinai K, Koda S, Miyazato M, Shimbara T, Tsuruta T, Niijima A, Kangawa K, Nakazato M. Peripheral interaction of ghrelin with cholecystokinin on feeding regulation. Endocrinology. 2005;146:3518–3525. doi: 10.1210/en.2004-1240. [DOI] [PubMed] [Google Scholar]
- Dockray GJ. The brain-gut axis. In: Yamada T, Alpers DH, Kaplowitz L, Laine L, Owyang C, Powell DW, editors. Textbook of gastroenterology. Philadelphia: Lipincott Williams and Wilkins; 2003. pp. 77–92. [Google Scholar]
- Dockray GJ. Gut endocrine secretions and their relevance to satiety. Curr Opin Pharmacol. 2004;4:557–560. doi: 10.1016/j.coph.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Dominguez G, Kuhar MJ. Transcriptional regulation of the CART promoter in CATH.a cells. Brain Res Mol Brain Res. 2004;126:22–29. doi: 10.1016/j.molbrainres.2004.02.027. [DOI] [PubMed] [Google Scholar]
- Douglass J, McKinzie AA, Couceyro P. PCR differential display identifies a rat brain mRNA that is transcriptionally regulated by cocaine and amphetamine. J Neurosci. 1995;15:2471–2481. doi: 10.1523/JNEUROSCI.15-03-02471.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellacott KL, Cone RD. The central melanocortin system and the integration of short- and long-term regulators of energy homeostasis. Recent Prog Horm Res. 2004;59:395–408. doi: 10.1210/rp.59.1.395. [DOI] [PubMed] [Google Scholar]
- Gonzalez MF, Deutsch JA. Vagotomy abolishes cues of satiety produced by gastric distension. Science. 1981;212:1283–1284. doi: 10.1126/science.7233218. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Holmberg K, Shi TJ, Broberger C. CCK-ergic mechanisms in sensory systems. Scand J Clin Lab Invest Suppl. 2001;234:69–74. [PubMed] [Google Scholar]
- Hunter RG, Philpot K, Vicentic A, Dominguez G, Hubert GW, Kuhar MJ. CART in feeding and obesity. Trends Endocrinol Metab. 2004;15:454–459. doi: 10.1016/j.tem.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Jones DC, Kuhar MJ. Cocaine-amphetamine-regulated transcript expression in the rat nucleus accumbens is regulated by adenylyl cyclase and the cyclic adenosine 5′-monophosphate/protein kinase a second messenger system. J Pharmacol Exp Ther. 2006;317:454–461. doi: 10.1124/jpet.105.096123. [DOI] [PubMed] [Google Scholar]
- Kirkup AJ, Brunsden AM, Grundy D. Receptors and transmission in the brain-gut axis: potential for novel therapies. I. Receptors on visceral afferents. Am J Physiol Gastrointest Liver Physiol. 2001;280:G787–G794. doi: 10.1152/ajpgi.2001.280.5.G787. [DOI] [PubMed] [Google Scholar]
- Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang N, Larsen PJ, Hastrup S. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature. 1998;393:72–76. doi: 10.1038/29993. [DOI] [PubMed] [Google Scholar]
- Lakatos A, Dominguez G, Kuhar MJ. CART promoter CRE site binds phosphorylated CREB. Brain Res Mol Brain Res. 2002;104:81–85. doi: 10.1016/s0169-328x(02)00321-2. [DOI] [PubMed] [Google Scholar]
- Lambert PD, Couceyro PR, McGirr KM, ll Vechia SE, Smith Y, Kuhar MJ. CART peptides in the central control of feeding and interactions with neuropeptide Y. Synapse. 1998;29:293–298. doi: 10.1002/(SICI)1098-2396(199808)29:4<293::AID-SYN1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Liddle RA. Cholecystokinin cells. Annu Rev Physiol. 1997;59:221–242. doi: 10.1146/annurev.physiol.59.1.221. [DOI] [PubMed] [Google Scholar]
- Luckman SM, Lawrence CB. Anorectic brainstem peptides: more pieces to the puzzle. Trends Endocrinol Metab. 2003;14:60–65. doi: 10.1016/s1043-2760(02)00033-4. [DOI] [PubMed] [Google Scholar]
- Moran TH, Kinzig KP. Gastrointestinal satiety signals II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286:G183–G188. doi: 10.1152/ajpgi.00434.2003. [DOI] [PubMed] [Google Scholar]
- Murphy KG, Bloom SR. Gut hormones in the control of appetite. Exp Physiol. 2004;89:507–516. doi: 10.1113/expphysiol.2004.027789. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, McHugh PR, Moran TH. Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. Am J Physiol. 1993;265:R872–R876. doi: 10.1152/ajpregu.1993.265.4.R872. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Smedh U, Moran TH. Peptides that regulate food intake: separable mechanisms for dorsal hindbrain CART peptide to inhibit gastric emptying and food intake. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1418–R1426. doi: 10.1152/ajpregu.00665.2002. [DOI] [PubMed] [Google Scholar]
- Smith GP, Jerome C, Norgren R. Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am J Physiol. 1985;249:R638–R641. doi: 10.1152/ajpregu.1985.249.5.R638. [DOI] [PubMed] [Google Scholar]
- Sutton GM, Patterson LM, Berthoud HR. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. J Neurosci. 2004;24:10240–10247. doi: 10.1523/JNEUROSCI.2764-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YH, Tache Y, Sheibel AB, Go VL, Wei JY. Two types of leptin-responsive gastric vagal afferent terminals: an in vitro single-unit study in rats. Am J Physiol. 1997;273:R833–R837. doi: 10.1152/ajpregu.1997.273.2.R833. [DOI] [PubMed] [Google Scholar]
- Woods SC. Gastrointestinal satiety signals I. An overview of gastrointestinal signals that influence food intake. Am J Physiol Gastrointest Liver Physiol. 2004;286:G7–G13. doi: 10.1152/ajpgi.00448.2003. [DOI] [PubMed] [Google Scholar]
- Zheng H, Patterson C, Berthoud HR. Fourth ventricular injection of CART peptide inhibits short-term sucrose intake in rats. Brain Res. 2001;896:153–156. doi: 10.1016/s0006-8993(00)03256-x. [DOI] [PubMed] [Google Scholar]