

Abstract
NMDA-type glutamate receptors (NMDARs) play a central role in the rapid regulation of synaptic transmission, but their contribution to the long-term stabilization of glutamatergic synapses is unknown. We find that, in hippocampal pyramidal neurons in rat organotypic slices, pharmacological blockade of NMDARs does not affect synapse formation and dendritic spine growth but does increase the motility of spines. Physical loss of synaptic NMDARs induced by RNA interference against the NR1 subunit of the receptor also increases the motility of spines. Furthermore, knock-down of NMDARs, but not their pharmacological block, destabilizes spine structure and over time leads to loss of spines and excitatory synapses. Maintenance of normal spine density requires the coexpression of two specific splice isoforms of the NR1 subunit that contain the C-terminal C2 cassette. Thus, although ionotropic properties of NMDARs induce synaptic plasticity, it is the physical interactions of the C-tail of the receptor that mediate the long-term stabilization of synapses and spines.
Keywords: NMDAR, AMPAR, synapse stability, dendritic spines, hippocampus, NR1 splice variants
Introduction
The formation of memories and the acquisition of new behaviors are thought to occur through the activity-dependent regulation of synaptic connections in the brain (Martin et al., 2000). Similarly, changes in synaptic connectivity permit the refinement of neuronal circuits during development, and perturbation of these processes may underlie several degenerative neurological disorders (Arendt, 2001; Hensch, 2004). Two opposing processes, the formation of new synapses and the elimination of existing contacts, occur throughout life, and the balance of these determines the net change in circuit connectivity.
Neuronal activity regulates both synapse formation and elimination and hence influences the plasticity and the pruning of neuronal circuits. Many forms of activity-dependent regulation of synapses demonstrated both in vitro and in vivo require activation of NMDA-type glutamate receptors (NMDARs) (Malenka and Nicoll, 1993; Fox et al., 1996; Huang and Pallas, 2001). These receptors are among the first molecules to accumulate at sites of nascent synapses (Washbourne et al., 2002) where NMDARs become stable synaptic components compared with other proteins such AMPA-type glutamate receptors (AMPARs) (Luscher et al., 1999; Roche et al., 2001). NMDARs are also found at “silent synapses,” a developmentally immature class of synapses that lacks AMPARs but that can rapidly acquire them through induction of an NMDAR-dependent form of long-term potentiation (LTP) (Liao et al., 1995; Malinow et al., 2000). Similarly, the removal of AMPARs from more mature synapses is seen quickly after induction of NMDAR-dependent long-term depression (LTD) (Xiao et al., 1994; Carroll et al., 1999). On a structural level, patterned electrical stimulation can drive the NMDAR-dependent formation or elimination of dendritic protrusions in the hippocampus (Maletic-Savatic et al., 1999; Nagerl et al., 2004). Similarly, in vivo, changes in sensory experience alter connectivity in sensory cortex by inducing synapse formation (Knott et al., 2002), accelerating the rate of spine elimination (Zuo et al., 2005b), and inducing NMDAR-dependent plasticity (Sawtell et al., 2003). These findings have led to the hypothesis that NMDARs, by regulating LTD and LTP induction, control the maturation and stabilization of synapses over longer time scales.
Here, we compare in rat organotypic hippocampal slices the effects of blocking NMDARs with pharmacological antagonists with those of physically eliminating the receptor from the synapse. We find that normal developmental increases in dendritic spine density occur in the absence of ionotropic NMDAR-dependent signaling. When physical loss of NMDARs is induced by RNA interference (RNAi) against the obligatory NR1 subunit of the receptor, morphologically mature dendritic spines containing AMPARs and postsynaptic density-95 (PSD-95) are transiently present. Knock-down of NMDARs increases motility of spines and leads to a progressive loss of excitatory synapses and spines. Thus, although NMDAR signaling is not necessary for initial synapse formation and spine growth, NMDARs are necessary for the stabilization of synapses. Maintenance of synaptic structure requires the cooperative action of NR1 splice variants 1 and 2, which contain an alternatively spliced C-terminal C2 cassette. Our results indicate that NMDARs regulate the number of dendritic spines and active synapses through a mechanism that requires structural features of its C-tail domain but is independent of ionotropic signaling.
Materials and Methods
Animals.
Sprague Dawley rats were purchased from Charles River Laboratories (Wilmington, MA). All procedures were approved by the Harvard Medical School Animal Care Committee and conducted in accordance with Harvard Medical School and federal guidelines.
Expression vectors.
RNAi was accomplished using a dual-promoter CMV-EGFP/U6-shRNA vector (pGUR) (Tavazoie et al., 2005) that ensures coexpression of green fluorescent protein (GFP) and short hairpin RNA (shRNA). shRNAs directed against NR1 were designed by searching the NR1 sequence for 19–21 bp regions beginning with G, with a G:C content of ∼50%, and no homology with other mammalian mRNAs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Reverse and forward oligonucleotides consisting of the target sequence, a spacer with a restriction site, the reverse complement of the target sequence, 5 Ts for U6 polymerase termination, and an EcoRI site for cloning were synthesized and SDS-PAGE purified (IDT, Coralville, IA). pGUR was digested with ApaI, Klenowed, digested with EcoRI, and treated with calf intestinal alkaline phosphatase. Oligonucleotides were annealed, phosphorylated with T4 polynucleotide kinase, and ligated into the prepared digested plasmid. Vectors expressing shRNA against NR1 (shNR1) were screened for off-target effects through activation of an interferon pathway using a luciferase reporter assay (Alvarez et al., 2006).
NR1 splice variants were kindly provided by R. Wenthold (National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Bethesda, MD), and PSD-95–GFP fusion protein was provided by M. Sheng (RIKEN–Massachusetts Institute of Technology, Cambridge, MA). For rescue construct preparation, an ∼2 kb fragment (TR-wt) containing the shNR1 target region was removed from NR1-1a by SacI and HincII digestion, blunt-ended using Klenow, and cloned into the AseI site of a pEGFP-N1 construct, the multiple cloning site of which had been removed by digestion with BglII and BamHI and religation. Two primers coding for the mutated target region, in which 8 of the 21 bp of the shNR1 target region were changed to confer resistance to shNR1 without changing the amino acid sequence, 5′ccggaattcaatgaggatggCgaTAggaaAttCgcTaaTtaCagtatcatgaacctgcagaaccg-3′ and 5′-ggggtaccattgtagatgcccacttgcaccagcttgcggttctgcaggttcatgatactgtaatt-3′, were annealed, digested with EcoRI and KpnI, and cloned into the similarly digested EGFP-TR-wt vector to generate EGFP-TRmut. The TRmut insert was then digested out of enhanced green fluorescent protein (EGFP) using AfeI and BmgBI and cloned into the various pcDNA::NR1 splice variants 1a, 2a, and 3a to generate NR1res variants. A dual-promoter CMV-mRFP/U6-shNR1 vector was constructed by replacing GFP in shNR1 with monomeric red fluorescent protein (mRFP) and used for coexpression with PSD-95–GFP.
Immunocytochemistry and analysis.
For NR1 immunostaining, dissociated hippocampal cultures were prepared from postnatal day 1 (P1) to P3 rats and plated (8 × 104 cells/12 mm coverslip) on confluent glial monolayers as described previously (Boyer et al., 1998). For PSD-95 staining, cultures were prepared from embryonic day 18 rat embryos and cultured (6 × 104) directly on coated coverslips (12 mm, poly-d-lysine). Neurons were transfected after 5–7 d in culture with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and cultured for 10 d. For NR1 staining, cultures were washed with PBS, lightly fixed in 0.5% paraformaldehyde/4% sucrose in PBS [15 min, room temperature (RT)], and fixed in methanol (20 min, −20°C). For PSD-95, cultures were washed and fixed in 3.7% paraformaldehyde/4% sucrose in PBS (15 min, RT). Cells were washed, permeabilized (PBS plus 0.1% Triton X-100), and blocked for 30 min at RT (10% goat serum, 0.1% Triton X-100 in PBS). Incubations with primary antibodies [mouse NR1, 1:500, clone 54.1 (PharMingen, BD Bioscience, San Diego, CA); rabbit mitogen-activated protein 2 (MAP2), 1:1000 (Chemicon, Temecula, CA); mouse PSD-95, 1:300, MA1-045 (Affinity BioReagents, Golden, CO)] were done for 2 h at RT or overnight at 4°C. Cultures were washed and incubated with secondary antibodies [goat Cy5-conjugated anti-mouse (1:500) and goat Alexa594-conjugated anti-rabbit (1:500); Jackson ImmunoResearch, West Grove, PA] for 30 min at RT. All antibodies were diluted in blocking solution. Coverslips were washed and mounted with AntiFade reagent (Invitrogen). Controls without primary antibody were run in parallel.
Images (1024 × 1024 pixels) of green fluorescent pyramidal neurons were collected using a confocal microscope (LSM 510; Zeiss, Jena, Germany) and 40× lens (numerical aperture, 1.3; Zeiss) at a 2.5 μm optical section. Image acquisition and analysis were performed blinded to the identity of the transfected vectors. Quantification of NR1 and MAP2 immunostaining was performed in Igor Pro software (Wavemetrics, Lake Oswego, OR) by averaging the fluorescence intensity from a selected region of the soma (nucleus excluded). Background fluorescence was measured in regions without cells and subtracted. This method was used to judge relative differences in knock-down efficacy of shRNA vectors but not to quantify protein loss. Quantification of PSD-95 immunostaining was performed using MetaMorph (Molecular Devices, Union City, CA): green fluorescence was used to define a mask of the transfected neuron, the soma was removed, and PSD-95-stained puncta were detected within the mask area by automated routines. The area and intensity of the GFP mask were similar in control and shNR1-transfected neurons (area: control, 4365 ± 91 μm2; shNR1, 4216 ± 113 μm2; gray value: control, 12,203 ± 240; shNR1, 13,103 ± 365). The density of PSD-95 puncta was calculated as the number of PSD-95 puncta divided by the area of the green mask. PSD-95–GFP puncta in organotypic slices were analyzed using a similar algorithm from projections of dendritic stacks acquired with a two-photon laser-scanning microscope (2PLSM) as described below. The length of the dendritic branches was measured in the red fluorescence image.
Organotypic cultures and transfection.
Hippocampal slices were prepared from P7 rat pups as described previously (Stoppini et al., 1991). After 2 d in culture, slices were biolistically transfected with a Helios Gene Gun (Bio-Rad, Hercules, CA), resulting in ∼1–10 transfected pyramidal neurons per slice. Bullets were prepared using 12.5 mg of 1.6 μm gold particles and 25–80 μg of plasmid DNA, depending on the experiment. When more than one vector was transfected, the same total amount of plasmid expressing GFP and/or shNR1 was used to maintain fluorescence intensity and the concentration of shRNA kept constant across conditions. Morphological and electrophysiological studies were performed at 10, 15, and 20 d post-transfection (DPT), and for each time point, experiments were performed at the indicated day ± 1 d.
Two-photon imaging and spine analysis.
Imaging of dendrites and spine morphology were acquired with a custom 2PLSM built as described previously (Carter and Sabatini, 2004) and using 720 nm (mRFP) or 910 nm (EGFP) excitation wavelength. Transfected pyramidal neurons throughout the CA field of the hippocampus were identified by green fluorescence, characteristic morphology, and the presence of a gold particle in the soma seen under laser scanning or video infrared differential interference contrast. Images (512 × 512) were acquired as z-stacks of 12–32 sections at 1 μm spacing. Each section was an average of two scans.
For time-lapse imaging, organotypic slices were cut out with the membrane and transferred to the chamber of the 2PLSM under constant perfusion with culture medium at 30°C. One reference image and three fields of view containing dendritic branches were taken repetitively from each cell such that each section was imaged every 5 min (total time, 90–120 min). The reference image was used to detect and correct for drifts in the location of the cell and slice in the chamber. For non-time-lapse imaging experiments, artificial CSF (ACSF) was perfused at RT. Images were systematically collected from each neuron: one low-power image of the entire cell (0.8× zoom; image field, 300 × 270 μm) and three to four images of apical dendrites imaged at 3× zoom (image field, 64 × 64 μm).
Morphometric analysis was performed blind to genotype using custom software (Holtmaat et al., 2005) written in MATLAB (Mathworks, Natick, MA). Spines were counted, and the total number was divided by the length of dendrite in the field of view to calculate the density. Spine length was measured from the tip of the protrusion to the junction with the dendritic shaft. For the time-lapse data analyses, spine width was estimated by defining a line across each protrusion and measuring the width of the fluorescence intensity distribution at 30% of its maximal intensity. The microscope point-spread function was measured with yellow-green fluorescent microspheres of 100 nm diameter (FluorSpheres; Invitrogen) and estimated as 450 nm in the x–y-axes. Spines that were present at the beginning and persisted for the duration of the imagining session were defined as persistent spines. Extensions or retractions of protrusions were counted, and the percentage of transient protrusions was calculated. For neurons chronically exposed to d-2-amino-5-phosphonovaleric acid (AP-5), time-lapse imaging was performed in the constant presence of the antagonist.
Electrophysiology.
Organotypic hippocampal cultures were placed in the recording chamber, perfused with ACSF [containing (in mm) 127 NaCl, 25 NaHCO3, 1.25 Na2HPO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, and 25 glucose], and saturated with 95% O2/5% CO2 at RT. Whole-cell voltage-clamp recordings were performed from transfected pyramidal neurons (green fluorescence and visible gold particle in the soma) and from control untransfected neighbors. Pipettes were pulled from borosilicate glass capillary tubing with filaments to yield tips of 3–5 MΩ resistance and filled with internal solution containing (in mm) 120 cesium methane-sulfonate, 10 HEPES, 10 EGTA, 4 MgCl2, 0.4 NaGTP, 4 MgATP, and 10 phosphocreatine, pH 7.3 (320 mOsm). The red fluorosphore Alexa Fluor-594 (20 μm) was included in the internal solution to fill the cell and confirm that a recording was obtained from the transfected green fluorescent neuron. An Axopatch 200B amplifier (Molecular Devices) was used, and data were filtered at 2 kHz and digitized at 10 kHz. Series resistance (8–19 MΩ), input resistance, and membrane capacitance were calculated from a 5 mV, 50 ms hyperpolarizing step.
For miniature current recordings, TTX (1 μm) was added to the ACSF in all experiments, and bicuculline (20 μm) or NBQX and d(−)-3-(2-carboxypiperazine-4-yl)-propyl-1-phosphonic acid (CPP) (10 and 1 μm) were included for miniature EPSC (mEPSC) and miniature IPSC (mIPSC) recordings, respectively. For mIPSCs recordings, cesium methane-sulfate was replaced with cesium chloride in the internal solution. Series resistance was monitored on-line, and recordings were discontinued if it increased over 20 MΩ. Analyses of frequency and amplitude of miniature events were performed in Igor Pro using custom software that identified events based on rise time and amplitude thresholds.
For recording of electrically evoked EPSCs, a bipolar stimulating electrode was placed in the stratum radiatum at 250–350 μm from the soma of the recorded neuron, and stimuli were delivered at 0.125 Hz. ACSF was as described above but contained 4 mm CaCl2 and 4 mm MgCl2. The AMPAR current amplitude was measured as the peak current at a holding potential of −60 mV. The NMDAR current amplitude was measured at a holding potential of +40 mV at 50 or 150 ms after the stimulus for uncaging evoked or electrically evoked currents, respectively.
Fifteen to 45 trials were averaged at each holding potential. The coefficients of variation (CV = SD/mean) of the electrically evoked EPSCs recorded at −60 mV were similar in control and shNR1 neurons (0.5 ± 0.05 and 0.52 ± 0.08, respectively). All experiments were analyzed blind to the identity of transfected plasmids.
Glutamate uncaging.
Simultaneous two-photon laser-scanning imaging and two-photon uncaging were performed with a custom-built microscope as described previously (Carter and Sabatini, 2004). A small-volume perfusion system was used with ACSF containing 5 mm MNI-glutamate (Tocris, Ellisville, MO), 10 μm serine, and blockers of Na and Ca channels (in μm: 1 TTX, 0.3 SNX-482, 1 ω-conotoxin-MVIIC, 20 nimodipine, and 20 mibefradil) as well as GABAA receptor and metabotropic glutamate receptor antagonists [20 μm bicuculline and 100 μm LY-341495 ((2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanic acid)] to isolate AMPAR- and NMDAR-mediated currents. Spines from apical dendrites located within 100 μm from the soma were selected for stimulation to minimize differences in voltage clamp because of variable passive properties of the cells. Glutamate was uncaged using a 720 nm light pulse of 0.2–0.5 ms duration. Image drift was corrected to ensure consistency in the location of the uncaging pulse (Carter and Sabatini, 2004). Fifteen to 20 trials of glutamate-evoked currents were averaged while holding cells at −60 and +40 mV for each spine.
Statistics.
Statistical differences were determined using an unpaired t test (http://www.physics.csbsju.edu/stats/ttest.html) or the Kolmogorov–Smirnov test for cumulative distributions (http://www.physics.csbsju.edu/stats/KS-test.html). ANOVA followed by a post hoc test, as indicated in the figure legends, was performed using Igor Pro. The nonparametric χ2 test was used for the bivariate tabular data obtained in the time-lapse experiments.
Results
RNAi-mediated knock-down of NR1 in hippocampal pyramidal neurons
To physically eliminate NMDARs from neurons, we used RNAi to target its NR1 subunit, which is necessary for assembly of functional receptors (McIlhinney et al., 2003; Schorge and Colquhoun, 2003) (Fig. 1). Because the half-life of surface NMDARs is ∼30 h (Huh and Wenthold, 1999), RNAi was performed with vector-driven expression of shRNA that allows for long-term knock-down of the targeted protein. Identification of shRNA-expressing cells was ensured by designing a dual-promoter vector expressing cytomegalovirus-driven EGFP and U6-driven shRNA (Tavazoie et al., 2005). Eight sequences within the NR1 coding sequence (GenBank accession number X63255) with a g/c content of 45–55% were chosen as targets, and dual-promoter vectors (shNR1a-h) were constructed. The shNR1 vectors were screened in cultures of dissociated hippocampal neurons for their ability to trigger an interferon response (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Alvarez et al., 2006) as well as for their efficiency for NR1 knock-down. A single construct, shNR1a, referred as shNR1 below, was chosen for subsequent experiments. Neurons expressing shNR1 did not show activation of a nonspecific interferon response and exhibited reduced NR1 immunostaining in the soma compared with neurons expressing a control vector encoding only GFP (Fig. 1A). In contrast, the levels of MAP2 immunostaining were similar in neurons expressing control and shNR1 vectors (n = 23–40) (Fig. 1B). In addition, control GFP-expressing neurons had similar levels of NR1 immunostaining as untransfected neighbors (n = 18; data not shown).
Figure 1.

shRNA against NR1 reduces protein levels and NMDAR-mediated synaptic currents. A, Images of dissociated pyramidal neurons transfected with GFP control plasmid (top) or a dual-promoter plasmid coexpressing GFP and shNR1 (bottom). Neurons were transfected at 7 DIV and immunostained for NR1 and MAP2 at 10 d DPT. The arrowhead and asterisk indicate a transfected and untransfected neuron, respectively. B, Cumulative distribution of NR1 (top) and MAP2 (bottom) fluorescence immunostaining for control and shNR1-expressing neurons (n = 18–43). Data are from one of three independent experiments with similar results. The distributions of NR1 immunostaining are statistically different (p < 0.01, Kormogorov–Smirnov test). C, Wide-field fluorescence image of hippocampal organotypic slice showing sparse transfection of pyramidal neurons with GFP. The positions of the recording electrode (rec) in the stratum pyramidale (s.pyr.) and of the stimulating electrode (stim) in the stratum radiatum (s. rad.) are shown. D, EPSCs evoked by stimulation of Schaffer collaterals recorded from GFP-expressing (top) and shNR1-expressing (bottom) pyramidal neurons (10 DPT) at holding potentials of −60 mV (fast inward currents) and +40 mV (slow outward currents). Amplitudes were measured at the peak of the current at −60 mV and 150 ms after the peak at 40 mV. E, Normalized synaptic currents from control (black) and shNR1-expressing (gray) neurons showing the differences in time courses and amplitudes of the currents at +40 mV. F, Cumulative distribution of current ratios at +40 and −60 mV, R+40/−60, in GFP (control; filled circles) and shNR1-expressing (open circles) neurons at 10 DPT. Distributions are statistically different (p < 0.01, Kormogorov–Smirnov test). G, Average R+40/−60 measured in control NR1 knock-down neurons and control neurons in the presence of the NMDAR antagonist CPP (1 μm) at 10 DPT (n = 10–12). *p < 0.05 compared with control. Scale bars: A, 10 μm; C, 100 μm.
Loss of NMDAR-mediated currents at synapses
To quantify the loss of synaptic NMDARs, synaptic currents evoked by extracellular stimulation were measured in hippocampal organotypic cultures transfected with GFP or shNR1 vectors. Whole-cell voltage-clamp recordings were obtained from transfected pyramidal neurons that were identified by green fluorescence, morphology, and location in the pyramidal cell layer (Fig. 1C). Using this approach, only a few pyramidal neurons in each slice are transfected, and essentially all of the presynaptic partners of the transfected voltage-clamped cell are expected to be untransfected.
EPSCs were measured after electrical stimulation of the Shaffer collaterals at holding potentials of −60 and +40 mV (Fig. 1D). In control neurons, synaptic stimuli evoked a short-lived current at −60 mV that is consistent with an AMPAR-dominated EPSC. Depolarization to +40 mV revealed a long-lived current that is sensitive to the NMDAR antagonist CPP (1 μm), consistent with the relief of the Mg block of NMDARs and the prolonged opening of these receptors. At 3 DPT, the ratio of the current amplitude at +40 mV to that at −60 mV (R+40/−60) was similar for control and shNR1 neurons (R+40/−60 = 0.27 ± 0.04 and 0.39 ± 0.08 in control and shNR1-expressing neurons, respectively; n = 4–6), suggesting a slow turnover of synaptic NMDARs. At 10 DPT, control and shNR1-transfected neurons had similar fast AMPAR-mediated currents at −60 mV, but the outward currents at +40 mV were of shorter duration in shNR1 neurons, consistent with a reduction in synaptic NMDARs (Fig. 1D,E). Furthermore, R+40/−60 was reduced in shNR1 neurons to a value similar to that measured for control neurons in the presence of the NMDAR antagonist CPP (R+40/−60 = 0.29 ± 0.04, 0.08 ± 0.01, and 0.04 ± 0.02 for control, shNR1, and CPP neurons, respectively; n = 10, 12, and 3, respectively) (Fig. 1F,G). Thus, at 10 DPT, shNR1-expressing neurons display a severe reduction in NMDAR-mediated synaptic currents but still receive functional synapses.
Increased motility of dendritic spines in NMDAR knock-down neurons
Glutamatergic synapses on hippocampal pyramidal neurons are formed onto the heads of dendritic spines, which can extend, retract, or change shape rapidly. The motility of dendritic spines is larger in young animals and declines with age (Holtmaat et al., 2005; Zuo et al., 2005a), in parallel with the ability to induce NMDAR-dependent LTP. To examine whether NMDARs stabilize synapses and spines, we repetitively imaged segments of apical dendrites of pyramidal neurons in organotypic slices over a period of 90–120 min (every 5 min) using two-photon laser-scanning microscopy. We compared the motility of dendritic protrusions at 10 DPT in control neurons expressing GFP to that in neurons expressing shNR1 (Fig. 2A) (n = 6–7 cells, 272–215 spines). In control neurons, the majority of spines present at the beginning of the imaging session remained for the duration of the experiment (82% persistent spines), and only a small percentage of protrusions was transient (18%) (Fig. 2B). However, in neurons expressing shNR1, only 60% of spines were persistent with a concomitant increase in the number of transient protrusions (40%) (p < 0.01 vs control, χ2 test). In each condition, the transient protrusions were equally divided between those that extended and those that retracted (10 and 8%, respectively, for control; 19 and 21%, respectively for shNR1). Thus, loss of synaptic NMDARs reduces spine stability and increases the rate of spine turnover.
Figure 2.

Increased motility of dendritic spines in shNR1 neurons. A, Time-lapse two-photon laser-scanning microscopic imaging of a spiny dendrite of a shNR1-expressing pyramidal neuron in a hippocampal organotypic slices at 10 DPT. Slices were bathed in slice culture medium at 30°C for the duration of the imaging session (90–120 min). The arrow marks a transient dendritic protrusion. Some spines change shape and size (arrowhead), whereas other spines preserved a relatively constant length and width (asterisk). Scale bar, 2.5 μm. B, Percentage of persistent spines (spines that are present for the entire imaging session) and transient dendritic protrusions (extensions and retractions) in control GFP (black) and shNR1 (gray) neurons at 10 DPT (n = 6–7 cells, 267–293 spines). The percentages in each category for control and shNR1 neurons are statistically different (p < 0.01, χ2 test). C, Dynamics indices of spine length and width of persistent spines in control (black) and shNR1 (gray) neurons (n = 6–7 cells, 214–235 spines). *p < 0.01.
In addition to retraction and extension, spines undergo more subtle structural rearrangements that include changes in length and width. To quantify these rearrangements, for each persistent spine we calculated the length dynamics index defined as the average change in length per 5 min interval expressed as a percentage of the mean length:
 |
A similar calculation was performed to determine Dwidth. We found Dlen = 13 ± 0.5% and Dwidth = 14 ± 0.4% in control neurons and Dlen = 16 ± 0.6% and Dwidth = 17 ± 0.5% in shNR1 neurons (p < 0.01 for each comparison relative to control) (Fig. 2C). Thus, knock-down of NMDARs increases the rate of spine formation and retraction as well as spine morphological plasticity, indicating that these receptors normally act to promote morphological and structural stability of spines.
Loss of dendritic spines in NMDAR knock-down neurons
Pyramidal neurons in organotypic cultures undergo synaptic maturation that parallels many aspects of neuronal development in vivo and are therefore well suited for studying the role of the NMDARs in the long-term stabilization of spines and synapses. During the first 3 weeks in vitro, the density of spines in control pyramidal neurons increased from 0.5 ± 0.02 μm−1 at 10 DPT to 0.8 ± 0.04 and 0.93 ± 0.03 μm−1 at 15 and 20 DPT, respectively (Fig. 3B,C) (n = 12–25 cells). Pyramidal neurons expressing shNR1 showed reduced spine density at 10 DPT (0.35 ± 0.02 μm−1), which declined to reach 0.18 ± 0.02 and 0.12 ± 0.01 μm−1 at 15 and 20 DPT, respectively (Fig. 3B,C) (n = 10–28 cells). NMDARs are also necessary for the maintenance of dendritic spines outside of this developmental period, because neurons transfected 5 d later than usual and maintained in culture until 10 DPT for comparison with the 15 DPT data showed a similar reduction in spine density (0.17 ± 0.01 μm−1; n = 10 cells) (Fig. 3D). Transfection with shNR1 had no effect on dendritic branch complexity (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 3.

Loss of dendritic spines in neurons with reduced synaptic NMDARs. A, Hippocampal pyramidal neurons in organotypic slices transfected with GFP (top) or shNR1 (bottom) imaged with a 2PLSM at 20 DPT. B, Higher-magnification images of dendrites from control (top) and shNR1 (bottom) neurons at 10 DPT (left) and 20 DPT (right). C, Developmental profile of spine density in control and shNR1 neurons (n = 10–28). D, Summary of spine density at 15 DPT in a variety of conditions. NR1res denotes the NR1-1-resistant clone with silent mutations in the region targeted by shNR1; shNR1 late summarizes data from neurons transfected with shNR1 at 8 DIV (instead of 3 DIV) and imaged at 10 DPT. Scale bars: A, 50 μm; B, 2 μm. *p < 0.05 compared with control; #p < 0.05 compared with shNR1 neurons; by ANOVA and Student–Newman–Keuls multiple comparisons test.
To confirm that spine loss was attributable to loss of NMDARs, silent mutations were introduced into the nucleotide sequence of NR1, and a shNR1-resistant form (NR1res) was generated. Expression of NR1res partially rescued the extent of dendritic spine loss by shNR1 (0.41 ± 0.04 μm−1; n = 9) (Fig. 3D). The inability to rescue completely likely reflects that RNAi triggers loss of all eight NR1 splice isoforms, whereas only a single isoform was used in the rescue (see Fig. 8).
Figure 8.

Splice variants NR1-1 and NR1-2 are required for the maintenance of dendritic spines. A, Schematic structure of the NR1 subunit showing the C-terminal C2 and C2′ cassettes. B, Schematic of the NR1 mRNA (top) showing the four exons (N, C1, C2, C2′) that are differentially spliced to give rise to the NR1 isoforms. The back line indicates the shNR1 target region (not to scale). Schematics of the NR1 rescue clones (NR1res) used for the analyses of structural requirement are shown at the bottom. Blue areas indicate the mutated shNR1 target region, and gray areas represent the transmembrane domains. C, Summary of spine density at 15 DPT of pyramidal neurons transfected with NR1 rescue splice isoforms together with shNR1 (black bars). Shaded horizontal bars show the average ± 2*SEM of spine density for control (gray) and shNR1 (red) neurons at 15 DPT. *p < 0.05 compared with shNR1 neurons; #p < 0.05 compared with control neurons; by ANOVA and Student–Newman–Keuls multiple comparisons test.
Loss of synapses in NR1 knock-down neurons
In hippocampal pyramidal neurons, each spine is typically associated with one and only one synapse, and changes in spine numbers are thought to reflect changes in the number of active synapses. To confirm that shNR1-induced spine loss reflects a loss of synapses, whole-cell voltage-clamp recordings were made from transfected pyramidal neurons, and mEPSCs were monitored (Fig. 4). At 20 DPT, shNR1-expressing neurons had greatly reduced membrane capacitance (Cm = 384 ± 29 and 172 ± 18 pF for control and shNR1 neurons, respectively; n = 21–26) and increased input resistance (Rin = 111 ± 16 and 183 ± 16 MΩ for control and shNR1 neurons, respectively; n = 21–26) (Fig. 4B, left), consistent with decreased membrane area. Furthermore, the frequency of mEPSCs was significantly reduced in shNR1 neurons relative to controls (0.5 ± 0.1 and 0.11 ± 0.03 Hz in control and shNR1 neurons, respectively; n = 15–18) without changes in the amplitude of the individual events (control, −21 ± 0.9 pA; shNR1, −20 ± 1.3 pA; n = 15–18) (Fig. 4C). Neither the frequency nor amplitude of mIPSCs was different in control and shNR1 neurons (control: 1.8 ± 0.4 Hz and −30 ± 4 pA, n = 12; shNR1: 1.2 ± 0.3 Hz and −28 ± 2 pA, n = 13) (Fig. 4C). Thus, postsynaptic expression of NMDARs is required for the stabilization of synaptic structure and the maintenance of functional excitatory synapses.
Figure 4.

Loss of active synapses in neurons lacking synaptic NMDARs. A, Representative recordings of mEPSCs from control and shNR1 neurons at 20 DPT. Calibration: 200 ms, 20 pA. B, Summary of input resistance (Rin) and membrane capacitance (Cm) for control (black) and shNR1 (gray) neurons at 20 DPT (n = 21–26). *p < 0.05. C, Summary of mEPSC and mIPSC amplitude (left) and frequency (right) for control (black) and shNR1 (gray) neurons at 20 DPT (n = 15–18 and 12–13 for mEPSC and mIPSC, respectively). *p < 0.05.
AMPAR and PSD-95 are maintained at spines of shNR1 neurons
Our finding that shNR1-expressing cells have a small number of dendritic spines and receive functional synapses suggests that spines and synapses can at least transiently exist and express AMPARs, despite nearly full loss of NMDARs. Conversely, it is possible that the remaining spines in the shNR1-expressing cells are ones in which the PSD is remarkably stable and still contain normal complement of NMDARs. Last, it is also possible that AMPAR-mediated mEPSCs are generated by synapses made directly onto the dendritic shaft.
To address these issues, two-photon laser-scanning microscopy and two-photon laser uncaging (2PLU) of caged glutamate (MNI-glutamate) were combined with electrophysiological recordings to probe for functional NMDARs and AMPARs on visualized, individual spines (Fig. 5). To maximize voltage control, spines close to the soma (<100 μm) were selected, and recordings were made in a mixture of Na, K, and Ca channel antagonists (see Materials and Methods). The power of the uncaging laser pulse (500 μs at 720 nm) was set to generate an ∼20 pA current with fast-rising kinetics at a holding potential of −60 mV. In control neurons at 10 DPT, uncaging of MNI-glutamate onto spines activated a rapidly decaying current at −60 mV, consistent with the activation of AMPARs, and a long-lived current at +40 mV mediated by AMPARs and NMDARs (IAMPA = 21 ± 2 pA; INMDA = 14.2 ± 1.8 pA; R+40/−60 = 0.89 ± 0.12; n = 6) (Fig. 5B–D). Glutamate uncaging onto individual spines of shNR1 neurons held at −60 mV evoked AMPAR-mediated currents of similar amplitude to spines of control neurons (−19 ± 2.5 pA; n = 9) (Fig. 5B,C). However, these spines showed little or no long-lived currents at +40 mV (2.9 ± 0.6 pA) and a severe reduction in R+40/−60 (0.25 ± 0.07) (Fig. 5D). Similarly, at 20 DPT, when <10% of the spines remain in shNR1 neurons, R+40/−60 was decreased to an average value of 0.33 ± 0.12 (n = 10) (Fig. 5D). Thus, spines that remain on shNR1 neurons have surface AMPARs, indicating that synaptic AMPARs and spines can exist despite loss of the majority of NMDARs.
Figure 5.

Functional AMPARs are expressed in spines lacking NMDAR-mediated currents. A, Image of a spiny dendrite of a pyramidal neuron filled with Alexa Fluor-594 showing site of glutamate uncaging (asterisk). B, Currents evoked by 2PLU of glutamate on spines of control (left) and shNR1 (right) neurons held at −60 mV (inward current) and +40 mV (outward current). The amplitude of the currents mediated by AMPAR and NMDAR were measured at the times indicated by the filled dots. C, Normalized currents evoked by glutamate uncaging onto single spines from control (black) and shNR1 (gray) neurons recorded at −60 and +40 mV. D, Cumulative histogram plots of R+40/−60 measured at individual spines of control neurons (black circle), control neurons in CPP (1 μm; gray circles), and shNR1 neurons (open circles) at 10 DPT (n = 12–18). E, Average R+40/−60 from individual spines of control, control plus CPP, shNR1 neurons, and shNR1 neurons cotransfected with the NR1-1-resistant clone (shNR1+NR1res) at 10 DPT (n = 12–18). *p < 0.05 compared with control; #p < 0.05 compared with shNR1.
Combined 2PLU and two-photon laser-scanning microscopy was also used to judge the recovery of NMDAR-mediated currents at spines of neurons coexpressing shNR1 and NR1-1res, the shNR1-resistant form of NR1. Spines from these neurons displayed a higher NMDAR/AMPAR current ratio than spines from shNR1 neurons alone (R+40/−60 = 0.52 ± 0.04; n = 5; 10 DPT), indicating the exogenously expressed NR1 subunits did incorporate into functional NMDARs within dendritic spines (Fig. 5D). However, the recovery of the current by this construct is incomplete, in agreement with the partial recovery of spine density observed by this NR1res construct (Fig. 3D).
The above results indicate the NMDARs are necessary for spine stability and suggest that they might not be required for spine growth and expression of synaptic AMPARs. Does spine instability in the absence of NMDARs result from loss of synaptic scaffolding proteins such as PSD-95 that are known to interact with NMDAR (Kornau et al., 1995)? To test this possibility, the distribution of PSD-95–GFP fusion proteins was examined in neurons expressing mRFP or a vector that coexpresses shNR1 and mRFP (shNR1mRFP). At 10 DPT, clusters of PSD-95–GFP were observed along the dendrites of both control and shNR1mRFP neurons in hippocampal slices cultures, but the density of the PSD-95 puncta was reduced in shNR1mRFP neurons (density per dendrite length was 0.36 ± 0.04 and 0.23 ± 0.02 μm−1 in control and shNR1mRFP neurons, respectively; p < 0.05) (Fig. 6A,C) (n = 6 cells). This 37 ± 5% reduction in PSD-95 puncta correlates well with the 30 ± 4% reduction in spine density observed in these neurons at this time point.
Figure 6.
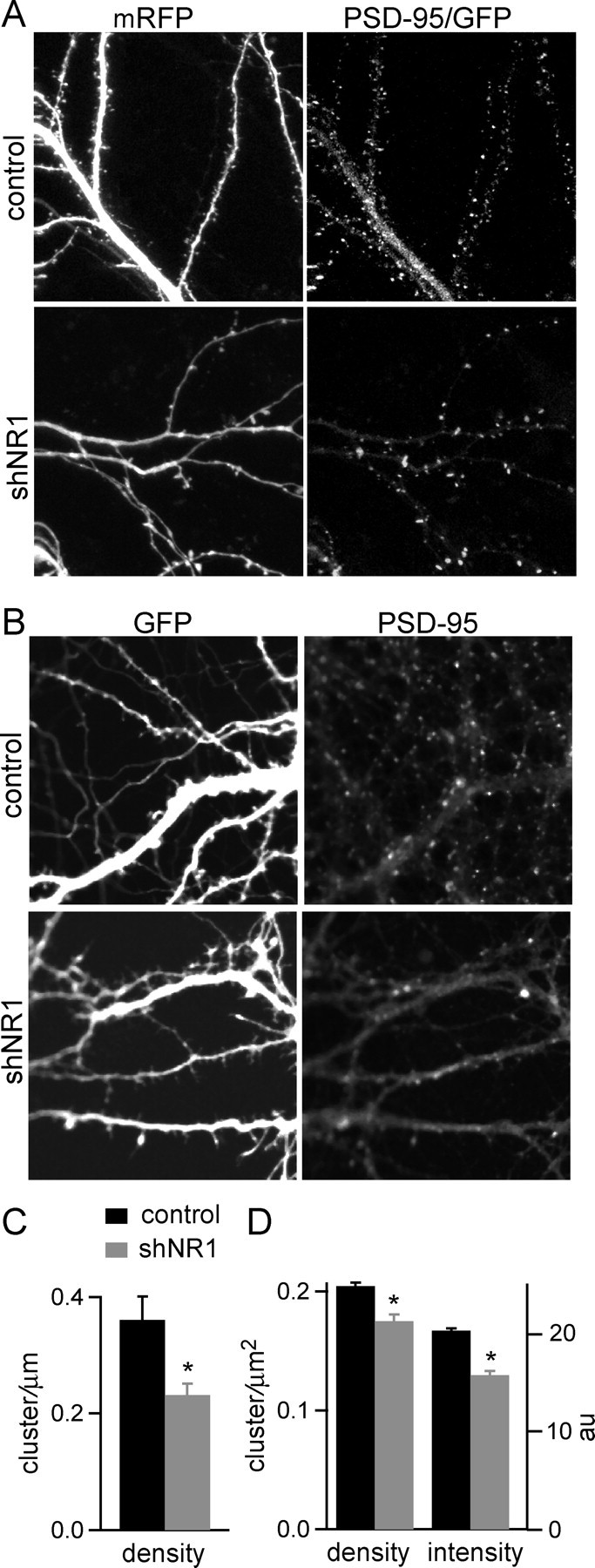
PSD-95 clusters form in remaining spines of shNR1 neurons. A, Images of dendritic branches of control (top) and shNR1 (bottom) pyramidal neurons in organotypic slices showing dendrite morphology (mRFP) and the distribution of PSD-95–GFP at 10 DPT. B, Dissociated pyramidal neurons expressing GFP (top) and shNR1 (bottom) showing GFP fluorescence and fluorescence immunostaining for endogenous PSD-95 at 10 DPT. C, Summary of linear density of PSD-95–GFP puncta in control (black) and shNR1 (gray) neurons in organotypic slices at 10 DPT (n = 6). *p < 0.05. D, Summary of immunostaining for endogenous PSD-95 puncta density and intensity for control (black) and shNR1 (gray) neurons in dissociated cultures at 10 DPT (n = 14–12). *p < 0.05.
Similar results were observed using fluorescence immunohistochemistry to label endogenous PSD-95 in cultures of dissociated hippocampal neurons (Fig. 6B,D). Clusters of PSD-95 staining were seen distributed along the dendrites of control and shNR1 neurons, but the density was reduced in neurons expressing shNR1 (control, 0.2 ± 0.003 μm−2; shNR1, 0.16 ± 0.005 μm−2; n = 14–12; p < 0.05), consistent with the reduction in spine and synapse density. When using antibodies to stain for endogenous PSD-95, all neurons in the culture are labeled, and dendrites from untransfected neurons contaminate the measurement, leading to an underestimate of the reduction in PSD-95 puncta in shNR1 neurons. The average intensity of PSD-95 puncta was reduced in shNR1 neurons (control, 20 ± 0.2; shNR1, 15.6 ± 0.4; n = 14–12; p < 0.05), suggesting reduced PSD-95 levels at synapses that lack NMDARs.
Ionotropic signaling is not necessary to maintain spine density
RNAi-induced loss of NMDARs eliminates NMDAR-dependent protein–protein interactions as well as current flux through NMDARs. To determine whether both the ionotropic and protein interaction components of NMDAR signaling are necessary to maintain spine density, the effects of the NMDAR antagonist AP-5 (50 μm) on spine density and dynamics were examined (Fig. 7). Neurons in organotypic slice cultures were transfected with GFP and chronically treated with AP-5. Time-lapse imaging experiments showed that chronic blockade of NMDAR activation for 1 week increased the percentage of spines that appeared or disappeared during the course of the experiment (transient spines: 16.7% in AP-5 treated vs 8.5% in control; n = 257–293 spines, 6 cells) (Fig. 7A). Concomitantly, a reduction was observed in the percentage of persistent spines in these neurons compared with control neurons (AP-5 treated, 83.3%; control, 91.5%; p < 0.05, χ2 test). However, the structural dynamics indices for persistent spines were similar in neurons experiencing chronic AP-5 treatment and in control untreated neurons (Dlen = 15 ± 0.7% for control and 14 ± 0.6% for AP-5 treated; Dwidth = 14 ± 0.4% for control and 15 ± 0.5% for AP-5 treated) (Fig. 7B). Thus, blocking NMDAR activation and eliminating NMDARs from synapses both increase the abundance of transient spines, but only physical elimination of NMDARs affects the rapid morphological plasticity of persistent spines.
Figure 7.

Ionotropic properties of NMDAR regulate spine dynamics but not spine numbers. A, Percentage of persistent and transient spines in control untreated GFP neurons (−AP-5; black) and GFP neurons treated with 50 μm AP-5 (+AP-5; gray) for 8 d (n = 6 cells, 257–293 spines). The distributions for control and AP-5-treated neurons are statistically different (p < 0.01, χ2 test). B, Dynamics indices for spine length and width of persistent spines in untreated control (black) and AP-5-treated (gray) neurons. C, Dendrites of control and shNR1 pyramidal neurons in untreated (left) and AP-5-treated (right) slices at 20 DPT. Treatment with 50 μm AP-5 began at 10 DPT. D, Spine density of control and shNR1 pyramidal neurons at 20 DPT in untreated slices (black) and slices treated with AP-5 (gray).
Moreover, long-term blockade of NMDARs did not trigger spine elimination (Fig. 7C,D) and spine density increased normally despite prolonged blockade of the receptor. This was the case whether the AP-5 treatment was started at 10 DPT and continued for 10 d (spine density: untreated controls at 20 DPT, 0.92 ± 0.03 μm−1; 10 d AP-5/20 DPT neurons, 0.90 ± 0.05 μm−1; n = 10–12 cells) or when AP-5 incubation was started on the day of transfection [3 d in vitro (DIV)] and maintained until imaging (spine density: untreated controls at 15 DPT, 0.80 ± 0.04 μm−1; 15 d AP-5/15DPT neurons, 0.76 ± 0.1 μm−1; n = 8–9 cells). The failure of the receptor antagonist treatment to induce spine loss demonstrates that the ionotropic properties of the NMDARs are not required to maintain normal density of dendritic spines throughout this period of synaptogenesis.
Neurons transfected with shNR1 were subject to the same chronic exposure to AP-5. Spine density in shNR1 neurons cultured in AP-5 (shNR1 10 d AP-5/20DPT, 0.1 ± 0.02 μm−1; n = 10 cells) was the same as that of untreated shNR1 neurons (0.12 ± 0.01 μm−1; n = 9 cells) (Fig. 7C,D). This result demonstrates that loss of dendritic spines in shNR1 neurons is not caused by a partial reduction in NMDAR ionotropic signaling during the slow process of RNAi-induced receptor knock-down. Furthermore, it demonstrates that loss of spines is not a consequence of shNR1 neurons being placed at a disadvantage in a competition for presynaptic boutons compared with neighboring NMDAR-expressing neurons.
Structural requirements for spine stability
Native, functional NMDARs are heteromeric assemblies composed of two NR1 subunits and two NR2 subunits (Monyer et al., 1992; Laube et al., 1998; Schorge and Colquhoun, 2003). The NR1 gene generates multiple splice isoforms that confer unique properties to the receptor assembly (Laurie and Seeburg, 1994). These range from inhibition by protons and Zn to protein–protein interactions with calmodulin, intermediate filaments, and PSD-95/Discs large/zona occludens-1 (PDZ)-containing proteins (Wenthold et al., 2003; Cull-Candy and Leszkiewicz, 2004). In addition, splicing of the C terminus of NR1 determines the inclusion or exclusion of the C1 cassette and expression of either the C2 or C2′ cassettes, which regulate NMDAR trafficking, surface expression, signaling to the nucleus, and PDZ-based interactions (Ehlers et al., 1995, 1998; Standley et al., 2000; Wenthold et al., 2003; Bradley et al., 2006).
To understand whether NR1 splice variants with different C-terminus tails differentially affect spine density, we tested the ability of different isoforms to rescue shNR1-induced spine loss (Fig. 8). Silent mutations were introduced in three NR1 splice isoforms that rendered them resistant to shNR1 (NR1-1res, NR1-2res, and NR1-3res) (Fig. 8B). Each construct was coexpressed with shNR1, and spine density was measured at 15 DPT and compared with GFP control neurons and shNR1 neurons (Fig. 8C). Expression of NR1-1res, which contains the C0, C1, and C2 cassettes, increased spine density approximately twofold but did not restore spine density to wild-type levels (0.41 ± 0.04 μm−1; n = 9 cells). Similar partial rescue of spine density was seen with expression of NR1-2res, which contains the C0 and C2 cassettes (0.47 ± 0.07 μm−1; n = 12 cells). Thus, the presence or absence of C1 did not affect the ability to rescue spine density. Expression of NR2A subunit together with NR1-1res, a condition shown to increase surface expression of NR1 under some circumstances (Barria and Malinow, 2002), did not improve the partial recovery of spine density (data not shown). Furthermore, expression of NR1-3res, which contains the C0, C1, and C2′ cassettes, failed to increase spine density above shNR1-expressing neurons (0.27 ± 0.03 μm−1; n = 10) (Fig. 8C). Because NR1-1res and NR1-3res differ only in the inclusion of the C2 or C2′ cassettes, these results suggest that expression of NR1 subunits with the C2 cassette, and not the C2′ cassette, is necessary for spine maintenance.
To examine whether multiple NR1 isoforms cooperatively promote spine growth, the rescue efficiency of coexpressing combinations of NR1 isoforms was tested. The total amount of constructs used for biolistic transfection was kept in the same narrow range (40–50 μg) used to test the ability of each single isoforms to rescue spine density. Therefore, in these experiments, the amount of each expression plasmid used was one-half to one-third of that used above. Coexpression of splice variants 1 and 2 returned spine density to near wild-type levels (0.65 ± 0.07 μm−1; n = 17 cells) (Fig. 8C), suggesting a cooperative effect on spine maintenance. Interestingly, splice variants 1 and 2 are the predominant forms of NR1 subunits in hippocampal pyramidal neurons (Laurie and Seeburg, 1994; Paupard et al., 1997). Conversely, coexpression of splice variant 3 with splice variant 2 alone or with splice variants 1 and 2 together did not increase spine density beyond those seen without the inclusion of variant 3. These experiments indicate that the assembly of isoforms 1 and 2 of the NR1 subunit are required for stable formation of dendritic spines and that this process is neither enhanced nor inhibited by the expression of isoform 3.
Discussion
NMDARs regulate neuronal function at many levels and through multiple pathways. First, opening of NMDARs contributes to postsynaptic depolarization and shapes circuit firing. Second, Ca influx through open receptors locally regulates synaptic properties and globally regulates gene transcription and cell survival. Third, NMDAR-dependent protein–protein interactions play a role in the assembly of the PSD. Here, we show that NMDARs, through ionotropic and structural signaling, regulate the rapid dynamics of spine morphology. In addition, non-ionotropic NMDAR-dependent signaling through specific C-tail splice isoforms regulates the long-term stability and density of dendritic spines as well as the numbers of excitatory synapses.
Stability of synaptic structure and function
Loss of NMDAR protein from excitatory synapses was accompanied by increased motility of dendritic spines, a larger number of transient protrusions, and a pronounced decrease in dendritic spine density. Functional excitatory synapses were also lost, as indicated by reduced mEPSCs frequency and PSD-95 cluster density. However, at 10 DPT, remaining spines and synapses were able to express functional AMPARs and cluster PSD-95, suggesting that many components of the excitatory synapse remain intact. Some of these spines displayed no NMDAR-mediated currents, suggesting that AMPAR-only synapses can at least transiently exist (Bekkers and Stevens, 1989). Because elimination of synaptic NMDARs occurs in certain forms of LTD (Gean and Lin, 1993; Morishita et al., 2005), our finding that synapses that are losing NMDARs have a short half-life and, over time, are eliminated suggests a mechanism for neuronal circuit remodeling in which loss of NMDARs is the final signal to trigger the retraction of the spine and elimination of the synapse.
Our study indicates that the presence of NMDARs makes synapses more stable and dendritic spines less motile; nevertheless, it also suggests that spines and synapses can form despite losing the majority of NMDARs. The spines and synapses that remain in shNR1 neurons at 20 DPT are unlikely to have formed before loss of NR1 given the high turnover rate of spines in organotypic slices during this period (Zito et al., 2004). Furthermore, if the spines that remain at 20 DPT in shNR1 cells had been formed before RNAi induction, we would have expected a higher density of spines when transfection with shNR1 was performed 5 d later than usual (8 DIV vs 3 DIV) (Fig. 3). Although spine density increases steeply in control cells during these 5 DIV, delayed transfection was equally efficacious in triggering spine loss. Alternatively, it is possible that the physical presence of NMDARs is strictly necessary for spine formation and that the small number of NMDARs remaining in shNR1 neurons is sufficient to support this process.
In this work, RNAi was used to induce sustained (up to several weeks) loss of NR1 subunit and a subsequent loss of functional synaptic NMDARs. NR1 knock-out mice die shortly after birth from respiratory failure (Forrest et al., 1994), and spine density has not been evaluated in these mice. Adult expression of Cre-recombinase in forebrain of knock-in mice carrying a conditional allele of NR1 has been reported to not affect spine density within CA1 (Rampon et al., 2000). However, in that study, Cre expression commenced approximately after P19, and it was not confirmed that the cells analyzed morphologically had lost synaptic NMDARs. Furthermore, work from several groups has shown that, because of compensation, analysis of neurons derived from knock-out mice for a synaptic protein can yield different results than those in which the same protein has been targeted by RNAi (Elias et al., 2006; Schluter et al., 2006).
Differential roles of NMDAR activation and structural interactions
Physical loss of NMDARs leads to a vast reduction (∼90%) in the density of dendritic spines and the number of excitatory synapses. Because spine loss is not seen with pharmacological blockade of NMDARs, the ionotropic properties of these receptors are not required for spine maintenance. Non-ionotropic actions of glutamate and glycine binding to NMDARs have been described previously (Vissel et al., 2001; Nong et al., 2003). However, the effects described here are also likely to be independent of glutamate binding to the receptor, because spine density was unaffected by the high-affinity competitive antagonists of glutamate used to block NMDARs.
Nevertheless, blockade of NMDARs for hours to days is known to trigger many synaptic changes. Several hours blockade of NMDARs triggers an increase in the number of synaptic AMPARs (Sutton et al., 2006), a process analogous to synaptic scaling (Turrigiano et al., 1998). Pharmacological blockade for days leads to increased clustering of synaptic NMDARs in dissociated hippocampal and cortical cultures (Rao and Craig, 1997). Similarly, prolonged incubation of organotypic slices in NMDAR antagonists increases the number of functional synapses by increasing dendritic branching without affecting spine density (Luthi et al., 2001), and in vivo application during cerebellar and cortical development prevents the retraction of exuberant synapses (Rabacchi et al., 1992; Zuo et al., 2005b).
NR1 domain analysis
The NR1 subunit of the NMDAR is necessary for the formation of functional receptors, which are tetrameric structures formed by the assembly of pairs of dimers of NR1 and NR2 subunits. The intracellular C terminus of the NR1 subunit is highly regulated by alternative splicing, which determines the presence or absence of the C1 cassette and the mutually exclusive expression of a terminal C2 or C2′ cassette. All NR1 isoforms contain a C0 cassette which, together with C1, mediates protein–protein interactions with calmodulin (Ehlers et al., 1996), α-actinin (Wyszynski et al., 1997), neurofilaments (Ehlers et al., 1998), and yotaio (Lin et al., 1998) as well as signaling to the nucleus (Bradley et al., 2006) and export from the endoplasmic reticulum (ER) (Wenthold et al., 2003). The C2′ cassette contains a terminal PDZ-binding motif that mediates interactions with PSD-95, PSD-93, synapse-associated protein-102 (SAP-102), and SAP-97 (for review, see O'Brien et al., 1998) as well as a divaline motif that promotes COPII-dependent exit from the ER (Standley et al., 2000). mRNA splicing that regulates C2/C2′ expression is activity regulated such that activity blockade leads to enhanced expression of C2′-containing NR1 subunits and promotes surface expression of NMDARs (Mu et al., 2003).
We find that expression of NR1 subunits containing the C2 cassette is necessary for maintenance of dendritic spines. Either C0/C1/C2-containing (NR1-1a) or C0/C2-containing (NR1–2a) receptors are able to preserve some levels of spine density. These receptor isoforms are the predominant forms in hippocampal pyramidal neurons (Laurie and Seeburg, 1994; Paupard et al., 1997), and we find that their coexpression supports normal spine density. This result suggests that native NMDARs in these neurons might be heteromeric assemblies of NR1-1a and NR1-2a or that NR1-1a- and NR1-2a-containing receptors each facilitate different but necessary steps in the growth and stabilization of dendritic spines. In contrast, C0/C1/C2′-containing receptors (NR1–3a) are insufficient to maintain spine density and do not enhance the spine recovery by C2-containing receptors. Thus, although the PDZ-binding domain of the C2′ domain is required for interactions with PSD-95 and may be important for anchoring to the PSD, it is not necessary for spine maintenance. The combination of RNAi-mediated knock-down and pharmacological blockade of NMDARs used here, coupled with the rescue of spine density by reintroduction of RNAi-resistant form NR1, reveal a central, structural role of NMDARs in regulating synapse and spine number.
Footnotes
This work was supported by National Institutes of Health Training Grant 5T32 NS07484 (V.A.A.), the Burroughs Wellcome Fund, the Searle Scholars Fund, and the McKnight Foundation (B.L.S.). We thank the members of the Sabatini laboratory for helpful discussions and critical reading of this manuscript, Michelle Ocana for technical assistance, and the Harvard Center for Neurodegeneration and Repair. We are grateful to Robert J. Wenthold and Morgan Sheng for gifts of reagents.
References
- Alvarez VA, Ridenour DA, Sabatini BL. Retraction of synapses and dendritic spines induced by off-target effects of RNA interference. J Neurosci. 2006;26:7820–7825. doi: 10.1523/JNEUROSCI.1957-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T. Alzheimer's disease as a disorder of mechanisms underlying structural brain self-organization. Neuroscience. 2001;102:723–765. doi: 10.1016/s0306-4522(00)00516-9. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- Boyer C, Schikorski T, Stevens CF. Comparison of hippocampal dendritic spines in culture and in brain. J Neurosci. 1998;18:5294–5300. doi: 10.1523/JNEUROSCI.18-14-05294.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Carter SR, Rao VR, Wang J, Finkbeiner S. Splice variants of the NR1 subunit differentially induce NMDA receptor-dependent gene expression. J Neurosci. 2006;26:1065–1076. doi: 10.1523/JNEUROSCI.3347-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, Zastrow MV, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Tingley WG, Huganir RL. Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science. 1995;269:1734–1737. doi: 10.1126/science.7569904. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Fung ET, O'Brien RJ, Huganir RL. Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J Neurosci. 1998;18:720–730. doi: 10.1523/JNEUROSCI.18-02-00720.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O'Leary DD. Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci USA. 1996;93:5584–5589. doi: 10.1073/pnas.93.11.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gean PW, Lin JH. D-2-amino-5-phosphonovaleate blocks induction of long-term depression of the NMDA receptor-mediated synaptic component in rat hippocampus. Neurosci Lett. 1993;158:170–172. doi: 10.1016/0304-3940(93)90256-k. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Huang L, Pallas SL. NMDA antagonists in the superior colliculus prevent developmental plasticity but not visual transmission or map compression. J Neurophysiol. 2001;86:1179–1194. doi: 10.1152/jn.2001.86.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh KH, Wenthold RJ. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-D-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- Knott GW, Quairiaux C, Genoud C, Welker E. Formation of dendritic spines with GABAergic synapses induced by whisker stimulation in adult mice. Neuron. 2002;34:265–273. doi: 10.1016/s0896-6273(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Laube B, Kuhse J, Betz H. Evidence for a tetrameric structure of recombinant NMDA receptors. J Neurosci. 1998;18:2954–2961. doi: 10.1523/JNEUROSCI.18-08-02954.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J Neurosci. 1994;14:3180–3194. doi: 10.1523/JNEUROSCI.14-05-03180.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci. 1998;18:2017–2027. doi: 10.1523/JNEUROSCI.18-06-02017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Luthi A, Schwyzer L, Mateos JM, Gahwiler BH, McKinney RA. NMDA receptor activation limits the number of synaptic connections during hippocampal development. Nat Neurosci. 2001;4:1102–1107. doi: 10.1038/nn744. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RGM. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- McIlhinney RA, Philipps E, Le Bourdelles B, Grimwood S, Wafford K, Sandhu S, Whiting P. Assembly of N-methyl-D-aspartate (NMDA) receptors. Biochem Soc Trans. 2003;31:865–868. doi: 10.1042/bst0310865. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Mu Y, Otsuka T, Horton AC, Scott DB, Ehlers MD. Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron. 2003;40:581–594. doi: 10.1016/s0896-6273(03)00676-7. [DOI] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Lau L-F, Huganir RL. Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Curr Opin Neurobiol. 1998;8:364–369. doi: 10.1016/s0959-4388(98)80062-7. [DOI] [PubMed] [Google Scholar]
- Paupard MC, Friedman LK, Zukin RS. Developmental regulation and cell-specific expression of N-methyl-D-aspartate receptor splice variants in rat hippocampus. Neuroscience. 1997;79:399–409. doi: 10.1016/s0306-4522(96)00677-x. [DOI] [PubMed] [Google Scholar]
- Rabacchi S, Bailly Y, Delhaye-Bouchaud N, Mariani J. Involvement of the N-methyl D-aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science. 1992;256:1823–1825. doi: 10.1126/science.1352066. [DOI] [PubMed] [Google Scholar]
- Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci. 2000;3:238–244. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF. NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron. 2003;38:977–985. doi: 10.1016/s0896-6273(03)00323-4. [DOI] [PubMed] [Google Scholar]
- Schluter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schorge S, Colquhoun D. Studies of NMDA receptor function and stoichiometry with truncated and tandem subunits. J Neurosci. 2003;23:1151–1158. doi: 10.1523/JNEUROSCI.23-04-01151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley S, Roche KW, McCallum J, Sans N, Wenthold RJ. PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron. 2000;28:887–898. doi: 10.1016/s0896-6273(00)00161-6. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Wigstrom H, Gustafsson B. Long-term depression in the hippocampal CA1 region is associated with equal changes in AMPA and NMDA receptor-mediated synaptic potentials. Eur J Neurosci. 1994;6:1055–1057. doi: 10.1111/j.1460-9568.1994.tb00600.x. [DOI] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005a;46:181–189. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Yang G, Kwon E, Gan WB. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 2005b;436:261–265. doi: 10.1038/nature03715. [DOI] [PubMed] [Google Scholar]