

Abstract
Cerebral palsy remains a public health priority. Recognition of factors of susceptibility to perinatal brain lesions is key for the prevention of cerebral palsy. In most cases, the pathophysiology of these lesions is thought to involve prior exposure to predisposing factors that make the developing brain more vulnerable to perinatal events. The present study tested the hypothesis that exposure to chronic minimal stress throughout gestation would sensitize the offspring to neonatal excitotoxic brain lesions, which mimic lesions observed in cerebral palsy. Pregnant mice were exposed to chronic, ultramild stress, applied throughout gestation. Neonatal brain lesions were induced by intracerebral injection of glutamate analogs. Excitotoxic lesions were significantly worsened in pups exposed to gestational stress. Stress induced a significant rise of circulating corticosterone levels both in pregnant mothers and in newborn pups. The deleterious effects of stress on excitotoxicity were totally suppressed in mice with reduced levels of glucocorticoid receptors. Stress induced a significant increase of neopallial NMDA binding sites in the offspring. At adulthood, animals exposed to stress and neonatal excitotoxic challenge showed a significant impairment in the Morris water maze test when compared with animals exposed to the excitotoxic challenge but not the gestational stress. These findings suggest that stress during gestation, which may mimic low-level stress in human pregnancy, could be a novel risk factor for cerebral palsy.
Keywords: cerebral palsy, neonatal, excitotoxicity, glucocorticoids, NMDA, sensitization
Introduction
The development of the CNS is dependent on interactions between genetic and epigenetic factors, some of which could affect the susceptibility of the developing brain to subsequent damaging insults (Bhutta and Anand, 2002). Supporting this hypothesis, exposure of the human fetal brain to chorioamnionitis-driven inflammation makes the developing brain exquisitely vulnerable to perinatal hypoxic-ischemic insults (Dommergues et al., 2000; Eklind et al., 2001). This two-hit mechanism seems to be key in the pathophysiology of cerebral palsy (CP) and other neurological impairments linked to term and preterm (23–32 weeks of gestation) birth (Dammann et al., 2002).
Gestational stress has been shown to be a potential factor associated with a higher risk to develop some neurological and psychiatric disorders (for review, see Weinstock, 2001). Similarly, in animal models, stress can induce lasting deficits in the offspring (for review, see Kofman, 2002). Long-lasting effects of stress have been linked to disturbances in the function of the hypothalamo–pituitary–adrenocortical axis (Avishai-Eliner et al., 2002; Van den Bergh et al., 2005). Moreover, stress during pregnancy may cause alteration in brain development, leading to altered synaptic connectivity and persistent deficits in brain function.
Based on these studies, we propose the hypothesis that maternal stress during gestation could be a risk factor for perinatal brain lesions. To test it, we used a well characterized mouse model of neonatal excitotoxic brain lesions (Marret et al., 1995; Gressens et al., 1997; Dommergues et al., 2000; Tahraoui et al., 2001; Husson et al., 2002), which mimic lesions observed in preterm and term human infants who will later develop CP. In this model, intracerebral administration of glutamate analogs acting on NMDA or non-NMDA receptors replicates the excess release of glutamate induced, among others, by hypoxic–ischemic insult (Volpe, 2001). This excitotoxic model was combined with an original chronic ultramild stress procedure (CUMS) applied during the entire gestation. From a mechanistic point of view, the effects of CUMS on circulating corticosterone levels and ionotropic glutamatergic receptor density were evaluated in this model.
Materials and Methods
Animals and mating procedure
Three types of virgin female mice were used: 64 C57BL/6 mice (Charles River, L'Arbresle, France), eight glucocorticoid receptor-impaired (GR-I) mice (Pepin et al., 1992), a transgenic B6C3F1 mouse strain homozygous for the presence of rat glucocorticoid receptor antisense RNA and eight wild-type B6C3F1 control mice. At their arrival in the laboratory, all mice were 8 weeks of age and were housed in groups of five per cage in our animal research facility in 37.5 × 21.5 × 18 cm cages and kept on standard conditions (12 h light/dark cycle, 22 ± 2°C, food and water ad libitum). All experimental protocols and procedures complied with the guidelines of the French Ministry of Agriculture.
All females were mated at 12 weeks with a sexually experienced male of the same genotype. Each female was paired with one male at 9:00 A.M., and the experimenter checked the plugged females at 2:00 P.M., which were immediately housed individually in cages for the whole gestation. Eight C57BL/6 plugged females were isolated to constitute the multigravid female group. The aim was to estimate the importance of parity. Although the link between age and parity is obvious, we tried, as far as possible, to treat the variable parity independently of the age of the mothers. Consequently, the second and third litters of the multigravid females were killed 24 h after birth, to compare multigravid and primigravid females of the same age (of note, it was impossible to obtain aged primigravid females, because they could not maintain their pregnancies until term). This protocol was justified by previous results (Cohen-Salmon et al., 1982; Cohen-Salmon, 1988), showing that quality of pup care behavior was not modified in multigravid mothers by the suppression of their first litters. Primigravid and multigravid gestating females were divided in two groups. The controls were undisturbed during the entire gestational period. The experimental groups (stressed) were subjected to a CUMS procedure from observation of the plug to parturition [postnatal day 0 (P0)] and placed in a stress room (22 ± 2°C, variable light/dark cycle, food and water ad libitum). If no plug was observed, females returned to their home cage until the next morning for a new mating procedure. Ten days after the beginning of the mating procedure, no gestating females were excluded from the study.
CUMS procedure
The CUMS procedure was described in detail by Lanfumey et al. (1999). This procedure was based solely on environmental and social stressors, without any food/water deprivation or any nociceptive events: (1) three ultramild diurnal stressors delivered over a period of 1 or 2 h with a 2 h stress-free time between two diurnal stressors: periods of cage tilt (30°), confinement in smaller cages (11 × 8 × 8 cm), and paired housing; (2) four ultramild nocturnal stressors: one overnight period of difficult access to food, one overnight period of permanent light, one overnight period of 30° cage tilt, and one overnight in a soiled cage (50 ml of water in sawdust bedding); (3) a reversed light/dark cycle during the weekend. This 1 week stress regimen was repeated three times during gestational time.
Experimental groups
Pups from at least two different litters were used in each experimental group, and data were obtained from two or more successive experiments. Sex of pups was determined by visual inspection at birth and was confirmed after death by both anatomical inspection and PCR for srY.
In a first set of experiments, first-litter and fourth-litter C57BL/6, first-litter GR-I, and wild-type control pups from stressed and control groups were submitted to an excitotoxic challenge on P5 before seizure activity determination during the first 24 h and lesion size determination on P10. In addition, first-litter GR-I and wild-type control pups from unstressed mothers were used to measure glucocorticoid receptor mRNA expression at P0 and P5.
In a second set of experiments, first-litter control C57BL/6 pups received a daily subcutaneous injection of 40 mg · kg−1 · d−1 corticosterone (Sigma-Aldrich, St. Quentin Fallavier, France) diluted in 5 μl of DMSO or DMSO alone either between P1 and P5 or once on P5. Some control pups did not receive any subcutaneous injection. Ibotenate was injected intracerebrally to some P5 pups, immediately after the last corticosterone injection and lesion size determination was performed on P10. Other corticosterone-exposed pups were killed at P5 8 h after the last corticosterone injection and used to determine circulating corticosterone levels.
In a third set of experiments, primigravid control C57BL/6 and B6C3F1 GR-I mothers received a daily subcutaneous injection of 3 mg · kg−1 · d−1 corticosterone diluted in 100 μl of DMSO or DMSO alone between embryonic day 13 (E13) and E18. Ibotenate was injected intracerebrally to pups on P5, and lesion size determination was performed on P10.
In a fourth set of experiments, first-litter stressed and control C57BL/6 pups were killed on P0 or P5 to measure circulating corticosterone levels, brain BDNF expression, and brain ionotropic glutamate receptor expression. These pups were not submitted to any excitotoxic challenge. In this set of experiments, corticosterone levels were also measured in the mothers on E15, P0, and P5.
In a fifth and last set of experiments, first-litter stressed and control C57BL/6 pups were submitted to an excitotoxic challenge on P5 before behavioral analysis at 2 months of age.
Excitotoxic lesions and lesion size determination
Excitotoxic brain lesions were induced by injecting 10 μg of ibotenate (NMDA and metabotropic receptor agonist; Tocris Bioscience, Bristol, UK), 5 μg of NMDA (NMDA receptor agonist; Tocris Bioscience), or 15 μg of S-bromowillardiine (AMPA and kainate receptor agonist; Tocris Bioscience) into P5 mouse brains. As described previously (Marret et al., 1995; Dommergues et al., 2000; Tahraoui et al., 2001), mouse pups anesthetized with isoflurane were kept under a warming lamp and were injected intracerebrally (into the neopallial parenchyma). Intraparenchymal injections were performed with a 25 gauge needle on a 50 μl Hamilton syringe mounted on a calibrated microdispenser. The needle was inserted 2 mm under the external surface of scalp skin in the frontoparietal area of the right hemisphere, 1 mm from the midline in the lateral–medial plane, and 1.5 mm anterior to bregma in the rostrocaudal plane. Two 1 μl boluses were injected at 20 s intervals.
On P10, pups were killed by decapitation. Brains were immediately fixed in 4% formalin. After embedding in paraffin, we cut 16-μm-thick coronal sections. Every third section was stained with cresyl violet. Previous studies (Gressens et al., 1997; Husson et al., 2002) have shown an excellent correlation between the maximal size of the lesion in the lateral–medial and fronto-occipital axes of the excitotoxic lesions. Based on these findings, we cut serial sections of the entire brain in the coronal plane for this study. This permitted an accurate and reproducible determination of the sagittal fronto-occipital diameter. We used this linear measure as an index of the lesion volume.
To further confirm the correlation between the sagittal diameter and the volume of the lesion, both the sagittal diameter and the total volume of the lesion were measured in a subset of experiments. Brains were serially sectioned as described above, and volumes were measured using the Neurolucida software-controlled computer system (MicroBrightField Europe, Magdeburg, Germany).
Circulating corticosterone level determination
Stressed and control C57BL/6 pups (on P0 and P5) and their mothers (on E15, P0, and P5) were decapitated at 8:30 A.M. to collect trunk blood in 1.5 ml of EDTA-coated microcentrifuge tubes. In addition, control C57BL/6 pups treated with a single injection of corticosterone on P5 or with daily (between P1 and P5) subcutaneous corticosterone were decapitated on P5 8 h after the corticosterone injection. Samples were kept on ice and later centrifuged for 20 min at 9000 rpm at 4°C. Plasma was transferred to clean 1.5 ml microcentrifuge tubes, and plasma samples were stored frozen at −20°C until the determination of corticosterone. Corticosterone levels were measured by RIA (MP Biomedicals, New York, NY).
Real-time reverse transcription-PCR for glucocorticoid receptor mRNA
Stressed and control C57BL/6 pups, B6C3F1 GR-I, and control pups were killed on P0 or P5. Ten animals were included in each treatment group. Brains were removed, and cortex and underlying white matter immediately adjacent to the normal site of excitotoxin injection (see above) were rapidly dissected from the different animals for RNA extraction. Total RNA was extracted as described previously (Lelievre et al., 2002), followed by DNaseI. For each sample, 600 ng were used in reverse transcription (iScript kit; Bio-Rad, Hercules, CA). As described previously (Husson et al., 2005), for the glucocorticoid receptor, the following pair of primers was designed to selectively amplify a 93 bp fragment spanning the 1234–1336 region of the published mouse sequence (NR3C1, NM008173): 5′-TTATGGATATTCAAGCCCTGG-OH and 5′AGGCAGAGTTTGGGAGGTAGT-OH. To standardize the experiments, we used a primer set (5-CCGGCTTGTATGCTATC and 5-TCAGTATGTTCGGCTTC, as sense and antisense, respectively) for the mouse β2-microglobulin gene. These primers amplified an 87 bp region encoding the nucleotides 99–185 of the published sequence (MMB2MR) of the mouse mRNA. Amplified BDNF variants and β2-microglobulin bands were cloned into PCRII and sequenced to confirm identity. Real-time PCR was set up using SYBR Green-containing supermix from Bio-Rad, for 58 cycles of a three-step procedure, including a 30 s denaturation at 96°C, a 30 s annealing at 64°C, and a 30 s extension at 72°C. Quantification used standard curves made from serial dilutions of control RNA sample or of the corresponding cDNA cloned into PCRII vector. Differences between samples were calculated as the difference between the specific ratios (BDNF variant/β2-microglobulin) calculated for each individual sample. Experiments were independently run three times. In each experiment, samples were performed in triplicates.
NMDA, AMPA, and kainate receptor binding sites
Stressed and control C57BL/6 pups were killed on P5. Ten animals were included in each treatment group. Brains were dissected, and hemispheres were used for membrane preparation. The density of NMDA receptor was studied by the binding of the tritiated channel blocker dizocilpine (3H-MK-801) (PerkinElmer, Zaventen, Belgium). Tissue preparation and incubations were performed according to Reynolds and Sharma (1999). The binding assay was performed for 2 h at room temperature in 1 ml of HEPES (10 mm; pH 7.4) buffer containing 100 μg of proteins, 0.5–1 nm 3H-MK-801, in the presence of l-glutamate (100 μm) and glycine (30 μm). The nonspecific binding (NSB) was evaluated by addition of 30 μm unlabeled MK-801 (ICN Biomedicals, Costa Mesa, CA) or 100 μm ketamine (Ketalar; Pfizer, Zurich, Switzerland). Incubations were stopped by rapid filtration through glass fiber filters (GF/B; Whatman; Sigma-Aldrich) presoaked with poly-ethyleneimine. The filters were rinsed four times by 4 ml of ice-cold HEPES buffer and counted for radioactivity. Experiments were independently run seven times.
The density of kainate receptors was studied by the binding of kainic acid (vinylidene-3H) (3H-kainic acid; PerkinElmer). Tissue preparation and incubations were performed according to Hawkins et al. (1995). The binding assays were performed for 90 min at room temperature in 0.3 ml of Tris (100 mm; pH 7.4) buffer containing 25–100 μg of proteins, in the presence of NaCl (100 mm) and MgCl2 (5 mm). The binding was assessed at two radioligand concentrations (2–15 and 20–70 nm). NSB was evaluated by addition of 1 mm l-glutamate. Incubations were stopped by rapid filtration on GF/B presoaked filters as described for 3H-MK-801 binding assay, using ice-cold Tris (100 mm), pH 7.4. Experiments were independently run three times. AMPA receptors were studied by the binding of amino-3-hydroxy-5-methyloxazole-4-propionic acid, dl-α[5-methyl-3H] (3H-AMPA; PerkinElmer) according to the same procedure, in the presence of the chaotropic agent KSCN (100 mm) in incubation buffer.
Seizure activity
Stressed and control C57BL/6 P5 pups received ibotenate intracerebrally. Seizures were recorded by video during the first 15 min of the first, fourth, eighth, and 24th hours after the excitotoxic insult. After each period of recording, pups were returned to dams. Seizures were defined as paroxysmal events characterized by clonic jerks of the contralateral limbs or of the whole body and were quantified by two blinded investigators. As there was no effect of the time elapsed after the insult on seizure occurrence (data not shown), data from the four time points were pooled in both experimental groups.
Behavioral testing
Behavioral tests and confirmation of brain lesions.
Control and stressed C57BL/6 adult males that had been exposed to an excitotoxic challenge on P5 were randomly coded to conduct a blind study. Animals were 42 ± 7 d of age at the onset of testing. Behavioral testing was conducted during 4 consecutive days using a Morris water maze habituation. Immediately after the end of the Morris water maze task, the animals were killed, and their brains were immediately fixed in 4% formalin. After embedding in paraffin, we cut 16-μm-thick coronal sections. Every third section was stained with cresyl violet, allowing confirmation of the presence of a brain lesion in all animals.
Morris water maze apparatus and procedure.
A circular pool (100 cm diameter) filled to a depth of 24 cm with opacified water maintained at 35 ± 2°C was used. Water was made opaque by the addition of a nontoxic product provided from the cosmetology industry. The pool was virtually divided into four quadrants of equal area: northeast, northwest, southeast, and southwest. A circular escape platform (8 cm diameter) was located in the center of the southeast quadrant and submerged ∼0.5 cm below the surface of the water. Around the pool, various extramaze cues served as visual reference points for the animals: black geometric images (squares, crow, and triangles) on the walls, the light, and shelves.
On the first day, mice were familiarized with water and swimming in a Morris water maze habituation session during which animals were allowed to swim ad libitum during two 60 s sessions in the pool without the escape platform. During the 3 test days, mice were submitted to three sessions separated by a 1 h period. A session was composed of four randomized trials so that the four cardinal start points were used three times every day. A trial was initiated by placing each mouse in the water facing the pool wall in one of the four start points. Mice were allowed to swim a maximum of 60 s to find the hidden platform. When it succeeded, it could stay on it during a 30 s period (intertrial time). The experimenter noted the time taken to reach the hidden platform [latency escape (LE)]. In case of failure, the mouse was given the maximum score (61 s), and the experimenter put the mouse in the hidden platform for the intertrial time. The next trial started just after the intertrial time.
Statistical analysis
Most of the neuropathological, biochemical, and molecular data were analyzed with a Student's t test or a one-way ANOVA. In ANOVA analyses, when a primary effect of treatment group was found to be significant, Dunnett's multiple-comparison tests were performed. In the subset of experiments in which lesion size was evaluated in males and females, results were studied using a three-way ANOVA with treatment (stress vs control), gender (female vs male), and gravidity (primigravid vs multigravid) as between-subjects factors. When a main effect of treatment, gender, or gravidity or their interaction was found to be significant, we conducted pairwise comparisons between treatment groups using a Student's t test.
In behavioral tests, for each mouse, mean LE was calculated for each testing day (D1, D2, and D3). These means were then analyzed using two-way repeated-measures ANOVA with day as the within-subject factor and treatment (CUMS vs control) as the between-subjects factor. When a main effect of treatment, day, or their interaction was found to be significant, post hoc comparisons between groups were made using the Tukey's correction. Differences with p < 0.05 were considered statistically significant.
Results
Mortality and epileptic manifestations
Overall mortality was low in the present study, with death seen in <3% of the animals injected with excitotoxin. No significant difference was observed in a test of contingency (exact Fisher test) when the CUMS and control groups were compared. Epileptic manifestations including clonic or tonic seizures and apneas were observed in all excitotoxin-treated animals. However, when compared with controls, CUMS did not induce any difference in frequency (53 ± 9 convulsions per 15 min in stressed pups vs 49 ± 10 in control pups) or severity (qualitatively assessed) of seizures during the first 24 h after the excitotoxic challenge.
Effects of CUMS on neonatal excitotoxic brain lesions in C57BL/6 mice
Control pups injected intracerebrally with ibotenate (an agonist of both NMDA and metabotropic glutamatergic receptors) at P5 developed cortical lesions and periventricular white matter cysts (Figs. 1A, 2A–D). The cortical lesion was typical of ibotenate-induced lesions, with severe neuronal loss in all neocortical layers and almost complete disappearance of neuronal cell bodies along the axis of ibotenate injection. In control pups, there was no significant effect of gravidity and gender on the size of the cortical plate and white matter lesions (Fig. 2A–D).
Figure 1.
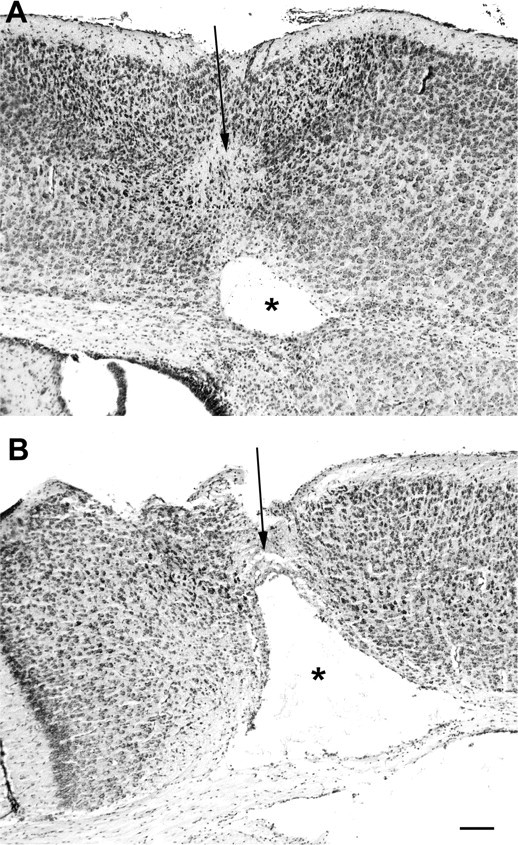
CUMS during gestation exacerbates ibotenate-induced brain lesions in C57BL/6 offspring. Shown are cresyl violet-stained sections showing brain lesions induced by ibotenate injected at P5 and studied at the age of P10. A, Brain from a control pup not exposed to CUMS during gestation and treated with intracerebral ibotenate, showing the typical neuronal loss in layers II–VI (arrow) and the white matter cystic lesion (*). B, Brain from a pup exposed to CUMS during gestation and treated with intracerebral ibotenate. Scale bar, 40 μm.
Figure 2.
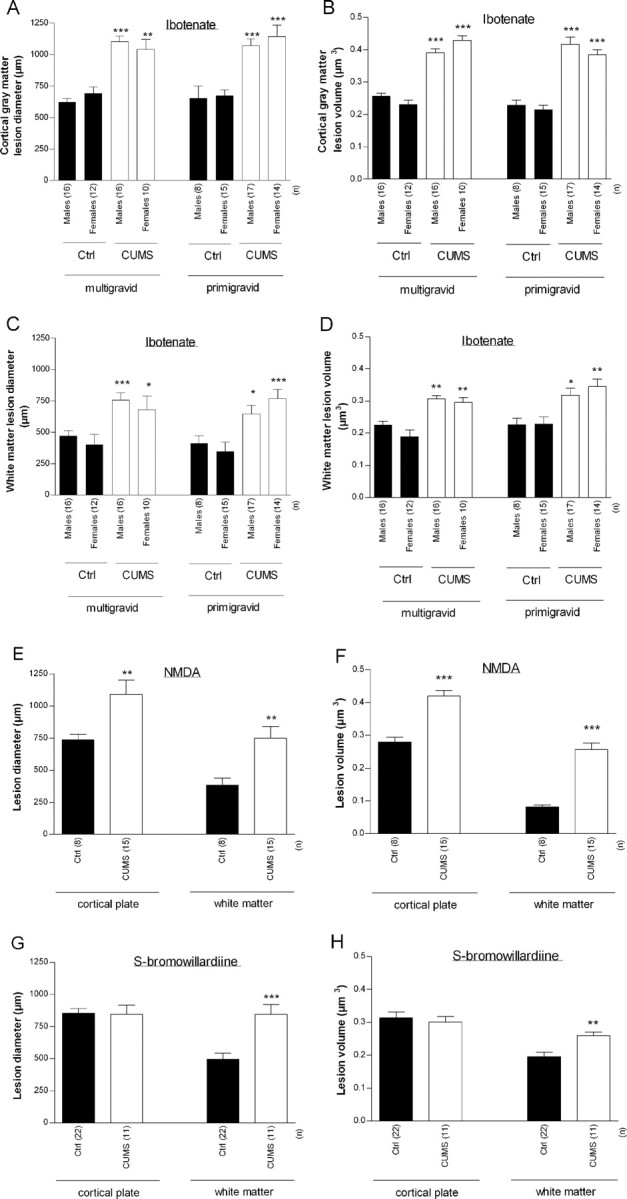
A–D, Effects of CUMS on excitotoxic brain lesions induced by ibotenate (A–D), NMDA (E, F), or S-bromowillardiine (G, H) in C57BL/6 mice. Excitotoxins were injected at P5, and pups were killed at P10. Bars represent mean sagittal diameter (A, C, E, G) or mean volume (B, D, F, H) of the lesions ± SEM. Asterisks indicate statistically significant difference from control males (A–D) or controls (E–H). *p < 0.05, **p < 0.01, ***p < 0.001 in three-way ANOVA with contrasts (A–G) or in Student's t test (E–H). Ctrl, Control.
CUMS exposure significantly increased the size of both cortical plate and underlying white matter lesions [up to 77% increase of cortical plate lesion size (p < 0.0001 vs unstressed animals) and up to 120% increase of white matter lesion size (p < 0.0001 vs unstressed animals)] (Figs. 1B, 2A–D). Again, there was no significant effect of gravidity and gender. Accordingly, in subsequent experiments using C57BL/6 mice, male and females pups from primigravid mothers were used and combined for analysis.
CUMS-induced exacerbation of ibotenate-induced lesions was replicated in pups injected with NMDA [an agonist of NMDA glutamatergic receptors; 60% increase of cortical plate lesion size (p < 0.001 vs unstressed animals) and 59% increase of white matter lesion size (p < 0.001 vs unstressed animals)] (Fig. 2E,F).
In pups injected with S-bromowillardiine (an agonist of AMPA and kainate glutamatergic receptors), CUMS exacerbated excitotoxic periventricular white matter lesions (71% increase of lesion size; p < 0.01 vs unstressed animals) without significantly modulating cortical plate lesions (Fig. 2G,H).
In this first set of experiments, measurement of the maximal sagittal fronto-occipital diameter of the lesion or of the volume of the lesion showed very similar results (Fig. 2). Accordingly, in the following sets of experiments, measurement of the maximal sagittal fronto-occipital diameter of the lesion was used as an index of the volume of the lesion.
Effects of CUMS on corticosterone levels in C57BL/6 mice
The effects of CUMS on corticosterone blood levels were determined both in pregnant and postdelivery mothers, as well as in newborn pups. CUMS significantly increased corticosterone levels in mothers at E14 (47.2 ± 1.0 vs 36.8 ± 1.4 μg/100 ml in unstressed controls; p < 0.0001) and P0 (22.4 ± 1.1 vs 9.9 ± 0.7 μg/100 ml; p < 0.0001) and in pups at P0 (4.6 ± 0.8 vs 1.8 ± 0.4 μg/100 ml; p = 0.039) and P5 (0.13 ± 0.04 vs 0.06 ± 0.01 μg/100 ml; p = 0.03).
Effects of CUMS on neonatal excitotoxic brain lesions in GR-I mice
To test the hypothesis that this CUMS-induced surge in corticosterone levels plays a role in the observed effects of CUMS exposure on excitotoxic lesions, we studied B6C3F1 GR-I mice, which have reduced levels of glucocorticoid receptors (Dijkstra et al., 1998). We further confirmed by real-time PCR the significantly reduced expression of glucocorticoid receptor in P0 and P5 B6C3F1 GR-I pups both in the cortical plate (p < 0.001 at P0 and p < 0.01 at P5 vs control B6C3F1 pups) and underlying white matter (p < 0.001 at P0 and p = 0.015 at P5 vs control B6C3F1 pups) (Fig. 3).
Figure 3.

Real-time PCR quantification of glucocorticoid receptor (GR) mRNA. Pups from control (Ctrl) and GR-I unstressed B6C3F1 mothers were killed at P0 or P5. Brains were removed, and cortical plate (A) and underlying white matter (B) immediately adjacent to the normal site of excitotoxin injection were collected for RNA extraction. Bars represent mean GR/β2-microglobulin ratios ± SEM. Asterisks indicate statistically significant differences between Ctrl pups from unstressed mothers and pups from mothers exposed to CUMS. *p < 0.05, **p < 0.01, ***p < 0.001 in ANOVA with Dunnett's test.
In control B6C3F1 mice, CUMS significantly exacerbated ibotenate-induced lesions both in cortical plate (p < 0.01 vs unstressed animals) and underlying white matter (p < 0.001 vs unstressed animals) (Fig. 4A). As in C57BL/6 mice, there was no significant effect of gender (Fig. 4A). In contrast, in B6C3F1 GR-I-deficient mice, there was no detectable effect of CUMS exposure on ibotenate-induced brain lesions (Fig. 4B).
Figure 4.
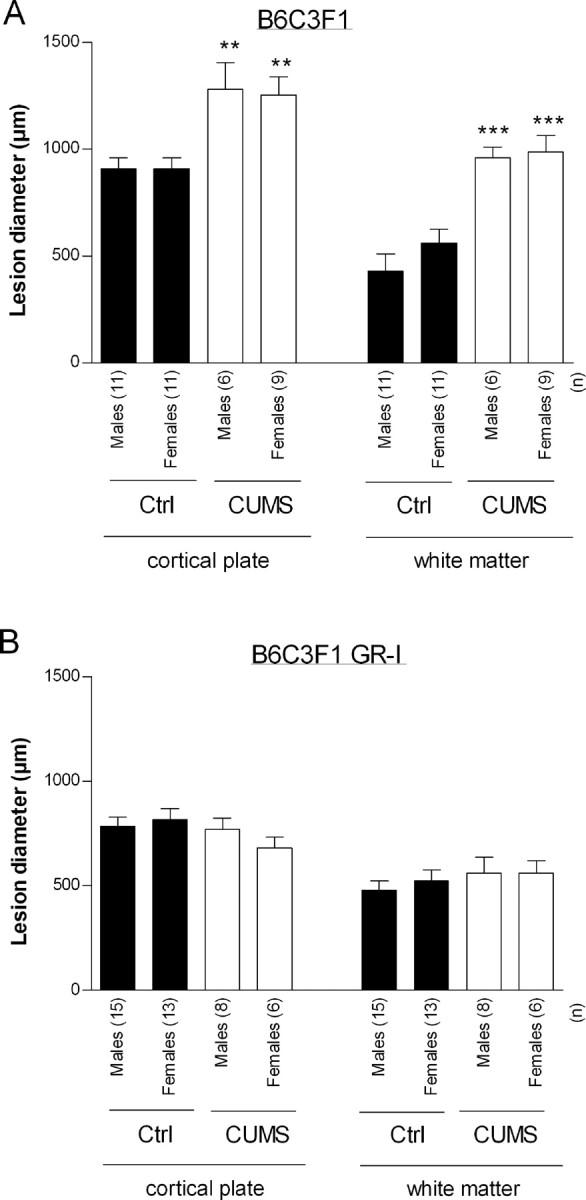
Effects of CUMS on excitotoxic brain lesions induced by ibotenate in B6C3F1 control (A) and B6C3F1 GR-I (B) mice. Ibotenate was injected at P5, and pups were killed at P10. Bars represent mean sagittal diameter of the lesions ± SEM. Asterisks indicate statistically significant difference from controls. **p < 0.01, ***p < 0.001 in two-way ANOVA with Student's t posttest. Ctrl, Control.
Effects of prenatal and postnatal corticosterone on excitotoxic brain lesions in C57BL/6 and B6C3F1 GR-I mice
To further elucidate the role of corticosterone in CUMS effects and, in particular, to discriminate the effects of in utero increased levels of corticosterone in mother and fetuses from the effects of increased levels of corticosterone in postnatal pups, pregnant unstressed mothers or pups from unstressed mothers were injected intraperitoneally with corticosterone before ibotenate injection.
In unstressed C57BL/6 pups, a single injection of 40 mg/kg corticosterone immediately before ibotenate injection had no detectable effect on excitotoxic brain lesions (Fig. 5A). This regimen induced a significant increase of circulating corticosterone when measured 8 h after the subcutaneous injection (0.63 ± 0.12 vs 0.36 ± 0.05 μg/100 ml in control pups; p = 0.049).
Figure 5.

Effects of corticosterone administration at P5 (A) between P1 and P5 (B) or between E13 and E18 (C, D) on ibotenate-induced brain lesions in C57BL/6 (A–C) or B6C3F1 GR-I (D) pups. Ibotenate was injected at P5, and pups were killed at P10. Bars represent mean sagittal diameter of the lesions ± SEM. Asterisks indicate statistically significant difference from controls. **p < 0.01 in ANOVA with Dunnett's test. Ctrl, Untreated control; Corticost, corticosterone.
In unstressed C57BL/6 pups, daily administration of 40 mg/kg corticosterone between P1 and P5 induced a significant protection of cortical plate against ibotenate insult (p < 0.01), whereas it had no detectable effect on the underlying white matter lesion (Fig. 5B). This regimen induced a significant increase of circulating corticosterone when measured 8 h after the last subcutaneous injection (1.53 ± 0.09 vs 0.49 ± 0.09 μg/100 ml in control pups; p < 0.0001).
In contrast, daily administration of 3 mg/kg corticosterone to unstressed pregnant C57BL/6 mothers between E13 and E18 significantly increased the size of both cortical plate (p < 0.001) and underlying white matter (p < 0.01) lesions (Fig. 5C). However, a similar corticosterone treatment of unstressed pregnant B6C3F1 GR-I mice did not significantly affect the size of cortical plate and underlying white matter lesions (Fig. 5D).
Effects of CUMS on neonatal ionotropic glutamate receptor expression in C57BL/6 mice
In all experiments, the binding of 3H-MK-801 was increased in membrane extracts from CUMS-exposed pups. The mean density was approximately twofold higher in extracts from CUMS-exposed pups than in controls (5.3 ± 0.8 vs 2.4 ± 0.3 fmol/100 μg of protein; p < 0.01). In contrast, the binding of 3H-AMPA and of 3H-kainate was not significantly affected by CUMS (130.2 ± 15.2 vs 113.1 ± 2.0 fmol/100 μg of protein for 3H-AMPA and 73.9 ± 14.4 vs 67.9 ± 10.8 fmol/100 μg of protein for 3H-kainate).
Effects of CUMS and excitotoxic brain lesions on long-term behavior
The results of ANOVA of latency escape revealed a significant effect of day (F(2,36) = 27.13; p < 0.0001) showing an evolution of performance along the three test periods as well as a day-by-treatment significant interaction (F(2,36) = 4.93; p = 0.0128). Post hoc comparisons of both group performances between day 1 and day 3 of testing showed a significant decrease of latencies in unstressed lesioned (excitotoxic challenge) animals (p < 0.0001) and an absence of evolution of performance with time in stressed lesioned animals. These results revealed that only the unstressed animals were able to learn this spatial memory task (Fig. 6).
Figure 6.

Mean values (in seconds) ± SEM of LE for the 3 d of testing in the Morris water maze. US-L, Unstressed lesioned; S-L, stressed lesioned. Asterisks indicate statistically significant difference from unstressed lesioned animals on day 1. ***p < 0.001 in ANOVA with Tukey's post hoc comparison.
Discussion
The present study showed that prenatal exposure to CUMS exacerbates neonatal excitotoxic brain lesions in murine offspring. This vulnerability to excitotoxicity induced by CUMS seems to be mediated by increased corticosterone levels during gestation and increased NMDA receptor density in the neonatal brain and has long-term functional consequences. These findings suggest that gestational CUMS, which may mimic low-level stress in human pregnancy, could be a novel risk factor for perinatal brain lesions, as may be seen in conditions such as CP.
CUMS is a factor of vulnerability to neonatal excitotoxic brain lesions in mice
One striking result of the present study is the demonstration that exposure of pregnant mice to CUMS is a factor of vulnerability to neonatal excitotoxic brain lesions in the offspring. To our knowledge, this is the first demonstration that gestational stress can exacerbate neonatal brain lesions mimicking some lesions observed in human infants with cerebral palsy.
The mouse brain is lissencephalic, its periventricular white matter thickness is much smaller, and brain maturation is different from in humans; these factors limit extrapolation of observations in rodents to humans. However, excitotoxic white matter lesions in newborn mice mimic several key aspects of human periventricular leukomalacia, a lesion most frequently observed in preterm infants (Marret et al., 1995; Tahraoui et al., 2001). Similarly, excitotoxic gray matter lesions mimic lesions observed in asphyxiated human term neonates (Marret et al., 1995). The fact that CUMS exacerbated both periventricular white matter and gray matter excitotoxic lesions suggests that gestational stress might be a risk factor for preterm and term human neonates at risk of perinatal brain lesions.
This exacerbating effect of gestational CUMS was independent from the gender of the offspring. The interaction between gender and gestational stress is rather complex. Although gestational stress has differential neuroendocrine effects in males and females (Weinstock et al., 1992; McCormick et al., 1995; Jezova et al., 2002), it also contributes to eliminate gender differences usually observed in neurochemical profiles of the prefrontal cortex, amygdala, and hippocampus (Bowman et al., 2004).
The exacerbating effect of gestational CUMS was also independent from the parity of the pregnant mother, suggesting that, in mice, experiencing one or several normal pregnancies does not protect against a subsequent stressed pregnancy in terms of vulnerability of offspring in the neonatal period.
The exacerbating effect of CUMS was observed for different types of glutamate receptor agonists. However, CUMS effects were present in both white matter and gray matter lesions when induced by an NMDA receptor agonist (ibotenate or NMDA), whereas this deleterious effect was observed only in white matter lesions induced by an AMPA-kainate receptor agonist (S-bromowillardiine). These differential effects of CUMS might be a result of the molecular mechanisms by which CUMS induces an increased vulnerability to neonatal excitotoxic lesions (see below) and/or of the specific cellular and molecular mechanisms by which these glutamatergic analogs damage the gray and white matters in the newborn brain (Tahraoui et al., 2001; Mesples et al., 2005; Sfaello et al., 2005). The fact that CUMS increased NMDA receptor binding sites could explain the specificity (see below).
Interestingly, gestational CUMS by itself did not induce any detectable brain lesion or disruption of cytoarchitecture on cresyl violet-stained sections in the offspring (data not shown). Although more sensitive techniques are necessary to fully exclude the lack of effects of gestational CUMS on the developing brain as shown in other models of gestational stress (for review, see Weinstock, 2001), the present data further support the ultramild character of this stress protocol (Pardon et al., 2000).
Rationale for choosing CUMS
Protocols for chronic multiple and unpredictable stress were initiated by Katz et al. (1981), later adapted by Willner et al. (1987), and more recently modified (CUMS) for gestating females by Pardon et al. (2000), who excluded the noxious and metabolic stressors and included long stress-free intervals between stressors. These studies have shown that rodents exposed to chronic, mild, and unpredictable stressors exhibited marked behavioral disturbances. When adapted to gestation, this type of stress regimen could be of interest for the study of biological and behavioral disorders triggered by daily life environmental factors during gestation (Pardon et al., 2000; Dole et al., 2003).
Mechanistic basis for the increased susceptibility conferred by CUMS to neonatal excitotoxic brain lesions
Role of glucocorticoids
Effects of gestational stress on neonatal excitotoxic lesions could be linked to disturbances in the function of the hypothalamo–pituitary–adrenal axis. Three lines of evidence strongly support this hypothesis: (1) as previously observed in other models of gestational stress (Zarrow et al., 1970; Barbazanges et al., 1996), in the present study, corticosterone levels were significantly increased in stressed mothers and in the offspring when compared with unstressed animals; (2) the effects of gestational stress on neonatal excitotoxic lesions were totally abolished in transgenic B6C3F1 GR-I mice, which have reduced levels of glucocorticoid receptors; and (3) administration of corticosterone to pregnant mothers mimicked the effects of gestational stress on neonatal excitotoxic lesions.
Corticosteroids have been largely used for inducing lung maturation in fetuses who are expected to be born preterm and for preterm infants with respiratory diseases. However, in recent years, several epidemiological and experimental studies have suggested the potential neurotoxic effects of perinatal steroid therapy on the immature brain (Baud, 2004a,b). Therefore, much effort has been directed toward finding the optimal regimen that may reduce lung disease without incurring significant brain injury in fetuses and preterm infants (Friedman and Shinwell, 2004).
Postnatal administration of exogenous corticosterone to newborn pups from unstressed mothers failed to mimic the deleterious effects of gestational stress on excitotoxic lesions. These results suggest that the level of circulating corticosterone at the time of the excitotoxic insult is not a key factor, but rather, exposure to abnormally increased levels of corticosterone during brain development perturbs some specific step(s) of neocorticogenesis, leading to an increased vulnerability to a secondary excitotoxic insult (double-hit hypothesis).
Accordingly, we first explored the potential implication of disturbances in BDNF production in response to gestational stress, because immobilization stress or exogenously applied corticosterone markedly reduce BDNF mRNA and protein levels in the adult hippocampus (Barbany and Persson, 1992; Smith et al., 1995; Schaaf et al., 1998). In contrast to these paradigms of stress in the adult, CUMS did not reduce BDNF expression in the neonatal brain (data not shown). We then studied the expression of ionotropic glutamate receptors in response to gestational stress.
Ionotropic glutamate receptor expression
Social stress and corticosterone have been previously shown to upregulate NMDA receptor subunits NR2A and NR2B in the adult lizard (Meyer et al., 2004), whereas adrenalectomy decreases NMDA receptors in the rat hippocampus (Mangat et al., 1998). In contrast, other studies performed in adult mice have shown that chronic restraint stress induces a reduction of NMDA receptors and dendritic spines in hippocampus (Watanabe et al., 1995; Pawlak et al., 2005), suggesting that different types of stressors can have differential effects on NMDA receptor expression. The present study showed that CUMS increases NMDA receptor binding sites in the neonatal brain. Although additional studies are necessary to unravel the precise underlying mechanism linking CUMS to the increased density of NMDA receptors, one can make the hypothesis that the CUMS-induced increased levels of corticosterone during gestation lead to changes in cortical NMDA receptor densities.
This observed increase in NMDA receptors in the offspring most likely participates in the CUMS-induced exacerbation of the excitotoxic lesions after ibotenate or NMDA injection. Furthermore, several studies have shown that glucocorticoids are able to activate NMDA receptors by different direct and indirect mechanisms, including activation of voltage-dependent calcium channels or modulation of calcium influx (Kim et al., 1996; Coussens et al., 1997). The potential implication of these mechanisms in the present model remains to be determined.
Although we did not detect significant changes in the neopallial expression of AMPA and kainate receptor after CUMS, we cannot exclude the hypothesis that CUMS affects AMPA and/or kainate receptor efficacy in the white matter. The CUMS-induced increase in NMDA receptor density could also participate in the exacerbation of white matter lesions after S-bromowillardiine injection. Indeed, we previously showed that neonatal white matter lesions, but not gray matter lesions, induced by AMPA-kainate receptor agonists are partly blocked by NMDA receptor antagonists, suggesting that activation of AMPA-kainate receptors leads to glutamate release, which will in turn activate all types of white matter glutamatergic receptors, including NMDA receptors (Tahraoui et al., 2001).
Conclusion
Altogether, our data show that maternal chronic stress applied during gestation can exacerbate excitotoxic neonatal brain damage in mice. Deleterious effects of gestational stress involve increased production of corticosterone during gestation and increased neonatal expression of NMDA receptors. Assuming that data obtained for the present murine model can be extrapolated to human neonates, maternal chronic stress should be considered as a factor of vulnerability for CP.
Footnotes
This work was supported by Inserm, the Centre National de la Recherche Scientifique, the Universités Paris 6 and 7, the Fondation Grace de Monaco, the association Société d'Etudes et de Soins pour les Enfants Paralysés et Polymalformés, the Assistance Publique-Hôpitaux de Paris, and the Académie Nationale de Médecine. We thank Leslie Scwhendimann for her excellent technical assistance.
References
- Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbany G, Persson H. Regulation of neurotrophin mRNA expression in the rat brain by glucocorticoids. Eur J Neurosci. 1992;4:396–403. doi: 10.1111/j.1460-9568.1992.tb00888.x. [DOI] [PubMed] [Google Scholar]
- Barbazanges A, Piazza PV, Le Moal M, Maccari S. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci. 1996;16:3943–3949. doi: 10.1523/JNEUROSCI.16-12-03943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud O. Antenatal corticosteroid therapy: benefits and risks. Acta Paediatr. 2004a;93(Suppl):6–10. doi: 10.1111/j.1651-2227.2004.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Baud O. Postnatal steroid treatment and brain development. Arch Dis Child Fetal Neonatal Ed. 2004b;89:F96–F100. doi: 10.1136/adc.2003.028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutta AT, Anand KJ. Vulnerability of the developing brain. Neuronal mechanisms. Clin Perinatol. 2002;29:357–372. doi: 10.1016/s0095-5108(02)00011-8. [DOI] [PubMed] [Google Scholar]
- Bowman RE, MacLusky NJ, Sarmiento Y, Frankfurt M, Gordon M, Luine VN. Sexually dimorphic effects of prenatal stress on cognition, hormonal responses, and central neurotransmitters. Endocrinology. 2004;145:3778–3787. doi: 10.1210/en.2003-1759. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon C. What role does sensory perception play in the onset and maintenance of pup care behavior in laboratory rodents? Eur Bull Cogn Psychol. 1988;8:53–94. [Google Scholar]
- Cohen-Salmon C, Carlier M, Roubertoux P. Differences in patterns of pup care in Mus musculus domesticus. II. Effects of parity on eight inbred strains. Biol Behav. 1982;7:337–346. doi: 10.1016/s0163-1047(82)91213-4. [DOI] [PubMed] [Google Scholar]
- Coussens CM, Kerr DS, Abraham WC. Glucocorticoid receptor activation lowers the threshold for NMDA-receptor-dependent homosynaptic long-term depression in the hippocampus through activation of voltage-dependent calcium channels. J Neurophysiol. 1997;78:1–9. doi: 10.1152/jn.1997.78.1.1. [DOI] [PubMed] [Google Scholar]
- Dammann O, Kuban KC, Leviton A. Perinatal infection, fetal inflammatory response, white matter damage, and cognitive limitations in children born preterm. Ment Retard Dev Disabil Res Rev. 2002;8:46–50. doi: 10.1002/mrdd.10005. [DOI] [PubMed] [Google Scholar]
- Dijkstra I, Tilders FJ, Aguilera G, Kiss A, Rabadan-Diehl C, Barden N, Karanth S, Holsboer F, Reul JM. Reduced activity of hypo-thalamic corticotropin-releasing hormone neurons in transgenic mice with impaired glucocorticoid receptor function. J Neurosci. 1998;18:3909–3918. doi: 10.1523/JNEUROSCI.18-10-03909.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dole N, Savitz DA, Hertz-Picciotto I, Siega-Riz AM, McMahon MJ, Buekens P. Maternal stress and preterm birth. Am J Epidemiol. 2003;157:14–24. doi: 10.1093/aje/kwf176. [DOI] [PubMed] [Google Scholar]
- Dommergues MA, Patkai J, Renauld JC, Evrard P, Gressens P. Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol. 2000;47:54–63. [PubMed] [Google Scholar]
- Eklind S, Mallard C, Leverin AL, Gilland E, Blomgren K, Mattsby-Baltzer I, Hagberg H. Bacterial endotoxin sensitizes the immature brain to hypoxic–ischaemic injury. Eur J Neurosci. 2001;13:1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- Friedman S, Shinwell ES. Prenatal and postnatal steroid therapy and child neurodevelopment. Clin Perinatol. 2004;31:529–544. doi: 10.1016/j.clp.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Gressens P, Marret S, Hill JM, Brenneman DE, Gozes I, Fridkin M, Evrard P. Vasoactive intestinal peptide prevents excitotoxic cell death in the murine developing brain. J Clin Invest. 1997;100:390–397. doi: 10.1172/JCI119545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins LM, Beaver KM, Jane DE, Taylor PM, Sunter DC, Roberts PJ. Binding of the new radioligand (S)-[3H]AMPA to rat brain synaptic membranes: effects of a series of structural analogues of the non-NMDA receptor agonist willardiine. Neuropharmacology. 1995;34:405–410. doi: 10.1016/0028-3908(94)00157-n. [DOI] [PubMed] [Google Scholar]
- Husson I, Mesples B, Bac P, Vamecq J, Evrard P, Gressens P. Melatoninergic neuroprotection of the murine periventricular white matter against neonatal excitotoxic challenge. Ann Neurol. 2002;51:82–92. doi: 10.1002/ana.10072. [DOI] [PubMed] [Google Scholar]
- Husson I, Rangon CM, Lelievre V, Bemelmans AP, Sachs P, Mallet J, Kosofsky BE, Gressens P. BDNF-induced white matter neuroprotection and stage-dependent neuronal survival following a neonatal excitotoxic challenge. Cereb Cortex. 2005;15:250–261. doi: 10.1093/cercor/bhh127. [DOI] [PubMed] [Google Scholar]
- Jezova D, Skultetyova I, Makatsori A, Moncek F, Duncko R. Hypothalamo-pituitary-adrenocortical axis function and hedonic behavior in adult male and female rats prenatally stressed by maternal food restriction. Stress. 2002;5:177–183. doi: 10.1080/1025389021000010512. [DOI] [PubMed] [Google Scholar]
- Katz RJ, Roth KA, Carroll BJ. Acute and chronic stress effects on open field activity in the rat: implications for a model of depression. Neurosci Biobehav Rev. 1981;5:247–251. doi: 10.1016/0149-7634(81)90005-1. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF. Behavioral stress modifies hippocampal plasticity through N-methyl-d-aspartate receptor activation. Proc Natl Acad Sci USA. 1996;93:4750–4753. doi: 10.1073/pnas.93.10.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofman O. The role of prenatal stress in the etiology of developmental behavioural disorders. Neurosci Biobehav Rev. 2002;26:457–470. doi: 10.1016/s0149-7634(02)00015-5. [DOI] [PubMed] [Google Scholar]
- Lanfumey L, Pardon MC, Laaris N, Joubert C, Hanoun N, Hamon M, Cohen-Salmon C. 5-HT1A autoreceptor desensitization by chronic ultramild stress in mice. NeuroReport. 1999;10:3369–3374. doi: 10.1097/00001756-199911080-00021. [DOI] [PubMed] [Google Scholar]
- Lelievre V, Hu Z, Byun JY, Ioffe Y, Waschek JA. Fibroblast growth factor-2 converts PACAP growth action on embryonic hindbrain precursors from stimulation to inhibition. J Neurosci Res. 2002;67:566–573. doi: 10.1002/jnr.10153. [DOI] [PubMed] [Google Scholar]
- Mangat HS, Islam A, Heigenskold C, Mustafa A, Winblad B, Adem A. Long-term adrenalectomy decreases NMDA receptors in rat hippocampus. NeuroReport. 1998;9:2011–2014. doi: 10.1097/00001756-199806220-00018. [DOI] [PubMed] [Google Scholar]
- Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- McCormick CM, Smythe JW, Sharma S, Meaney MJ. Sex-specific effects of prenatal stress on hypothalamic-pituitary-adrenal responses to stress and brain glucocorticoid receptor density in adult rats. Brain Res Dev Brain Res. 1995;84:55–61. doi: 10.1016/0165-3806(94)00153-q. [DOI] [PubMed] [Google Scholar]
- Mesples B, Plaisant F, Fontaine RH, Gressens P. Pathophysiology of neonatal brain lesions: lessons from animal models of excitotoxicity. Acta Paediatr. 2005;94:185–190. doi: 10.1111/j.1651-2227.2005.tb01888.x. [DOI] [PubMed] [Google Scholar]
- Meyer WN, Keifer J, Korzan WJ, Summers CH. Social stress and corticosterone regionally upregulate limbic N-methyl-d-aspartatereceptor (NR) subunit type NR2A and NR2B in the lizard Anolis carolinensis. Neuroscience. 2004;128:675–684. doi: 10.1016/j.neuroscience.2004.06.084. [DOI] [PubMed] [Google Scholar]
- Pardon M, Gerardin P, Joubert C, Perez-Diaz F, Cohen-Salmon C. Influence of prepartum chronic ultramild stress on maternal pup care behavior in mice. Biol Psychiatry. 2000;47:858–863. doi: 10.1016/s0006-3223(99)00253-x. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci USA. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin MC, Pothier F, Barden N. Antidepressant drug action in a transgenic mouse model of the endocrine changes seen in depression. Mol Pharmacol. 1992;42:991–995. [PubMed] [Google Scholar]
- Reynolds IJ, Sharma TA. The use of ligand binding in assays of NMDA receptor function. Methods Mol Biol. 1999;128:93–102. doi: 10.1385/1-59259-683-5:93. [DOI] [PubMed] [Google Scholar]
- Schaaf MJ, de Jong J, de Kloet ER, Vreugdenhil E. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–120. doi: 10.1016/s0006-8993(98)01010-5. [DOI] [PubMed] [Google Scholar]
- Sfaello I, Baud O, Arzimanoglou A, Gressens P. Topiramate prevents excitotoxic damage in the newborn rodent brain. Neurobiol Dis. 2005;20:837–848. doi: 10.1016/j.nbd.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kim SY, Kvetnansky R. Stress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitary. Endocrinology. 1995;136:3743–3750. doi: 10.1210/endo.136.9.7649080. [DOI] [PubMed] [Google Scholar]
- Tahraoui SL, Marret S, Bodenant C, Leroux P, Dommergues MA, Evrard P, Gressens P. Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol. 2001;11:56–71. doi: 10.1111/j.1750-3639.2001.tb00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh BR, Mulder EJ, Mennes M, Glover V. Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: links and possible mechanisms. A review. Neurosci Biobehav Rev. 2005;29:237–258. doi: 10.1016/j.neubiorev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Perinatal brain injury: from pathogenesis to neuroprotection. Ment Retard Dev Disabil Res Rev. 2001;7:56–64. doi: 10.1002/1098-2779(200102)7:1<56::AID-MRDD1008>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Weiland NG, McEwen BS. Effects of adrenal steroid manipulations and repeated restraint stress on dynorphin mRNA levels and excitatory amino acid receptor binding in hippocampus. Brain Res. 1995;680:217–225. doi: 10.1016/0006-8993(95)00235-i. [DOI] [PubMed] [Google Scholar]
- Weinstock M. Alterations induced by gestational stress in brain morphology and behaviour of the offspring. Prog Neurobiol. 2001;65:427–451. doi: 10.1016/s0301-0082(01)00018-1. [DOI] [PubMed] [Google Scholar]
- Weinstock M, Matlina E, Maor GI, Rosen H, McEwen BS. Prenatal stress selectively alters the reactivity of the hypothalamic-pituitary adrenal system in the female rat. Brain Res. 1992;595:195–200. doi: 10.1016/0006-8993(92)91049-k. [DOI] [PubMed] [Google Scholar]
- Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology (Berl) 1987;93:358–364. doi: 10.1007/BF00187257. [DOI] [PubMed] [Google Scholar]
- Zarrow MX, Philpott JE, Denenberg VH. Passage of 14C-4-corticosterone from the rat mother to the foetus and neonate. Nature. 1970;226:1058–1059. doi: 10.1038/2261058a0. [DOI] [PubMed] [Google Scholar]