

Abstract
Previous studies demonstrated that cross-linking of GM1 ganglioside with multivalent ligands, such as B subunit of cholera toxin (CtxB), induced Ca2+ influx through an unidentified, voltage-independent channel in several cell types. Application of CtxB to undifferentiated NG108-15 cells resulted in outgrowth of axon-like neurites in a Ca2+ influx-dependent manner. In this study, we demonstrate that CtxB-induced Ca2+ influx is mediated by TRPC5 channels, naturally expressed in these cells and primary neurons. Both Ca2+ influx and neurite induction were blocked by TRPC5 small interfering RNA (siRNA). Pretreatment of NG108-15 cells with neuraminidase increased cell-surface GM1 and greatly enhanced the signal. GM1 was not directly associated with TRPC5 but rather with α5β1 integrin, which opened the channel through a signaling sequence after cross-linking of the GM1/integrin complex. This cascade included autophosphorylation of focal adhesion kinase and subsequent activation of phospholipase Cγ (PLCγ) and phosphoinositide-3 kinase [PI(3)K]. Pharmacological blockers that inhibited tyrosine kinase, PLC, and PI(3)K suppressed both CtxB-induced Ca2+ influx and neurite outgrowth. These were also suppressed by SK&F96365, a nonspecific transient receptor potential channel blocker. Confocal immunocytochemistry revealed that GM1 cross-linking induced colocalization of GM1 with these signaling elements in sprouting regions of plasma membrane. In primary cerebellar granular neurons (CGNs), TRPC5 was detected at 2 d in vitro (2 DIV), a stage corresponding to CtxB-stimulated Ca2+ influx. Neurite outgrowth in CGNs, determined at 3 DIV, was accelerated by CtxB and suppressed by TRPC5 siRNA and the above blockers. The crucial role of GM1 was indicated with CGNs from ganglio-series null mice, in which growth of axons was significantly retarded.
Keywords: TRPC5 channel, GM1 ganglioside, calcium signaling, integrin, neuritogenesis, focal adhesion kinase
Introduction
The subgroup of glycosphingolipids containing sialic acid, collectively termed gangliosides, exerts diverse functions through interaction with a large and growing list of proteins. In particular, GM1 ganglioside, the monosialosyl member of the gangliotetraose (“ganglio-series”) family that predominates in the nervous system, has been shown to modulate activity of various membrane-associated enzymes, receptors, ion transporters, integrins, adhesion molecules, etc. (for review, see Yates and Rampersaud, 1998) (Ledeen et al., 1998; Hakomori, 2000; Mocchetti, 2005). Some GM1-modulated proteins are functionally involved in regulation of Ca2+, including transport, homeostasis, and signaling activities of this ion (Ledeen and Wu, 1992, 2002; Wu and Ledeen, 1994). In many cases, GM1 asserts a modulatory effect through direct association with the protein; one example is the high-affinity association of this ganglioside with the Trk protein in facilitating translocation and functional expression of the latter (Mutoh et al., 2002).
A somewhat different, indirect type of modulatory function commences with cross-linking of membrane-bound GM1 by multivalent ligands, such as B subunit of cholera toxin (CtxB), certain antibodies (Abs), and lectins; this leads to channel activation and Ca2+ influx as a downstream event. This property of CtxB was observed in various non-neuronal cells (Dixon et al., 1987; Buckley et al., 1990; Gabellini et al., 1991; Guoy et al., 1994) as well as in some neuronal cell lines (Carlson et al., 1994; Fang et al., 2000) and primary neurons (Milani et al., 1992; Wu et al., 1996). The chief consequence of this induced Ca2+ influx, in the case of N18 and NG108-15 neural cell lines, was outgrowth of axon-like processes (Wu et al., 1998a; Fang, 2000). This mode of Ca2+ influx was initially proposed to involve L-type Ca2+ channels, with GM1 postulated to function as a constitutive negative modulator (Carlson et al., 1994). However, a subsequent study (Fang et al., 2002) revealed the relevant channel to be voltage independent, and the present study demonstrates this to be a TRPC5 channel. TRPC5 is an isoform of the canonical subgroup of mammalian genes homologous to the transient receptor potential (TRP) family in Drosophila (Clapham et al., 2001; Montell, 2004). These ion channels are permeant to cations, with Ca2+ entry as a common and critical feature. TRPC5 was shown to be a negative regulator of neurite length (Greka et al., 2003; Bezzerides et al., 2004), representing a different mechanism at a later stage of neuritogenesis than that revealed in the present study involving TRPC5 activation of incipient neurite sprouting.
Studies have shown that TRPC channels are operated by membrane receptors, including tyrosine kinase receptors that activate intracellular signaling cascades. Our findings reveal that activation of TRPC5 channels by GM1 cross-linking requires α5β1 integrin with which GM1 directly associates. α5β1 integrin responds to GM1 cross-linking by inducing autophosphorylation of its associated focal adhesion kinase (FAK), which in turn activates phospholipase Cγ (PLCγ) and phosphatidylinositol 3-kinase [PI(3)K]. The resulting Ca2+ influx was shown to initiate neurite outgrowth in NG108-15 cells as well as primary cerebellar granule neurons (CGNs). Some of the preliminary results were presented previously (Wu et al., 2004a).
Materials and Methods
Materials.
The following Abs were obtained from the sources indicated: goat anti-TRPC5, rabbit anti-TRPC6, rabbit and goat anti-β1 integrin, rabbit anti-FAK, rabbit anti-PLCγ, rabbit anti-PI(3)K, and rabbit anti-phosphorylated tyrosine 508 in 85 subunit of PI(3)K [p85 PI(3)Kpy508] from Santa Cruz Biotechnology (Santa Cruz, CA); hamster anti-α5 integrin from BD Biosciences (San Diego, CA); rabbit anti-TRPC5 from Alomone Labs (Jerusalem, Israel); rabbit anti-phosphorylated tyrosine 397 in FAK (FAKpy397) from Biosource (Camarillo, CA); PT66 monoclonal mouse anti-phosphorylated tyrosine and agarose beads conjugated with PT66 from Sigma (St. Louis, MO); SMI-31 monoclonal Ab (mAb) against phosphorylated high-molecular-weight neurofilament (pNF-H) from Sternberger (Baltimore, MD); and secondary Abs linked to HRP, FITC, and Texas Red from Jackson ImmunoResearch (West Grove, PA). The following materials were obtained from the sources indicated: RNAeasy kit and QIAquick gel extraction kit from Qiagen (Valencia, CA); One-Step RT-PCR kit, Lipofectamine and Plus reagents, DMEM, PBS, fetal bovine serum (FBS), and other materials for cell culture from Invitrogen (Carlsbad, CA); CtxB and CtxB linked to FITC or HRP from List (Campbell, CA) or Sigma; M-PER mammalian protein extraction reagent from Pierce (Rockford, IL); 1-(6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U73122) and 1-(6-((17b-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-2,5-pyrrolidine-dione (U73343) from Biomol (Plymouth, PA); aluminum-backed silica gel G plate for high-performance thin-layer chromatography (HPTLC) and Permount from Fisher Scientific (Pittsburgh, PA); Blue BIO film from Denville Scientific (Edison, NJ); ECL reagent from Amersham Biosciences (Piscataway, NJ); Complete protease inhibitor cocktail tablet from Roche (Mannheim, Germany); and Brij 98, neuraminidase (N′ase, Clostridium perfringens, type V), 1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole·HCl (SK&F96365), 2-(morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), wortmannin, genistein, ATP, phosphatase inhibitor cocktail 2, and other chemicals from Sigma.
Cell culture.
Three neuroblastoma cell lines were used: NG108-15 (neuroblastoma × glioma hybrid), NG-CR72 (a mutant of the former that lacks GM1 synthase) (Wu et al., 2001), and Neuro-2A. They were grown as described previously (Wu et al., 1998a, 2001). NG108-15 cells were differentiated with N′ase (0.2 U/ml) plus CtxB (5 μg/ml) or with KCl (30 mm) plus dibutyryl cAMP (db-cAMP; 1 mm); both treatments stimulated outgrowth of axon-like neurites in these cells (Wu et al., 1998a). To test involvement of tyrosine phosphorylation, PLC, PI(3)K, and the TRPC5 channel in CtxB-induced neuritogenesis, tyrosine kinase inhibitor genistein, PLC blocker U73122 (and its noneffective analog, U73343), PI(3)K inhibitors wortmannin and LY294002, and TRP channel blocker SK&F96365 were individually applied. After 48–72 h of treatment, neurite outgrowth was quantified as percentage of neurite-bearing cells (Wu et al., 1998a); neurites were defined as processes of length more than twofold the soma diameter Counting included 200–300 cells in several randomly chosen fields of each well, using three wells per experiment (n = 1). Data were collected from three independent experiments, and statistical difference was assessed with the two-tailed Student's t test.
Primary cultures of cerebellar granular neurons (CGNs) were prepared as described previously (Wu et al., 1996, 2004b). Cells were dissociated from 7-d-old pup cerebella of normal and ganglio-series null [GM2/GD2 synthase knock-out (KO)] C57BL/6 mice. Some cells were transfected with DNA cassettes encoding TRPC5 small interfering RNA (siRNA) (0.2 μg/104 cells) by using Lipofectamine and Plus reagents (see below) before they were seeded. On the second day in vitro (2 DIV) before process outgrowth, cells were replaced in fresh DMEM containing N2 supplement and 1% FBS and treated with CtxB (5 μg/ml) in the presence or absence of the above inhibitors. They were photographed on 3 DIV, and the length of the longest neurite (axon) in each granular neuron was measured by UltraView software (PerkinElmer, Wellesley, MA). A total of ≥300 cells in three independent cultures was measured, and the data were statistically analyzed with GraphPad (San Diego, CA) Prism statistics software; differences were determined by one-way ANOVA with Dunnett's post-test. The term DIV represents the initial day of plating (day 1) plus additional days in culture.
Expression and knock-down of TRPC5 gene.
Gene expression of the TRPC family was analyzed with reverse transcription (RT)-PCR. Total RNA was extracted, using an RNAeasy kit, from undifferentiated and KCl/db-cAMP differentiated NG108-15 cells, undifferentiated NG-CR72 cells, and Neuro-2A cells. During the extraction, DNase was used to remove residual DNA. RT-PCR was performed following previous protocol (Xie et al., 2002) using the One-Step RT-PCR kit and appropriate primers (Table 1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for comparison. The resulting DNA products were resolved on a 1% agarose gel containing ethidium bromide, isolated from the gel with the QIAquick gel extraction kit, and sequenced in the Molecular Resource Facility of this institute. The resulting sequences were identified by the Basic Local Alignment Search Tool (BLAST) protocol from the National Institutes of Health–National Center for Biotechnology Information (NIH–NCBI) website.
Table 1.
Primers for TRPC mRNA
| Gene and direction | Primer sequence | Base pairs | Product identitya or accession numberb |
|---|---|---|---|
| TRPC1 forward | ATGGGACAGATGTTACAA | 400 | NM_011643.1a |
| TRPC2 forward | CGCTGGGCACTCTGCAGA | 370 | U40981b |
| TRPC3 forward | AGGACATATTCAAGTTCA | 340 | NM_019510.1a |
| TRPC4 forward | CAGATATCTCTGGGAAGGATG | 400 | NM_016984b |
| TRPC5 forward | TTCTCTTTATCTACTGCC | 360 | NM_009428.1a |
| TRPC6 forward | TTCATGGTCATATTCATC | 320 | NM_013838.1a |
| All TRPC reverse | TGGAGCRAAYTTCCAYTC (R = G or A and Y = C or T) | ||
| GAPDH | |||
| Forward | ACCACAGTCCATGCCATCAC | 450 | NM_199472b |
| Reverse | TCCACCACCCTGTTGCTGTA |
All primers are referenced to Tesfai et al. (2001).
aIdentity numbers of product sequences for isoforms detected in cells, which were obtained from the NIH–NCBI website using the BLAST protocol.
bAccession numbers for isoforms absent in cells, from which primers were designed.
To suppress TRPC5 expression in NG108-15 cells, four DNA cassettes containing a U6 promoter and a sequence encoding designated siRNA (Table 2) were synthesized by GenScript (Scotch Plans, NJ). Each cassette (0.2 μg) was transfected into ∼104 NG108-15 cells by using Lipofectamine and Plus reagents according to the manufacturer's instructions. As a control, similar transfection was performed with an siRNA mixture against TRPC6 (sc-42673; Santa Cruz Biotechnology); the precise composition of this mixture was not revealed by the supplier. Twenty-four hours after transfection, cells were reseeded onto plates or glass coverslips in regular culture medium and allowed to grow for another 24–48 h (total time of siRNA treatment was 48–72 h). Inhibition of TRPC5 or TRPC6 by siRNA was confirmed by RT-PCR (see above), immunocytochemistry (IC) (see below), and immunoblotting (IB) (see below).
Table 2.
siRNA sequence against mouse TRPC5 gene (accession number NM_009428.1)
| siRNA sequence | Start point | |
|---|---|---|
| 1 | GTCGTCGAGCCAACGGTCCT | 2895 |
| 2 | GCTTCTCAACATATCGTCAG | 1552 |
| 3 | CCAACAATTGCTAGCCACCT | 1323 |
| 4 | TGAGTGGAAGTTTGCGAGAA | 2313 |
Specificity of siRNAs was identified by the BLAST protocol from the NIH–NCBI website.
Intracellular calcium ([Ca2+] i) measurement and electrophysiological study.
[Ca2+]i measurements were performed as described previously (Fang et al., 2000, 2002) and applied to the following: (1) undifferentiated NG108-15 cells untransfected or transfected with TRPC5 or TRPC6 siRNA; (2) NG108-15 cells differentiated in KCl/db-cAMP; (3) undifferentiated NG-CR72 cells; and (4) undifferentiated Neuro-2A cells. Cells pretreated with N′ase (0.5 U/ml) or GM1 (20 μm; NG-CR72 only) at 37°C for 2 h were loaded with 5 μm fura-2 AM and 0.25 mm sulfinpyrazone for 30 min. Aliquots of 1–1.5 × 105 cells were suspended in 4-morpholinepropanesulfonic acid (20 mm, pH 7.2)-buffered physiological saline solution (PSS) consisting of 140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm glucose, 0.25 mm sulfinpyrazone, and 1% bovine serum albumin. Measurements were run 600–800 s, and Ca2+ (5 mm), CtxB (5 μg/ml), and ATP (2 mm) were applied at designated times. [Ca2+]i levels were expressed as the fluorescent ratio between the two excitation wavelengths (R340/380). For inhibition studies, cells in suspension were preincubated 15–30 min with the indicated inhibitor (see above) in PSS containing 2 mm Ca2+ and replaced in Ca2+-free PSS for [Ca2+]i determination.
The inward current in NG108-15 cells was recorded according to previously described procedures (Obukhov and Nowycky, 2004, 2005). Cells that were vehicle transfected or transfected with TRPC5 siRNA were grown on coverslips for 48 h. They were treated with N′ase for 2 h and replaced in a solution containing (in mm) 10 HEPES, pH 7.2, 150 NaCl, 1.2 CaCl2, 1 MgCl2, and 10 glucose. Cells were patched with an electrode filled with an intracellular solution containing (in mm) 20 HEPES, pH 7.2, 135 Cs-methylSO3, 10 CsCl, 1 MgCl2, and 0.5 EGTA. Whole-cell currents were recorded using an Axopatch 200B integrating patch-clamp amplifier and the episodic mode of pClamp 9 software. Data were sampled at 1–5 kHz with a Digidata 1322A 16-bit data acquisition system. Every 3 s, currents recorded at −60 mV were averaged over 1 s intervals, followed by a 150 ms voltage ramp from −100 to +100 mV to determine the current–voltage relationship.
Immunocytochemistry.
Colocalization of GM1 with functionally related molecules was studied in NG108-15 cells. Cells grown on coverslips were pretreated with N′ase (0.5 U/ml) in culture at 37°C for 2 h and subjected to CtxB–FITC (5 μg CtxB/ml) in ice-cold Ca2+-free Hanks' solution for 15 min. Some were fixed immediately, and others were replaced in fresh Hanks' solution at 37°C for 15 min before fixation. This was followed by IC staining with various Abs according to protocols we used previously (Wu et al., 1998a). Primary Abs included rabbit anti-TRPC5 (1:50), rabbit anti-β1 integrin (1:100), hamster anti-α5 integrin (1:200), mouse PT66 monoclonal anti-phosphorylated tyrosine (1:500), mouse anti-FAKpy397 (1:200), and rabbit anti-p85 PI(3)Kpy508 (1:200); corresponding secondary Abs were conjugated with Texas Red. Cells were observed with a Radiance2100MP two-photon confocal microscope (Bio-Rad, Hercules, CA) with a 60× lens; Z-sections close to the bottom surface of the cells were photographed. The colocalization index of each molecule with GM1 was measured in 4–10 images with UltraView software (PerkinElmer) and expressed as percentage relative to GM1. Similar colocalization of GM1 and TRPC5 was also performed with CGNs at 2 and 5 DIV. In addition, CGNs at 3 DIV were stained with SMI-31 mAb (1:1000) against pNF-H to identify axons (Sternberger and Sternberger, 1983).
To detect knock-down of TRPC5 or TRPC6 protein, NG108-15 cells transfected with or without siRNA were seeded onto coverslips and allowed to grow 1–2 d. After fixation and permeabilization, cells were incubated overnight with rabbit polyclonal Ab against TRPC5 or TRPC6 (1:100), followed by goat anti-rabbit Ab conjugated with HRP. Cells were developed with diaminobenzidine/H2O2, dehydrated in alcohol/xylene gradient, and mounted in Permount (Wu et al., 1995).
Immunoprecipitation and IB.
To determine association of GM1 with TRPC5 or α5β1 integrin, cells were treated with N′ase (0.5 U/ml) for 2 h. After washing two times with cold PBS, cells were scraped into cold lysis buffer consisting of 1% Brij 98, 25 mm HEPES, pH 7.5, 150 mm NaCl, and 5 mm EDTA plus protease inhibitor mixture (Kawakami et al., 2002). This was agitated 20 min at 4°C, homogenized well, and centrifuged at 15,000 × g for 10 min. Approximately 100 μl of supernatant containing ∼300 μg of protein was precleared with 50 μl of agarose beads for 2 h at 4°C. After brief centrifugation at 1300 × g for 5 min, the supernatant was transferred to a fresh vial and incubated with primary Ab overnight at 4°C, followed by the addition of 30 μl of protein A–agarose beads at room temperature for 3 h. After centrifugation at 1300 × g for 5 min, bead sediment was washed two times with cold PBS and boiled in 50 μl of sample buffer. Precipitation using protein A–agarose beads alone was done in parallel as a negative control. Precipitates were separated by SDS-PAGE with a 7% nonreducing gel and electrophoretically transferred to polyvinylidene difluoride membrane. Blotting was performed with CtxB–HRP (1 μg CtxB/ml) or primary Ab followed by secondary Ab conjugated to HRP as described previously (Xie et al., 2002). The Ab combinations used for immunoprecipitation (IP)/IB were as follows: goat anti-TRPC5 (1:100)/rabbit anti-TRPC5 (1:50); hamster anti-α5 integrin (1:200)/goat anti-β1 integrin (1:250); goat anti-β1 integrin (1:100)/hamster anti-α5 integrin (1:200) Ab. Blots were visualized on Blue BIO film using ECL reagent. The precipitates with anti-α5 or -β1 integrin Ab were also extracted with methanol/chloroform (1:1, v/v) for 1 h at room temperature for GM1 determination. The lipid extract was evaporated to dryness under nitrogen stream and applied to an HPTLC plate that was developed in chloroform/methanol/aqueous KCl (2 m) (50:40:10, v/v/v), followed by N′ase treatment and CtxB–HRP overlay as described previously (Wu and Ledeen, 1988). Gangliotetraose bands were visualized using ECL reagent on film.
To determine activation (tyrosine phosphorylation) of the integrin signaling pathway after GM1 cross-linking by CtxB, NG108-15 cells treated with N′ase were reacted with CtxB (5 μg/ml) for 0, 5, 10, and 15 min. Cells were washed two times in PBS and lysed in M-PER mammalian protein extraction reagent containing mixtures of protease and phosphatase inhibitors at room temperature for 5 min. After centrifugation at 13,000 × g, supernatant containing ∼300 μg of protein was immunoprecipitated with 50 μl of agarose beads linked with PT66 mAb against phosphorylated tyrosine and subjected to IB analysis as above. Abs used for IB included goat anti-β1 integrin (1:250), rabbit anti-PLCγ (1:200), and goat anti-FAK (1:200). In addition, tyrosine phosphorylation of FAK and PI(3)K was directly detected in the lysate (50–100 μg of protein separated on a 7% polyacrylamide nonreducing gel) without IP by using anti-FAKpy397 (1:100) and anti-p85 PI(3)Kpy508 (1:200) Abs. Direct IB was also performed to detect TRPC5 protein with goat anti-TRPC5 Ab (1:100) in NG108-15 cells treated with TRPC5-specific siRNA for 48 h.
Results
CtxB-induced Ca2+ influx and neurite initiation are mediated by TRPC5 channels
[Ca2+]i changes resulting from CtxB-induced cross-linking of GM1 were determined by fura-2 spectrofluorimetry. Our previous studies showed that such [Ca2+]i elevation in undifferentiated NG108-15 cells is greatly enhanced by N′ase pretreatment, because of GM1 elevation in the plasma membrane (Fang et al., 2000, 2002); hence, N′ase-treated NG108-15 cells were used in this study. A typical response involving elevation of [Ca2+]i after CtxB application is shown in Figure 1A. CtxB failed to alter [Ca2+]i in NG-CR72 (lacking GM1) and Neuro-2A (lacking TRPC5) cells; bath incubation of NG-CR72 cells with exogenous GM1 resulted in partial restoration of the signal. CtxB exerted only a limited effect on differentiated NG108-15 cells. The fact that CtxB induced virtually no [Ca2+]i elevation in Ca2+-free buffer (Fig. 1B) indicated Ca2+ influx as the major contributor to [Ca2+]i elevation. Dose-dependent blockage of the CtxB effect in undifferentiated NG108-15 cells with SK&F96365, a general inhibitor of TRP channels (Fig. 1C), suggested the functional channel to be of the TRP type. SK&F96365 also inhibited CtxB-induced neurite outgrowth in these cells; after 48 h treatment with 50 μm SK&F96365, neurite-bearing cells were reduced from 55.4 ± 6.1 to 24.3 ± 4.1% of total cells, and with 100 μm SK&F96365 to 15.3 ± 4.3% (n = 3; p < 0.01). Because the TRPC family is expressed in developing neurons, RT-PCR was performed to identify which of these isoforms is expressed in undifferentiated NG108-15 cells; these proved to be TRPC1, TRPC5, and TRPC6 (Fig. 2A). These products were verified by sequencing, and the BLAST identification numbers based on these sequences are indicated in Table 1. In contrast, differentiated NG108-15 cells had decreased TRPC5, along with the disappearance of TRPC6 and slight elevation in TRPC1 compared with the undifferentiated cells (Fig. 2B). Undifferentiated NG-CR72 cells showed the same pattern as undifferentiated NG108-15 cells (Fig. 2C). Neuro-2A cells, which did not respond to CtxB, expressed TRPC3 and TRPC6 but not TRPC5 (Fig. 2D). These RT-PCR products were also verified by sequencing (Table 1). These results point to TRPC5 as the likely channel responsible for the CtxB effect in undifferentiated NG108-15 cells. Additional evidence came from IP/IB and IC analyses showing reduction in the TRPC5 protein after differentiation (see below).
Figure 1.

CtxB-induced Ca2+ influx in neuroblastoma cells. After N′ase treatment, NG108-15 and Neuro-2A cells were subjected to [Ca2+]i measurement with fura-2 indicator, monitored ratiometrically (R340/380) first in Ca2+-free PSS, followed by addition of Ca2+ (5 mm) and CtxB (5 μg/ml). A, CtxB induced [Ca2+]i elevation in undifferentiated NG108-15 cells but not NG-CR72 cells (lacking GM1 and not treated with N′ase); bath incubation of NG-CR72 cells with GM1 (20 μm) induced partial response. Neuro-2A cells, which contain GM1 but lack TRPC5, did not respond. B, Compared with undifferentiated (Undiff.) NG108-15 cells, CtxB induced much smaller [Ca2+]i elevation in differentiated (Diff.) cells. No [Ca2+]i increase occurred in the absence of extracellular Ca2+. C, Dose-dependent inhibition of CtxB-induced Ca2+ influx by SK&F96365 in NG108-15 cells. Each trace in A–C is an average of three independent runs.
Figure 2.

Gene expression of TRPC isoforms. RNA extracted from indicated cells was subjected to RT-PCR to determine TRPC1–TRPC6 gene expression; primers of each gene are listed in Table 1. M, Oligonucleotide marker; G, GAPDH; N, negative control (without template RNA). A, TRPC1, TRPC5, and TRPC6 were detected in undifferentiated (Undiff.) NG108-15 cells. B, In differentiated (Diff.) NG108-15 cells, TRPC5 decreased, TRPC6 disappeared, and TRPC1 slightly increased. C, NG-CR72 cells (undifferentiated) showed the same pattern as undifferentiated NG108-15. D, Neuro-2A cells, which failed to respond to CtxB, expressed TRPC3 and TRPC6 but not TRPC5.
Using the “knock-down” strategy, four different siRNA fragments specific for TRPC5 were transfected into NG108-15 cells to diminish expression of TRPC5. RT-PCR analysis revealed that three of these siRNAs (1, 2, and 4) (Table 2) inhibited expression of TRPC5 mRNA only and one (3) deleted both TRPC1 and TRPC5 in cells treated for 48 h (Fig. 3A). This was consistent with IB and IC evidence showing blocked expression of TRPC5 protein 48 h after transfection (Fig. 3B,Ca,Cc). Partial recovery of TRPC5 expression was found for both protein (Fig. 3Cb) and mRNA (data not shown) at 72 h after transfection. Using NG108-15 cells transfected with TRPC5 siRNA for 48 h, CtxB-induced Ca2+ influx determined by fura-2 was essentially eliminated (Fig. 4A). When TRPC5 currents were measured directly by whole-cell patch clamping (Fig. 4B) in N′ase-pretreated NG108-15 cells, CtxB application induced a small inward current at −60 mV that activated over ∼2 min and deactivated very slowly over several minutes (Fig. 4Ba). The current–voltage relationship elicited by voltage ramps from −100 to +100 mV was strongly outwardly rectifying (Fig. 4Bb). All inward currents were eliminated by substitution of NMDG+ for extracellular Na+, indicating that the current was carried by a nonspecific cation channel (data not shown). The kinetics of activation and deactivation are reminiscent of TRPC5 properties in transfected HEK293 cells (Obukhov and Nowycky, 2004) and correspond well to the kinetics of the fura-2 data (Figs. 1, 4A). The current–voltage relationship resembles previously published data from both mouse and rat TRPC5 at specific stages of the activation–deactivation cycle (Plant and Schaefer, 2003; Obukhov and Nowycky, 2004, 2005). However, a slight contribution of TRPC1–TRPC5 heteromers to the I–V shape cannot be excluded (Strubing et al., 2001; Obukhov and Nowycky, 2005). The inward current was significantly reduced in cells transfected with TRPC5 siRNA (Fig. 4Bc,Bd). TRPC5 knock-down after 24 h also inhibited neurite outgrowth induced by CtxB in N′ase-treated cells, as seen for siRNA #1 and #2 (Fig. 4Cb,Cc). The percentage of neurite-bearing cells was significantly reduced after treatment with each of the four siRNAs (Fig. 4Ce). This was in contrast to neurite outgrowth induced by KCl/db-cAMP that promoted Ca2+ influx through L-type voltage-operated Ca2+ channels and was not significantly affected by siRNA (Fig. 4Cd). On the other hand, NG108-15 cells transfected with TRPC6 siRNA, which showed loss of TRPC6 but not TRPC5 expression (Fig. 3D), showed no inhibition of CtxB-induced Ca2+ influx (Fig. 4A) or neurite outgrowth (neurite-bearing cells, 44.2 ± 4.7% vs vehicle-transfected cells, 45.4 ± 6.7%).
Figure 3.
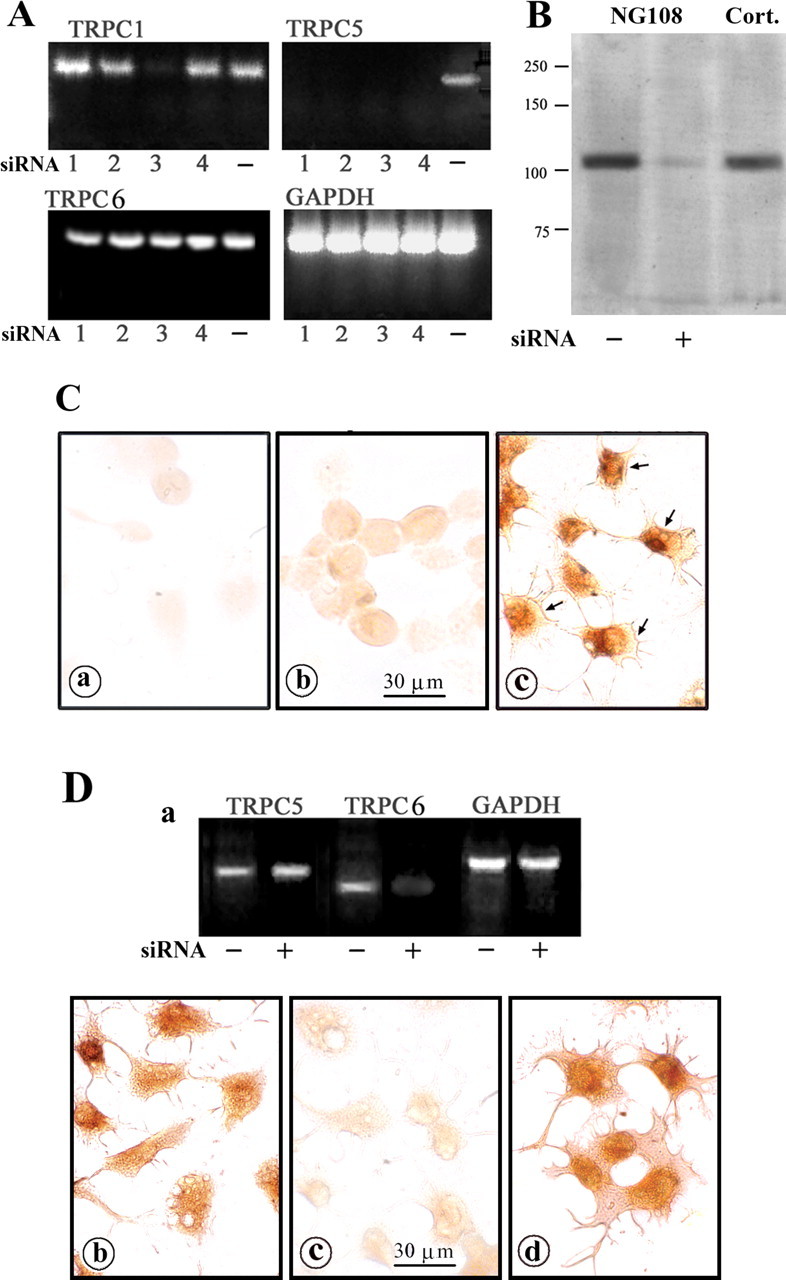
Inhibition of TRPC5 expression by siRNA in NG108-15 cells. Four designated TRPC5 siRNA sequences (Table 2) were transfected into undifferentiated NG108-15 cells; control transfection used TRPC6 siRNA sequences. A, RT-PCR assay 48 h after transfection. siRNA 1, 2, and 4 sequences knocked down TRPC5 only, whereas siRNA 3 reduced both TRPC5 and TRPC1. TRPC6 was not affected by this siRNA treatment. The designation (−) indicates vehicle transfection. B, Immunoblot 48 h after transfection with TRPC5 siRNA 2, showing inhibition of TRPC5 protein synthesis. Cort., Positive control with protein extract from cortical tissue of a 7-d-old rat pup. C, IC images in cells transfected with TRPC5 siRNA 2. After 48 h (a) and 72 h (b) treatment, cells were stained with anti-TRPC5 Ab and HRP-linked secondary Ab. TRPC5 expression was inhibited 48 h after transfection and partially recovered 72 h after transfection. Cells treated with vehicle alone (c) expressed TRPC5 in both cytosol and plasma membrane (arrows). D, NG108-15 cells were treated with TRPC6 siRNA for 48 h and subjected to RT-PCR, showing selective loss of TRPC6 mRNA (a). b, c, TRPC6 siRNA-treated cells were strained with TRPC5 Ab (b) and TRPC6 Ab (c), which verified TRPC6 knock-down. d, Vehicle-transfected cells were stained with TRPC6 Ab.
Figure 4.

Suppression of CtxB-induced Ca2+ influx and neuritogenesis by TRPC5 knock-down. Undifferentiated NG108-15 cells were transfected with siRNA targeting TRPC5 or TRPC6 as a control; some cells were transfected with vehicle alone. A, Cells were treated with N′ase, and [Ca2+]i changes were determined at 48 h after transfection, as described in Figure 1. CtxB-induced Ca2+ influx was nearly totally inhibited by all four TRPC5 siRNAs but unaffected by TRPC6 siRNA. B, Whole-cell current recordings at 48 h after transfection. Cells were treated with N′ase, and inward currents were recorded with a membrane potential held at −60 mV. a, Typical traces induced by CtxB (5 μg/ml) in vehicle-transfected cells. Traces resemble the slow deactivation (or inactivation) lasting ≥1 min and asymmetrical current–voltage relationships of TRPC5 channels recorded in overexpression systems (Obukhov and Nowycky, 2004). b, Two current traces elicited by rapid voltage ramps from −100 to +100 mV taken at times indicated in a. The inward current recorded in TRPC5 siRNA-transfected cells is shown in c. d, Average values (±SEM; n = 5) of peak current activated by CtxB, in which data are normalized to the surface area of the cells and averaged; the current in TRPC5 siRNA-treated cells was significantly suppressed (p < 0.05, two-tailed Student's t test). C, Neuritogenic assay. After 24 h TRPC5 siRNA treatment, cells were reseeded and treated with N′ase/CtxB (a–c) or KCl/db-cAMP (d) for another 24 or 48 h. b, c, Blockade of CtxB-induced neurite outgrowth by siRNA 1 and 2 compared with vehicle-transfected cells (a). The same siRNA 2 treatment did not affect KCl/db-cAMP-induced neuritogenesis (d). e, Quantification of CtxB-induced neurite outgrowth in the absence and presence of all four TRPC5 siRNAs. Data are averages (±SD) of three independent experiments; #p < 0.001 versus CtxB untreated; *p < 0.05 and **p < 0.01 versus CtxB-treated, siRNA untreated cells (2-tailed Student's t test).
These results demonstrate that neuritogenesis induced by CtxB/GM1 interaction was mediated by Ca2+ influx through TRPC5 channels. The crucial role of GM1 was illustrated with NG-CR72 cells that lack GM1 and other a-series gangliosides (Wu et al., 2001) and did not respond to CtxB with Ca2+ uptake (Fig. 1A), although these mutant cells possess the same TRPC isoforms as NG108-15 cells (Fig. 2C). As indicated above, supplementation of NG-CR72 cells with exogenous GM1 resulted in partial restoration of CtxB-promoted [Ca2+]i elevation (Fig. 1A).
Membrane association of GM1 with α5β1 integrin but not TRPC5
To determine whether GM1 in the plasma membrane associates directly with TRPC5, IP/IB was performed with anti-TRPC5 Ab in N′ase-pretreated NG108-15 cells. GM1 did not coprecipitate (Fig. 5A), indicating absence of such association. This was supported by confocal IC analysis (see below). Parallel IP/IB analyses were performed with anti-α5 and anti-β1 integrin. These showed that in addition to its partner isoform (Fig. 5Ba), both anti-α5 and anti-β1 Abs coprecipitated GM1, as seen in migration of the latter with the SDS-PAGE solvent front (Fig. 5Bb). This was confirmed by HPTLC after extraction of GM1 (along with other lipids) from the immunoprecipitates with chloroform–methanol (Fig. 5C). The specific HPTLC overlay method using CtxB (Wu and Ledeen, 1988) applied to these lipid extracts permitted clear identification of GM1.
Figure 5.

Coimmunoprecipitation of GM1 with α5β1 integrin but not TRPC5. A, N′ase-treated undifferentiated (Undiff.) and differentiated (Diff.) NG108-15 cells were lysed in 1% Brij 98 and precipitated with rabbit anti-TRPC5 Ab plus protein A–agarose beads. a, b, IB was performed with goat anti-TRPC5 Ab plus secondary Ab linked with HRP (a) or CtxB–HRP (b). Consistent with the RT-PCR result (Fig. 2), TRPC5 expressed in undifferentiated cells was depressed after differentiation. CtxB–HRP blot failed to detect coprecipitation of GM1 with TRPC5. B, The above lysate from undifferentiated cells was subjected to IP with anti-β1 or anti-α5 integrin Ab and IB with Ab against partner integrin plus HRP-linked secondary Ab (a) or CtxB–HRP (b). The latter blot showed GM1 in the migration front (arrow), indicating coprecipitation of GM1 with both integrins. Bands at ∼250 kDa in Ba represent α5β1 dimers. C, The precipitates were also extracted with chloroform/methanol (1:1, v/v), and the extracts were subjected to HTPLC and reaction with CtxB–HRP after N′ase treatment on the plate (Wu and Ledeen, 1988). PM, Lipids from the plasma membrane of NG108-15 cells; BBG, bovine brain gangliosides mixture; none, IP with beads alone. These results provide evidence for GM1 association with α5β1 integrin but not TRPC5.
Confocal IC analysis applied to NG108-15 cells in culture also suggested non-association of GM1 with TRPC5. Both before and after GM1 cross-linking by CtxB (4°C vs 37°C), GM1 was primarily in the plasma membrane, including what appeared to be regions of incipient sprouting (Fig. 6a,a′), whereas TRPC5 was mainly in the cytosol with some expression in plasma membrane (Fig. 6b,b′). The latter showed a modest increase after GM1 cross-linking but did not appear to coincide with GM1. With regard to integrin, cross-linking enhanced colocalization of both α5 (Fig. 6e,e′) and β1 (Fig. 6h,h′) with GM1 in the plasma membrane. Colocalization indices before and after GM1 cross-linking were as follows: α5 integrin, 74.3 and 78.6%; β1 integrin, 75.2 and 86.7% (Table 3). These results demonstrate membrane association of GM1 with α5β1 integrin.
Figure 6.

Colocalization of GM1 with α5β1 integrin. Undifferentiated NG108-15 cells treated with N′ase were incubated with CtxB–FITC (5 μg B unit/ml) in Ca2+-free buffer (4°C) for 15 min. One portion was fixed with paraformaldehyde, and another was incubated in the same buffer at 37°C for an additional 15 min to cross-link GM1, followed by fixation. Cells were stained with Ab against TRPC5, α5 integrin, or β1 integrin plus secondary Ab linked to Texas Red. Confocal images showed localization of GM1 in the plasma membrane at 4°C (a, d, g), which was enhanced at that locus with suggested sprouting after cross-linking at 37°C (a′, d′, g′). b, b′, TRPC5 was expressed throughout the cell body with a small portion in the plasma membrane. TRPC5 distribution appeared distinct from GM1. In contrast, α5 integrin (e vs e′) and β1 integrin (h vs h′) were recruited into same membrane regions as GM1 after cross-linking.
Table 3.
Colocalization of TRPC5, α5β1 integrin, and related signaling molecules with GM1 in NG108-15 cells
| Percentage relative to GM1 |
||
|---|---|---|
| Before cross-linking | After cross-linking | |
| TRPC5 | 9.7 ± 1.5 (4) | 17.2 ± 2.3 (7) |
| α5 integrin | 74.3 ± 4.9 (6) | 78.6 ± 5.6 (10) |
| β1 integrin | 75.2 ± 9.4 (4) | 86.7 ± 6.2 (10) |
| Phosphotyrosine | 38.0 ± 4.2 (5) | 79.7 ± 6.9 (7) |
| FAKpy397 | 48.5 ± 9.4 (6) | 83.0 ± 6.2 (6) |
| P85 PI(3)Kpy508 | 31.0 ± 6.9 (5) | 80.1 ± 6.0 (5) |
Cells were doubly stained with CtxB–FITC and indicated Abs plus secondary Abs conjugated with Texas Red, and confocal images were taken as described in Figures 6 and 7. Immunocolocalization was analyzed with UltraView, taking the GM1-positive region as 100%. Data are the average (±SEM) from the image number in parentheses.
Activation of integrin signaling cascade by CtxB
To determine whether CtxB activation of TRPC5 channels proceeds via integrin-controlled signaling, we focused on activation of an integrin-coupled nonreceptor tyrosine kinase, FAK, and its downstream effectors. NG108-15 cells were subjected to IP with PT66 mAb against phosphotyrosine, and IB analysis was performed using Abs to FAK, PLCγ, and β1 integrin. This revealed a significant increase in tyrosine phosphorylation in FAK and PLCγ and a less pronounced increase in β1 integrin phosphorylation after 10–15 min exposure to CtxB (Fig. 7A). Direct IB analysis using site-specific phosphorylation Abs demonstrated that both FAKpy397 and p85 PI(3)Kpy508 were significantly increased after 10–15 min of CtxB treatment (Fig. 7B). This was supported by IC confocal images showing tyrosine phosphorylation (Fig. 7C, top row) and, more specifically, tyrosine of FAKpy397 (Fig. 7C, middle row) and p85 PI(3)Kpy508 (Fig. 7C, bottom row); the latter two were recruited to the same membrane regions in which GM1 aggregated (colocalization indices, ∼ 80%) (Table 3). Because these experiments were performed in the absence of extracellular Ca2+, activation of integrin signaling appeared to precede Ca2+ influx through TRPC5 channels.
Figure 7.

Activation of FAK, PLCγ, and PI(3)K and colocalization with GM1. A, IP/IB evidence. N′ase-treated NG108-15 cells were reacted with CtxB for up to 15 min, and proteins were extracted with M-PER reagent. IP was performed with mouse anti-phosphotyrosine PT66 mAb linked to agarose bead and IB with Ab against β1 integrin (a), PLCγ (b), and FAK (c), showing an increase in tyrosine phosphorylation in these proteins over time. B, Direct IB evidence. The above lysate was directly blotted (without IP) after SDS-PAGE and electrophoretic transfer, with phospho-specific Ab against FAKpy397 (a) and P85 PI(3)Kpy508 (b), showing an increase over time after GM1 cross-linking. C, Colocalization of tyrosine-phosphorylated proteins, FAKpy397, and p85 PI(3)Kpy508 with GM1. NG108-15 cells were treated with CtxB–FITC in Ca2+-free buffer as described in Figure 6, and IC was performed with Ab against phosphotyrosine (top), FAKpy397 (middle), and P85 PI(3)KIPY508 (bottom) plus secondary Ab linked to Texas Red. These confocal images show a significant increase in membrane levels of the tyrosine-phosphorylated proteins that colocalized with GM1 after CtxB-induced cross-linking.
To determine whether PLCγ, PI(3)K, and protein tyrosine kinase were involved in activation of TRPC5 channels leading to neurite outgrowth, pharmacological blockers for tyrosine kinase (genistein), PLC (U73122), and PI(3)K (wortmannin, LY294002) were used. These widely used inhibitors were reported to show specificity for the indicated reactions (Akiyama et al., 1987; Bleasdale et al., 1990; Powis et al., 1994; Vlahos et al., 1994; Laurino et al., 2005; Li et al., 2005; Jia et al., 2007). In some cases, ATP, known to activate TRPC5 channels via PLC through stimulation of G-protein-controlled signaling (Shimizu et al., 2006), was also applied subsequent to CtxB. Genistein at 100 μm or U73122 at 1 μm inhibited both CtxB- and ATP-stimulated [Ca2+]i responses (Fig. 8Aa). CtxB-induced [Ca2+]i elevation was also effectively blocked by PI(3)K inhibitors wortmannin (at 2.5 μm) and LY294002 (at 3 μm) (Fig. 8Ab). Significantly, CtxB-induced neuritogenesis was also effectively blocked by these inhibitors at the same doses (Fig. 8B); U73343, an inactive analog of U73122, produced no inhibition. These results support a sequence beginning with activation of α5β1 integrin by cross-linking of integrin-associated GM1, followed by activation of FAK and downstream PLCγ and PI(3)K, leading to opening of TRPC5 channels. The resulting elevation of [Ca2+]i is seen as triggering neurite outgrowth, previously shown to be axonogenic in nature (Wu et al., 1998a).
Figure 8.

Blockade of CtxB activities by inhibitors of signaling reactions. A, Ca2+ influx. N′ase-treated NG108-15 cells were incubated with indicated inhibitors and subjected to [Ca2+]i measurement. In a, ATP (2 mm) was applied after CtxB (5 μg/ml), showing that tyrosine kinase inhibitor genistein (100 μm) and PLC inhibitor U73122 (1 μm) inhibited Ca2+ influx induced by both agents. b, PI(3)K inhibitors, wortmannin (2.5 μm) and LY294002 (3 μm), also suppressed CtxB reaction. B, Neuritogenesis. NG108-15 cells were stimulated with N′ase plus CtxB for 48 h in the presence or absence of the indicated inhibitor at the same concentration as above; U73343, an inactive analog of U73122, was also used. Quantification results in e show that all reagents, except U73343, inhibited neurite formation stimulated by CtxB. Data are averages (±SD) of three independent experiments (n = 3). #p < 0.001 versus CtxB-untreated (control) cells; *p < 0.01 versus cells treated with CtxB alone (2-tailed Student's t test).
Acceleration of axon outgrowth in primary CGNs by cross-linking of GM1
Our previous study of CGNs in primary culture showed a neurotrophic effect of CtxB leading to enhanced survival related to [Ca2+]i elevation (Wu et al., 1996). In keeping with a previous study (Carlson et al., 1994), this was initially viewed as reflecting Ca2+ influx through L-type channels. However, this interpretation was subsequently modified when channel activity of this type in other cells was shown to be voltage independent (Fang et al., 2002). The present results extend this conclusion by indicating TRPC5 as the operative channel. In the present study, 1 d after seeding CGNs (2 DIV), before process outgrowth, GM1 was concentrated in the plasma membrane (Fig. 9a), and TRPC5 was dispersed throughout the cell body (Fig. 9b). TRPC5 localization changed radically after initiation of process outgrowth (5 DIV), expression now occurring only in developing neurites (Fig. 9d). At that time, GM1 continued to show strong expression in the plasma membrane of soma and processes (Fig. 9c). Similar developmental change of TRPC5 involving depletion of that protein from the soma was seen in NG108-15 cells differentiated with KCl/db-cAMP (Fig. 9f,h), whereas GM1 was elevated in both loci (Fig. 9e,g).
Figure 9.

Expression of GM1 and TRPC5 in primary CGNs and NG108-15 cells. a–d, CGNs grown 2 and 5 DIV were costained with CtxB–FITC (a, c) and anti-TRPC5 Ab, followed by a secondary Ab linked to Texas Red (b, d). e–h, NG108-15 cells (NG) were grown 6 d in differentiating medium with KCl/db-cAMP and similarly stained with CtxB–FITC (e, g) and anti-TRPC5 (f, h). TRPC5 in less differentiated CGN (b) and NG108-15 (f) cells was localized primarily in the cell body, whereas in more differentiated cells (d, h), TRPC5 was depleted in cell body and localized in processes. GM1 was expressed in soma and processes and became stronger at the later stage.
We next focused on CGN cultures 48 h after seeding (3 DIV), the period of initial neuritogenesis when TRPC5 channel activity appeared most active. This showed enhanced process outgrowth in the presence of CtxB (Fig. 10Aa,Ab,Ba) in normal CGNs, and neurite length increased from 199 ± 6 to 292 ± 7 μm (mean ± SEM; p < 0.05). In contrast, neurite outgrowth in similar cultures from ganglio-series null mice lacking GM1 was significantly delayed and not affected by CtxB (Fig. 10Ac,Ad,Bd); neurite lengths were 132 ± 4 and 149 ± 5 μm in the absence and presence of CtxB, respectively (p < 0.05 and 0.01 compared with normal CGNs with the same treatment). That the processes promoted by CtxB were axonal in character was shown by IC with SMI-31 mAb against pNF-H protein (Sternberger and Sternberger, 1983) (Fig. 10Af). Such CtxB-induced and even “spontaneous” axonogenesis was effectively inhibited by siRNA to TRPC5 (Fig. 10Ae,Ag,Bb). Axonal outgrowth was also significantly inhibited by SK&F96365 (Fig. 10Bb), a relatively nonspecific inhibitor of TRP channels (Li et al., 1999), as well as wortmannin and U73122 (Fig. 10Bc) compared with untreated CGNs (Fig. 10Ba). Neurite lengths achieved with these various treatments are summarized in Fig. 10Be. The finding that TRPC5 siRNA inhibited spontaneous axon outgrowth in normal CGNs (Fig. 10Be) suggests the presence of an endogenous GM1-binding agent analogous to CtxB that promotes axonogenesis.
Figure 10.

Effect of CtxB on axon formation in primary CGN cultures from normal and ganglio-series null (KO) mice. A, CGN prepared from normal (a, b, e–g) and KO (c, d) mice were grown for 48 h (3 DIV) in the presence (b, d–g) or absence (a, c) of CtxB (5 μg/ml). Cells in e and g were treated with TRPC5-specific siRNA. a–e, Phase images of living cells. f, g, Fluorescent images with SMI-31 mAb against pNF-H (axonal marker). Retarded spontaneous axon growth was seen in KO cells compared with normal cells (c vs a). Axon formation was enhanced by CtxB in normal but not KO cells (b vs d) and inhibited by TRPC5-specific siRNA (e vs a, g vs f). B, Axon length was statistically analyzed with GraphPad Prism software. More than 300 cells in each group were assayed. a–d, Histograms fitted with Gaussian curves. e, Average axon length (±SEM) of each group. ▵p < 0.05 compared with control (CtxB-untreated) normal cells; *p < 0.01 compared with CtxB-treated cells; #p < 0.05 and ##p < 0.01 compared with normal cells with the same treatment (1-way ANOVA with Dunnett's post-test). SKF, SK&F96365; Wort., wortmannin.
Discussion
The early finding that GM1 cross-linking agents such as CtxB induced Ca2+ influx in certain cell lines (Dixon et al., 1987; Buckley et al., 1990; Guoy et al., 1994) commanded interest when it was found that such elevation of [Ca2+]i in neuroblastoma cells resulted in prolific process outgrowth (Masco et al., 1991; Fang et al., 2000). Physiological relevance was further suggested by the fact that CtxB induced [Ca2+]i elevation in primary cultures of dorsal root ganglia neurons with alteration of neurite morphology (Milani et al., 1992) and in CGN cultures with enhancement of cell survival (Wu et al., 1996). Anti-GM1 Abs of the cross-linking IgM type were shown to induce similar Ca2+ changes (Quattrini et al., 2001) and neurite outgrowth in neuroblastoma cells (O'Hanlon et al., 2003).
Efforts were subsequently undertaken to elucidate the channel type and mechanism mediating Ca2+ influx by this means. The fact that CtxB induced little or no Ca2+ influx in certain cells, such as Neuro-2A (Fig. 1A) (Fang et al., 2000) or NG108-15 cells after differentiation (Fang et al., 2002) (Fig. 1B) argued against a nonspecific membrane phenomenon as in bilayer models (Krasilnikov et al., 1991). The proposal that CtxB induced Ca2+ influx through L-type Ca2+ channels, with GM1 functioning as the constitutive negative modulator (Carlson et al., 1994), was later questioned by the finding that the channel functioned in a voltage-independent manner (Fang et al., 2002). This was consistent with a previous report that CtxB-induced elevation of [Ca2+]i in dorsal root ganglia neurons was not inhibited by L-type Ca2+ channel blockers (Milani et al., 1992). The present study indicates the relevant channel is TRPC5, a member of the canonical subfamily belonging to the TRP superfamily of signal-transduction-gated ion channels first discovered in Drosophila (Montell et al., 1985). Mammals contain >20 TRP channel isoforms, each structured with six-transmembrane domains that are thought to assemble into tetramers to form nonselective ion channels that allow passage of Ca2+ and other cations (Clapham et al., 2001). TRPC5 has sequence homology with TRPC1 and TRPC4 and is expressed in mammalian brain, where it forms homomeric and heteromeric channels with these two isoforms (Strubing et al., 2001).
A principal finding of the present study is the regulatory role of GM1 ganglioside, which, when cross-linked by CtxB, triggers a signaling cascade that culminates in TRPC5 channel opening. The resulting Ca2+ influx results in neurite initiation in its earliest stage. A key result linking these events was inhibition of both Ca2+ influx and neuritogenesis by TRPC5-specific siRNA. Our finding of abundant TRPC5 in the soma of primary neurons and neuroblastoma cells at an early stage of differentiation (Figs. 6, 9) parallels that of Bezzerides et al. (2004) and Greka et al. (2003), but whereas those studies revealed TRPC5 in vesicles undergoing transport to newly forming growth cones and synapses, the present study focused on TRPC5 in the soma membrane, the function of which is to initiate the early stage of neurite sprouting. These processes, although involving the same ion channel, represent distinct stages of differentiation and are likely subject to different signaling and regulatory mechanisms. Additional functional diversity can arise from TRPC5 forming not only homomers but also heteromeric structures with TRPC1 or TRPC4, as demonstrated in embryonic brain (Strubing et al., 2001, 2003). It was noteworthy that TRPC5 expression in the soma declined sharply as differentiation progressed in both NG108-15 cells (Figs. 2B, 5Aa, 9) and CGNs (Fig. 9), in parallel with the decreasing ability of CtxB to elevate [Ca2+]i in these cells (Fig. 1B) (Wu et al., 1996). The results of the present study also showed that GM1 is not directly associated with TRPC5 but rather with α5β1 integrin heterodimer, which, as a result of co-cross-linking, caused autophosphorylation of associated FAK and activation of subsequent signaling.
A key element in the opening of TRPC5 channels of NG108-15 cells was pretreatment with N′ase, an enzyme that converts oligosialo-gangliosides to GM1 (Ledeen, 1989). This elevates the normally low level of GM1 on the surface of undifferentiated NG108-15 cells, thereby facilitating co-cross-linking with integrin. A similar process appears to occur naturally in primary neurons, as revealed in the existence of a ganglioside-specific N′ase (sialidase) in the hippocampus at an early developmental stage that promotes axonogenesis and is downregulated in adulthood (Rodriguez et al., 2001; Da Silva et al., 2005). This type of ganglioside-specific N′ase has been observed in other neural cell loci (Hasegawa et al., 2000; Monti et al., 2000).
The pivotal role of integrin α5β1 in initiating the signaling cascade through cross-linking, a process dependent, in this case, on association with GM1, supports the idea that integrin clustering is necessary, and often sufficient, for activation of the crucial enzyme FAK through tyrosine phosphorylation (Rodriguez-Fernandez, 1999). Integrins are often aggregated in transmembrane complexes known as focal contacts, structures that are highly enriched in cytoskeletal proteins and signaling molecules (including FAK) (Clark and Brugge, 1995). FAK activation is generally initiated with autophosphorylation at Tyr 397, as was the case here (Fig. 7). This resulted in assembly of a multicomponent signaling complex that included PLCγ and PI(3)K (and possibly additional proteins). TRPC5 channels are described as generally responsive to signals mediated by PLC (Desai and Clapham, 2005), which is consistent with our finding that Ca2+ influx and neurite outgrowth were blocked by U73122; similar blockade by wortmannin and LY294002 confirmed participation by PI(3)K (Figs. 8, 10). Although FAK has been closely associated with such cellular functions as migration and cell spreading, it is known to have many additional roles such as control of apoptosis and cell-cycle reactions (Schlaepfer and Hunter, 1998). Importantly, FAK was shown to regulate neurite formation through interaction with integrin and growth factor receptors (Ivankovic-Dikic et al., 2000).
Parallel results were obtained in this study with NG108-15 cells and CGN in primary culture. A previous report demonstrated CtxB-induced elevation of [Ca2+]i in cultured CGN up to the first 7 DIV, after which the phenomenon reversed and CtxB reduced [Ca2+]i (Wu et al., 1996). Although the first phase likely represented the same CtxB/TRPC5-mediated events depicted here, the mechanism of the second phase is not known. The present study, in addition to demonstrating coexpression of TRPC5 and GM1 in the CGN at 2 DIV (Fig. 9), showed accelerated neuritogenesis with CtxB and inhibition of this effect by the same factors that blocked neuritogenesis in NG108-15 cells (e.g., TRPC5 siRNA, SK&F96365, U73122, and wortmannin) (Fig. 10). The fact that neuritogenesis occurs in ganglio-series null mice, albeit at a retarded rate, likely indicates an alternate parallel pathway(s) for neurite initiation. As mentioned, the observation that TRPC5 rapidly disappeared from the soma after neurite initiation (Fig. 9) does not preclude a continuing (but likely different) role for this channel in other parts of the neuron at later stages of development. The resultant effects of Ca2+ on neurite behavior and axon/growth cone activity are known to vary significantly depending on mode and topography of stimulation (Henley and Poo, 2004).
Many studies by many laboratories over the years have implicated gangliosides in general and GM1 in particular as trophic agents that influence neuronal differentiation and function (Hakomori and Igarashi, 1993; Ledeen et al., 1998; Yates and Rampersaud, 1998; Mocchetti, 2005). Some experiments suggested Ca2+ modulation as a crucial element in some of these systems (Wu and Ledeen, 1994; Ledeen and Wu, 2002; Wu et al., 2005), an idea supported by the example of GM1 potentiating a Na+/Ca2+ exchanger in the nuclear membrane (Xie et al., 2002). Additional examples are known in which GM1 asserts its modulatory action through direct association with a membrane protein, as with the δ-opioid receptor (Wu et al., 1997, 1998b) and the Trk protein (Mutoh et al., 2002). The present study depicts a different kind of modulatory effect in which GM1 associates with an upstream integrin dimer in the plasma membrane and effects channel opening through signaling initiated by cross-linking of the GM1/integrin complex. The amphiphilic properties of GM1, together with its negative charge on sialic acid that is resistant to N′ase, render it an extremely versatile molecule capable of interacting with a variety of proteins through hydrophobic, hydrophilic, and electrostatic associations.
In summary, this study has revealed a new regulatory mechanism for TRPC5 channels and clarified a long-standing mystery concerning CtxB-induced Ca2+ influx. In view of the ability of CtxB to inhibit neurite outgrowth in some systems, such as Neuro-2A cells (Fang et al., 2000), one can speculate that GM1 cross-linking may activate other forms of signaling in a manner that limits neuritogenesis, perhaps in those cells (e.g., interneurons, amacrine cells) destined to develop limited or no axons. Further speculation concerns the identity of the natural agonist that induces GM1 cross-linking. In that regard, it seems worth considering the large family of galectins, some of which have been shown to bind and even cross-link membrane-bound gangliosides and which are known to occur in the nervous system (Gabius et al., 2002). Whether or not such molecules have an active role in neurite initiation is a subject for future study.
Footnotes
This work was supported by National Institutes of Health Grants NS033912 (R.W.L.) and NS040167 (M.C.N.), an American Heart Association Scientist Development Grant (A.G.O.), and National Multiple Sclerosis Society Pilot Award PP1217 (G.W.).
References
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- Bezzerides VJ, Ramsey S, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol. 2004;6:709–720. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, Bunting S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther. 1990;255:756–768. [PubMed] [Google Scholar]
- Buckley NE, Matyas GR, Spiegel S. The bimodal growth response of Swiss 3T3 cells to the B subunit of cholera toxin is independent of the density of its receptor, ganglioside GM1. Exp Cell Res. 1990;189:13–21. doi: 10.1016/0014-4827(90)90250-e. [DOI] [PubMed] [Google Scholar]
- Carlson RO, Masco D, Brooker G, Spiegel S. Endogenous ganglioside GM1 modulates L-type calcium channel activity in N18 neuroblastoma cells. J Neurosci. 1994;14:2272–2281. doi: 10.1523/JNEUROSCI.14-04-02272.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strubing C. The TRP ion channel family. Nat Rev Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, Abad-Rodriguez J. Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat Neurosci. 2005;8:606–615. doi: 10.1038/nn1442. [DOI] [PubMed] [Google Scholar]
- Desai BN, Clapham DE. TRP channels and mice deficient in TRP channels. Pflügers Arch. 2005;451:11–18. doi: 10.1007/s00424-005-1429-z. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Stewart D, Grinstein S, Spiegel S. Transmembrane signaling by the B subunit of cholera toxin: increased cytoplasmic free calcium in rat lymphocytes. J Cell Biol. 1987;105:1153–1161. doi: 10.1083/jcb.105.3.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Wu G, Xie X, Lu Z-H, Ledeen RW. Endogenous GM1 ganglioside of the plasma membrane promotes neuritogenesis by two mechanisms. Neurochem Res. 2000;25:931–940. doi: 10.1023/a:1007596223484. [DOI] [PubMed] [Google Scholar]
- Fang Y, Xie X, Ledeen RW, Wu G. Characterization of cholera toxin B subunit-induced Ca2+ influx in neuroblastoma cells: evidence for a voltage independent GM1-associated Ca2+ channel. J Neurosci Res. 2002;57:1–10. doi: 10.1002/jnr.10333. [DOI] [PubMed] [Google Scholar]
- Gabellini N, Facci L, Milani D, Negro A, Callegaro L, Skaper SD, Leon A. Differences in induction of c-fos transcription by cholera toxin-derived cyclic AMP and Ca2+ signals in astrocytes and 3T3 fibroblasts. Exp Cell Res. 1991;194:210–217. doi: 10.1016/0014-4827(91)90356-y. [DOI] [PubMed] [Google Scholar]
- Gabius HJ, Andre S, Kaltner H, Siebert HC. The sugar code: functional lectinomics. Biochim Biophys Acta. 2002;1572:165–177. doi: 10.1016/s0304-4165(02)00306-9. [DOI] [PubMed] [Google Scholar]
- Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- Guoy H, Detterre P, Debre P, Bismuth G. Cell calcium signaling via GM1 cell surface gangliosides in the human Jurkat T cell line. J Immunol. 1994;152:3271–3281. [PubMed] [Google Scholar]
- Hakomori S-I. Traveling for the glycosphingolipid path. Glycoconj J. 2000;17:627–647. doi: 10.1023/a:1011086929064. [DOI] [PubMed] [Google Scholar]
- Hakomori S-I, Igarashi Y. Gangliosides and glycosphingolipids as modulators of cell growth, adhesion, and transmembrane signaling. Adv Lipid Res. 1993;25:147–162. [PubMed] [Google Scholar]
- Hasegawa T, Yamaguchi K, Wada T, Takeda A, Itoyama Y, Miyagi T. Molecular cloning of mouse ganglioside sialidase and its increased expression in Neuro2a cell differentiation. J Biol Chem. 2000;275:8007–8015. doi: 10.1074/jbc.275.11.8007. [DOI] [PubMed] [Google Scholar]
- Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–330. doi: 10.1016/j.tcb.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivankovic-Dikic I, Gronroos E, Blaukat A, Barth B-U, Dikic I. Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol. 2000;2:574–581. doi: 10.1038/35023515. [DOI] [PubMed] [Google Scholar]
- Jia Y, Zhou J, Tai Y, Wang Y. TRPC channels promote cerebellar granule neuron survival. Nat Neurosci. 2007;10:559–567. doi: 10.1038/nn1870. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Kawakami K, Steelant WF, Ono M, Baek RC, Handa K, Withers DA, Hakomori S. Tetraspanin CD9 is a “proteolipid,” and its interaction with alpha 3 integrin in microdomain is promoted by GM3 ganglioside, leading to inhibition of laminin-5-dependent cell motility. J Biol Chem. 2002;277:34349–34358. doi: 10.1074/jbc.M200771200. [DOI] [PubMed] [Google Scholar]
- Krasilnikov OV, Muratkhodjaev JN, Voronov SE, Yezepchuk YV. The ionic channels formed by cholera toxin in planar bilayer lipid membranes are entirely attributable to its B-subunit. Biochim Biophys Acta. 1991;1067:166–170. doi: 10.1016/0005-2736(91)90039-b. [DOI] [PubMed] [Google Scholar]
- Laurino L, Wang XX, de la Houssaye BA, Sosa L, Dupraz S, Caceres A, Pfenninger KH, Quiroga S. PI3K activation by IGF-1 is essential for the regulation of membrane expansion at the nerve growth cone. J Cell Sci. 2005;118:3653–3663. doi: 10.1242/jcs.02490. [DOI] [PubMed] [Google Scholar]
- Ledeen RW. Biosynthesis, metabolism, and biological effects of gangliosides. In: Margolis RU, Margolis RK, editors. Neurobiology of glycoconjugates. New York: Plenum; 1989. pp. 43–83. [Google Scholar]
- Ledeen RW, Wu G. Ganglioside function in the neuron. Trends Glycosci Glycotechnol. 1992;4:174–187. [Google Scholar]
- Ledeen RW, Wu G. Ganglioside function in calcium homeostasis and signaling. Neurochem Res. 2002;27:637–647. doi: 10.1023/a:1020224016830. [DOI] [PubMed] [Google Scholar]
- Ledeen RW, Wu G, Lu Z-H, Kozireski-Chuback D, Fang Y. The role of GM1 and other gangliosides in neuronal differentiation. Ann NY Acad Sci. 1998;845:161–175. doi: 10.1111/j.1749-6632.1998.tb09669.x. [DOI] [PubMed] [Google Scholar]
- Li HS, Xu XZ, Montell C. Activation of a TRPC3-dependent cation current through the neurotrophin BDNF. Neuron. 1999;24:261–273. doi: 10.1016/s0896-6273(00)80838-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia Y-C, Cui K, Li N, Zheng Z-Y, Wang Y-Z, Yuan X-B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Masco D, Van de Walle M, Spiegel S. Interaction of ganglioside GM1 with the B subunit of cholera toxin modulates growth and differentiation of neuroblastoma N18 cells. J Neurosci. 1991;11:2443–2452. doi: 10.1523/JNEUROSCI.11-08-02443.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani D, Minozzi M-C, Petrelli L, Guidolin D, Skaper SD, Spoerri PE. Interaction of ganglioside GM1 with the B subunit of cholera toxin modulates intracellular free calcium in sensory neurons. J Neurosci Res. 1992;33:446–475. doi: 10.1002/jnr.490330313. [DOI] [PubMed] [Google Scholar]
- Mocchetti I. Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell Mol Life Sci. 2005;62:2283–2294. doi: 10.1007/s00018-005-5188-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C. Exciting trips for TRPs. Nat Cell Biol. 2004;6:690–692. doi: 10.1038/ncb0804-690. [DOI] [PubMed] [Google Scholar]
- Montell C, Jones K, Hafen E, Rubin G. Rescue of the Drosophila phototransduction mutation trp by germline transformation. Science. 1985;230:1040–1043. doi: 10.1126/science.3933112. [DOI] [PubMed] [Google Scholar]
- Monti E, Bassi MT, Papini N, Riboni M, Manzoni M, Venerando B, Croci G, Preti A, Ballabio A, Tettamanti G, Borsani G. Identification and expression of NEU3, a novel human sialidase associated to the plasma membrane. Biochem J. 2000;349:343–351. doi: 10.1042/0264-6021:3490343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh T, Hamano T, Yano S, Koga H, Yamamoto H, Furukawa K, Ledeen RW. Stable transfection of GM1 synthase gene into GM1-deficient NG108-15 cells, CR-72 cells, rescues the responsiveness of TRK-neurotrophin receptor to its ligand, NGF. Neurochem Res. 2002;27:801–806. doi: 10.1023/a:1020209008169. [DOI] [PubMed] [Google Scholar]
- Obukhov AG, Nowycky MC. TRPC5 activation kinetics are modulated by the scaffolding protein ezrin/radixin/moesin-binding phosphoprotein-50 (EBP50) J Cell Physiol. 2004;201:227–235. doi: 10.1002/jcp.20057. [DOI] [PubMed] [Google Scholar]
- Obukhov AG, Nowycky MC. A cytosolic residue mediates Mg2+ block and regulates inward current amplitude of a transient receptor potential channel. J Neurosci. 2005;25:1234–1239. doi: 10.1523/JNEUROSCI.4451-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hanlon GM, Hirst TR, Willison HJ. Ganglioside GM1 binding toxins and human neuropathy-associated IgM antibodies differentially promote neuritogenesis in a PC12 assay. Neurosci Res. 2003;47:383–390. doi: 10.1016/s0168-0102(03)00239-6. [DOI] [PubMed] [Google Scholar]
- Plant TD, Schaefer M. TRPC4 and TRPC5: receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium. 2003;33:441–450. doi: 10.1016/s0143-4160(03)00055-1. [DOI] [PubMed] [Google Scholar]
- Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J, Grindey G, Vlahos CJ. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–2423. [PubMed] [Google Scholar]
- Quattrini A, Lorenzetti I, Sciorati C, Corbo M, Previtali SC, Feltri ML, Canal N, Wrabetz L, Nemni R, Clementi E. Human IgM anti-GM1 autoantibodies modulate intracellular calcium homeostasis in neuroblastoma cells. J Neuroimmunol. 2001;114:213–219. doi: 10.1016/s0165-5728(00)00452-5. [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Piddini E, Hasegawa T, Miyagi T, Dotti CG. Plasma membrane ganglioside sialidase regulates axonal growth and regeneration in hippocampal neurons in culture. J Neurosci. 2001;21:8387–8389. doi: 10.1523/JNEUROSCI.21-21-08387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fernandez JL. Why do so many stimuli induce tyrosine phosphorylation of FAK? BioEssays. 1999;21:1069–1075. doi: 10.1002/(SICI)1521-1878(199912)22:1<1069::AID-BIES13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Integrin signaling and tyrosine phosphorylation. Just the FAKs? Trends Cell Biol. 1998;8:151–157. doi: 10.1016/s0962-8924(97)01172-0. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Yoshida T, Wakamori M, Ishii M, Okada T, Takahashi M, Seto M, Sakurada K, Kiuchi Y, Mori Y. Ca2+-calmodulin-dependent myosin light chain kinase is essential for activation of TRPC5 channels expressed in HEK293 cells. J Physiol (Lond) 2006;570:219–235. doi: 10.1113/jphysiol.2005.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and non-phosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci USA. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- Tesfai Y, Brereton HM, Barritt GJ. A diacylglycerol-activated Ca2+ channel in PC12 cells (an adrenal chromaffin cell line) correlates with expression of the TRP-6 (transient receptor potential) protein. Biochem J. 2001;358:717–726. doi: 10.1042/0264-6021:3580717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Wu G, Ledeen RW. Quantification of gangliotetraose gangliosides with cholera toxin. Anal Biochem. 1988;173:368–375. doi: 10.1016/0003-2697(88)90201-1. [DOI] [PubMed] [Google Scholar]
- Wu G, Ledeen RW. Gangliosides as modulators of neuronal calcium. Prog Brain Res. 1994;101:101–112. doi: 10.1016/s0079-6123(08)61942-1. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu Z, Ledeen RW. Induced and spontaneous neuritogenesis are associated with enhanced expression of ganglioside GM1 in the nuclear membrane. J Neurosci. 1995;15:3739–3746. doi: 10.1523/JNEUROSCI.15-05-03739.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu Z-H, Nakamura K, Spray DC, Ledeen RW. Trophic effect of cholera toxin B subunit in cultured cerebellar granule neurons: modulation of intracellular calcium by GM1 ganglioside. J Neurosci Res. 1996;44:243–254. doi: 10.1002/(SICI)1097-4547(19960501)44:3<243::AID-JNR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu Z-H, Ledeen RW. Interaction of δ-opioid receptor with GM1 ganglioside: conversion from inhibitory to excitatory mode. Mol Brain Res. 1997;44:341–346. doi: 10.1016/s0169-328x(96)00281-1. [DOI] [PubMed] [Google Scholar]
- Wu G, Fang Y, Lu Z-H, Ledeen RW. Induction of axon-like and dendrite-like processes in neuroblastoma cells. J Neurocytol. 1998a;27:1–14. doi: 10.1023/a:1006910001869. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu Z-H, Wei TJ, Howells RD, Christoffers K, Ledeen RW. The role of GM1 ganglioside in regulating excitatory opioid effects. Ann NY Acad Sci. 1998b;845:126–138. doi: 10.1111/j.1749-6632.1998.tb09666.x. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu Z-H, Xie X, Li L, Ledeen RW. Mutant NG108-15 cells (NG-CR72) deficient in GM1 synthase respond aberrantly to axonogenic stimuli and are vulnerable to calcium-induced apoptosis: they are rescued with LIGA-20. J Neurochem. 2001;76:690–702. doi: 10.1046/j.1471-4159.2001.00036.x. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu Z, Ledeen RW. Role of GM1 ganglioside in operation of TRPC5 calcium channels [abstract] J Neurochem. 2004a;90:90. [Google Scholar]
- Wu G, Lu Z, Xie X, Ledeen RW. Susceptibility of cerebellar granule neurons from GM2/GD2 synthase-null mice to apoptosis induced by glutamate excitotoxicity and elevated KCl: rescue by GM1 and LIGA20. Glycoconj J. 2004b;21:303–311. doi: 10.1023/B:GLYC.0000046273.68493.f7. [DOI] [PubMed] [Google Scholar]
- Wu G, Lu Z-H, Wang J, Wang Y, Xie X, Meyenhofer MF, Ledeen RW. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane-permeant analog of GM1. J Neurosci. 2005;25:11014–11022. doi: 10.1523/JNEUROSCI.3635-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Wu G, Lu Z, Ledeen RW. Potentiation of sodium-calcium exchanger in the nuclear envelope by nuclear GM1 ganglioside. J Neurochem. 2002;81:1185–1195. doi: 10.1046/j.1471-4159.2002.00917.x. [DOI] [PubMed] [Google Scholar]
- Yates AJ, Rampersaud A. Sphingolipids as receptor modulators. Ann NY Acad Sci. 1998;845:57–71. doi: 10.1111/j.1749-6632.1998.tb09662.x. [DOI] [PubMed] [Google Scholar]