

Abstract
Visual adaptation is thought to occur partly at retinal synapses that are subject to plastic changes. However, the locus and properties of this plasticity are not well known. Here, we studied short-term plasticity at the reciprocal synapse between bipolar cell terminals and amacrine cells in goldfish retinal slices. Depolarization of a single bipolar cell terminal for 100 ms triggers the release of glutamate onto amacrine cell processes, which in turn leads to GABAergic feedback from amacrine cells onto the same terminal. We find that this release of GABA undergoes paired-pulse depression (PPD) that recovers in <1 min (single exponential time constant, τ ≅ 12 s). This disynaptic PPD is independent of mGluR-mediated plasticity and depletion of glutamatergic synaptic vesicle pools, because exocytosis assayed via capacitance jumps (ΔCm) recovered completely after 10 s (τ ≅ 2 s). Fast application of GABA (10 mm) onto outside-out patches excised from bipolar cell terminals showed that the recovery of GABAA receptors from desensitization depends on the duration of the application [fast recovery (<2 s) for short applications; slow (τ ≅ 12 s) for prolonged applications]. We thus blocked GABAA receptors and retested the GABAergic response mediated by nondesensitizing GABAC receptors to two rapid glutamate puffs onto the bipolar cell terminal. These responses consistently displayed PPD. Furthermore, blocking AMPA-receptor desensitization with cyclothiazide, or evoking GABA release with NMDA receptors, did not reduce PPD. We thus suggest that depletion of synaptic vesicle pools in GABAergic amacrine cells is a major contributor to PPD.
Keywords: retina, inner plexiform layer, microcircuits, bipolar cell terminals, amacrine cells, adaptation, membrane capacitance, paired-pulse depression, short-term plasticity
Introduction
Signals from photoreceptors are passed onto retinal ganglion cell dendrites via excitatory synapses formed by intervening bipolar cells (Slaughter and Miller, 1983; Wässle, 2004). However, before these signals are passed onto ganglion cells they are often processed in local microcircuits formed between bipolar cell terminals and amacrine cells in the inner plexiform layer (IPL) of the vertebrate retina. These IPL microcircuits can form complex nested feedback loops involving reciprocal and serial inhibitory synapses (Zhang et al., 1997; Marc and Liu, 2000), and they probably play a crucial role in several aspects of vision, including adaptation to changes in visual scene contrast and ambient light level.
Some bipolar and amacrine cell synaptic output is capable of undergoing both fast (<2 s) and slow (10 s timescale; recovery over 5–20 s) contrast adaptation (Kim and Rieke, 2001; Baccus and Meister, 2002), and the slow form of contrast adaptation on the inputs to ganglion cells likely results from short-term synaptic depression at bipolar cell terminals caused by vesicle pool depletion (Manookin and Demb, 2006; Singer and Diamond, 2006). More recently, Hosoya et al. (2005) found that responses to a fixed orientation of lines diminishes with a time course of ∼10 s, whereas a response to a perpendicular set of lines remains undiminished. They thus suggested that the retina adjusts its visual information processing dynamically to changes in visual statistics, and they attributed this ability to plastic synapses made by amacrine cells. However, how these plastic changes occur at the synaptic level and the properties of the underlying mechanism is mostly unknown.
The basic architecture of reciprocal synapses between bipolar cell terminals and amacrine cells is well preserved from fish to mammals (Dowling and Boycott, 1966; Witkovsky and Dowling, 1969; Masland, 2001). Namely, depolarization of the bipolar cell terminal releases glutamate onto amacrine cells, which then triggers the release of GABA back onto the same bipolar cell terminal (Dong and Werblin, 1998; Protti and Llano, 1998; Hartveit, 1999; Singer and Diamond, 2003; Chavez et al., 2006). Previously, we found a form of long-term plasticity at this disynaptic contact whereby prolonged glutamate release from bipolar cell terminals could activate mGluR1 on amacrine cells, which leads to enhanced GABA release for several minutes (Vigh et al., 2005).
Here, we set out to investigate short-term plasticity at this reciprocal synapse. Patch-clamp recordings were made directly from single axotomized Mb-type bipolar cell terminals in goldfish retinal slices (Palmer et al., 2003b). We found that the reciprocal feedback response was depressed for up to 1 min after a 100 ms prepulse stimulus. We aimed to dissect the underlying synaptic mechanisms that contribute to this paired-pulse depression (PPD) of evoked GABA release. Is bipolar cell vesicle pool depletion, or GABAA or AMPA receptor desensitization necessary for generating PPD? We thus studied the recovery of exocytosis from the bipolar cell terminal with membrane capacitance (Cm) measurements (Heidelberger et al., 2005), the recovery from desensitization of GABAA receptors on bipolar cell terminals, and the effects on PPD of blocking AMPA-receptor desensitization.
Materials and Methods
Retinal slice preparation.
Retinal slices (∼250 μm in thickness) were prepared from goldfish (Carassius auratus; 8∼14 cm) according to the procedures described previously (Palmer et al., 2003b). Slices were cut freshly from one piece of retina (3∼5 mm square) and transferred to a recording chamber. The recording chamber, together with slices, was moved to an upright microscope (Axioskop 2; Zeiss, Oberkochen, Germany) where slices were perfused continuously (2–3 ml/min) with Ringer's solution and viewed with infrared differential interference contrast optics through a 40× water-immersion objective coupled with a 1.6 premagnification and a CCD camera (C79; Hamamatsu, Tokyo, Japan). Bipolar cell terminals were identified by their size (∼10 μm diameter), shape, and position in the slice, as well as by a depolarization evoked L-type Ca2+ current and capacitance jump (ΔCm) response.
Electrophysiology.
For whole-cell recordings, the Ringer's solution contained (in mm) 100 NaCl, 2.5 KCl, 1.0 MgCl2, 2.5 CaCl2, 25 NaHCO3, and 12 glucose, pH 7.45 (set with NaOH), bubbled continuously with 95% O2 and 5% CO2. For outside-out patch recordings the Ringer's solution contained (in mm) 120 NaCl, 2.5 KCl, 1.0 MgCl2, 2.5 CaCl2, 10 HEPES, and 12 glucose, pH 7.45 (set with NaOH).
Patch pipettes were pulled from thick-walled borosilicate glass (World Precision Instruments, Sarasota, FL) using a Narishige (Tokyo, Japan) puller (model PP-830). They had a resistance of ∼10 MΩ when filled with an internal solution containing (in mm) 104 Cs-gluconate, 25 HEPES, 1 tetraethylammonium (TEA)-Cl, 10 CH3NH2Cl (methylamine), 3 Mg-ATP, 0.5 Na-GTP, and 0.5 EGTA, adjusted to pH 7.2 with CsOH, and a resistance of ∼7 MΩ when filled with a internal solution containing (in mm) 105 CsCl, 25 HEPES, 10 TEA-Cl, 3 Mg-ATP, 0.5 Na-GTP, and 0.5 EGTA, adjusted to pH 7.2 with CsOH. For capacitance measurement, pipettes were coated with dental wax (Cavex, West Chester, PA) to reduce pipette capacitance and electrical noise.
Whole-cell voltage-clamp recordings were made by means of an EPC-9/2 (HEKA Elektronik, Lambrecht/Pfalz, Germany) patch-clamp amplifier and Pulse software (HEKA Elektronik). Cells were held at −60 mV and the current signal was low-pass filtered at 2 kHz and sampled at 20 kHz. Capacitance measurements were performed using the “sine+DC” method with the Lockin extension in Pulse software, and using a 1 kHz sinusoidal voltage command (30 mV peak to peak) added to the holding potential. The resulting current was analyzed at two orthogonal phase angles (Gillis, 2000).
Drugs were bath applied into the perfusing Ringer's solution. NBQX, CNQX, 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid (SR95531), and AP-5 were obtained from Tocris (Bristol, UK). All other chemicals, toxins, and salts were obtained from Sigma (St. Louis, MO). Off-line analysis of the data were performed with Igor Pro 5.0 software (WaveMetrics, Lake Oswego, OR).
Outside-out patches and fast application.
Outside-out patches were obtained from axotomized bipolar cell terminals by slowly lifting the recording pipette after break-in to obtain an excised patch of membrane (Hull et al., 2006). GABA was applied to patches using a borosilicate, two-barreled theta glass pipette (outer diameter, 1.5 mm; inner diameter, 1.0 mm; septum, 0.2 mm; Warner Instruments, Hamden, CT) pulled to a tip diameter of ∼200 μm. For rapid translation, the theta glass pipette was attached to a piezo bending actuator (Piezo Systems, Cambridge, MA) driven by a 10 V command voltage from the amplifier. An adjustable resistor (1∼10 MΩ) was placed in series with the piezo actuator, and it works with the capacitor in the piezo actuator as a filter to slow down the charging process so that the movement of the piezo actuator is smooth. The time required for exchanging solutions, estimated by the open-tip recording resulting from a junction potential between normal Ringer's solution and 10% diluted Ringer's solution, was <1 ms. Patch currents were recorded with a high-chloride internal solution at a holding potential of −60 mV. No voltage dependence was observed for the kinetics of GABA currents in the range of −60 to +60 mV and currents reversed at the Nernst Cl− potential ECl (data not shown).
Results
PPD at reciprocal retinal synapses
ON-type Mb bipolar cell terminals were identified in the IPL of goldfish retinal slices, and whole-cell voltage-clamp recordings were directly made from these terminals (Palmer et al., 2003b). Time-resolved membrane capacitance (Cm) measurements were used to monitor exocytosis and glutamate release (Neher and Marty, 1982; von Gersdorff et al., 1998). To isolate the reciprocal feedback from amacrine cells, 10 mm methylamine was included in the pipette solution to minimize the reduction of calcium current (ICa) by protons released together with glutamate (Palmer et al., 2003a; Vigh et al., 2005).
Reciprocal feedback from amacrine cells was evoked by depolarizing bipolar cell terminals from a holding potential of −60 to 0 mV for 100 ms. Before and after the depolarization, a 100 ms sinusoidal wave (1 kHz, 30 mV peak to peak) was added to the holding potential (Fig. 1A), and the resulting current responses were used to calculate Cm (Gillis, 2000). A representative current response, and the corresponding Cm trace, are shown in Figure 1A. With a Cs+-based pipette solution, the current response to depolarization contained an inward calcium current (ICa) that triggered an increase of Cm (ΔCm). This indicates that synaptic vesicles containing glutamate were exocytosed in response to the depolarization. The released glutamate activated AMPA and NMDA receptors on amacrine cells, triggering GABA release onto the bipolar cell terminal (Vigh and von Gersdorff, 2005). Therefore, superimposed on ICa, an evoked reciprocal IPSC was present, and it was mediated by both GABAA and GABAC receptors on the bipolar cell terminal (Vigh and von Gersdorff, 2005).
Figure 1.

PPD and mGluR1-mediated potentiation of GABAergic synapses. A, A Mb-type bipolar cell terminal current (black trace) is shown together with the membrane capacitance (Cm, blue) and the stimulus protocol (red). The Cm jump (ΔCm) indicates that exocytosis and glutamate release occurred in response to the depolarizing pulse. The current response to the step depolarization consisted of a Ca2+ current (ICa) and a superimposed evoked reciprocal feedback IPSC. B, To isolate the RFR, the current response from A was leak-subtracted (black trace). The blue trace is the scaled pure ICa waveform (averaged from 29 recordings in 100 μm picrotoxin, scaled to the peak of ICa in A). The subtracted trace (shown in the inset, in red) represents the isolated RFR, and the area between the black and blue traces was used to quantify RFR (Fig. 2C). C, RFR in response to paired-pulse stimuli, was depressed at 10 s (blue) and potentiated at 78 s (red) when compared with that evoked by the prepulse (black). D, In the presence of 100 μm LY367385 (mGluR1 blocker), paired-pulse depression of RFR remained unaffected at 10 s (blue), whereas the potentiation of RFR was blocked at times >1 min.
The reciprocally evoked IPSCs displayed considerable variability from trial to trial and among different terminal recordings (Hull and von Gersdorff, 2004; Vigh and von Gersdorff, 2005). To study their average properties and isolate them from ICa, we first recorded the pure ICa in 100 μm picrotoxin (a blocker of both GABAA and GABAC receptors). There was no significant rundown of ICa within 2 min of recording time (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material) and the peak amplitude of ICa ranged from 200 to 600 pA among different terminals (n = 29). However, after normalizing to the peak ICa amplitude and averaging these scaled pure ICa traces, the average waveform of ICa was very similar to individual traces of ICa (n = 29) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material, Fig. 1B, blue trace). We thus subtracted this averaged ICa waveform from each individual current response with IPSCs superimposed. We took the resulting trace (Fig. 1B, inset) as the isolated GABAergic reciprocal feedback response (RFR), and we used the area of the RFR trace (i.e., the total chloride charge transfer QCl) (Vigh et al., 2005) to quantify the amount of GABAergic feedback (Fig. 2C). The capacitive transient artifacts were omitted for clarity and display purposes.
Figure 2.

PPD and glutamate vesicle pool depletion. A, For RFRs evoked by paired-pulse stimuli with an interpulse interval of 1 s, RFR was depressed and ΔCm has not fully recovered. B, For paired-pulse stimuli with an interval of 10 s, RFR was depressed, but ΔCm has fully recovered. C, For a paired-pulse stimulus with an interval of 1 min, both RFR and ΔCm recovered fully. D, Recovery time courses of both RFR and ΔCm (both normalized to the prepulse values) were constructed. Both were fitted well with a single exponential equation, with a time constant (τ) of 12.19 and 2.02 s, respectively. All data for RFR recovery were obtained in the presence of 100 μm LY367385; recordings obtained >2 min after break-in were not included to minimize the slowing down of vesicle cycling caused by washout of exocytosis-related proteins. The red data point shows that at 10 s the percentage of PPD without LY367385 is slightly larger than that with LY367385 (but this difference is not statistically significant). Error bars indicate SE.
In the previous study (Vigh et al., 2005), we found that the reciprocally evoked IPSCs are potentiated after one or two stimuli. This potentiation could last up to 10 min, and can be blocked by the mGluR1 antagonist (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385) (Bruno et al., 1999). In the present study, we confirmed this finding by using a paired-pulse protocol of depolarizing stimuli. For two 100 ms pulses with >1 min apart, RFR was potentiated. However, RFR was depressed for paired-pulses with an interval of <1 min (Fig. 1C), suggesting the existence of a form of short-term depression. Because a strong potentiation can occur for interpulse times >1 min, the short-term depression is probably not caused by run-down of GABAergic reciprocal feedback. Similar results were obtained from eight control Mb terminals (namely, potentiation for interpulse times >1 min and depression for interpulse times <1 min). In the presence of 100 μm LY367385, the potentiation of RFR was blocked, whereas the depression remained unaffected (Fig. 1D) (n = 6). Together, these results suggested that the reciprocal synapse underwent PPD at short interpulse time ranges, and that this PPD was independent to mGluR1-mediated long-term potentiation. Nevertheless, in the following experiments, in which we investigated the recovery of RFR, we included 100 μm LY367385 in the Ringer's solution. We next studied the underlying mechanisms of the PPD at this reciprocal synapse.
PPD and glutamatergic vesicle-pool depletion
After a single 100 ms prepulse stimulus, the availability of synaptic vesicles that are ready for release may become lower. This vesicle pool depletion is usually thought to be the major cause of PPD at many CNS synapses (Regehr and Stevens, 2001). Could reduced availability of primed synaptic vesicles containing glutamate underlie the PPD of the reciprocal synapse? To address this possibility, we applied a paired-pulse stimuli with variable interpulse intervals and built recovery curves for RFR and for ΔCm.
At 1 s after a 100 ms prepulse, the RFR of the second pulse was depressed, and the ΔCm of the second pulse was also reduced compared with that in the prepulse (Fig. 2A). At 10 s after a 100 ms prepulse, RFR was still depressed whereas ΔCm had recovered completely (Fig. 2B). However, both RFR and ΔCm recovered completely after a 1 min interpulse interval (Fig. 2C). The recovery curves are shown in Figure 2D. Both can be fitted with a single exponential equation with a time constant (τ) of 12.19 and 2.02 s, respectively. These data were all obtained in the presence of 100 μm LY367385. Without LY367385 at 10 s, the percentage of PPD was slightly smaller when compared with that in 100 μm LY367385, but this difference was not statistically significant (Fig. 2C, red point) (t test, p = 0.276).
We note also that the time constant (τ) for ΔCm recovery could be underestimated here because ΔCm recovery slowed down gradually after break-in. For instance, ΔCm recovered completely at 10 s immediately after break-in (99.6 ± 2.6%; n = 8). But 1 min after break-in, it recovered only to 72.6 ± 3.5% (n = 7) at a 10 s interpulse interval, indicating that the cycling of synaptic vesicles was slowed down because of the dilution or washout of soluble factors in the cytoplasm that are important for exocytosis (Hull and von Gersdorff, 2004). To minimize this effect, all data concerning ΔCm and RFR in the present study were obtained within 2 min after break-in.
Recovery from desensitization of GABAA receptors
We had shown that both desensitizing GABAA and nondesensitizing GABAC receptors are involved in the reciprocal feedback response (Vigh and von Gersdorff, 2005). Receptor desensitization is a mechanism that contributes to PPD at many synapses (Regehr and Stevens, 2001). To address this possibility, we directly measured the time course of GABAA recovery from desensitization in outside-out patches excised from bipolar cell terminals.
After exocytosis, it is estimated that neurotransmitters reach a very high peak concentration in the synaptic cleft, which may transiently saturate the postsynaptic receptors at CNS synapses (Clements, 1996). Therefore, to simulate conditions in the synaptic cleft, we applied 10 mm GABA to outside-out patches via a piezo-driven fast application system (Hull et al., 2006). As shown in Figure 3A, the current responses contained a nondesensitizing GABAC component, and a fully desensitizing GABAA component, which can be blocked by bicuculline (data not shown). We were unable to dissect the GABAA component pharmacologically, because low concentrations of the GABAC receptor blocker (1,2,5,6,-tetrahydropyridin-4-yl)methylphosphinic acid (TPMPA) cannot block the GABAC current induced by high concentration of GABA. However, high concentrations of TPMPA start to block GABAA receptors. Therefore, we took the desensitizing part of the current response (i.e., that fast decaying component) as the GABAA component, because the GABAC component does not desensitize (Hull et al., 2006).
Figure 3.

The recovery of GABAA receptors from desensitization depends on the duration of GABA application. GABA (10 mm) was applied to outside-out patches excised from Mb-type bipolar cell terminals after a whole-cell recording. The current responses consisted of a nondesensitizing GABAC component, and a fully desensitizing GABAA component (Hull et al., 2006) A, A preapplication of GABA for 5 s (as indicated by the black bar) was used to fully desensitize GABAA receptors, and then GABA (10 mm) was applied for 2 s (as indicated by gray arrows) to induce current after a variable interpulse interval. The longer the interval, the more the peak of the current response recovered. B, The recovery time course of peak current from A was constructed and fitted with a single exponential equation (τ = 11.9 s; gray trace). C, In this example, for a 100 ms GABA preapplication, GABAA current recovered 73.6% at 10 s (gray trace; the arrow indicates the peak of the red trace) of the preapplication control (black). D, For 5 ms GABA preapplications, the peak of the current response recovered completely within 2 s. Error bars indicate SE.
Our first objective was to determine the kinetics of recovery of GABAA receptors from desensitization after these receptors were fully desensitized. To answer this question, we preapplied GABA for 5 s (Fig. 3A, black bar) to fully desensitize the GABAA receptors (Hull et al., 2006). We then waited for increasing lengths of time, and reapplied GABA for 2 s (as indicated by gray arrows) to induce a second current response. As shown in Figure 3A, the longer we waited, the more the peak of the second current response recovered. The recovery time course was fit very well with a single exponential function (time constant, τ = 11.9 s) (Fig. 3B). It took almost 1 min for GABAA receptors to completely recover from full desensitization.
Second, we determined whether the recovery kinetics varied when shorter GABA applications were used so as to better simulate the synaptic conditions of our paired-pulse stimuli of 100 ms (Figs. 1, 2). Therefore, we applied GABA for 100 ms as a preapplication, and the second GABA application for 100 ms was delivered after 10 s. The GABAA response to the second GABA application was now 81.3 ± 2.4% (n = 6) of that induced by the first prepulse (Fig. 3C). We then did similar experiments with very short prepulse applications of GABA (5 ms). Under these conditions, the GABAA response recovered very quickly, and complete recovery took only 1∼2 s (Fig. 3D) (n = 5). The recovery kinetics thus vary strongly with the duration of the saturating GABA application. However, it is highly unlikely that GABA released from amacrine cells in the synaptic cleft will have a concentration as high as 10 mm for 100 ms. These results thus suggest that GABAA receptor desensitization is an unlikely mechanism to mediate PPD of the reciprocal feedback response at 10 s, because PPD is ∼0.4 at 10 s (Fig. 2D) whereas GABAA receptors already recover 81.3% at 10 s for a 100 ms GABA application (Fig. 3C).
Despite the above argument, we emphasize that for the paired-pulse stimuli we used to induce RFR the GABA concentration in the synaptic cleft will vary quickly and stochastically, as exemplified by the IPSCs shown in Figures 1 and 2. Thus, we cannot mimic in a realistic manner the synaptic GABA release during RFR with our fast and constant application system. Therefore, we cannot rule out completely that desensitization of GABAA receptors on the bipolar cell terminal is capable of contributing to the PPD observed at the reciprocal synapse.
GABAA desensitization may not contribute to PPD
As we mentioned above, the cycling of glutamate containing synaptic vesicles in bipolar cell terminals slows down gradually after whole-cell break-in. This wash-out effect complicates studies of the mechanisms of PPD. To overcome this obstacle, we by-passed the need to first depolarize the bipolar cell terminal by directly puffing glutamate onto the bipolar cell terminal and its closely attached amacrine cell boutons. This glutamate puff will activate AMPA and NMDA receptors on the amacrine cells and thus evoke a GABAergic feedback response on bipolar cell terminals. Note, however, that not all of the boutons on the bipolar cell terminals will be engaged in the reciprocal response (it is estimated that in the goldfish, Mb bipolar cell terminals are completely surrounded by ∼350 GABAergic boutons and 60% of these contain reciprocal synapses) (Marc and Liu, 2000).
With a CsCl-based internal pipette solution, the current response to a 2 mm glutamate puff was an inward current. Most of the current was mediated by picrotoxin-sensitive GABA receptors and a small portion was mediated by glutamate transporters on the bipolar cell terminal (Palmer et al., 2003b) (Fig. 4C). For the paired puff with an interval of 10 s, the response to the second test puff was smaller than that to the first prepuff (Fig. 4A). This result further confirmed that PPD was not caused by reduced glutamate release, because it occurs even with two identical glutamate puffs. The strength of the paired-puff depression was dependent on the glutamate concentration and puffer pipette location (data not shown). We used a puffer pipette with a small tip (∼1 μm) and localized ∼3–5 μm from the bipolar cell terminal in an attempt to depolarize only the amacrine cell boutons on that particular bipolar cell terminal.
Figure 4.

GABAA desensitization and PPD at 10 s. A, The feedback responses (GABA currents) to 2 mm focal glutamate puffs were recorded from Mb bipolar cell terminals. The feedback response was depressed at 10 s. B, In the presence of 50 μm SR95531 (GABAA blocker), the feedback response, which is mediated exclusively by nondesensitizing GABAC receptors, was increased and the paired-pulse (or puff) depression also increased. The total feedback response (black) was even larger than that without SR95531 (black trace in A), probably because of the disinhibition of amacrine cells. The response was completely recovered in 4 min (red trace). C, In the presence of 100 μm picrotoxin, a small current (probably mediated by glutamate transporters) remained for the same paired-puff application. Recordings in A–C were obtained from the same terminal. D, Pooled data from six terminals. Error bars are ±SE. ***p < 0.001; **p < 0.01. PTX, Picrotoxin. E, In the presence of 25 μm SR95531, the nondesensitizing synaptic GABAC-mediated RFR showed depression at 10 s.
In the presence of the GABAA blocker SR95531 (50 μm), the current response to the glutamate puff, which was mediated by nondesensitizing GABAC receptors, became larger than that without SR95531, and the amount of depression was also larger (Fig. 4B). The probable explanation is that GABAergic amacrine cells are disinhibited in the presence of SR95531 (Zhang et al., 1997; Watanabe et al., 2000; Vigh et al., 2005). These disinhibited amacrine cells will release more GABA containing synaptic vesicles in response to the same amount of glutamate, and this may lead to a more rapid vesicle pool depletion and less available GABAergic vesicles. This then leads to a stronger paired-puff depression (Fig. 4A,B). The averaged data from these experiments is shown in Figure 4D (n = 6). Furthermore, PPD of RFR at 10 s remained with 50 μm SR95531 (Fig. 4E). Thus, PPD is still present even when it is mediated solely by nondesensitizing GABAC receptors. All of these results suggested that GABAA desensitization is not necessary for paired-pulse depression to occur. Furthermore, these results suggest that the slow replenishment rate of depleted GABAergic synaptic vesicle pools is the major mechanism that contributes to PPD at 10 s.
PPD remains with either AMPA or NMDA receptor blockade
Glutamate release from bipolar cell terminals activates both AMPA and NMDA receptors on amacrine cells (Vigh and von Gersdorff, 2005). The feedback response consists of an AMPA-mediated fast and transient portion, and an NMDA-mediated slow and sustained portion. Retinal AMPA receptors are known to desensitize in the presence of glutamate (Lukasiewicz et al., 1995). Can PPD at the reciprocal synapse be attributed specifically to the distinct properties of AMPA or NMDA receptors on amacrine cells? To answer this question, we applied paired-pulse stimuli with an interval of 10 s to cells in the presence of AMPA and NMDA receptor antagonists CNQX (25 μm) and AP-5 (50 μm), respectively.
With 50 μm AP-5 in the external solution, the RPR was more transient. However, PPD was still present (Fig. 5A) (n = 7). In 25 μm CNQX, RFR showed a delayed and relatively sustained response, which is supposed to be mediated by NMDA receptors, which desensitize very slowly (Fig. 5B) (Vigh and von Gersdorff, 2005). However, PPD remained unaffected (n = 6). Here, we removed Mg2+ from the Ringer's solution to enhance the NMDA-mediated reciprocal feedback. Together, these results demonstrate that PPD at the reciprocal synapse could not be attributed to either AMPA or NMDA receptor properties.
Figure 5.
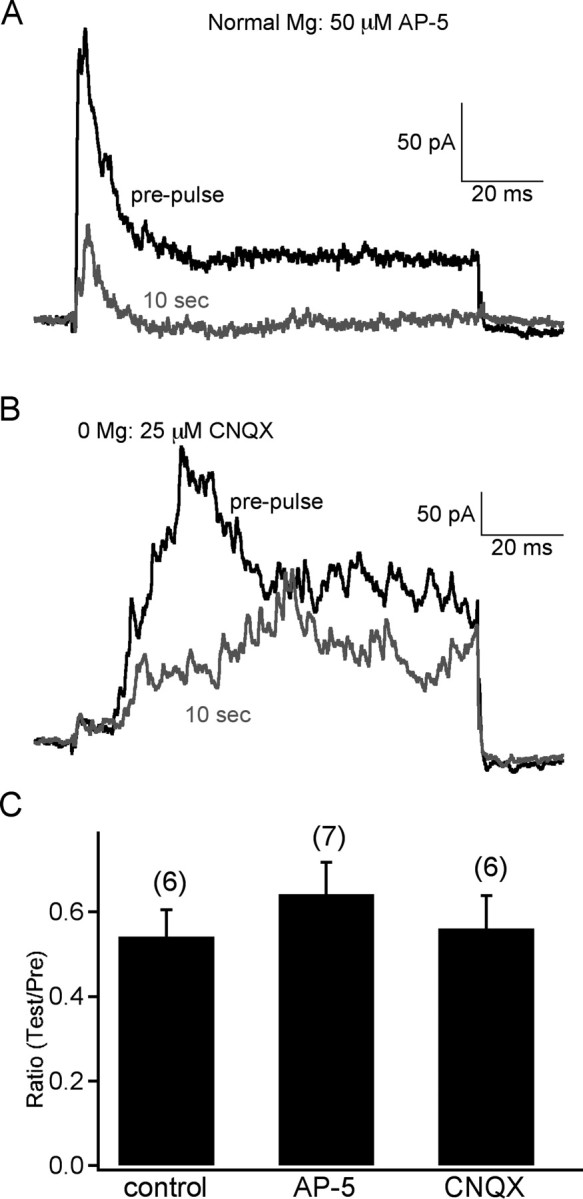
Synaptic PPD cannot be attributed specifically to NMDA or AMPA receptors. A, Blockade of NMDA receptors with 50 μm AP-5 did not block PPD of RFR at 10 s. Notice that RFR was more transient in AP-5. B, In 25 μm CNQX (blocker of AMPA receptors) and 0 Mg2+ Ringer's solution, the RFR was delayed and increased (NMDA receptors were relieved from Mg2+ blockade), but this did not block the PPD at 10 s. C, The pooled data of PPD at 10 s shows that PPD did not change under these conditions (error bars are ±SE).
To further study the possible contribution of AMPA receptor desensitization to PPD, we applied glutamate focally in the presence of cyclothiazide, which is widely used to reduce AMPA-receptor desensitization (Matsui et al., 2001; Singer and Diamond, 2003). As shown in Figure 6, A (control) and B, in the presence of 50 μm, cyclothiazide PPD still occurred. Note that the total feedback response was increased by cyclothiazide compared with control, indicating that it had an effect on the responses. However, PPD was not blocked by cyclothiazide. In fact, PPD was even more pronounced, probably because more GABA-containing vesicles fused with the first glutamate puff and vesicle pool depletion was thus more severe. Even the simultaneous presence of both 50 μm cyclothiazide and 50 μm SR95531 could not abolish PPD (Fig. 6C).
Figure 6.

AMPA receptor desensitization cannot account for glutamate puff-induced PPD at 10 s. A, The PPD of the feedback response to a 2 mm glutamate puff onto the Mb bipolar cell. B, This PPD was not blocked by 50 μm cyclothiazide. C, It was also not blocked by the presence of both 50 μm cyclothiazide and 50 μm SR95531. Traces in A–C are successively obtained traces from the same Mb-type bipolar cell terminal. D, The pooled data from five terminals are shown (error bars are ±SE; ***p < 0.001; **p < 0.01). CTZ, Cyclothiazide.
We also performed experiments with similar results with the synaptic evoked RFR. In Figure 7, A and B, we show that synaptic PPD of RFR remained either with 50 μm cyclothiazide (n = 6 terminals) or with both 50 μm cyclothiazide and 25 μm SR95531 (n = 5 terminals). These results further confirmed that neither AMPA receptor desensitization nor GABAA receptor desensitization is necessary to induce PPD at a 10 s interpulse interval.
Figure 7.
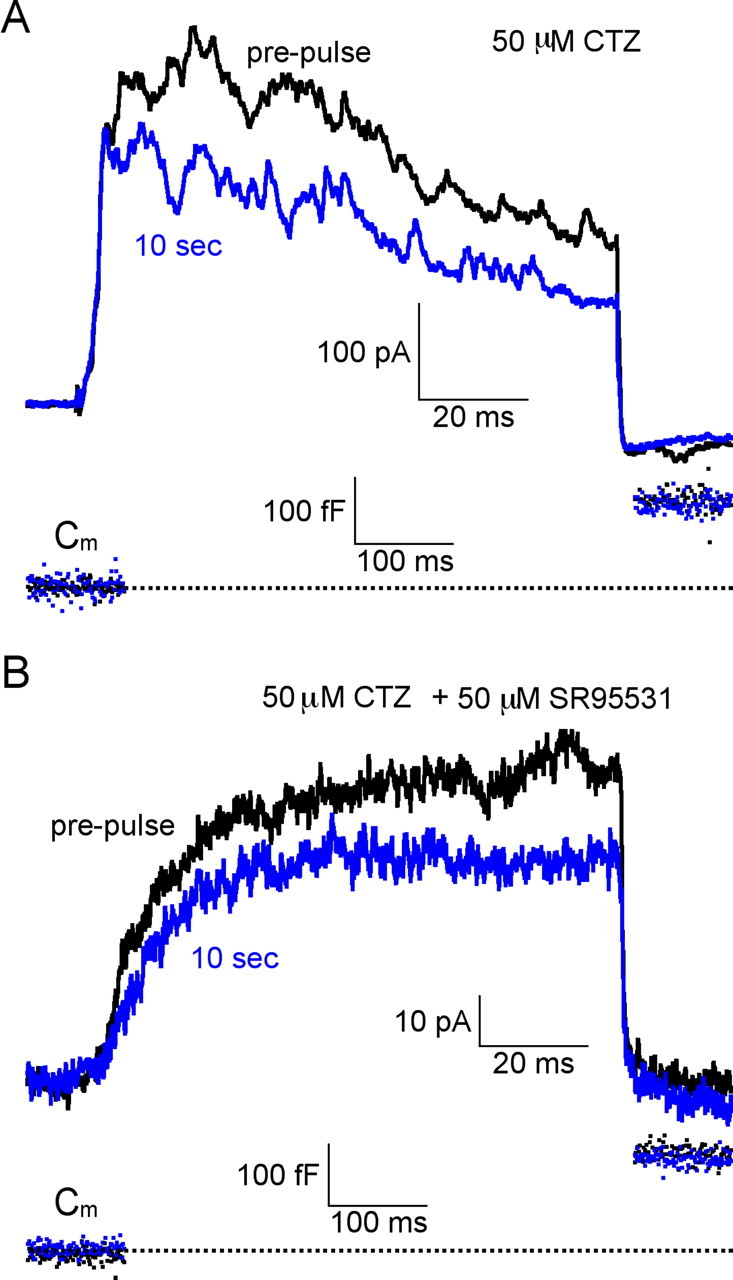
Synaptic AMPA receptor or GABAA receptor desensitization is not involved in PPD of RFR at 10 s. A, The synaptic PPD of RFR at 10 s was not blocked by 50 μm cyclothiazide (n = 6). B, Synaptic PPD of RFR at 10 s was also not blocked by the presence of both 50 μm cyclothiazide and 50 μm SR95531 (n = 5). Notice that for both cases in A and B, ΔCm at 10 s fully recovered to the prepulse level. CTZ, Cyclothiazide.
We next checked whether calcium-permeable AMPA receptors contribute to RFR. With 50 μm d-AP-5 blocking the NMDA-mediated sustained component of RFR, leaving only an isolated AMPA-mediated transient RFR, we observed that 1 μm philanthotoxin (a specific calcium-permeable AMPA receptor blocker) (Chavez et al., 2006) did not block the GABAergic RFR (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (n = 4 terminals) after 4–5 min of application of the toxin. In contrast to rat retina (Chavez et al., 2006), we thus conclude that in goldfish retina the Mb-type bipolar cell terminal reciprocal GABAergic response is not mediated by calcium-permeable AMPA receptors sensitive to 1 μm philanthotoxin. However, we note that prolonged synaptic activity and longer applications may be necessary to see a complete block of calcium-permeable AMPA receptors with philanthotoxin (Tóth and McBain, 1998). Given the sometimes rapid rundown of capacitance jumps during whole-cell recordings of Mb-type bipolar cell terminals, prolonged applications of philanthotoxin are technically not feasible on a routine basis. Future experiments with the perforated patch mode of recording may be able to address this issue in a more definitive manner.
Finally, we note that calcium current inactivation in nerve terminals may also contribute to short-term depression (von Gersdorff and Matthews, 1996). However, paired recordings of cultured amacrine cells reveal that presynaptic calcium current inactivation is minimal during a depolarization that causes significant depletion of the readily releasable pool of GABAergic vesicles (Gleason et al., 1994).
Discussion
Short-term forms of plasticity such as paired-pulse depression occur at both excitatory and inhibitory synapses and can be mediated by multiple mechanisms. Here, we studied simultaneously the short-term plasticity of two separate synapses that are connected in a reciprocal manner by making recordings directly from a single Mb-type bipolar cell terminal embedded in a retinal slice.
In situ GABAergic synapses display paired-pulse depression
Because excitatory synapses and inhibitory feedback synapses are interacting with each other at these reciprocal synapses their, state of facilitation and/or depression depends on their mutual past history. The synapse between bipolar cell terminals and amacrine cells is a ribbon-type glutamatergic excitatory synapse that displays PPD (von Gersdorff and Matthews, 1997; Gomis et al., 1999; Singer and Diamond, 2006). Here, we determined that the feedback GABAergic synaptic input to bipolar cells also displays PPD (Fig. 2). We have done this by showing that after a 10 s interpulse interval, the two excitatory 100 ms pulses produce identical ΔCm jumps (recovery of ΔCm has a fast τ = 2 s), but the resulting evoked GABAergic RFR still displays PPD (Fig. 2B). We found that for paired-pulse stimuli, this disynaptic connection was depressed for up to 1 min, and it recovered from depression with a τ = 12.2 s (Fig. 2D). Additional analysis of experiments that tested the role of GABAA and AMPA receptor desensitization, and of experiments that used nondesensitizing GABAC and slowly desensitizing NMDA receptors, strongly suggests that this PPD is mainly caused by the depletion of a pool of GABA-containing vesicles in amacrine cells. Furthermore, we suggest that the rate of replenishment of this pool is relatively slow (τ = 12.2 s). However, we note that desensitization of GABAA receptors on bipolar cell terminals may also contribute to some forms of synaptic depression if GABA is present in the synaptic cleft for prolonged periods of time at high concentrations (Fig. 3A,B).
Recovery from depression at ribbon-type synapses
The high rates of sensory information that needs to be continuously and faithfully transmitted by the retina may have promoted the evolution of a specialized synaptic ribbon-type active zone. Ribbon synapses, compared with conventional synapses, are thought to be specialized for the continuous release of neurotransmitter. They are located in the first order sensory synapses of cochlea hair cells and retinal photoreceptors, as well as in second-order retinal bipolar cells. In acutely isolated goldfish Mb-type bipolar cell terminals, a 250 ms depolarization to 0 mV depletes a readily releasable pool of synaptic vesicles, which recovers with τ = 8 s (von Gersdorff and Matthews, 1997), and this recovery can be accelerated by elevated [Ca2+]i (Gomis et al., 1999). For axotomized bipolar cell terminals in slices, ΔCm jumps induced by short 2 ms depolarizations recovered fully with a double-exponential time course (τfast = 1.0 s and τslow = 11.8 s) (Palmer et al., 2003a). Here, we show that ΔCm recovered with a τ = 2 s for 100 ms depolarizations. Similarly, using paired recordings between rat rod bipolar cells and AII amacrine cells, Singer and Diamond (2006) found that paired-pulse depression of EPSCs has a mean recovery time constant that varied slightly with stimulus protocol (τ = 2.6 or 3.9 s). This short-term form of synaptic depression is thus present in rat and goldfish retinal bipolar cell synapses.
At conventional synapses, the readily releasable pool of vesicles can be depleted even after a very short train of action potentials and recovery can take several seconds (Regehr and Stevens, 2001). Here, we showed for a reciprocal synapse, which included both synaptic ribbon-aided release of glutamate and conventional active zone release of GABA, that PPD of GABA release is primarily attributable to the slow recovery of GABA-containing synaptic vesicle pools, whereas glutamate-containing vesicle pools recover much more quickly. Differences in the recovery rates of excitatory and inhibitory synapses that are linked in a microcircuit can have interesting consequences (Kirischuk et al., 2002). For example, in the hippocampus, differences in short-term plasticity at excitatory and feedforward inhibitory synapses can determine the degree of synaptic gain that enhances the transmission of burst-like impulses (Klyachko and Stevens, 2006). So the interplay of excitatory and inhibitory synapses can form a dynamic filter that selects for certain frequencies of action potentials. Within our reciprocal synapse, inhibitory GABAergic inputs may reduce the amount of initial glutamate release so that a subsequent depolarization of the bipolar cell can trigger more release of glutamate. Amacrine cells may thus protect the bipolar cell terminal from premature vesicle pool depletion after a strong stimulus or during prolonged depolarizations (Vigh and von Gersdorff, 2005). This reduction in bipolar cell vesicle pool depletion may allow ganglion cells to better follow high-frequency light stimuli, improving the time resolution of visual perception and contributing to the detection of moving objects.
Recovery from desensitization: AMPA and GABAA receptors
Glutamate and/or GABA receptor desensitization is another possible mechanism for PPD. Indeed, AMPA receptor desensitization has been shown to contribute to PPD at retinogeniculate synapses in mouse (Chen et al., 2002). However, at our retinal reciprocal synapse, blockade of AMPA receptors with cyclothiazide left PPD unchanged, suggesting AMPA desensitization does not contribute to short-term plasticity. Accordingly, reciprocal synapses mediated solely by nondesensitizing GABAC receptors and NMDA receptors still displayed PPD (Fig. 7B). In rat retina, PPD of rod bipolar cells is also not caused by AMPA receptor desensitization (Singer and Diamond, 2006). Likewise, paired-pulse depression in rod and cone photoreceptors is not influenced by glutamate receptor desensitization (Rabl et al., 2006).
The complete recovery of GABAA receptors from desensitization induced by brief pulses can take several hundred milliseconds to seconds (Jones and Westbrook, 1996). Our study shows that in situ GABAA receptors in outside-out patches excised from bipolar cell terminals recover from desensitization at a relatively fast rate for a brief 5 ms preapplication of 10 mm GABA (Fig. 3D). In fact, the synaptically released GABA concentration transients at each of the 350 GABA synapses on the goldfish Mb bipolar cell terminal is expected to individually last ∼1 ms (Jones and Westbrook, 1996). So this fast recovery also suggests that desensitization of GABAA receptors is not involved in synaptic PPD. However, recovery becomes slower with a longer preapplication of GABA. Even assuming that synaptic GABA reaches constant levels of 10 mm for 100 ms (which surely does not occur physiologically at the synaptic cleft), recovery from desensitization is much faster (Fig. 3C) (81% recovered by 10 s) than recovery from depression (40% recovered by 10 s). Only after a prolonged application (5 s) that fully desensitizes the GABAA receptors do we reach recovery times comparable with those for synaptic PPD (i.e., τ = 11.9 s for desensitization vs τ = 12.2 for PPD) (Figs. 2D, 3B). Thus, under our conditions of paired-pulse stimuli (100 ms), it is very unlikely that GABAA receptor desensitization will contribute to PPD at 10 s.
Visual adaptation and short-term depression
The vertebrate retina does not work like a passive camera that captures and transmits images. Within seconds, the retina is capable of adapting to the statistical changes of visual images, both spatially and temporally. These adaptations are probably achieved in part through plastic changes at the synaptic level. In a previous study, we showed that the GABAergic responses can be enhanced for up to 10 min after a strong and prolonged bipolar cell depolarization (Vigh et al., 2005). Here, at the same synapse, we provided evidence suggesting that GABAergic responses can be depressed for up to 1 min after a 100 ms depolarizing pulse. Both the previous long-term enhancement and the PPD in the present study may thus help to implement different adaptational changes of visual information processing in the retina, and, in combination, both can make the retina even more versatile for visual information processing.
Frequency-dependent short-term depression promotes neuronal firing in response to sudden changes in presynaptic firing rate (Abbott et al., 1997). Short-term depression is thus a plausible mechanism for mediating part of the contrast adaptation in the neural circuits of the inner retina. Moreover, the timescale of recovery from slow contrast adaptation is ∼5–20 s (Baccus and Meister, 2002) and is independent of GABAergic inhibition (Manookin and Demb, 2006). This matches the time scale of recovery of EPSCs and ΔCm jumps from short-term PPD (see above; τ = 2–12 s).
Finally, we note that the microcircuits of the IPL also play a crucial role in several other aspects of vision, including determining bipolar cell light sensitivity (Euler and Masland, 2000), changing ganglion cell and amacrine cell receptive fields (Cook and McReynolds, 1998; Völgyi et al., 2002; Lukasiewicz et al., 2004), truncating ganglion cell light responses (Nirenberg and Meister, 1997; Roska et al., 1998), modulating the dynamic range of ganglion cell output (Sagdullaev et al., 2006), and segregating object and background motion (Ölveczky et al., 2003). Detailed studies of the properties of different IPL synapses will thus be needed for a better understanding of signal processing and neuronal coding in the retina.
Footnotes
This work was supported by grants from the Human Frontier Science Program and the National Eye Institute (EY14043). We thank Court Hull (University of California at San Diego, La Jolla, CA) and Mary J. Palmer (Keele University, Keele, UK) for discussions on synaptic transmission in the retina.
References
- Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- Baccus SA, Meister M. Fast and slow contrast adaptation in retinal circuitry. Neuron. 2002;36:909–919. doi: 10.1016/s0896-6273(02)01050-4. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Kingston A, O'Neill MJ, Catania MV, Di Grezia R, Nicoletti F. Neuroprotective activity of the potent and selective mGlu1a metabotropic glutamate receptor antagonist, (+)-2-methyl-4 carboxyphenylglycine (LY367385): comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology. 1999;38:199–207. doi: 10.1016/s0028-3908(98)00159-2. [DOI] [PubMed] [Google Scholar]
- Chavez AE, Singer JH, Diamond JS. Fast neurotransmitter release triggered by Ca influx through AMPA-type glutamate receptors. Nature. 2006;443:705–708. doi: 10.1038/nature05123. [DOI] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron. 2002;33:779–788. doi: 10.1016/s0896-6273(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Clements JD. Transmitter time course in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- Cook PB, McReynolds JS. Lateral inhibition in the inner retina is important for spatial tuning of ganglion cells. Nat Neurosci. 1998;1:714–719. doi: 10.1038/3714. [DOI] [PubMed] [Google Scholar]
- Dong CJ, Werblin FS. Temporal contrast enhancement via GABAC feedback at bipolar terminals in the tiger salamander retina. J Neurophysiol. 1998;79:2171–2180. doi: 10.1152/jn.1998.79.4.2171. [DOI] [PubMed] [Google Scholar]
- Dowling JE, Boycott BB. Organization of the primate retina: electron microscopy. Proc R Soc Lond B Biol Sci. 1966;166:80–111. doi: 10.1098/rspb.1966.0086. [DOI] [PubMed] [Google Scholar]
- Euler T, Masland RH. Light-evoked responses of bipolar cells in a mammalian retina. J Neurophysiol. 2000;83:1817–1829. doi: 10.1152/jn.2000.83.4.1817. [DOI] [PubMed] [Google Scholar]
- Gillis KD. Admittance-based measurement of membrane capacitance using the EPC-9 patch-clamp amplifier. Pflügers Arch. 2000;439:655–664. doi: 10.1007/s004249900173. [DOI] [PubMed] [Google Scholar]
- Gleason E, Borges S, Wilson M. Control of transmitter release from retinal amacrine cells by Ca2+ influx and efflux. Neuron. 1994;13:1109–1117. doi: 10.1016/0896-6273(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Gomis A, Burrone J, Lagnado L. Two actions of calcium regulate the supply of releasable vesicles at the ribbon synapse of retinal bipolar cells. J Neurosci. 1999;19:6309–6317. doi: 10.1523/JNEUROSCI.19-15-06309.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartveit E. Reciprocal synaptic interactions between rod bipolar cells and amacrine cells in the rat retina. J Neurophysiol. 1999;81:2923–2936. doi: 10.1152/jn.1999.81.6.2923. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Thoreson WB, Witkovsky P. Synaptic transmission at retinal ribbon synapses. Prog Retin Eye Res. 2005;24:682–720. doi: 10.1016/j.preteyeres.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya T, Baccus SA, Meister M. Dynamic predictive coding by the retina. Nature. 2005;436:71–77. doi: 10.1038/nature03689. [DOI] [PubMed] [Google Scholar]
- Hull C, von Gersdorff H. Fast endocytosis is inhibited by GABA-mediated chloride influx at a presynaptic terminal. Neuron. 2004;44:469–482. doi: 10.1016/j.neuron.2004.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull C, Li GL, von Gersdorff H. GABA transporters regulate a standing GABAC receptor-mediated current at a retinal presynaptic terminal. J Neurosci. 2006;26:6979–6984. doi: 10.1523/JNEUROSCI.1386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Rieke F. Temporal contrast adaptation in the input and output signals of salamander retinal ganglion cells. J Neurosci. 2001;21:287–299. doi: 10.1523/JNEUROSCI.21-01-00287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Clements JD, Grantyn R. Presynaptic and postsynaptic mechanisms underlie paired pulse depression at single GABAergic boutons in rat collicular cultures. J Physiol (Lond) 2002;543:99–116. doi: 10.1113/jphysiol.2002.021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko VA, Stevens CF. Excitatory and feed-forward inhibitory hippocampal synapses work synergistically as an adaptive filter of natural spike trains. PLoS Biol. 2006;4:1187–1200. doi: 10.1371/journal.pbio.0040207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, Lawrence JE, Valentino TL. Desensitizing glutamate receptors shape excitatory synaptic inputs to tiger salamander retinal ganglion cells. J Neurosci. 1995;15:6189–6199. doi: 10.1523/JNEUROSCI.15-09-06189.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiewicz PD, Eggers ED, Sagdullaev BT, McCall MA. GABAC receptor-mediated inhibition in the retina. Vision Res. 2004;44:3289–3296. doi: 10.1016/j.visres.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Manookin MB, Demb JB. Presynaptic mechanism for slow contrast adaptation in mammalian retinal ganglion cells. Neuron. 2006;50:453–464. doi: 10.1016/j.neuron.2006.03.039. [DOI] [PubMed] [Google Scholar]
- Marc RE, Liu W. Fundamental GABAergic amacrine cell circuitries in the retina: nested feedback, concatenated inhibition, and axosomatic synapses. J Comp Neurol. 2000;425:560–582. doi: 10.1002/1096-9861(20001002)425:4<560::aid-cne7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Masland RH. The fundamental plan of the retina. Nat Neurosci. 2001;4:877–886. doi: 10.1038/nn0901-877. [DOI] [PubMed] [Google Scholar]
- Matsui K, Hasegawa J, Tachibana M. Modulation of excitatory synaptic transmission by GABAC receptor-mediated feedback in the mouse inner retina. J Neurophysiol. 2001;86:2285–2298. doi: 10.1152/jn.2001.86.5.2285. [DOI] [PubMed] [Google Scholar]
- Neher E, Marty A. Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci USA. 1982;79:6712–6716. doi: 10.1073/pnas.79.21.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg S, Meister M. The light response of retinal ganglion cells is truncated by a displaced amacrine circuit. Neuron. 1997;18:637–650. doi: 10.1016/s0896-6273(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Ölveczky BP, Baccus SA, Meister M. Segregation of object and background motion in the retina. Nature. 2003;423:401–408. doi: 10.1038/nature01652. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J, von Gersdorff H. Synaptic cleft acidification and modulation of short-term depression by exocytosed protons in retinal bipolar cells. J Neurosci. 2003a;23:11332–11341. doi: 10.1523/JNEUROSCI.23-36-11332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Taschenberger H, Hull C, Tremere L, von Gersdorff H. Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J Neurosci. 2003b;23:4831–4841. doi: 10.1523/JNEUROSCI.23-12-04831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti DA, Llano I. Calcium currents and calcium signaling in rod bipolar cells of rat retinal slices. J Neurosci. 1998;18:3715–3724. doi: 10.1523/JNEUROSCI.18-10-03715.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl K, Cadetti L, Thoreson WB. Paired-pulse depression at photoreceptor synapses. J Neurosci. 2006;26:2555–2563. doi: 10.1523/JNEUROSCI.3667-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF. Physiology of synaptic transmission and short-term plasticity. In: Cowan WM, Südhof TC, Stevens CF, editors. Synapses. Baltimore: Johns Hopkins UP; 2001. pp. 135–175. [Google Scholar]
- Roska B, Nemeth E, Werblin FS. Response to change is facilitated by a three-neuron disinhibitory pathway in the tiger salamander retina. J Neurosci. 1998;18:3451–3459. doi: 10.1523/JNEUROSCI.18-09-03451.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagdullaev BT, McCall MA, Lukasiewicz PD. Presynaptic inhibition modulates spillover, creating distinct dynamic response ranges of sensory output. Neuron. 2006;50:923–935. doi: 10.1016/j.neuron.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003;23:10923–10933. doi: 10.1523/JNEUROSCI.23-34-10923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Vesicle depletion and synaptic depression at a mammalian ribbon synapse. J Neurophysiol. 2006;95:3191–3198. doi: 10.1152/jn.01309.2005. [DOI] [PubMed] [Google Scholar]
- Slaughter MM, Miller RF. Bipolar cells in the mudpuppy retina use an excitatory amino acid neurotransmitter. Nature. 1983;303:537–538. doi: 10.1038/303537a0. [DOI] [PubMed] [Google Scholar]
- Tóth K, McBain CJ. Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat Neurosci. 1998;1:572–578. doi: 10.1038/2807. [DOI] [PubMed] [Google Scholar]
- Vigh J, von Gersdorff H. Prolonged reciprocal signaling via NMDA and GABA receptors at a retinal ribbon synapse. J Neurosci. 2005;25:11412–11423. doi: 10.1523/JNEUROSCI.2203-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigh J, Li GL, Hull C, von Gersdorff H. Long-term plasticity mediated by mGluR1 at a retinal reciprocal synapse. Neuron. 2005;46:469–482. doi: 10.1016/j.neuron.2005.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völgyi B, Xin D, Bloomfield SA. Feedback inhibition in the inner plexiform layer underlies the surround-mediated responses of AII amacrine cells in the mammalian retina. J Physiol (Lond) 2002;539:603–614. doi: 10.1113/jphysiol.2001.013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Calcium-dependent inactivation of calcium current in synaptic terminals of retinal bipolar neurons. J Neurosci. 1996;16:115–122. doi: 10.1523/JNEUROSCI.16-01-00115.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Sakaba T, Berglund K, Tachibana M. Submillisecond kinetics of glutamate release from a sensory synapse. Neuron. 1998;21:1177–1188. doi: 10.1016/s0896-6273(00)80634-0. [DOI] [PubMed] [Google Scholar]
- Wässle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–757. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Koizumi A, Matsunaga S, Stocker JW, Kaneko A. GABA-mediated inhibition between amacrine cells in the goldfish retina. J Neurophysiol. 2000;84:1826–1834. doi: 10.1152/jn.2000.84.4.1826. [DOI] [PubMed] [Google Scholar]
- Witkovsky P, Dowling JE. Synaptic relationships in the plexiform layers of carp retina. Z Zellforsch Mikrosk Anat. 1969;100:60–82. doi: 10.1007/BF00343821. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jung CS, Slaughter MM. Serial inhibitory synapses in retina. Vis Neurosci. 1997;14:553–563. doi: 10.1017/s0952523800012219. [DOI] [PubMed] [Google Scholar]