

Abstract
Drug addiction places an enormous burden on society through its repercussions on crime rate and healthcare. Repeated exposure to drugs of abuse causes cellular adaptations in specific neuronal populations that ultimately can lead to a state of addiction. In the present study, we have identified a novel molecule “shati” from the nucleus accumbens (NAc) of mice treated with methamphetamine (METH) using the PCR-select complementary DNA subtraction method. Moreover, we investigated whether shati is involved in METH-induced hyperlocomotion, sensitization, and conditioned place preference (CPP). METH induced expression of shati mRNA dose dependently via dopamine (DA) receptors. We prepared antibodies against shati and, using them, found shati to be expressed in neuronal cells of the mouse brain. Treatment with the shati antisense oligonucleotide (shati-AS), which significantly inhibited the expression of shati mRNA, enhanced the acute METH response, METH-induced behavioral sensitization, and CPP. Blockage of shati mRNA by shati-AS potentiated the METH-induced increase of DA overflow in the NAc and the METH-induced decrease in synaptosomal and vesicular DA uptake in the midbrain. These results suggest that a novel molecule shati is involved in the development of METH-induced hyperlocomotion, sensitization, and CPP. The functional roles of shati in METH-regulated behavioral alternations are likely to be mediated by its inhibitory effects on the METH-induced increase of DA overflow in the NAc and the METH-induced decrease in DA uptake in the midbrain.
Keywords: shati, methamphetamine, behavioral sensitization, conditioned place preference, dopamine, addiction
Introduction
In terms of lost lives and productivity, drug dependence remains one of the most serious threats to the public health of a nation (Nestler, 2002). Drugs of abuse, including methamphetamine (METH), modulate the activity of mesolimbic dopaminergic neurons, projecting from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) (Koob, 1992; Wise, 1996b; Koob et al., 1998). The psychostimulatory effects of METH are associated with an increase in extracellular dopamine (DA) levels in the brain, by facilitating the release of DA from presynaptic nerve terminals and inhibiting reuptake (Heikkila et al., 1975; Seiden et al., 1993; Giros et al., 1996). In rodent, augmentation of behavioral responses to psychostimulants is observed during and after their repeated administration. Therefore, it has been proposed that activity-dependent synaptic plasticity and remodeling of the mesolimbic dopaminergic system may play a crucial role in drug dependence (Nestler, 2001; Yamada and Nabeshima, 2004).
Using cDNA microarrays, changes in the mRNA expression profile in relevant brain regions (e.g., NAc) have been assessed after chronic administration of abused drugs (Douglass and Daoud, 1996; Cha et al., 1997; Wang et al., 1997). Evidence from this line of research has implicated nuclear factor-κB (Ang et al., 2001) and ΔFosB (Zachariou et al., 2006) in signal transduction pathways that modulate behavioral effects induced by drugs and contribute to long-term neuronal changes associated with dependence (Laakso et al., 2002). To elucidate the mechanism, caused by chronic drug abuse, of stable changes in the brain that play a role in the long-lasting behavioral abnormalities of dependent subjects, the candidates for drug-dependence-related genes whose expression was altered by repeated administration of METH or morphine (MOR) were screened by using cDNA microarray. Recently, there are many studies that showed that cytokines/neurotrophic factors and extracellular matrix/proteases play critical roles in activity-dependent synaptic plasticity and remodeling of the mesocorticolimbic dopaminergic system (Horger et al., 1999; Messer et al., 2000; Mizoguchi et al., 2007). We found that tumor necrosis factor-α (TNF-α) plays a neuroprotective role in METH-induced dependence and neurotoxicity (Nakajima et al., 2004) and reduces MOR-induced rewarding effects and behavioral sensitization (Niwa et al., 2007a,d). Furthermore, the rewarding effects and sensitization induced by METH and MOR are attenuated by Leu-Ile, an inducer of TNF-α, and glial cell line-derived neurotrophic factor (GDNF) (Niwa et al., 2007a–d). The tissue plasminogen activator (tPA)–plasmin system potentiates the rewarding and locomotor-stimulating effects of METH, MOR, and nicotine by regulating release of DA (Nagai et al., 2004, 2005a,b, 2006). However, the exact neuronal circuits and molecular cascade essential for drug dependence remain unclear. Therefore, we attempt to explore the novel molecules that play more critical roles in drug dependence, because the functions of molecules targeted by DNA microarray screening have been already well known.
In the present study, we identified a novel molecule “shati” from the NAc of mice treated with METH using the PCR-select cDNA subtraction method, which is a differential and epochal cloning technique. Moreover, we demonstrated that shati is involved in the METH-induced hyperlocomotion, sensitization, and conditioned place preference (CPP).
Materials and Methods
Animals.
The male C57BL/6J inbred mice were obtained from SLC Japan (Hamamatsu, Japan). Animals were housed in plastic cages and kept in a temperature-, humidity-, and light-controlled room (23 ± 1°C; 50 ± 5% humidity; 12 h light/dark cycle starting at 8:00 A.M.) and had ad libitum access to food and water, except during behavioral experiments. All animal care and use was in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Nagoya University School of Medicine. Animals were treated according to the Guidelines of Experimental Animal Care issued from the Office of the Prime Minister of Japan.
PCR-select cDNA subtraction.
Mice were administered METH (2 mg/kg, s.c.) or saline for 6 d and took NAc 2 h after the last injection of METH. PCR-select cDNA subtraction (Clontech, Palo Alto, CA) was performed using a previously established procedure (Diatchenko et al., 1996; Gurskaya et al., 1996) to detect the genes in the NAc affected by METH treatment. Briefly, they involve hybridization of cDNA from one population (tester; METH-treated NAc) to excess of mRNA (cDNA) from other population (driver; saline-treated NAc) and then separation of the unhybridized fraction (target) from hybridized common sequences. Total RNAs were extracted by RNeasy Max (Qiagen, Hilden, Germany). For each subtraction, we performed two PCR amplifications. Products from the secondary PCRs were inserted into pCRII using a T/A cloning kit (Invitrogen, Carlsbad, CA). Plasmid or cosmid DNAs were prepared using QIAwell 8 Plus kit (Qiagen) according to the protocol of the manufacturer. Nucleic acid homology searches were performed using the BLAST (basic local alignment search tool) program through e-mail servers at the National Center for Biotechnology Information (NCBI) (National Institutes of Health, Bethesda, MD).
Structure models.
Homology modeling for C-terminal domain of shati was established using Molecular Operating Environment (MOE) software (Chemical Computing Group, Montreal, Quebec, Canada). Molecular mechanics calculations were performed by using an MMFF94x force field. Docking simulations of acetyl-CoA or ATP with shati protein were also examined using MOE software (Chemical Computing Group) to calculate the interactive potential energy of molecules.
Reverse transcription-PCR and real-time reverse transcription-PCR.
Mice were administered METH (0.3, 1, and 2 mg/kg, s.c., once a day for 3 or 6 d) and decapitated 2 h after the last injection of METH. In the real-time reverse transcription (RT)-PCR experiment on the antagonism of METH-induced shati mRNA expression, mice were treated with the DA D1-like receptor antagonist R(+)-SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine] (0.1 mg/kg, i.p.) or DA D2-like receptor antagonist raclopride (2 mg/kg, i.p.) 30 min before METH (2 mg/kg, s.c.) once a day for 6 d. Functionally, R(+)-SCH23390 (0.1–0.5 mg/kg) is a potent blocker of stereotyped behaviors and increased locomotion induced by amphetamine or apomorphine (Christensen et al., 1984; Napier et al., 1986). The increase in TNF-α or tPA mRNA expression in the NAc induced by METH is inhibited by pretreatment with either R(+)-SCH23390 (0.1 or 0.5 mg/kg, i.p.) or raclopride (2 mg/kg, i.p.) (Nakajima et al., 2004; Nagai et al., 2005a). R(+)-SCH23390 at the dose of 0.1 mg/kg, not 0.03 mg/kg, significantly inhibits the hyperphosphorylation of extracellular signal-regulated kinase 1/2 in the NAc and striatum evoked by METH-induced CPP as well as the expression of CPP in METH-treated animals (Mizoguchi et al., 2004). Depending on these evidences, we selected the doses of R(+)-SCH23390 at 0.1 mg/kg and raclopride at 2 mg/kg.
Total RNA was isolated using an RNeasy kit (Qiagen) and converted into cDNA using a SuperScript First-Strand System for RT-PCR kit (Invitrogen). The primers used for RT-PCR were as follows: 5′-CTTGCCTCCCCAGCCCATCA-3′ (forward-1; base pairs 1987–2006) and 5′-CTGGGGGCCAGGGTTCTGCT-3′ (reverse-1; base pairs 2147–2166) for set of sequences 1; 5′-GGGTGGCCGGGTAGGTGGAA-3′ (forward-2; base pairs 2909–2928) and 5′-GGCAGTGCCCAGCCCTTCCT-3′ (reverse-2; base pairs 3073–3092) for set of sequences 2; and 5′-TGTACATTCCTCCCTGGTGGTG-3′ (forward-3; base pairs 3521–3542) and 5′-AAATCTGAGAGCTGCAAGAAAATAGGG-3′ (reverse-3; base pairs 3594–3620) for set of sequences 3 (Table 1). The amplification consisted of an initial step (95°C for 5 min) and then 35 cycles of denaturation for 30 s at 94°C and annealing for 1 min at 70, 71, and 65°C in a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA). The levels of shati and TNF-α mRNA were determined by real-time RT-PCR using a TaqMan probe. The 18S ribosomal RNA was used as the internal control (PE Applied Biosystems, Foster City, CA). The mouse shati primers used for real-time RT-PCR were as follows: 5′-TGTAAACACCCCTAAAGTGCCCT-3′ (forward; base pairs 2967–2989) and 5′-TCAATCCTGCATACAAGGAATCAA-3′ (reverse; bare pairs 3022–3045); and TaqMan probe, 5′-CACAGTCTGTGAGGCTCAGGTTGCCC-3′ (probe; base pairs 2995–3020). The amplification consisted of an initial step (95°C for 5 min) and then 40 cycles of denaturation for 30 s at 95°C and annealing for 1 min at 59°C in an iCycle iQ Detection System (Bio-Rad, Hercules, CA). The expression levels were calculated as described previously (Wada et al., 2000).
Table 1.
Primers sequences and their targets for RT-PCR
| Primer | Sequence | Target (bp) |
|---|---|---|
| 1 | ||
| Forward | 5′-CTTGCCTCCCCAGCCCATCA-3′ | 1987–2006 |
| Reverse | 5′-CTGGGGGCCAGGGTTCTGCT-3′ | 2147–2166 |
| 2 | ||
| Forward | 5′-GGGTGGCCGGGTAGGTGGAA-3′ | 2909–2928 |
| Reverse | 5′-GGCAGTGCCCAGCCCTTCCT-3′ | 3073–3092 |
| 3 | ||
| Forward | 5′-TGTACATTCCTCCCTGGTGGTG-3′ | 3521–3542 |
| Reverse | 5′-AAATCTGAGAGCTGCAAGAAAATAGGG-3′ | 3594–3620 |
The amplification consisted of an initial step (95°C for 5 min) and then 35 cycles of denaturation for 30 s at 94 °C and annealing for 1 min at 70, 71, and 65°C.
Immunohistochemistry.
Two antibodies against the peptide of the hypothetical protein, CNTAFRGLRQHPRTQLL (S-3) and CMSVDSRFRGKGIAKALG (S-4), unique to shati were generated. These peptides were conjugated to the keyhole limpet hemocyanin and injected into rabbits six times at 1 week intervals. Serum was taken from the rabbits 1 week after the final injection of these peptides. The serum was diluted 200 times used for the immunostaining.
For immunohistochemical analysis, mice were killed 24 h after repeated treatment with METH (2 mg/kg, s.c., once a day for 6 d). The brains were sliced at 20 μm in the cryostat. Polyclonal rabbit anti-S-3 or S-4 antibody (1:200), monoclonal mouse anti-neuron-specific nuclear antigen (NeuN) antibody (1:200; Chemicon, Temecula, CA), and monoclonal mouse anti-glial fibrillary acidic protein (GFAP) antibody (1:200; Chemicon) served as primary antibodies. Goat anti-mouse Alexa Fluor 546 (1:1000; Invitrogen) and goat anti-rabbit Alexa Fluor 488 (1:1000; Invitrogen) were used as secondary antibodies. Each stained slice was observed under a fluorescence microscope (Axioskoop 2 plus; Zeiss, Jena, Germany) and checked with Axiovision 3.0 systems (Zeiss).
Shati-antisense oligonucleotide treatment.
Mice were anesthetized with pentobarbital (40 mg/kg, i.p.) and placed in a stereotaxic apparatus. The infusion cannula was connected to a miniosmotic pump (total capacity was 90 μl, Alzet 1002; Alza, Palo Alto, CA) filled with shati-antisense oligonucleotide (shati-AS) and -scramble oligonucleotide (shati-SC) and was implanted into the right ventricle [anteroposterior (AP) −0.5 mm, mediolateral (ML) +1.0 mm from the bregma, and dorsoventral (DV) −2.0 mm from the skull, according to the atlas of Franklin and Paxinos (1997)]. No histological or mechanical disruption was produced by implantation of the infusion cannula (data not shown). Phosphorothionate oligonucleotides were custom synthesized at Nisshinbo Biotechnology (Tokyo, Japan) and dissolved in artificial CSF (in mm: 147 NaCl, 3 KCl, 1.2 CaCl2, and1.0 MgCl2, pH 7.2). We used shati-SC as a control of shati-AS, because we should deny the secondary effects on other genes or toxic effects, and we selected the design of shati-AS, which does not affect the other genes and already have been identified. The oligonucleotides were phosphorothionated at the three bases of both 5′ and 3′ ends, which results in increased stability and less toxicity. The sequences of shati-AS and shati-SC were 5′-TCTTCGTCTCGCAGACCATGTCG-3′ and 5′-GGTCTGCTACACTGCTGCTAGTC-3′, respectively. Shati-AS and shati-SC were continuously infused into the cerebral ventricle at a dose of 1.8 nmol/6 μl per day (flow rate, 0.25 μl/h). Additionally, shati-SC was used as a control. Three days after the start of oligonucleotide infusion, mice were subjected to METH treatment for sensitization.
Locomotor activity.
Locomotor activity was measured using an infrared detector (Neuroscience Company, Tokyo, Japan) in a plastic box (32 × 22 × 15 cm high) and determined as described previously (Nakajima et al., 2004; Niwa et al., 2007b,d). One day after the start of oligonucleotide infusion, mice were habituated for 3 h in the box for 2 d and then administered METH (1 mg/kg, s.c.) or saline once a day for 5 d. Locomotor activity was measured for 2 h immediately after the METH or saline administration.
In vivo microdialysis.
Mice were anesthetized with sodium pentobarbital, and a guide cannula (AG-8; EICOM, Kyoto, Japan) was implanted into the NAc (AP +1.7 mm, ML +0.8 mm mediolateral from the bregma, and DV −4.0 mm from the skull) according to the atlas of Franklin and Paxinos (1997) and secured to the skull using stainless steel screws and dental acrylic cement. Mice were administered METH (1 mg/kg, s.c.) 3 d after implantation of the guide cannula and the start of oligonucleotide infusion. One day after METH treatment for 2 d, a dialysis probe (AI-8-1, 1 mm membrane length; EICOM) was inserted through the guide cannula and perfused continuously with CSF (in mm: 147 NaCl, 4 KCl, and 2.3 CaCl2) at a flow rate of 1.0 μl/min. Dialysate was collected in 20 min fractions and injected into the HPLC system (EICOM) for the measurement of DA levels. Three samples were used to establish baseline levels of DA before the administration of METH (1 mg/kg, s.c.).
Synaptosomal [3H]DA uptake.
Three days after the start of oligonucleotide infusion, mice were subjected to METH treatment once a day for 3 d. Mice were decapitated 1 h after the final METH treatment. Midbrain synaptosomal [3H]DA uptake was determined as described previously (Fleckenstein et al., 1997; Nakajima et al., 2004; Niwa et al., 2007b). The final concentration of [3H]DA (PerkinElmer, Wellesley, MA) was 5 nm. Samples were incubated at 37°C for 4 min, and then ice-cold Krebs–Ringer's solution containing 10 μm GBR12909 [1-(2[bis(4-fluorophenyl-)methoxy]ethyl)-4-(3-phenylpropyl)piperazine] bimesylate hydrate] (Sigma, St. Louis, MO), a specific DA uptake inhibitor, was added. Nonspecific values were determined in the presence of 100 μm GBR12909 during the incubation. The radioactivity trapped on filters was measured with a liquid scintillation counter (Beckman Coulter, Fullerton, CA).
Vesicular [3H]DA uptake.
Vesicular [3H]DA uptake was determined as described by Erickson et al. (1990). Synaptosomes were prepared as described by Nakajima et al. (2004). Vesicular [3H]DA uptake was performed by incubating synaptic vesicle samples (15 μg protein/100 μl) at 30°C for 4 min in assay buffer (in mm: 25 HEPES, 100 potassium tartrate, 1.7 ascorbic acid, 0.05 EGTA, 0.1 EDTA, and 2 ATP-Mg2+, pH 7.0) in the presence of 30 nm [3H]DA (PerkinElmer). The reaction was terminated by the addition of 1 ml of cold wash buffer (assay buffer containing 2 mm MgSO4 substituted for the ATP-Mg2+, pH 7.0) and rapid filtration. Nonspecific values were determined by measuring vesicular [3H]DA uptake at 4°C. The radioactivity was measured with a liquid scintillation counter (Beckman Coulter).
Conditioned place preference.
The apparatus used for the place conditioning task consisted of two compartments: a transparent Plexiglas box and a black Plexiglas box (both 15 × 15 × 15 cm high). To enable mice to distinguish easily the two compartments, the floors of the transparent and black boxes were covered with white plastic mesh and black frosting Plexiglas, respectively. Each box could be divided by a sliding door (10 × 15 cm high). The place conditioning paradigm was performed by using a previously established procedure with a minor modification (Noda et al., 1998; Schechter and Calcagnetti, 1998; Niwa et al., 2007a,b,d). In the preconditioning test, the sliding door was opened, and the mouse was allowed to move freely between both boxes for 15 min once a day for 3 d. On the third day of the preconditioning test, we measured the time that the mouse spent in the black and transparent boxes by using a Scanet SV-20 LD (Melquest, Toyama, Japan). The box in which the mouse spent the most time was referred to as the “preferred side” and the other box as the “nonpreferred side.” Conditioning was performed during 6 successive days. Mice were given METH or saline in the apparatus with the sliding door closed. That is, a mouse was subcutaneously given METH and put in its nonpreferred side for 20 min. On the next day, the mouse was given saline and placed opposite the drug conditioning site for 20 min. These treatments were repeated for three cycles (6 d). In the postconditioning test, the sliding door was opened, and we measured the time that the mouse spent in the black and transparent boxes for 15 min, using the Scanet SV-20 LD. Place conditioning behavior was expressed by Post-Pre, which was calculated as: [(postvalue) − (prevalue)], where postvalue and prevalue were the difference in time spent at the drug conditioning and the saline conditioning sites in the postconditioning and preconditioning tests, respectively.
Statistical analysis.
All data were expressed as means ± SE. Statistical differences between two groups were determined with Student's t test. Statistical differences among more than three groups were determined using a one-way ANOVA, two-way ANOVA, or an ANOVA with repeated measures (two or three-factor), followed by the Bonferroni's multiple comparison test (Bonferroni's correction; 3, 6, 15, and 36 comparisons in 3, 4, 6, and 9 groups, respectively). p < 0.05 was regarded as statistically significant.
Nucleotide sequences.
The DNA Data Bank of Japan/GenBank/European Molecular Biology Laboratory accession number for the primary nucleotide sequence of shati is DQ174094.
Results
Identification of shati
The reasons why we pursued shati for intensive investigation arose from our preliminary findings with the PCR-select cDNA subtraction method to detect the genes in the NAc affected by METH treatment: mice were administrated METH (2 mg/kg, s.c.) or saline for 6 d, and shati mRNA production in the NAc was found to increase by 640% in METH-treated mice with robust behavioral sensitization compared with saline-treated mice (data not shown). The sequence of cDNA was completely matched to accession number NM_001001985 of NCBI gene bank (the gene record was replaced by accession number NM_001001985.2 on April, 10, 2005). The sequence has been identified by the Mammalian Gene Collection Program Team (Strausberg et al., 2002). Blackshaw et al. (2004) has demonstrated the extended cDNA sequence by serial analysis of gene expression methods, which provides an unbiased and nearly comprehensive readout of gene expression and that the gene was for one of the proteins related to the retina development. We named this novel molecule shati after the symbol at Nagoya castle in Japan. The sequence is translated to a protein LOC269642 (accession number is NP_001001985.1 and 2; 001001985.1 was a part of 001001985.2) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Characterization of shati
Homology modeling for C-terminal domain of shati was established using MOE software (Chemical Computing Group) (Fig. 1A,B). Red character in Figure 1A showed homology modeling of shati. From motif analysis of shati, shati contained the sequence of GCN5-related N-acetyltransferase (GCAT) (Fig. 1C). Underlined character in Figure 1A showed GNAT motif. Docking simulations of acetyl-CoA or ATP with shati protein were also examined using MOE software (Chemical Computing Group) to calculate the interactive potential energy of molecules. Shati also contained acetyl-CoA binding or ATP binding site, because the analysis showed the lowest interactive potential energy of shati with acetyl-CoA or ATP, −301 and −322 kcal, respectively (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Docking simulations of shati with DA, DNA binding site, and nuclear localization signals showed too high interactive potential energy of molecules or no domain.
Figure 1.

Characterization of shati. A, The sequence of shati. The red character showed homology modeling of shati. The underlined character showed GCN5-related N-acetyltransferase motif. B, Homology modeling for C-terminal domain of shati. C, Homology modeling and motif analysis of shati. Shati has the sequence of GCAT. Red ribbon, Homology model of shati; sphere, acetyl-CoA analyzed by x-ray crystallography; green ribbon, N-acetyltransferase.
Expression of shati mRNA
As shown in Figure 2A, RT-PCR analysis revealed that shati is expressed at high levels in the cerebrum, cerebellum, liver, kidney, and spleen. We amplified and analyzed its three different target sequences by RT-PCR (Table 1). Similar results of RT-PCR were obtained with three different sets of primers (Fig. 2A).
Figure 2.
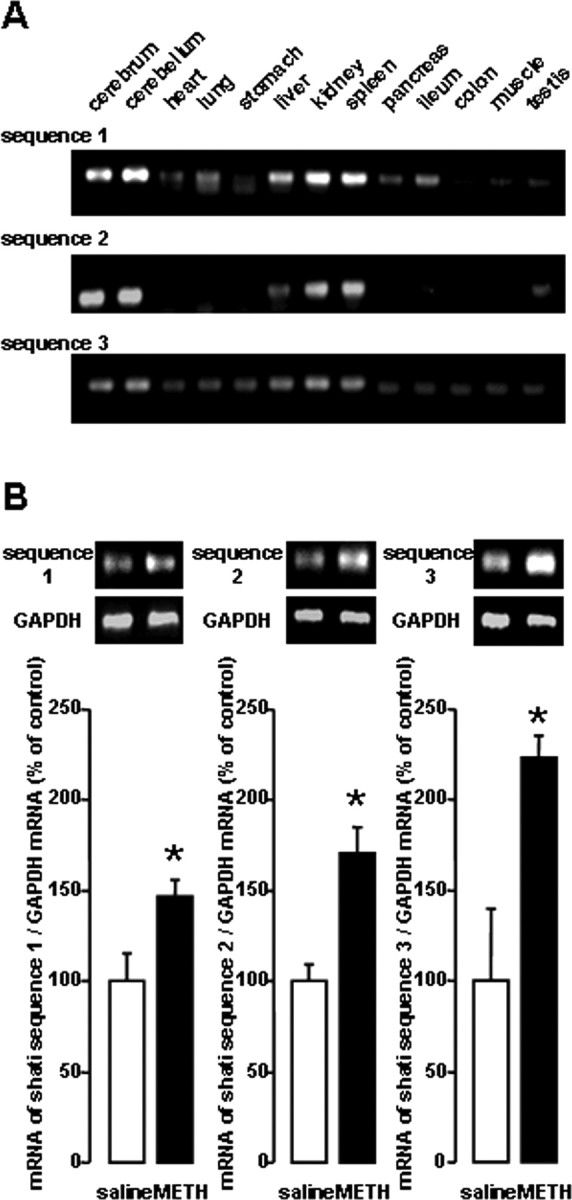
Expression of shati mRNA in the various organs of mice. A, RT-PCR analysis of shati in the various organs in mice. Mice were decapitated without any treatment, and the brains were quickly removed. The sets of primers used for PCR are listed in Table 1. B, Increase in the production of the three sets of target sequences of shati induced by repeated METH treatment in the NAc of mice. Mice were administered METH (2 mg/kg, s.c.) for 6 d and decapitated 2 h after the last METH treatment. Values are means ± SE (n = 5). *p < 0.05 versus saline-treated mice. The sets of primers used for PCR are listed in Table 1. To standardize the PCR products, we used primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control.
We performed a series of experiments to validate the results of cDNA subtraction. Repeated METH treatment (2 mg/kg, s.c.) for 6 d significantly elevated the mRNA levels of the target sequences of shati in the NAc (Fig. 2B).
METH-induced expression of shati mRNA in the brain
As an initial step in assessing the relationship between shati and METH-induced sensitization and dependence, we examined whether single and repeated METH treatment altered the expression of shati mRNA in the mouse brain using the real-time RT-PCR method. The effects of repeated METH treatment (0.3, 1 and 2 mg/kg, s.c. for 3 d) on shati mRNA expression in the NAc were dose dependent (F(3,28) = 5.503; p < 0.01, one-way ANOVA) (Fig. 3A). The levels of shati mRNA were significantly increased 2, 6, and 24 h after the last METH treatment and then returned to control value 1 week after the treatment (F(6,41) = 4.444; p < 0.01, one-way ANOVA) (Fig. 3B). Single METH treatment (2 mg/kg, s.c.) remarkably induced shati mRNA expression in the NAc and hippocampus (Hip). METH (2 mg/kg, s.c.) or saline challenge on day 6 after repeated administration of METH (2 mg/kg, s.c.) for 5 d remarkably induced shati mRNA expression in the frontal cortex (Fc), NAc, and caudate–putamen (CPu) (repeated drug administration, F(1,32) = 20.368, p < 0.01 for Fc; single administration, F(1,32) = 0.005, p = 0.942 for Fc; repeated drug administration × single administration, F(1,32) = 1.643, p = 0.209 for Fc; repeated drug administration, F(1,31) = 14.436, p < 0.01 for NAc; single administration, F(1,31) = 4.917, p < 0.05 for NAc; repeated drug administration × single administration, F(1,31) = 10.545, p < 0.01 for NAc; repeated drug administration F(1,32) = 8.023, p < 0.01 for CPu; single administration, F(1,32) = 4.833, p < 0.05 for CPu; repeated drug administration × single administration, F(1,32) = 1.669, p = 0.206 for CPu; repeated drug administration, F(1,32) = 0.628, p = 0.434 for Hip; single administration, F(1,32) = 6.464, p < 0.05 for Hip; repeated drug administration × single administration, F(1,32) = 2.496, p = 0.124 for Hip; two-way ANOVA) (Fig. 3C). The increase caused by METH in the NAc was inhibited by pretreatment with either the DA D1-like receptor antagonist R(+)-SCH23390 (0.1 mg/kg, i.p.) or the D2-like receptor antagonist raclopride (2 mg/kg, i.p.) (agonist, F(1,34) = 18.649, p < 0.01; antagonist, F(2,34) = 5.554, p < 0.01; agonist × antagonist, F(2,34) = 5.382, p < 0.01; two-way ANOVA) (Fig. 3D), although neither antagonists had an effect on shati mRNA expression in the saline-treated mice. These results indicate that METH induces the expression of shati mRNA in the brain through the activation of both DA D1 and D2 receptors.
Figure 3.
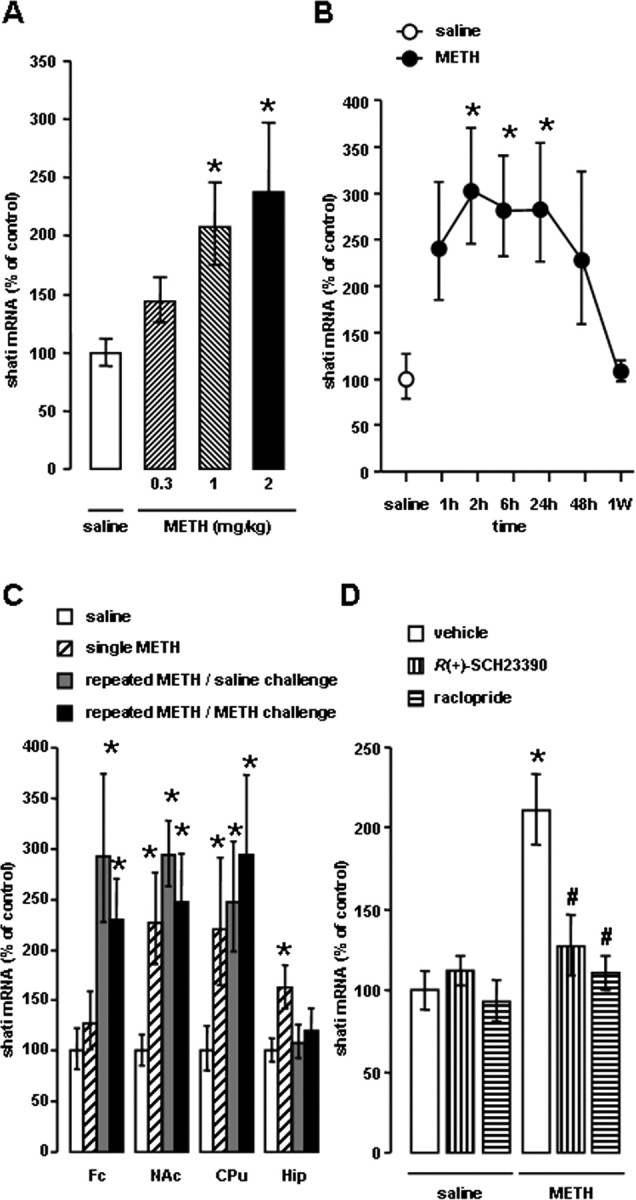
METH induced expression of shati mRNA in the brain. A, Dose-dependent effect of repeated METH treatment on shati mRNA expression in the NAc. Mice were administered METH (0.3, 1, and 2 mg/kg, s.c.) for 3 d. Mice were decapitated 2 h after the last METH treatment. Values are means ± SE (n = 8). *p < 0.05 versus saline-treated mice. B, Time course changes in the expression of shati mRNA after repeated METH treatment in the NAc. Mice were administered METH (2 mg/kg, s.c.) for 6 d and decapitated 1, 2, 6, 24, and 48 h and 1 week after the last METH treatment. Values are means ± SE (n = 6–7). *p < 0.05 versus saline-treated mice. C, Changes in the expression of shati mRNA in the various brain regions (Fc, NAc, CPu, and Hip) of the mice after single and repeated METH treatment. Mice were administered METH (2 mg/kg, s.c.) for 5 d and challenged with METH (2 mg/kg, s.c.) or saline on day 6. Mice were decapitated 2 h after last treatment of METH (2 mg/kg, s.c.) or saline challenge. Values are means ± SE (n = 8–10). *p < 0.05 versus saline-treated mice. D, The effects of the DA D1-like receptor antagonist R(+)-SCH23390 or D2-like receptor antagonist raclopride on METH-induced expression of shati mRNA in the NAc. Mice were treated with R(+)-SCH23390 (0.1 mg/kg, s.c.) or raclopride (2 mg/kg, s.c.) 30 min before daily METH (2 mg/kg, s.c. for 6 d) treatment. Mice were decapitated 2 h after the last METH treatment. Values are means ± SE (n = 6–8). *p < 0.05 versus vehicle/saline-treated mice. #p < 0.05 versus vehicle/METH-treated mice.
Localization of shati in the brain of mice treated with METH
There were few shati-immunopositive cells in saline-treated mouse brain (Fig. 4Aa,Ah). METH (2 mg/kg, s.c. for 6 d) increased the number of shati-immunopositive cells in the NAc compared with that in saline-treated mice (Fig. 4A). The shati-immunopositive cells were diminished when the antibodies were absorbed by S-3 or S-4 antigen (data not shown). The shati-immunopositive cells were colocalized with the cells that were immunopositive for NeuN, a neuronal marker, but not for GFAP, an astroglial marker, in the NAc of mice (Fig. 4Ab–Ag,Ai–An). The repeated METH treatment-induced increase in the numbers of shati-immunopositive cells in the NAc was abolished by shati-AS treatment, although shati-SC had no effect (Fig. 4Ba–Bf).
Figure 4.
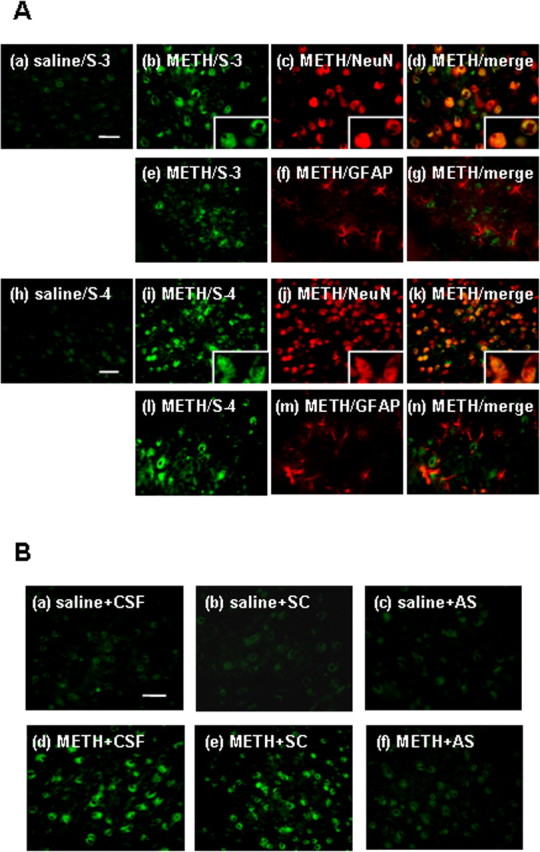
Immunostaining of shati in the NAc after repeated treatment with METH. Mice were administered METH (2 mg/kg, s.c.) for 6 d and decapitated 24 h after the last treatment. A, Double-labeling fluorescence photomicrographs for shati and NeuN or GFAP. The shati-immunopositive cells (green) were colocalized with NeuN-immonopositive cells (red). Double immunostaining for S-3 or S-4 and NeuN in the NAc reveals shati expression in neuronal cells. Scale bars, 20 μm. B, Effect of shati-AS on METH-induced increase in shati expression. METH-induced increase in shati expression in the NAc was inhibited by shati-AS. Scale bar, 20 μm.
Roles of shati in METH-induced hyperlocomotion and sensitization
To examine the role of shati in the behavioral and neurochemical phenotype in response to METH, we used an AS strategy, which widely used to manipulate gene expression in the brain via intracerebroventricular infusion (Taubenfeld et al., 2001; Bowers et al., 2004). The experimental schedules are shown in Figure 5, A and C. The AS downregulated the expression of shati mRNA in the NAc (Fig. 5B). The increase in the levels of shati mRNA expression evoked by repeated METH treatment in the NAc was significantly and completely abolished by shati-AS, although shati-SC had no effect. Moreover, shati mRNA expression in the NAc of saline-treated mice was also reduced by shati-AS, whereas shati-SC did not affect the expression in saline-treated mice (drug, F(1,42) = 72.765, p < 0.01; intracerebroventricular treatment, F(2,42) = 14.104, p < 0.01; drug × intracerebroventricular treatment, F(2,42) = 0.092, p = 0.912; two-way ANOVA) (Fig. 5B), indicating that shati-AS has an ability to reduce effectively the expression of shati mRNA. We also examined the effect of shati-AS on tPA expression as one of drug-dependence-related other proteins, because tPA–plasmin system potentiates the rewarding and locomotor-stimulating effects of METH, MOR, and nicotine by regulating release of DA (Nagai et al., 2004, 2005a,b, 2006). The increase in the levels of tPA mRNA expression in the NAc was not abolished by shati-AS (drug, F(1,47) = 62.530, p < 0.01; intracerebroventricular treatment, F(2,47) = 0.148, p = 0.862; drug × intracerebroventricular treatment, F(2,47) = 0.803, p = 0.454; two-way ANOVA). Moreover, tPA mRNA expression in the NAc of saline-treated mice was not also reduced by shati-AS, indicating that shati-AS has no ability to reduce effectively the expression of tPA mRNA (data not shown). Therefore, shati-AS is considered to have no secondary effects.
Figure 5.
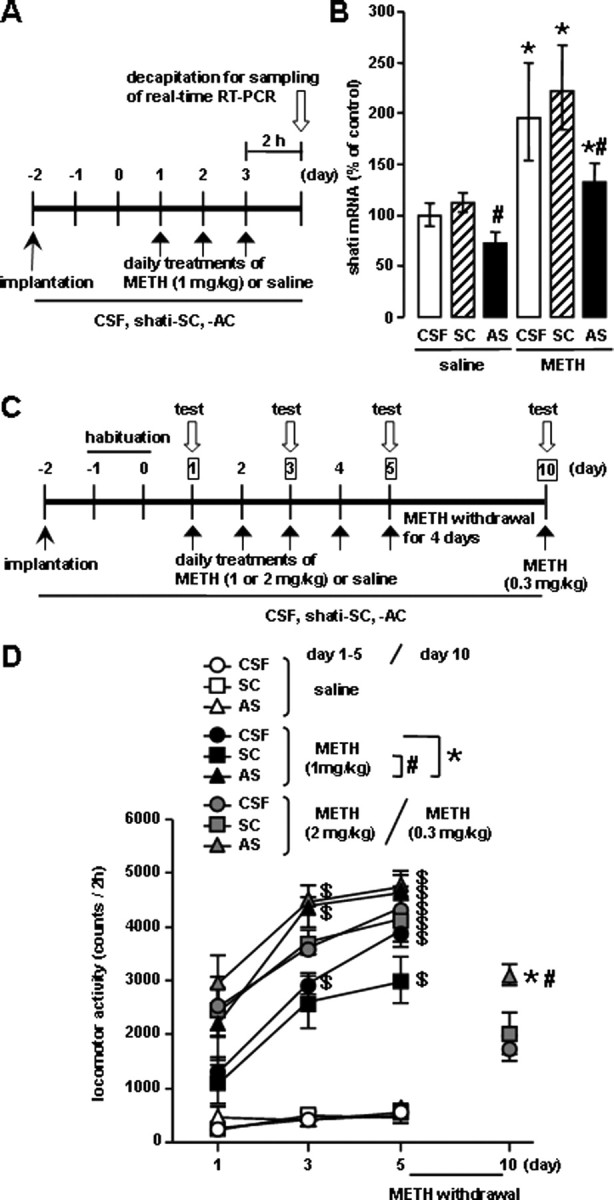
Roles of shati in METH-induced hyperlocomotion and sensitization. An osmotic minipump was used to deliver a continuous infusion of shati-AS (1.8 nmol/6 μl per day), shati-SC (1.8 nmol/6 μl per day), or CSF into the right ventricle (AP −0.5 mm, ML +1.0 mm from bregma, and DV −2.0 mm from the skull). A, Experimental schedule for the real-time RT-PCR using shati-AS. B, Effect of shati-AS on shati mRNA expression. Mice were administered METH (1 mg/kg, s.c.) for 3 d and decapitated 2 h after METH treatment on the day 3. Values are means ± SE (n = 8). *p < 0.05 versus saline-treated mice. #p < 0.05 versus shati-SC-treated mice. C, Experimental schedule for measurement of locomotor activity using shati-AS. D, Effect of shati-AS on repeated METH-induced behavioral sensitization. Mice were administered METH (1 or 2 mg/kg, s.c.) or saline for 5 d and challenged with METH (0.3 mg/kg, s.c.) on day 10. Locomotor activity was measured for 2 h on the days 1, 3, 5, and 10. Values are means ± SE (n = 5–7). ANOVA with repeated measures revealed significant differences in METH-induced sensitization (group, F(8,47) = 51.238, p < 0.01; day, F(2,94) = 68.423, p < 0.01; group × day, F(16,94) = 4.412, p < 0.01). *p < 0.05 versus METH plus CSF-treated mice. #p < 0.05 versus METH plus shati-SC-treated mice. $p < 0.05 versus the locomotor activity on day 1 in the same group.
Repeated METH administration leads to a progressive augmentation of many behavioral effects of the drug (behavioral sensitization). Sensitization is of interest as a model for drug-induced neuroplasticity in neuronal circuits important for addiction. It is well established that the induction of sensitization involves complex neuronal circuitry (Wolf, 1998). In rodent, sensitization is observed as a progressive augmentation of locomotor activity that may relate to an increase in the incentive to obtain drugs (Robinson and Berridge, 1993; Lorrain et al., 2000). There is also evidence of sensitization in human drug users (Satel et al., 1991) and normal subjects (Strakowski and Sax, 1998). Repeated METH treatment (1 and 2 mg/kg, s.c.) for 5 d produced behavioral sensitization [F(2,12) = 7.404 for METH (1 mg/kg) plus shati-AS-treated mice; F(2,18) = 5.593 for METH (1 mg/kg) plus shati-SC-treated mice; F(2,18) = 30.917 for METH (1 mg/kg) plus CSF-treated mice; F(2,12) = 7.453 for METH (2 mg/kg) plus shati-AS-treated mice, F(2,12) = 4.243 for METH (2 mg/kg) plus shati-SC-treated mice; F(2,15) = 8.569 for METH (2 mg/kg) plus CSF-treated mice; p < 0.05, one-way ANOVA] (Fig. 5D). As shown in Figure 5D, the shati-AS treatment potentiated the METH (1 mg/kg, s.c.)-induced hyperlocomotion and sensitization compared with shati-SC- or CSF-treated mice (drug, F(2,141) = 291.696, p < 0.01; intracerebroventricular treatment, F(2,141) = 28.223, p < 0.01; time, F(2,141) = 17.154, p < 0.01; drug × intracerebroventricular treatment, F(4,141) = 12.432, p < 0.01; drug × time, F(4,141) = 12.913, p < 0.01; intracerebroventricular treatment × time, F(4,141) = 0.156, p = 0.960; drug × intracerebroventricular treatment × time, F(8,141) = 0.427, p = 0.903; three-factor repeated ANOVA), whereas the shati-AS, shati-SC, or CSF treatment had no effect on spontaneous locomotor activity (Fig. 5D). The sensitization was observed on day 10 after challenge administration of METH (0.3 mg/kg, s.c.). Shati-AS-treated mice showed a marked potentiation of METH (0.3 mg/kg, s.c.)-induced sensitization on day 10 compared with shati-SC- or CSF-treated mice (F(2,13) = 6.974, p < 0.05, one-way ANOVA), although shati-AS-treated mice did not show a potentiation of METH (2 mg/kg, s.c.)-induced hyperlocomotion and sensitization compared with shati-SC- or CSF-treated mice on days 1–5 (Fig. 5D).
Roles of shati in METH-induced dopaminergic responses
The pharmacological effects of METH are linked to its capacity to elevate extracellular DA levels by releasing DA from presynaptic nerve terminals and inhibiting its reuptake (Heikkila et al., 1975; Seiden et al., 1993). In addition, METH and the amphetamines redistribute DA from synaptic vesicles to the cytosol and promote reverse transport (Seiden et al., 1993). Therefore, we examined the effect of shati-AS on the METH-induced increase in overflow of DA in the NAc using an in vivo microdialysis technique. The experimental schedule is shown in Figure 6A. METH caused a marked increase in overflow of DA in the NAc of the CSF-treated mice on the day 3 (Fig. 6B). The peak of overflow of DA was increased by METH treatment to ∼360% of the baseline level in the CSF-treated mice. In shati-AS-treated mice, the METH-induced increase in overflow of DA was significantly potentiated compared with that in the shati-SC- or CSF-treated mice (intracerebroventricular treatment, F(2,14) = 5.662, p < 0.05; time, F(10,140) = 35.646, p < 0.01; intracerebroventricular treatment × time, F(20,140) = 1.927, p < 0.05; repeated ANOVA) (Fig. 6B). The levels of basal DA did not differ among the three groups.
Figure 6.
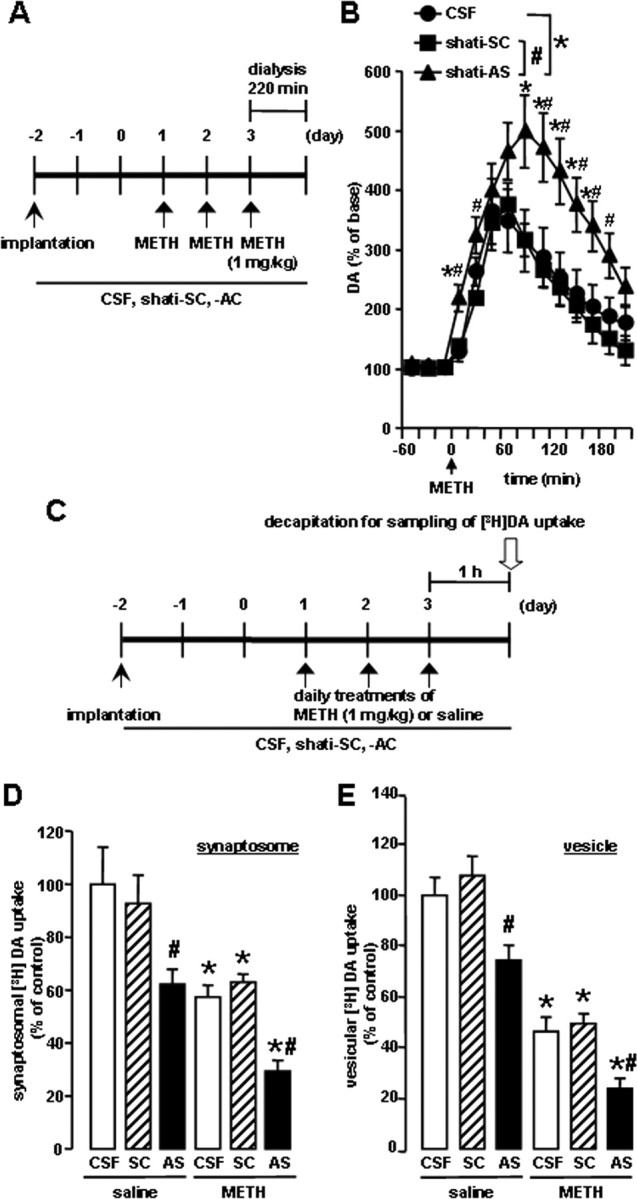
Effects of shati-AS on METH-induced dopaminergic responses. A, Experimental schedule for the measurement of overflow of DA using in vivo microdialysis using shati-AS. B, Effect of shati-AS on METH-induced increase in overflow of DA in the NAc. Mice were administered METH (1 mg/kg, s.c.) for 3 d. On day 3, levels of DA were measured in the NAc (AP +1.7 mm, ML −0.8 mm from bregma, and DV −4.0 mm from the skull) for 220 min after METH treatment by in vivo microdialysis. Basal levels of DA were 0.30 ± 0.08, 0.31 ± 0.05, and 0.30 ± 0.04 nm for the CSF-treated, shati-SC-treated, and shati-AS-treated mice, respectively. ANOVA with repeated measures revealed significant differences in METH-induced increase in overflow of DA (group, F(2,14) = 5.662, p < 0.05; time, F(10,140) = 35.646, p < 0.01; group × time, F(20,140) = 1.927, p < 0.05). Values are means ± SE (n = 5–6). *p < 0.05 versus CSF-treated mice. #p < 0.05 versus shati-SC-treated mice. C, Experimental schedule for the [3H]DA uptake assay using shati-AS. D, Effect of shati-AS on METH-induced decrease of synaptosomal [3H]DA uptake. Mice were administered METH (1 mg/kg, s.c.) for 3 d and decapitated 1 h after the last injection. The synaptosomal [3H]DA uptake was 0.32 ± 0.04, 0.29 ± 0.03, 0.20 ± 0.02, 0.18 ± 0.01, 0.20 ± 0.01, and 0.09 ± 0.01 pmol/mg protein per 4 min for the saline plus CSF-treated, saline plus shati-SC-treated, saline plus shati-AS-treated, METH plus CSF-treated, METH plus shati-SC-treated, and METH plus shati-AS-treated mice, respectively. The final concentration of [3H]DA was 5 nm. Values are means ± SE (n = 7–8). *p < 0.05 versus saline-treated mice. #p < 0.05 versus shati-SC-treated mice. E, Effect of shati-AS on METH-induced decrease of vesicular [3H]DA uptake. Mice were administered METH (1 mg/kg, s.c.) for 3 d and decapitated 1 h after the last injection. The vesicular [3H]DA uptake was 3.76 ± 0.25, 4.05 ± 0.29, 2.80 ± 0.20, 1.74 ± 0.21, 1.85 ± 0.14, and 0.90 ± 0.14 pmol/mg protein per 4 min for the saline plus CSF-treated, saline plus shati-SC-treated, saline plus shati-AS-treated, METH plus CSF-treated, METH plus shati-SC-treated, and METH plus shati-AS-treated mice, respectively. The final concentration of [3H]DA was 30 nm. Values are means ± SE (n = 8). *p < 0.05 versus saline-treated mice. #p < 0.05 versus shati-SC-treated mice.
Next, we examined the in vivo effect of shati-AS on [3H]DA uptake into synaptosomes in the midbrain. The experimental schedule is shown in Figure 6C. METH decreased [3H]DA uptake compared with the saline-treated mice. In shati-AS-treated mice, the METH-induced decrease in [3H]DA uptake was significantly potentiated compared with that in the shati-SC- or CSF-treated mice. Moreover, [3H]DA uptake in the saline-treated group was also decreased by shati-AS compared with that in the shati-SC- or CSF-treated mice, although shati-SC had no effect on [3H]DA uptake (drug, F(1,40) = 30.447, p < 0.01; intracerebroventricular treatment, F(2,40) = 12.576, p < 0.01; drug × intracerebroventricular treatment, F(2,40) = 0.392, p = 0.678; two-way ANOVA) (Fig. 6D).
We also examined the in vivo effect of shati-AS on [3H]DA uptake into synaptic vesicle preparations in the midbrain, because the redistribution of DA from synaptic vesicles to cytoplasmic compartments through interaction with vesicular monoamine transporter-2 has been postulated to be primarily responsible for DA terminal injury by METH or amphetamines (Liu and Edwards, 1997; Uhl, 1998). METH decreased vesicular [3H]DA uptake compared with the saline-treated mice. In shati-AS-treated mice, the METH-induced decrease in vesicular [3H]DA uptake was significantly potentiated compared with that in the shati-SC- or CSF-treated mice. Moreover, [3H]DA uptake in the saline-treated group was also decreased by shati-AS compared with that in the shati-SC- or CSF-treated mice, although shati-SC had no effect on [3H]DA uptake (drug, F(1,42) = 137.229, p < 0.01; intracerebroventricular treatment, F(2,42) = 15.087, p < 0.01; drug × intracerebroventricular treatment, F(2,42) = 0.240, p = 0.788; two-way ANOVA) (Fig. 6E).
Different results were obtained from in vivo microdialysis and DA uptake studies, only in the basal conditions. These studies were performed in quite different situations. Living mice were used in vivo microdialysis study, and basal overflow of endogenous DA was measured 24 h after the last METH treatment in the NAc (Fig. 6B). Therefore, other factors (other neurotransmitters, neuroplasticity, and neuronal input from other brain regions) might affect basal DA overflow and compensate the dysfunction of DA uptake induced by repeated treatment of METH. Conversely, the experiment of [3H]DA uptake was ex vivo study by using the midbrain tissue (Fig. 6D,E). High-concentration and exogenous [3H]DA was used for the investigation of functional changes of DA uptake 1 h after the last METH treatment in the midbrain. The ex vivo method could more directly measure the changes of DA uptake in the midbrain, comparing in vivo microdialysis study.
Roles of shati in METH-induced conditioned place preference
The effect of shati-AS on METH-induced CPP was examined in a place conditioning paradigm, in which animals learn the association of an environment paired with drug exposure. This paradigm involves sensory perception of external stimuli, association of stimuli, and the approach-inducing actions of a drug, as well as the rewarding effects of a drug. The experimental schedule is shown in Figure 7A. As shown in Figure 7B, METH (0.3 mg/kg, s.c.) produced place preference in mice. In shati-AS-treated mice, the development of METH-induced CPP was significantly potentiated compared with that in the shati-SC- or CSF-treated mice (drug, F(1,46) = 78.202, p < 0.01; intracerebroventricular treatment, F(2,46) = 4.950, p = 0.011; drug × intracerebroventricular treatment, F(2,46) = 5.046, p = 0.010; two-way ANOVA) (Fig. 7B), indicating that downregulation of shati expression was sufficient to confer the enhanced METH-induced CPP. Shati-AS, shati-SC, or CSF treatment had no effect on CPP in saline-treated mice (Fig. 7B, left three columns), suggesting that the procedure in CPP might not reflect anxiolytic actions. These results suggest that shati participates in the repeated METH treatment-induced development of behavioral sensitization and CPP by regulating DA uptake.
Figure 7.
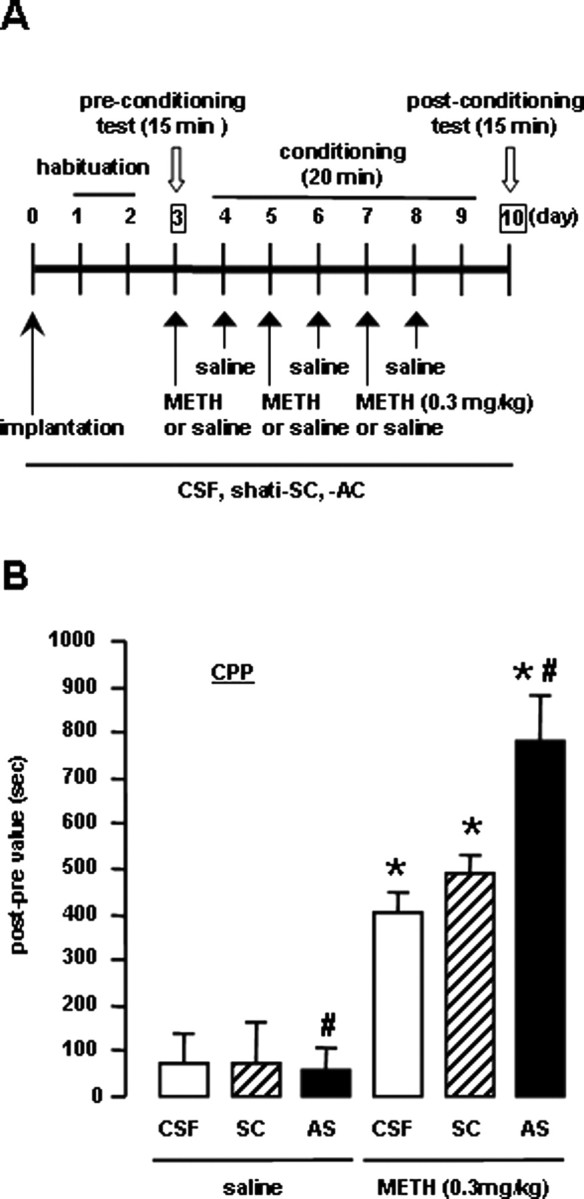
Effects of shati-AS on METH-induced conditioned place preference. A, Experimental schedule for the conditioned place preference task using shati-AS. B, Effect of shati-AS on METH-induced place preference. Mice were administered METH (0.3 mg/kg, s.c.) or saline during the conditioning for place preference. Values are means ± SE (n = 5–12). *p < 0.05 versus saline-treated mice. #p < 0.05 versus shati-SC-treated mice.
Discussion
In the present study, we identified a novel molecule shati from the NAc of mice treated with METH for the first time using the PCR-select cDNA subtraction method, which is a differential and epochal cloning technique.
From motif analysis of shati, shati contained the sequence of GCAT (Fig. 1C). Shati might have physiological action in producing acetylcholine or metabolic action of ATP, because the analysis showed the lowest interactive potential energy of shati with acetyl-CoA or ATP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Accordingly, we have to investigate the mechanism by which shati regulates production of acetylcholine or metabolic roles of ATP in subsequent studies.
Because shati expression was detected at high levels in not only the brain regions related to drug dependence but also the liver, kidney, and spleen (Fig. 2A), it is plausible that shati is involved in the regulation of pathophysiological function. Single METH treatment induced the expression of shati mRNA in the NAc and Hip (Fig. 3C). Repeated METH treatment produces an enhancement of the locomotor-stimulating effects of METH (data not shown). Remarkable induction of shati mRNA expression was detected in the Fc, NAc, and CPu of the mice that showed behavioral sensitization to METH (Fig. 3A–C). There is strong evidence that the dopaminergic system, which projects from the VTA of the midbrain to the NAc and to other forebrain sites, including the Fc, dorsal striatum, and Hip, is a major substrate of reward and reinforcement for both natural rewards and addictive drugs (Di Chiara and Imperato, 1988; Robbins and Everitt, 1996; Wise, 1996a). Therefore, shati may be involved in the rewarding effects and reinforcement of addictive drugs. Because the dopaminergic neuronal system is involved primarily in the pharmacological effects of METH (Melega et al., 1995; Larsen et al., 2002; Sulzer et al., 2005), we examined whether the METH-induced increase in shati mRNA levels is mediated by the activation of dopaminergic neurotransmission. The METH-induced increase in the expression of shati mRNA in the NAc was completely inhibited by pretreatment with the DA D1-like receptor antagonist R(+)-SCH23390 and the DA D2-like receptor antagonist raclopride (Fig. 3D), suggesting that the activation of DA D1 and D2 receptors is attributable to METH-induced expression of shati. Behavioral studies have suggested that both DA D1 and D2 receptors mediate reinforcing signals for drug of abuse, because amphetamine-induced CPP and METH-induced sensitization are blocked by either DA D1-like or D2-like receptor antagonist (Ujike et al., 1989; Hiroi and White, 1991). Therefore, it likely that activation of DA transmission is necessary for METH-induced shati expression in neurons, in which shati specifically acts (Fig. 4A).
The contribution of dopaminergic transmission to behavioral sensitization and CPP has been well documented (Nakajima et al., 2004; Nagai et al., 2005a,b; Niwa et al., 2007a,b,d). Shati expression was upregulated by repeated administration of METH (Figs. 2B, 3, 4A), and downregulation of shati by AS (Figs. 4B, 5B) led to an elevated synaptic DA concentration in the NAc and major behavioral manifestations in mice: heightened locomotor activity (Fig. 5D), the rate of development of sensitization (Fig. 5D), and CPP (Fig. 7B) responding to METH. Furthermore, downregulation of shati expression by AS (Figs. 4B, 5B) potentiated the effects of METH on overflow of DA in the NAc (Fig. 6B) and DA uptake (Fig. 6D,E). These findings strongly suggest that the overexpression of shati elicited by METH may serve as a homeostatic mechanism that prevents hyperlocomotion, sensitization, and CPP, by promoting plasmalemmal and vesicular DA uptake as well as attenuating the METH-induced increase in overflow of DA in the NAc.
We used METH at the dose of 2 mg/kg for 6 d in the experiments of RT-PCR, real-time RT-PCR, and immunohistochemistry (Figs. 3, 4B–D, 5), because expression of shati was induced in the NAc of mice by METH (2 mg/kg for 6 d), which was detected by using a PCR-select cDNA subtraction method (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In AS experiments, shati-AS-treated mice tended to show a potentiation of METH (2 mg/kg)-induced hyperlocomotion and sensitization on days 1–5, but there were no statistically significant differences among the three groups (Fig. 5D). The dose–response effects of METH on the locomotor activity may reflect a shift to the left, but these effects were reached the plateau. Conversely, shati-AS-treated mice showed a marked potentiation of METH (1 mg/kg)-induced hyperlocomotion and sensitization on days 1–5 compared with shati-SC- or CSF-treated mice (Fig. 5D). On day 3, the potentiation of the METH-induced hyperlocomotion by shati-AS reached the maximum and plateau. Therefore, in the experiments of AS, we selected METH at the dose of 1 mg/kg for 3 d (Figs. 5B, 6B,D,E). We also confirmed that shati mRNA was increased by METH at the doses of 1 and 2 mg/kg for 3 d (Fig. 3A). Then, in the AS study, we selected METH at the dose of 1 mg/kg to investigate effects of AS.
We selected the time point of 2 h after the last METH treatment for the time when the animals were to be killed in the experiments of RT-PCR and real-time RT-PCR (Figs. 2, 3A,C,D), because expression of shati mRNA showed peak in the NAc of mice 2 h after the last METH treatment (Fig. 3B). We prepared the brain samples 24 h after the last METH treatment for immunohistochemical study (Fig. 4). The levels of shati mRNA in the NAc of mice treated with repeated METH were significantly increased 2, 6, and 24 h after the last METH treatment (Fig. 3B). At 24 h after the METH treatment, both transcription and translation of shati protein could be induced in the brain. Therefore, we considered that 24 h after the METH treatment is the best time point for investigation of shati protein expression.
Changes in mRNA and protein expression caused by drugs are of particular interest. The expression of certain mRNAs and proteins appears to be a compensatory adaptation to excessive DA signaling, which could be biologically significant adaptive mechanisms contributing to dependence. Nevertheless, some proteins play a reverse role. For example, we previously demonstrated that tPA potentiates METH- or MOR-induced rewarding and locomotor-stimulating effects (Nagai et al., 2004, 2005a,b), whereas TNF-α and its inducer inhibit them (Nakajima et al., 2004; Niwa et al., 2007a,b,d). The development of sensitization to amphetamine is prevented when an antibody that neutralized basic fibroblast growth factor (bFGF) is infused into the VTA before amphetamine treatment (Flores et al., 2000). Infusion of brain-derived neurotrophic factor (BDNF) into the NAc enhances the stimulation of locomotor activity by cocaine in rats, whereas the development of sensitization and CPP is delayed in heterozygous BDNF knock-out mice compared with wild-type littermates (Horger et al., 1999; Hall et al., 2003). Infusion of GDNF into the VTA blocks certain biochemical adaptations to chronic cocaine treatment (induction of tyrosine hydroxylase, NR1 subunit of NMDA receptors, ΔFosB, and protein kinase A catalytic subunit) as well as cocaine-induced rewarding effects (Messer et al., 2000). Conversely, responses to cocaine are enhanced in rats by intra-VTA infusion of anti-GDNF antibody and in GDNF heterozygous knock-out mice (Messer et al., 2000). A partial reduction in the expression of GDNF potentiates METH self-administration, enhances motivation to take METH, increases vulnerability to drug-primed reinforcement, and prolongs cue-induced reinforcement of extinguished METH-seeking behavior (Yan et al., 2007). cAMP response element-binding protein (CREB) overexpression in the NAc reduces the rewarding properties of cocaine, whereas expression of a dominant-negative form of CREB in this region has the opposite effect (Carlezon et al., 1998; Walters and Blendy, 2001; McClung and Nestler, 2003). Furthermore, FosB mutant mice shows exaggerated locomotor activation in response to initial cocaine exposures as well as robust CPP to a lower dose of cocaine compared with wild-type littermates (Hiroi et al., 1997). Changes in the balance of levels between proaddictive factors, such as bFGF, tPA, and BDNF, and anti-addictive factors, such as TNF-α, GDNF, CREB, and FosB, induced by drugs of abuse seems to be important to the development of drug dependence. In the present study, the facilitation of METH-induced behavioral sensitization in mice with a targeted downregulation of shati highlights the opposing role of shati in drug-dependent behavioral plasticity. Therefore, upregulation of shati expression may represent a homeostastatic response of dopaminergic neurons in the NAc to excessive dopaminergic transmission, resulting in attenuation of hypersensitivity and CPP induced by METH-like drugs. Our findings, together with others, suggest that there are molecules in the brain that normally inhibit the behavioral actions of addictive substances. The mechanism underlying the upregulation of shati caused by METH remains to be elucidated; nevertheless, inhibitory feedback of the excessive DA signaling is likely to be a plausible candidate.
In conclusion, the present study established a functional interaction between shati and METH. Our findings suggest that shati is involved in the development of METH-induced hyperlocomotion, sensitization, and CPP, by promoting plasmalemmal and vesicular DA uptake as well as attenuating the METH-induced increase in overflow of DA in the NAc.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research and Special Coordination Funds for Promoting Science and Technology, Target-Oriented Brain Science Research Program; a Grant-in-Aid for Scientific Research (B), Exploratory Research, and Young Scientists (A); the 21st Century Center of Excellence Program “Integrated Molecular Medicine for Neuronal and Neoplastic Disorders” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; a Grant-in-Aid for Health Science Research on Regulatory Science of Pharmaceuticals and Medical Devices, and Comprehensive Research on Aging and Health from the Ministry of Health, Labor, and Welfare of Japan; a Smoking Research Foundation Grant for Biomedical Research; a grant from the Brain Research Center from the 21st Century Frontier Research Program funded by the Ministry of Science and Technology, Republic of Korea; by the Japan Canada Joint Health Research Program; and by a grant from Takeda Science Foundation. We are grateful to Drs. Kenji Kadomatsu, Yoshifumi Takei, and Hanayo Kawai (Department of Biochemistry, Nagoya University Graduate School of Medicine, Nagoya, Japan) for technical assistance, critical comments, and helpful discussions. We also thank Dr. Noboru Ogiso, Yasutaka Ohya, Yuuki Ushiro, and Kazumi Kawai (Division for Research of Laboratory Animals, Center for Research of Laboratory Animals and Medical Research Engineering, Nagoya University Graduate School of Medicine) and Nobuyoshi Hamada and Yoshiyuki Nakamura (Radioisotope Center Medical Branch, Nagoya University Graduate School of Medicine) for technical assistance.
References
- Ang E, Chen J, Zagouras P, Magna H, Holland J, Schaeffer E, Nestler EJ. Induction of nuclear factor-κB in nucleus accumbens by chronic cocaine administration. J Neurochem. 2001;79:221–224. doi: 10.1046/j.1471-4159.2001.00563.x. [DOI] [PubMed] [Google Scholar]
- Blackshaw S, Harpavat S, Trimarchi J, Cai L, Huang H, Kuo WP, Weber G, Lee K, Fraioli RE, Cho SH, Yung R, Asch E, Ohno-Machado L, Wong WH, Cepko CL. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:E247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, Lanier SM, Kalivas PW. Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron. 2004;42:269–281. doi: 10.1016/s0896-6273(04)00159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Cha XY, Pierce RC, Kalivas PW, Mackler SA. NAC-1, a rat brain mRNA, is increased in the nucleus accumbens three weeks after chronic cocaine self-administration. J Neurosci. 1997;17:6864–6871. doi: 10.1523/JNEUROSCI.17-18-06864.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AV, Arnt J, Hyttel J, Larsen JJ, Svendsen O. Pharmacological effects of a specific dopamine D-1 antagonist SCH 23390 in comparison with neuroleptics. Life Sci. 1984;34:1529–1540. doi: 10.1016/0024-3205(84)90607-6. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD. Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci USA. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglass J, Daoud S. Characterization of the human cDNA and genomic DNA encoding CART: a cocaine- and amphetamine-regulated transcript. Gene. 1996;169:241–245. doi: 10.1016/0378-1119(96)88651-3. [DOI] [PubMed] [Google Scholar]
- Erickson JD, Masserano JM, Barnes EM, Ruth JA, Weiner N. Chloride ion increases [3H]dopamine accumulation by synaptic vesicles purified from rat striatum: inhibition by thiocyanate ion. Brain Res. 1990;516:155–160. doi: 10.1016/0006-8993(90)90912-u. [DOI] [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther. 1997;282:834–838. [PubMed] [Google Scholar]
- Flores C, Samaha AN, Stewart J. Requirement of endogenous basic fibroblast growth factor for sensitization to amphetamine. J Neurosci. 2000;20:1–5. doi: 10.1523/JNEUROSCI.20-02-j0003.2000. RC55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain: in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman PM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Gurskaya NG, Diatchenko L, Chenchik A, Siebert PD, Khaspekov GL, Lukyanov KA, Vagner LL, Ermolaeva OD, Lukyanov SA, Sverdlov ED. Equalizing cDNA subtraction based on selective suppression of polymerase chain reaction: cloning of Jurkat cell transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Anal Biochem. 1996;240:90–97. doi: 10.1006/abio.1996.0334. [DOI] [PubMed] [Google Scholar]
- Hall FS, Drgonova J, Goeb M, Uhl GR. Reduced behavioral effects of cocaine in heterozygous brain-derived neurotropic factor (BDNF) knockout mice. Neuropsychopharmacology. 2003;28:1485–1490. doi: 10.1038/sj.npp.1300192. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Orlansky H, Cohen G. Studies on the distinction between uptake inhibition and release of 3H-dopamine in rat brain tissue slices. Biochem Pharmacol. 1975;24:847–852. doi: 10.1016/0006-2952(75)90152-5. [DOI] [PubMed] [Google Scholar]
- Hiroi N, White NM. The amphetamine conditioned place preference: differential involvement of dopamine receptor subtypes and two dopaminergic terminal areas. Brain Res. 1991;552:141–152. doi: 10.1016/0006-8993(91)90672-i. [DOI] [PubMed] [Google Scholar]
- Hiroi N, Brown JR, Haile CN, Ye H, Greenberg ME, Nestler EJ. FosB mutant mice: loss of chronic cocaine induction of Fos-related proteins and heightened sensitivity to cocaine's psychomotor and rewarding effects. Proc Natl Acad Sci USA. 1997;94:10397–10402. doi: 10.1073/pnas.94.19.10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotropic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Laakso A, Mohn AR, Gainetdinov RR, Caron MG. Experimental genetic approaches to addiction. Neuron. 2002;36:213–228. doi: 10.1016/s0896-6273(02)00972-8. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Edwards RH. The role of vesicular transporter proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci. 1997;20:125–156. doi: 10.1146/annurev.neuro.20.1.125. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Arnold GM, Vezina P. Previous exposure to amphetamine increases incentive to obtain the drug: long-lasting effects revealed by the progressive ratio schedule. Behav Brain Res. 2000;107:9–19. doi: 10.1016/s0166-4328(99)00109-6. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Melega WP, Williams AE, Schmitz DA, DiStefano EW, Cho AK. Pharmacokinetic and pharmacodynamic analysis of the actions of d-amphetamine and d-methamphetamine on the dopamine terminal. J Pharmacol Exp Ther. 1995;274:90–96. [PubMed] [Google Scholar]
- Messer CJ, Eisch AJ, Carlezon WA, Jr, Whisler K, Shen L, Wolf DH, Westphal H, Collins F, Russell DS, Nestler EJ. Role for GDNF in biochemical and behavioral adaptations to drugs of abuse. Neuron. 2000;26:247–257. doi: 10.1016/s0896-6273(00)81154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi H, Yamada K, Mizuno M, Mizuno T, Nitta A, Noda Y, Nabeshima T. Regulations of methamphetamine reward by extracellular signal-regulated kinase 1/2/ets-like gene-1 signaling pathway via the activation of dopamine receptors. Mol Pharmacol. 2004;65:1293–1301. doi: 10.1124/mol.65.5.1293. [DOI] [PubMed] [Google Scholar]
- Mizoguchi H, Yamada K, Niwa M, Mouri A, Mizuno T, Noda Y, Nitta A, Itohara S, Banno Y, Nabeshima T. Reduction of methamphetamine-induced sensitization and reward in matrix metalloproteinase-2 and -9-deficient mice. J Neurochem. 2007;100:1579–1588. doi: 10.1111/j.1471-4159.2006.04288.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Yoshimura M, Ishikawa K, Miyamoto Y, Hashimoto K, Noda Y, Nitta A, Nabeshima T. The tissue plasminogen activator-plasmin system participates in the rewarding effect of morphine by regulating dopamine release. Proc Natl Acad Sci USA. 2004;101:3650–3655. doi: 10.1073/pnas.0306587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Noda Y, Ishikawa K, Miyamoto Y, Yoshimura M, Ito M, Takayanagi M, Takuma K, Yamada K, Nabeshima T. The role of tissue plasminogen activator in methamphetamine-related reward and sensitization. J Neurochem. 2005a;92:660–667. doi: 10.1111/j.1471-4159.2004.02903.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kamei H, Ito M, Hashimoto K, Takuma K, Nabeshima T, Yamada K. Modification by the tissue plasminogen activator-plasmin system of morphine-induced dopamine release and hyperlocomotion, but not anti-nociceptive effect in mice. J Neurochem. 2005b;93:1272–1279. doi: 10.1111/j.1471-4159.2005.03117.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ito M, Nakamichi N, Mizoguchi H, Kamei H, Fukakusa A, Nabeshima T, Takuma K, Yamada K. The rewards of nicotine: regulation by tissue plasminogen activator-plasmin system through protease activated receptor-1. J Neurosci. 2006;26:12374–12383. doi: 10.1523/JNEUROSCI.3139-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima A, Yamada K, Nagai T, Uchiyama T, Miyamoto Y, Mamiya T, He J, Nitta A, Mizuno M, Tran MH, Seto A, Yoshimura M, Kitaichi K, Hasegawa T, Saito K, Yamada Y, Seishima M, Sekikawa K, Kim HC, Nabeshima T. Role of tumor necrosis factor-α in methamphetamine-induced drug dependence and neurotoxicity. J Neurosci. 2004;24:2212–2225. doi: 10.1523/JNEUROSCI.4847-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier TC, Givens BS, Schulz DW, Bunney BS, Breese GR, Mailman RB. SCH23390 effects on apomorphine-induced responses of nigral dopaminergic neurons. J Pharmacol Exp Ther. 1986;236:838–845. [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. From neurobiology to treatment: progress against addiction. Nat Neurosci. 2002;5:1076–1079. doi: 10.1038/nn945. [DOI] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Shen L, Noda Y, Nabeshima T. Involvement of glial cell line-derived neurotrophic factor in inhibitory effects of a hydrophobic dipeptide Leu-Ile on morphine-induced sensitization and rewarding effects. Behav Brain Res. 2007a;179:167–171. doi: 10.1016/j.bbr.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Yamada Y, Nakajima A, Saito K, Seishima M, Shen L, Noda Y, Furukawa S, Nabeshima T. An inducer for glial cell line-derived neurotrophic factor and tumor necrosis factor-α protects against methamphetamine-induced rewarding effects and sensitization. Biol Psychiatry. 2007b;61:890–901. doi: 10.1016/j.biopsych.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Yamada K, Nabeshima T. The roles of glial cell line-derived neurotrophic factor, tumor necrosis factor-α, and an inducer of these factors in drug dependence. J Pharmacol Sci. 2007c doi: 10.1254/jphs.cp0070017. in press. [DOI] [PubMed] [Google Scholar]
- Niwa M, Nitta A, Yamada Y, Nakajima A, Saito K, Seishima M, Noda Y, Nabeshima T. Tumor necrosis factor-α and its inducer inhibit morphine-induced rewarding effects and sensitization. Biol Psychiatry. 2007d doi: 10.1016/j.biopsych.2006.10.009. in press. [DOI] [PubMed] [Google Scholar]
- Noda Y, Miyamoto Y, Mamiya T, Kamei H, Furukawa H, Nabeshima T. Involvement of dopaminergic system in phencyclidine-induced place preference in mice pretreated with phencyclidine repeatedly. J Pharmacol Exp Ther. 1998;286:44–51. [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ. Neurobehavioural mechanisms of reward and motivation. Curr Opin Neurobiol. 1996;6:228–236. doi: 10.1016/s0959-4388(96)80077-8. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Satel SL, Southwick SM, Gawin FH. Clinical features of cocaine-induced paranoia. Am J Psychiatry. 1991;148:495–498. doi: 10.1176/ajp.148.4.495. [DOI] [PubMed] [Google Scholar]
- Schechter MD, Calcagnetti DJ. Continued trends in the conditioned place preference literature from 1992 to 1996, inclusive, with a cross-indexed bibliography. Neurosci Biobehav Rev. 1998;22:827–846. doi: 10.1016/s0149-7634(98)00012-8. [DOI] [PubMed] [Google Scholar]
- Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;33:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- Strakowski SM, Sax KW. Progressive behavioral response to repeated d-amphetamine challenge: further evidence for sensitization in humans. Biol Psychiatry. 1998;44:1171–1177. doi: 10.1016/s0006-3223(97)00454-x. [DOI] [PubMed] [Google Scholar]
- Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPβ. Nat Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- Uhl GR. Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson's disease. Ann Neurol. 1998;43:555–560. doi: 10.1002/ana.410430503. [DOI] [PubMed] [Google Scholar]
- Ujike H, Onoue T, Akiyama K, Hamamura T, Otsuki S. Effects of selective D-1 and D-2 dopamine antagonists on development of methamphetamine-induced behavioral sensitization. Psychopharmacology. 1989;98:89–92. doi: 10.1007/BF00442011. [DOI] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters CL, Blendy JA. Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J Neurosci. 2001;21:9438–9444. doi: 10.1523/JNEUROSCI.21-23-09438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XB, Funada M, Imai Y, Revay RS, Ujike H, Vandenbergh DJ, Uhl GR. rGβ1: a psychostimulant-regulated gene essential for establishing cocaine sensitization. J Neurosci. 1997;17:5993–6000. doi: 10.1523/JNEUROSCI.17-15-05993.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996a;19:319–340. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996b;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Yamada K, Nabeshima T. Pro- and anti-addictive neurotrophic factors and cytokines in psychostimulant addiction: mini review. Ann NY Acad Sci. 2004;1025:198–204. doi: 10.1196/annals.1316.025. [DOI] [PubMed] [Google Scholar]
- Yan Y, Yamada K, Niwa M, Nagai T, Nitta A, Nabeshima T. Enduring vulnerability to reinstatement of methamphetamine-seeking behavior in glial cell line-derived neurotrophic factor mutant mice. FASEB J. 2007 doi: 10.1096/fj.06-7772com. in press. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, Shaw-Lutchman T, Berton O, Sim-Selley LJ, Dileone RJ, Kumar A, Nestler EJ. An essential role for ΔFosB in the nucleus accumbens in morphine action. Nat Neurosci. 2006;9:205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]