

Abstract
Long-haul transport along microtubules is crucial for neuronal polarity, and transport defects cause neurodegeneration. Tau protein stabilizes microtubule tracks, but in Alzheimer's disease it aggregates and becomes missorted into the somatodendritic compartment. Tau can inhibit axonal transport by obstructing motors on microtubules, yet tau itself can still move into axons. We therefore investigated tau movement by live-cell fluorescence microscopy, FRAP (fluorescence recovery after photobleaching), and FSM (fluorescence speckle microscopy). Tau is highly dynamic, with diffusion coefficients of ∼3 μm2/s and microtubule dwell times of ∼4 s. This facilitates the entry of tau into axons over distances of millimeters and periods of days. For longer distances and times, two mechanisms of tau transport are observed. At low near-physiological levels, tau is cotransported with microtubule fragments from cell bodies into axons, moving at instantaneous velocities ∼1 μm/s. At high concentrations, tau forms local accumulations moving bidirectionally at ∼0.3 μm/s. These clusters first appear at distal endings of axons and may indicate an early stage of neurite degeneration.
Keywords: Alzheimer's disease, axonal transport, microtubules, tau, mitochondria, synapse
Introduction
Neurons are polarized cells with two types of cell extensions, dendrites and axons, whose establishment depends on the microtubule cytoskeleton. Microtubules (MTs) are used as tracks for long-distance transport of vesicles, organelles, and nucleoprotein complexes (Baas and Buster, 2004; Hirokawa and Takemura, 2005; Hollenbeck and Saxton, 2005; Miller and Sheetz, 2006). Tau, the main axonal MT-associated protein (MAP), regulates microtubule stability, functions as a spacer between MTs (Chen et al., 1992), and serves as an anchor for enzymes (Sontag et al., 1999). Tau can regulate MT-dependent transport by interfering with motor proteins (Stamer et al., 2002). In Alzheimer's disease (AD), the hyperphosphorylation of tau, aggregation into neurofibrillary tangles, and missorting into the somatodendritic compartment are hallmarks of the disease (Garcia and Cleveland, 2001; Lee et al., 2001).
Tau can be subdivided into a C-terminal “assembly domain,” which promotes microtubule assembly, and an N-terminal “projection domain,” which projects away from the microtubule surface (see Fig. 1a). The human CNS contains mainly six isoforms (352–411 residues), which are generated by alternative splicing (Andreadis, 2005; Goedert and Jakes, 2005). The microtubule-binding site of tau comprises the repeat domain (three or four semiconserved sequences of ∼31 residues) and the flanking proline-rich domains (Butner and Kirschner, 1991; Gustke et al., 1994). Phosphorylation of tau, especially of the KXGS motifs in the repeats, tends to detach tau from microtubules and thus destabilizes them (Biernat et al., 1993; Stoothoff and Johnson, 2005). Tau mutants, which cause neurodegeneration in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), have a reduced ability to bind to microtubules in vitro and in cells (Hong et al., 1998; Barghorn et al., 2000; Lu and Kosik, 2001).
Figure 1.
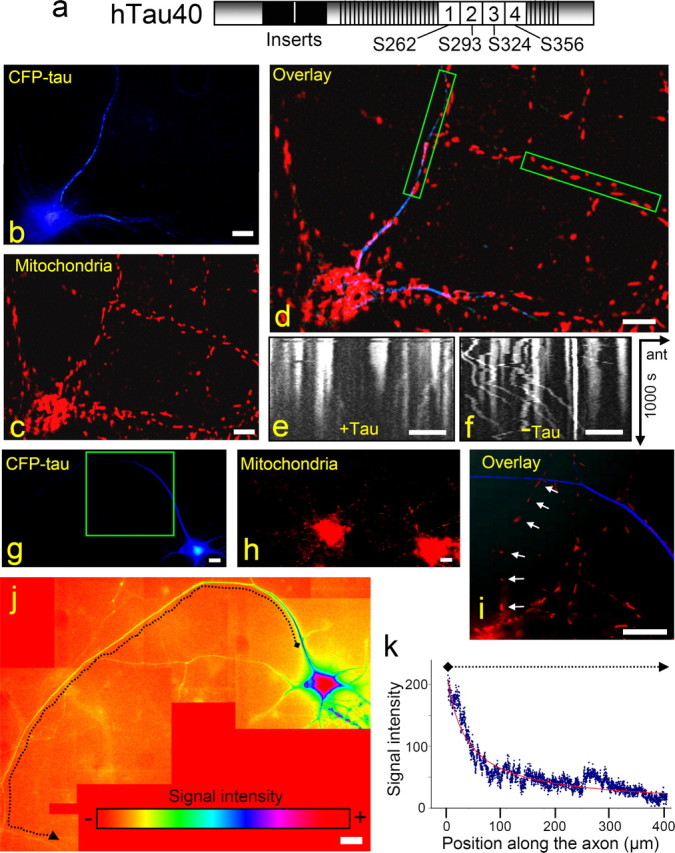
Tau expression inhibits anterograde transport of organelles and vesicles but not of tau itself. a, Diagram of tau441, the longest isoform in human CNS, with repeat domain (R1–R4), the flanking proline-rich regions (which together constitute the microtubule-binding domain), and the N-terminal projection domain (alternatively spliced inserts in black). Phosphorylation sites in the repeat domain that control MT binding are indicated. b–f, Cortical neurons 20 h after transfection with CFP-hTau40. CFP-hTau40 is shown in blue (b); mitochondria are labeled with MitoTracker Red (c). In the overlay (d), two neurites are boxed in green (left box, with tau expression; right, without tau). The movements of mitochondria during a 1000 s period are visualized in kymographs derived from the boxed neurites with Tau expression (e) or without (f). The anterograde direction is indicated by an arrow from left to right (“ant”). g–i, Cortical neurons expressing CFP-hTau40 for 21 h. CFP-hTau40 is pseudocolored in blue (g), and mitochondria are pseudocolored in red (h). In the overlay, mitochondria are visible in the axon without tau expression but not in the axon with tau (i). j, k, Quantitation of tau distribution along an axon (same as in g; highlighted by dotted line) shown by a color gradient (j) and plotted versus axon length (k; plot extends between positions marked by diamond and arrowhead). Note that the highest apparent level is in the cell body (red, blue edges) and extends into the axon (blue, proximal; green, distal). Occasional shifts in intensity (e.g., around L = 280 μm) are caused by stronger background intensity in this area. Scale bars, 20 μm.
The basis for the sorting of tau into axons (or missorting into dendrites during neurodegeneration) is still not well understood. In embryonic brain, fetal tau (the smallest isoform) and other MAPs are initially homogeneously distributed. Tau expression increases sharply during brain development, concomitant with neurite outgrowth, diversification of tau (from one isoform to six in humans) and sorting into axons (Drubin and Kirschner, 1986; Kosik and Finch, 1987). Several mechanisms could contribute to axonal sorting, e.g., tau protein in axons could bind more tightly to microtubules than in dendrites, where it might be degraded more easily; tau protein could be targeted into axons but not into dendrites; and tau mRNA could be targeted into axons for local translation (Hirokawa et al., 1996; Aronov et al., 2002; Utton et al., 2002). Tau is transported along the axon with the rate of slow axonal transport, similar to tubulin and most other cytoskeletal proteins (0.1–8 mm/d, or ∼0.001–0.08 μm/s), whereas vesicles move at rates of fast axonal transport (50–400 mm/d, ∼0.5–4 μm/s) (Mercken et al., 1995). In the case of MTs and neurofilaments, the slow axonal transport rate is the result of fast, bidirectional, and intermittent transport along axons (Roy et al., 2000; Wang et al., 2000; Wang and Brown, 2002). The transport mechanism of MAPs and cytosolic proteins in the axon is still a matter of debate (Baas and Buster, 2004). For vesicles, mitochondria, and neurofilaments, the long-haul transport uses mainly MTs as tracks and motor proteins kinesin for anterograde and dynein for retrograde movement. In contrast, actin filaments and their corresponding myosin motors are used for short-haul transport of mitochondria and vesicles to achieve exact positioning and membrane fusion (Hollenbeck and Saxton, 2005).
The present study was motivated by the apparent contradiction between the ability of tau to inhibit anterograde transport of vesicles and organelles, whereas tau itself is able to distribute along axons (Stamer et al., 2002). To understand how tau is transported along the axon and what changes occur when tau becomes missorted, we analyzed diffusion, MT binding, and the transport behavior of different cyan fluorescent protein (CFP)-tagged tau mutants in primary neurons by live cell microscopy, fluorescence recovery after photobleaching (FRAP), and fluorescence speckle microscopy (FSM). The results show that tau diffuses unexpectedly fast, considering its nature as an MT-bound protein. This is explained by the highly dynamic interaction between tau and MT in the axon. The high exchange rate of tau is observed even when tau is not phosphorylated. The fast diffusion of tau contributes to its dispersion along the axon. The main transport mechanism in near-physiological conditions appears to be the piggybacking of tau on MT fragments, which are transported along the axon rapidly at typical velocities of motor proteins and with saltatory characteristics. At elevated tau concentrations, reminiscent of an AD-like situation, globular accumulations of tau appear, which move bidirectionally along the axon and build up from the distal end of axons.
Materials and Methods
Plasmid construction, transfection, and adenovirus production.
For expression in Vero cells and adenovirus production, the SfiI–NheI fragments containing parts of the cDNAs of htau40-ΔK280, htau40-P301L (both FTDP-17 mutants), pseudophosphorylated tau-KXGE (Ser-to-Glu at all four KXGS motifs, positions 262, 293, 324, 356), and eight repeat mutants of tau (tau without N-terminal inserts and including a duplicated insertion of the four repeats) (Gustke et al., 1994) were subcloned into the respective restriction sites of the pShuttle cytomegalovirus (CMV) vector containing CFP-htau40. A fusion of three enhanced green fluorescent protein (EGFP) molecules to the N terminus of htau40 in the pShuttle vector was performed by PCR TOPO cloning (Invitrogen, Carlsbad, CA) of three EGFPs from the 3GFP-myo5 plasmid (kindly provided by G. Steinberg, Max-Planck Institute, Marburg, Germany), followed by subcloning into CMV-tau-pShuttle. For transfection into Vero cells, DNA was prepared by an endotoxin-free Maxiprep kit (Qiagen, Hilden, Germany) and 1 μg of DNA was transfected into 106 cells with an electroporation system (Amaxa, Cologne, Germany) according to the manual. After transfection, the cells were directly plated in self-made glass-bottom chambers and incubated for 6–24 h before imaging. Recombinant adenoviruses for transfection of primary neurons were generated according to the study by He et al. (1998).
Transfection of retinal ganglion and cortical cultured neurons.
Cortical neurons were obtained from embryonic day 18 (E18) rat embryos and cultured according to the study by Banker and Goslin (1988). Cells were transfected 6–8 d after plating. For adenoviral transfection of primary cortical and retinal neurons with the vectors encoding the tau fusion proteins a 100-fold multiplicity of infection was used. Retinal ganglion neurons were obtained from white leghorn chicken eyes at E6 and cultured in glass-bottom dishes. The dishes were previously incubated with 4 μg/ml laminin overnight (Sigma-Aldrich, St. Louis, MO) and washed twice. Retina explants were centered in the dishes and incubated for 24 h in DME-F-12 media (Invitrogen) with 10% FCS and 0.4% methyl cellulose at 37°C, 5% CO2, and 100% relative humidity. The transfection procedure was performed as described above.
Live-cell imaging and FSM.
For live-cell imaging and FSM, cells were imaged in glass-bottom dishes with an Axiovert 200 (Zeiss, Oberkochen, Germany) equipped with a planapochromatic 63× (numerical aperture, 1.4) oil-immersion objective lens and a 50 ms filterwheel (Sutter Instrument, Novato, CA) for fast dual-color imaging. The complete microscope was integrated in an incubation system (Harnischmacher, Hannover, Germany) to image cells at 37°C and 5% CO2. Images were captured with a CCD camera (CoolSNAP HQ; Roper Scientific, Tucson, AZ) and MetaMorph imaging software (Universal Imaging, Buckinghamshire, UK). Time-lapse series were obtained at 0.5–3 s intervals. Image processing and measurements were done with MetaMorph, ImageJ, and Photoshop (Adobe Systems, San Jose, CA). For FSM, the cells were only incubated for 4–6 h before imaging instead of 1–2 d. Mitochondria were visualized by changing the medium against fresh prewarmed medium with a final concentration of 12 nm MitoTracker Red (Invitrogen, Karlsruhe, Germany) for 2 h, followed by exchange of MitoTracker medium for normal medium before imaging. CFP quantification was performed by reference to calibrated solutions.
Photobleaching.
Two different types of bleaching experiments were done: (1) bleaching of CFP-tau in a large axonal region to obtain the diffusion coefficient and (2) bleaching of small regions of CFP-tau in neurons and on microtubules in Vero cells to study the microtubule binding behavior. In the first method, an LSM510 confocal microscope (Zeiss) was used to bleach 30- to 50-μm-long axonal regions of retinal ganglion cell (RGC) axons with 100% laser intensity of the 458 nm line of a 100 mW argon laser. The microscope was inside an incubation system (Harnischmacher) to image cells at 37°C and 5% CO2. For imaging, the same laser was used with 10% laser intensity. To calculate the apparent diffusion coefficient, the signals of the central region of the bleached area containing the whole width of the axon were averaged. The background obtained from areas outside the fluorescent axon was subtracted. Bleaching by imaging was nearly avoided by applying a low laser power and long imaging intervals; it was corrected by calculating the loss of signals in unbleached axons and from the signal intensities in the image series before bleaching. The apparent diffusion coefficient, Dapp, was calculated from the half-time of recovery, t1/2, at the center of the bleached region using the equation Dapp = 0.275 L2/t1/2, valid for diffusion in a narrow cylinder open at both ends, with L equaling the length of the bleached area. For the second photobleaching procedure, a confocal microscope Olympus (Tokyo, Japan) FV1000 was used. To obtain faster bleaching and faster images after bleaching, a second scanner for simultaneous bleaching was used, equipped with a 405 nm UV-laser diode (Melles Griot, Irvine, CA). A 2- to 4-μm-diameter circle was bleached with 50% laser intensity, with the Tornado scanning function and imaging of the bleached region started directly after bleaching ended (bleaching time, 0.1–0.3 s). To avoid bleaching by imaging the time interval was adapted to the half-time of the analyzed protein (0.3–2 s). The signals of a small linear region along the microtubule containing the whole width of the microtubule and the length of the bleached area were averaged and background subtracted. The background was measured in microtubule-free areas of the same cell. If general bleaching appeared, it was subtracted as explained above. The intensity was plotted as a function of time and fitted with a single-exponential function to generate the half-time of microtubule binding (Bulinski et al., 2001).
Results
Tau moves along the axon, whereas anterograde transport of organelles and vesicles is inhibited
The adenovirus vector encoding CFP-htau40 fusion protein (CFP-tau) was added to 8-d-old rat cortical neurons. Twenty hours after transfection CFP-tau became visible in the cell body and in proximal parts of the neurites (Fig. 1b). Time-lapse imaging of CFP-tau-expressing neurons showed a strong decrease in the number of moving mitochondria, in particular the net anterograde transport was reduced (Fig. 1c–f). Similar observations applied to other axonally transported vesicles and organelles (data not shown). When the expression rate was increased, the CFP-tau signals moved further along the axon in the same time period (20 h) (Fig. 1g–i), indicating that the entry of tau into neurites is concentration dependent. At the same time, extended axonal regions were already cleared of mitochondria because of the strong inhibition of anterograde transport by tau. Therefore, the question arises how tau manages to travel down the axons despite its negative effect on microtubule-based traffic in general.
CFP-tau expression leads to a gradient of tau distribution along the axon
To understand the movement of tau along the axon, series of z-stack images were collected from cortical neurons. Ten to twenty z-stacks from the cell body to the distal end of a given neuron were obtained. The images with highest signal intensities were joined up and pseudocolored to show the variations in the CFP-tau concentrations. CFP-tau clearly shows a smoothly decreasing concentration from the cell body toward the distal end of the axon (Fig. 1j–k). Over the distances and time periods of our experiments, we did not observe a sharply defined advancing front, nor did we observe vesicle-like movements. The decreasing signal intensity up to 400 μm away from the cell body can be explained by diffusion, without the need to assume active transport. Likewise, in primary neurons, tubulin showed no moving front or vesicle-like localization along the axon, although it is actively transported by motor proteins (He et al., 2005). One explanation of this observation is that diffusion is a major determinant of the distribution, at least over distances of up to ∼1, and that tau is not transported in well defined packets.
We investigated the formation of the tau gradient by time-lapse analysis. For long-time imaging, we used an adenovirus expressing mono-red fluorescent protein (mRFP)-tau instead of CFP-tau to reduce bleaching and photodamage. In strongly expressing cells, the first mRFP-tau signals were visible 3 h after transfection and were followed by neurodegeneration within 12 h. A milder expressing neuron is illustrated in Figure 2a–b. The signal intensity developed at the same rate in the cell body and the initial part of the axon, indicating rapid distribution by diffusion. At 100 μm into the axon, the tau signal increased more slowly because the protein has to move out along the axon. To understand how the appearance of tau depends on active transport and/or diffusion, we analyzed diffusion, MT binding, and transport of tau in this system using FRAP and FSM (Waterman-Storer et al., 1998).
Figure 2.

Tau binds to MTs and stains the axon smoothly. a, Time-lapse images showing the distribution of mRFP-htau40 in cortical neurons. b, Intensity versus time at different positions. c, d, Chicken RGC neurons transfected with CFP-htau40. Tau localization along the axon is visualized in a black and white image (c). d, Tau localization on single MTs in the growth cone, extending from the central domain to the left and to the bottom (MTs in the shaft are not resolved). e, In mouse cortical neurons expressing CFP-htau40, tau stains MTs in the cell body, axon, and dendrites smoothly. f, In Vero cells, tau stains the MTs. g, h, In contrast, the tau-KXGE mutant lacks strong axonal localization (g) and shows no MT binding in Vero cells (h). Scale bars, 20 μm.
Axonal tau accumulation depends on MT binding
We generated adenoviruses encoding CFP-tagged fusion proteins of different tau mutants and analyzed their localization in primary cortical neurons, RGC neurons, and Vero cells. Because high levels of tau can cause cell defects, we expressed CFP-tau only briefly (<24 h) and chose cells with low signal intensities, barely observable at sensitive settings. Chicken RGC neurons (Fig. 2c) showed smoothly decaying tau gradients along axons from the proximal to the distal end without major discontinuities (gaps or local clusters). In rat cortical neurons and Vero cells, CFP-tau also localized to microtubules, as indicated by the pattern in the growth cone and the smooth staining along the axon (Fig. 2d–f). A strong localization to the axon and on MTs in Vero cells was also observed for a tau mutant with a duplicated repeat domain (CFP-tau-8R) (data not shown). Two FTDP-17 mutants of full-length four-repeat tau were studied as well, CFP-tauΔK280 and CFP-tauP301L, which bind to MT with a somewhat reduced affinity (Barghorn et al., 2000; Lu and Kosik, 2001). In both cases, there was a smooth axonal localization, analogous to wild-type tau, and the proteins were localized on microtubules in Vero cells (data not shown). These mutants did not differ in their localization on microtubules in non-neuronal cells or in neuronal cell bodies and along axons (first 100 μm), compared with wild-type tau.
Of particular interest was the question of tau phosphorylation at the KXGS motifs of the repeats because this causes the detachment of tau from microtubules (Biernat et al., 1993). In normal conditions, tau showed a low extent of phosphorylation at these sites, consistent with its microtubule-bound state. Likewise, the CFP-tau-4KXGA mutant showed the same localization as the wild-type protein (data not shown), as expected because the phosphorylation sites were eliminated in all KXGS motifs. In contrast, the mutant CFP-tau-4KXGE is in a pseudophosphorylated state; it showed weak microtubule binding and a weaker axonal localization (Fig. 2g–h). In other words, tau mutants that bind less tightly to MTs are less localized in the axon. These data suggest that spreading of tau into the axon depends not only on its diffusion but also on the equilibration between free and microtubule-bound tau.
To verify differences between wild-type and mutants, the diffusion coefficients and MT-binding kinetics were analyzed and a mathematical model of the dispersion of the different tau mutants was calculated and will be described below (Fig. 6d and supplemental material, available at www.jneurosci.org).
Figure 6.

FRAP of CFP-tau on MTs in axons of RGC neurons. a, Photobleaching of CFP-tau8R in a 3 μm area of an axon. Because diffusion over this short distance is fast, the half-time of recovery represents the off-rate of tau from the MTs. b, Mean signal intensities along the axon shown in a before the bleach (black), directly after the bleach (red) and 15.4 s after the bleach (light blue). c, Mean recovery time ± SD of different tau mutants and of tau at different positions along the axon, showing only small variations (3–4 s). wt, Wild type. d, Comparison of tau distribution along the axon by diffusion and transport (computed on the basis of observed and published rates of transport and diffusion; details in supplemental material, available at www.jneurosci.org). The advance of the “front” of tau (defined as half of the initial tau concentration) is plotted versus time. Top, Long time axis (30 d). Bottom, Initial short time axis (16 h). The modeling shows that diffusion can move tau in 4 d by 1 mm along the axon (top, solid black line) before it becomes too slow to account for the published transport rate of ∼0.003 μm/s (solid blue line) (Mercken et al., 1995). At longer distances (beyond the intersection between the black and blue solid curves), diffusion is too inefficient to distribute tau, and active transport is needed for axons longer than ∼1 mm. The spreading of tau into the axon reported by Utton et al. (2002) (bottom, short pink line, marked by cross) could be fully explained by diffusion alone.
Quantification of CFP-tau in neurons
To understand how the tau transport in axons is correlated with the level of tau expression, we quantified the amount of CFP-tau in the axons. Recombinant CFP was cloned and purified, and a calibration curve of intensity versus concentration was generated using confocal microscopy. We compared the signal intensity of CFP in solution directly with the fluorescence intensity of CFP-tau in the axon to define the CFP-tau concentration. Image stacks of 16 different RGC axons expressing different amounts of CFP-tau in four separate experiments were imaged under the same microscope settings. The concentration of CFP-tau was calculated by correlating the signal intensity of up to 50-μm-long areas of axons with the intensities of purified CFP in solution. We found two different modes of tau localization in axons, one with a smooth tau distribution (type-1 axons) and axons with clusters or inhomogeneities in the tau distribution (type-2 axons). The mean tau concentrations in type-1 axons were relatively low (∼1–2 μm), whereas type-2 axons had higher tau concentrations (up to 4 μm). In the example of Figure 3, the tau concentrations along two axons were plotted over 40 μm to visualize the large variation of tau concentrations in a type-2 axon, compared with the more even distribution along type-1 axons (Fig. 3c). The transition from a smooth to an inhomogeneous distribution begins at a mean tau concentration of approximately ∼1 μm.
Figure 3.

Quantification of CFP-tau in neurons. a, CFP-tau image with two transfected axons. b, Same axons as in a in color spectrum to visualize the different signal intensities in the two axons. The protein concentration was calculated from a calibration against a CFP solution. c, Tau concentrations along two representative axons. Gray, Background intensity; blue, the tau concentration in an axon with low and uniformly dispersed tau, red, axon containing inhomogeneities. The line shows the moving average (50 pixels). Note the “bumpy” trace and higher tau concentration in an axon containing tau clusters. d, Scheme of a neuron. The concentration of smoothly distributed tau (red) decreases from the cell body to the growth cone, whereas local tau accumulations emanate from the distal end of the axon first. Scale bar, 10 μm.
FRAP experiments of CFP-tau reveals rapid diffusion
To determine tau distribution at near-physiological conditions of low tau expression (type-1 axons), we performed FRAP experiments on primary RGC axons growing from an explant of a chick retina. The bleaching eliminates the background of total tau (mostly microtubule-bound) in the bleached zone, and CFP-tau diffuses into the zone after bleaching. Thirty- to fifty-micrometer-long areas of axons were bleached and observed by confocal microscopy. The fluorescence in the bleached zone was filled up rapidly from both ends so that in ∼3 min, ∼50% of the initial fluorescence was recovered at the center (Fig. 4). We did not detect a preferential direction of tau recovery through the bleached zone, either in the form of a moving front or as tau particles. The recovery of the signal intensity could be fitted to a curve consistent with rapid diffusion in a thin axonal cylinder. In this model, the recovery time to 50% at the midpoint is given by t50% = 0.27 L2/D, yielding an apparent diffusion coefficient D ≈ 3 μm2/s for CFP-tau. This high value was unexpected considering that most tau is bound to MTs. In contrast, bleaching and photoactivation experiments with labeled tubulin lead to long-lived gaps or activated areas (200–400 min), indicating that tubulin does not diffuse readily (Lim et al., 1989; Sabry et al., 1995).
Figure 4.

FRAP of RGC neurons expressing CFP-tau. a, Time-lapse fluorescence images of axon (t = 0 at end of bleaching). a, b, Red vertical lines indicate the bleached region. The boxed area (a) defines the region used for the kymograph (b). Scale bars, 10 μm. c, Normalized signal intensities along the axon before bleaching (black), directly after bleaching (red), and 194 s after bleaching (blue), showing ∼50% recovery. Red lines in center delineate the area for measuring the intensity recovery and hence the diffusion coefficient, shown in d as a percentage of initial level (half time of recovery, ∼180 s). e, Apparent diffusion coefficients of GFP, CFP-tau, and 3 CFP-tau mutants derived from FRAP.
To understand the fast diffusion of tau, we performed FRAP experiments to define the diffusion coefficients of different CFP-tau mutants and GFP as a control. The experiments were done at a distance of at least 300 μm from the cell body and the growth cone. GFP diffuses at D ∼ 25 μm2/s, approximately twice as fast as CFP-tau-4KXGE, which cannot bind to MT (D ∼11 μm2/s) (Fig. 4e). Both proteins diffuse freely because their observed diffusion coefficients are consistent with theoretical expectations calculated from their molecular weights and hydrodynamic radii (see supplemental material, available at www.jneurosci.org) (GFP, Dcalc ∼ 28; CFP-tau-4KXGE, Dcalc∼12 μm2/s). In contrast, CFP-tau diffuses approximately four times more slowly (D ∼ 3 μm2/s) than CFP-tau-4KXGE because ∼75% of it is MT-bound (Table 1 and supplemental material, available at www.jneurosci.org). The enhancement of the microtubule affinity of the eight-repeat mutant results in a 12-fold reduced diffusion compared with CFP-tau-4KXGE (D ∼ 1 μm2/s).
Table 1.
Diffusion coefficients and microtubule-binding parameters
| Tau construct | Dapp (μm2/s) | Dtheor (μm2/s) | Fraction of free tau in axon |
Rate of detachment [koff (s−1)] | MT dwell time [t1/2 (s)] | |
|---|---|---|---|---|---|---|
| observed | Dapp/Dtheor | |||||
| GFP | 25 | 28 | ||||
| CFP-htau40 | 3 | 12 | 25% | 25% | 0.17 | 4 |
| CFP-htau40–4KXGA | 3 | 12 | n.d. | 25% | 0.17 | 4 |
| CFP-htau40–4KXGE | 11 | 12 | 100% | 92% | 1.4 | 0.5 |
| CFP-htau40–8R | 1 | 9 | n.d. | 11% | 0.046 | 15 |
| YFP-tubulin | n.d. | 14 | n.d. | n.d. | 0.008 | 90 |
| GFP-tau23a | 0.25 | 2.8 | ||||
| GFP-tau24a | 0.28 | 2.5 | ||||
| GFP-ensconsinb | 0.195 | 3.56 | ||||
n.d., Not determined.
aSamsonov et al., 2004 (values for axonal shaft, single MT).
The relatively fast recovery of CFP-tau after bleaching is suggestive of a highly dynamic interaction with microtubules. However, theoretically one might argue that a similar result could be achieved by phosphorylation of tau at sites which influence MT binding. To test this hypothesis, the diffusion of the nonphosphorylatable mutant CFP-tau-4KXGA was measured. Its diffusion (D ∼ 3 μm2/s) did not differ significantly from that of CFP-tau, arguing that the exchange rate of tau on the microtubule surface does not require phosphorylation.
Tau–microtubule interaction is highly dynamic in the axon
Although the above FRAP experiments over long axonal segments report on the overall diffusion of tau, FRAPping a small volume (4 μm long) reveals the dynamics of the tau–microtubule interaction because in this case the diffusion of tau is fast, and recovery of the fluorescence signal is mainly determined by the off-rate koff (Bulinski et al., 2001) (see supplemental material, available at www.jneurosci.org). Vero cells were transfected either with CFP-tau, CFP-8Rtau, or yellow fluorescent protein (YFP)-tubulin. Areas of 2–4 μm length along single microtubules were bleached, and the recovery of the fluorescence was measured (Fig. 5). The recovery time of CFP-tau was ∼3 s. By comparison, the tight binding CFP-8Rtau has a recovery half-time of 20 s, whereas YFP-tubulin requires a long time to recover (90 s), because tubulin is trapped within the microtubule lattice. Thus the recovery time of tau in Vero cells is extremely fast compared with MAP2 and MAP4 (15–30 times) (Olmsted et al., 1989; Samsonov et al., 2004). This result is consistent with the fast diffusion seen in axons.
Figure 5.

Photobleaching and recovery of CFP-tau bound to microtubules. a, FRAP of CFP-tau8R on MT in a Vero cell. Fluorescence recovers evenly in the entire bleached region rather than starting from the borders of the bleached zone (in contrast to large bleached areas in axons; see Fig. 4). b, Time-lapse imaging of cell shown in a in 2 s intervals (black and white; left), color spectrum view (right). Scale bar, 2 μm. c, Fluorescence recovery of b at center of bleached zone, reaching ∼50% of initial value with a half-time of 20 s. d, Mean recovery rate ± SEM of CFP-tau, YFP-tubulin, and CFP-tau8R. Note that CFP-tau recovers rapidly (half-time, ∼3 s), tightly binding tau8R slowly (∼20 s), and tubulin very slowly (∼90 s) because it is mostly integrated into the MT wall.
Next we wanted to quantify the rates of tau exchange on microtubules for the special geometry of an axon. Areas of 2–4 μm length along RGC axons were bleached with a 405 nm laser. In such small areas of the axon, the rate limiting step is the release of tau from the microtubule, rather than diffusion, resulting in a half-time that depends on microtubule binding (see supplemental material, available at www.jneurosci.org). The time-lapse video of the 8R-tau mutant (Fig. 6a) shows the clear borders after bleaching. The half-time of MT rebinding is ∼15 s, similar to the results in Vero cells. CFP-tau (wild type) has a rebinding half-time of 3.8 s, close to the 4KXGA mutant at 4.2 s (Fig. 6c). This confirms the results from the diffusion experiments that under these conditions, the fast dynamics of tau–MT binding is not dependent on tau phosphorylation. The t1/2 values of the FTDP-17 mutants ΔK280 and P301L (3.1 and 3.0 s) are marginally smaller than the t1/2 of tau, but these differences are hardly significant. Thus the FTDP-17 pathology of these tau mutants cannot be explained by large differences in the MT binding behavior in axons. Furthermore, the diffusion properties of tau were homogeneous throughout the axons, i.e., the microtubule binding in proximal axon segments, far away at the growth cone or in between were similar. By comparison, in the bleaching experiment of tau on MTs in glia cells of RGC explants, tau shows a recovery time t1/2 of 2.7 s, close to the recovery in Vero cells. The t1/2 values are somewhat shorter than in axons (3.8 s), indicating that diffusion cannot be neglected completely in these experiments, especially in the case of axons (see supplemental material, available at www.jneurosci.org).
We used FSM with the CFP-tau construct in Vero cells. The binding of tau to MTs is more dynamic than that of all other published MAPs so far but close to the t1/2 of ensconsin (MAP2, t1/2 = 60 s; MAP4, t1/2 = 44 s; ensconsin, t1/2 = 4 s) (Olmsted et al., 1989; Bulinski et al., 2001). Because our results were all derived from FRAP experiments, we wanted to confirm them with a second independent method such as FSM (Waterman-Storer et al., 1998). In this method, a low concentration of CFP-tau should lead to an uneven distribution of tau on MTs, resulting in a speckled appearance (Bulinski et al., 2001). The dynamics of these speckles can be used to study tau dynamics on MTs. Unexpectedly, low concentration of CFP-tau in Vero cells showed a uniform localization along microtubules (Fig. 7a,c). To visualize lower tau concentrations, we enhanced the signal intensity of single tau molecules by generating derivatives carrying up to three EGFP molecules at the N terminus. This revealed a speckled distribution of tau along MTs, which was followed over time (Fig. 7b,d,e). MT binding and diffusion was still rapid because most of the speckles were visible only over a short time period (2–5 s), confirming the results from the FRAP measurements. Thus, no movement of dots along microtubules was visible so that evidence for transport could not be obtained in this experiment.
Figure 7.
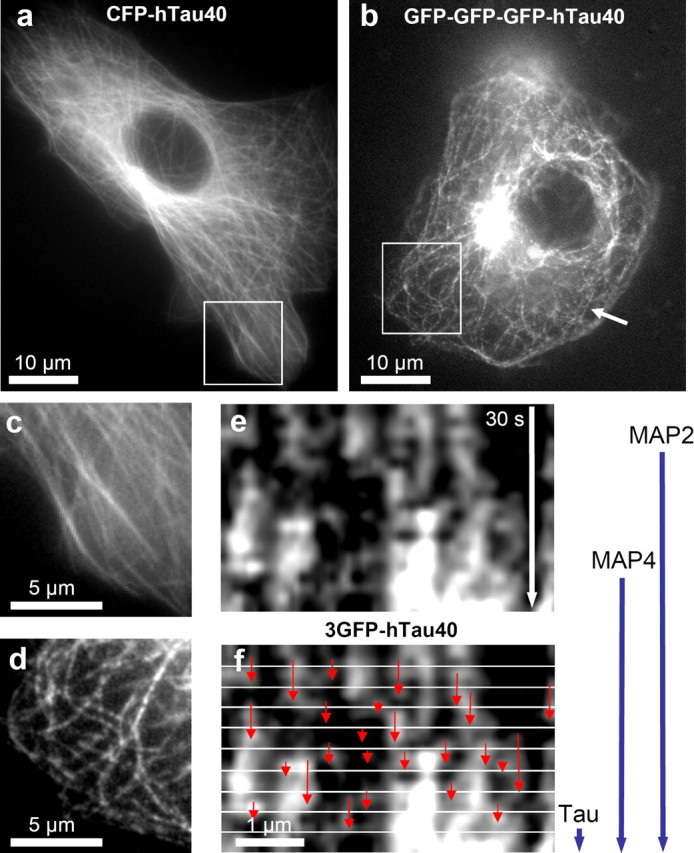
FSM of 3GFP-tau on microtubules. a, CFP-tau in Vero cells stains MTs evenly. b, 3GFP-tau shows a speckled distribution along MTs. c, d, Magnifications of marked areas in a and b. e, Kymograph of a MT from the cell in b. The time lapse consists of 20 pictures spaced 1.5 s (30 s total), illustrating the appearance and disappearance of speckles of fluorescent 3GFP-tau. f, In the same kymograph, 3 s intervals are separated by white lines. Red arrows indicate the residence time of single 3GFP-tau molecules on the MT. This is consistent with the half-time of tau-MT binding defined by the FRAP experiment (∼3 s) (Fig. 5), indicated by the blue vertical arrows on the right (this study and Olmsted et al., 1989; Samsonov et al., 2004).
Rapid movement of eight-repeat tau through bleached zones of RGC axons in short filament-like structures
The bleaching experiments did not reveal a transport mechanism of tau but showed that tau can distribute rapidly by diffusion because of the short-lived MT interaction, although on average, most of tau is bound to MTs. The results argue against the possibility that tau is transported along the axon as part of moving vesicles or protein complexes, because this kind of transport would have been visible in the bleaching experiments. A possible mechanism could be the transport as single proteins or in small accumulations not detectable with our microscopic system. A second possibility is that tau is transported along with a moving particle to which it attaches in a dynamic manner. Because tau has a dynamic interaction with MTs, we considered the possibility that tau binds to moving MT pieces, which have been postulated as the transport units of tubulin in neurons (He et al., 2005). To test this hypothesis, the CFP-8Rtau mutant and CFP-tau were analyzed more carefully. If tau were transported on MT fragments, the bleached area should be longer visible with CFP-8Rtau because it binds to MT more tightly and diffuses more slowly. The FRAP experiment of large regions of the axon (30–60 μm) was repeated with an Olympus FV1000 microscope, which is more sensitive, generates bleached regions more efficiently, and allows image collection during bleaching. The sensitivity and laser intensity were chosen so that the signal before the bleach was strongly oversaturated to be able to detect very weak signals. Under these conditions, it was possible to visualize CFP-8Rtau in 2- to 6-μm-long filament-like structures moving through the bleached zone (Fig. 8a,b). Movement events were observed at a frequency of 0.2 events per minute in an axon stretch of 100 μm. Instantaneous velocities ranged between 0.2–2 μm/s in both directions, with mean velocities ∼0.7 μm/s and pausing frequencies ∼2/min (Fig. 8c,d). The velocities are typical for motor protein-driven transport.
Figure 8.

Bidirectional axonal transport of tau in small filamentous structures in RGC neurons. a, Time lapse of a FRAP experiment showing anterograde CFP-tau movement in a 5 μm filamentous structure at a velocity of 0.4 μm/s. The image sequence starts before the bleach event. The recovery by diffusion is visible by the appearance of the CFP signal along the axon and the anterograde entry of a tau containing structure into the bleached region is indicated by arrows. In the right column the same time-lapse series was strongly contrast enhanced and blurred, and in every image, the fluorescence was subtracted by the signal of a later image (3 positions) to visualize tau movement without the background of recovery. b, Analogous time-lapse series but with a retrograde movement event of tau. The tau-containing structure is visible as a 4-μm-long filament moving rapidly (0.8 μm/s) along the axon. c, Anterograde displacement of tau structure along the axon with time and instantaneous velocities (average speed, ∼0.6 μm/s; instantaneous velocities range, ∼0.2–1.3 μm/s). d, Retrograde displacement of a tau structure and instantaneous velocity (average speed, ∼0.4 μm/s; instantaneous velocities, ∼0.2–0.9 μm/s).
Anterograde transport of tau on fast moving microtubules in cortical neurons
In bleaching experiments, the tau mutant with a duplicated repeat domain and enhanced MT affinity (8R-tau) was the only tau species visible as part of a moving filament-like structure. The results suggest that tau can be transported on small MT fragments through the axon, but the images from wild-type tau become blurred because of the short dwell time on MTs. To test whether wild-type four-repeat tau can be observed piggybacking on MT fragments from the cell body into axons, we applied the protocol of Ahmad and Baas (1995). Primary cortical rat neurons were grown for 3 d and transfected with CFP-tau adenovirus, and 24 h later, tau was visible on the MTs in the cell body (Fig. 9a). The neurons were incubated for 6 h with 10 μg/ml nocodazole to destroy the MT cytoskeleton (Fig. 9b). After nocodazole washout, the cells were incubated for 10 min at 37°C. During this time, the MTs resume growth from the centrosomes and reach lengths of ∼5 μm (Fig. 9c). The addition of 40 nm vinblastine stops MT polymerization, and subsequently, it was possible to observe MTs with CFP-tau being severed from the centrosome and moving to the periphery (Fig. 9d–h). Because the MTs are limited to the close proximity of the centrosome in this experiment, the moving MT fragments have to move to the periphery of the cell body and into axons in an actin-dependent manner (Ahmad and Baas, 1995). A fraction of these tau-containing MT fragments entered the axon (Fig. 9d). These MTs are moving at rates up to 1 μm/s (Fig. 9d,h). Together these results show that axonal transport of tau occurs at least in part by piggybacking on fast-moving MT fragments.
Figure 9.

Anterograde transport of tau on small MT filaments. a, Cortical mouse neurons transfected with CFP-htau40 and imaged after 24 h. The scheme (top row) shows MTs in red and the nucleus in blue; the images in the second row show CFP-tau. Images were processed with NIH Image (contrast, smooth, and shadow) to enhance contrast. b–d, The shape of the cell is indicated by a dashed line in orange color. b, After 24 h microtubules were depolymerized by nocodazole for 6 h. c, Image 10 min after nocodazole washout. Tau accumulates on small MTs growing out of the centrosome (arrow). d, After blocking MT dynamics by adding 40 nm vinblastine, tau localizes on small anterogradely moving MT fragments (length, ∼2–8 μm) detached from the centrosome. The boxed area was enlarged and signal enhanced in the top right corner. The orange arrow points to an MT moving into an axon, and white arrows point to moving MTs. e, Time-lapse series of tau cotransported with anterogradely moving MTs. f, Small MTs (white arrows) build up the usual MT array for neuronal cell bodies lying along the plasma membrane instead of forming an aster. Orange arrows indicate MTs already moved far away from the centrosome. g, Overlay of pictures 1 (red), 3 (green), and 4 (blue) of time-lapse series (e). Arrows indicate the movement of 3 MTs. h, Distances traveled by the three MTs in g versus time. Scale bar, 10 μm.
Rapid bidirectional movement of tau clusters from the distal end of the axon
At higher tau concentrations and longer expression times (>2 d), the pattern of tau localization changes its character in cortical and RGC neurons. Instead of a distribution with a smooth gradient (type-1 distribution), CFP-tau begins to accumulate in particles along the axon. In the two crossing RGC neurons of Figure 10a, one has a low CFP-tau intensity and a smooth tau distribution, and the other has stronger tau signals with a smooth background and additional tau “particles.” To visualize complete cortical neurons from the cell body to the distal end, they were imaged in multiple series of z-stacks after 2 d of tau expression, revealing a similar accumulation of tau into globular structures as in RGC neurons. Interestingly, in all neurons, these particles begin to appear near the distal end of the axon, not near the cell body, where tau was first visible after transfection (Fig. 10b). This indicates that the clustering of tau is a secondary effect that may result from tau-induced transport problems (Stamer et al., 2002). Time-lapse studies of the tau particles showed rapid but intermittent bidirectional movements at instantaneous velocities of ∼0.3 μm/s. They largely ceased during treatment with nocodazole, indicating that movements are dominated by MT-dependent transport (Fig. 10c–f).
Figure 10.

Tau forms dynamic axonal clusters when highly overexpressed. a, c, d, RGC neurons 2 d after transfection with CFP-htau40. Two axons are shown in a: black and white (left) and color spectrum image (right) to enhance the resolution of the signal intensities. Axon 1 has a low expression of CFP-htau40 and therefore a smooth distribution. Axon 2 has a threefold higher CFP-htau40 concentration, leading to an uneven tau distribution. The boxed area of axon 1 is signal enhanced to verify that the tau distribution is uniform. b, Cortical neurons 2 d after transfection with CFP-tau. The top shows an axon with an even tau distribution in the proximal part and a clustered tau distribution in the distal part. Note that after 2 d of expression the tau gradient is no longer visible in the 1-mm-long axon. The magnified image (inset; boxed) shows that the transition between smooth and granular tau distribution is quite abrupt. The axon in the bottom shows a clustered tau distribution along the whole visible part of the axon at a higher magnification. c, d, Time-lapse studies of CFP-tau clusters in axons of RGC neurons. Top left, Frame time in seconds; arrows mark moving particles. Left frames show anterograde movement (v = 0.4 μm/s), and right frames show retrograde (0.2 μm/s). d, Moving tau clusters in the same conditions but after treatment with 10 μm nocodazole for 6 h to destroy all MTs (v = 0.1 μm/s). e, Life history of moving tau cluster (position vs time). Red, pink, and white in bar indicate phases of anterograde or retrograde movements, and pausing. Note that the movement is intermittent and bidirectional. Fast phases in both direction have speeds of ∼0.5 μm/s, but the net movement during the observation period (180 s) is much slower (0.02 μm/s anterogradely). f, Histograms depict mean (left) and instantaneous (right) velocity distributions of moving tau clusters.
Discussion
Tau, one of the major MAPs in the brain, is important for axonal outgrowth and for stabilizing microtubules as tracks for axonal transport (Drubin and Kirschner, 1986). Tau is normally sorted into the axonal compartment; however, in disease states such as Alzheimer's, it becomes missorted into the somatodendritic compartment and forms abnormal aggregates (Lee et al., 2001). In previous studies, we noticed that tau has the potential of inhibiting MT-based transport by interfering with the attachment of motors to MTs. Both kinesin and dynein are inhibited, but the effect is more pronounced with kinesin. This leads to a predominance of retrograde flow and net inhibition of anterograde flow, so that vesicles and organelles tend to accumulate in the cell body. The consequence is the starvation and decay of cell processes (Trinczek et al., 1999; Stamer et al., 2002; Vershinin et al., 2006). Surprisingly, tau itself is able to enter cell processes despite the general inhibition of anterograde transport, suggesting some distinct mode of distribution. This prompted us to investigate the transport of tau in neurons. Previous radiolabeling studies had shown that tau propagates overall at the rate of slow axonal transport (∼0.2–0.4 mm/d, equivalent to ∼0.003 μm/s), close to that of tubulin (Mercken et al., 1995). This was confirmed by fluorescence microscopy using GFP-labeled tau transfected into neuronal cultures (Utton et al., 2002). These authors also provided evidence that tau forms local accumulations (particles) that move rapidly at instantaneous rates of ∼0.3 μm/s, close to the rate of fast axonal transport, but with extended pauses (Utton et al., 2005). This would make tau transport akin to the slow axonal transport described for neurofilaments (Wang et al., 2000).
We approached the tau transport problem by adenoviral transfection of different XFP-labeled tau variants into differentiated primary neuronal cultures and observing them by several time-resolved microscopic methods, including confocal, deconvolution, FRAP, and FSM. This enabled us to distinguish three modes of tau propagation into axons and their dependence on microtubule interactions. The main results are discussed below, and the main observations on diffusion and photobleach recovery are summarized in Table 1.
The major mechanism determining the tau distribution in the short-to-intermediate range is its rapid diffusion. The apparent diffusion constant of CFP-tau (D ≈ 3 μm2/s) is surprisingly high, only fourfold less than the diffusion constant of the free cytosolic protein and ensures that tau can distribute evenly throughout the cytosolic volume. This means, for example, that tau can move significantly into axons (millimeter range) without active transport (Fig. 6d). In fact, the rates of spreading by diffusion agree well with the rates considered as active transport by Utton et al. (2002). Because only a low tau/tubulin ratio is required for MT stabilization, diffusion of tau would suffice, at least for short segments of axons.
At first glance, the high mobility of tau appears contradictory to the view of tau as a microtubule-associated protein. Indeed, the majority of tau is concentrated on the microtubule surface, but because the exchange rates are high (t1/2 ≈ 4 s), this does not prevent tau from redistributing along the microtubule or between different microtubules.
The mechanism of diffusion can only explain some aspects of tau distribution, and active transport makes the major contribution over long distances (as part of slow axonal transport at rates of 0.2–0.4 mm/d) (Mercken et al., 1995). Axonal microtubules are mostly stationary (Sabry et al., 1995), but short microtubule fragments or tubulin oligomers moving along the axon would provide such a mechanism (for review, see Baas and Buster, 2004). The movement of such MT fragments has been observed by tubulin fluorescence (Ahmad and Baas, 1995; Wang and Brown, 2002), but the movement of tau has been difficult to detect. This can be explained by the loose coupling between MT and tau, which tends to blur the fluorescence of labeled tau. It requires tightly binding tau variants (such as 8R-tau) to visualize the movement of the tau component in such oligomeric complexes. In this context, we note that there are conflicting interpretations regarding the nature of the short tubulin-containing structures. They have been regarded either as MT fragments/tubulin oligomers (Wang and Brown, 2002; He et al., 2005), or alternatively as tubulin-containing vesicles (Ma et al., 2004). The binding of tau to these structures provides strong support for the first option, because tubulin contained in a vesicle would not be readily accessible to cytosolic tau.
The relationship between MT affinity, diffusion, and visualization is well illustrated by different tau variants: when critical sites within the repeat domain (KXGS motifs) are pseudophosphorylated (to KXGE), the MT affinity is strongly reduced. These variants are no longer “MT-centered” but “volume-centered,” because they diffuse almost freely (D ≈ 11 μm2/s) and thus fill the available space, regardless of MTs. However, they fail to reach a high concentration at far distances, where diffusion becomes slow because they are not tied to moving carriers (MT fragments). Conversely, tightly binding variants such as 8R-tau spend nearly all their time on a microtubule surface (t1/2 ≈ 15 s), hence their diffusion is much slower (D ≈ 1 μm2/s). Thus, complexes of MT fragments with 8R-tau contain a sufficient number of well localized fluorophores so that their movement becomes visible.
Tau phosphorylation and tau mutations are important issues in Alzheimer's disease and frontotemporal dementias. As mentioned above, tau pseudophosphorylated in the repeat domain (KXGE mutant) behaves essentially as a free molecule, consistent with the detachment of tau from MTs by this type of phosphorylation (Drewes et al., 1997). In contrast, the nonphosphorylatable protein (KXGA mutant) behaves identically with wild-type tau in terms of distribution and diffusion, arguing that phosphorylation affects only a small fraction of the expressed tau in these cells. Thus the highly dynamic behavior of tau can be explained without invoking phosphorylation, in contrast to previous proposals (Samsonov et al., 2004). The two FTDP-17 mutants tested, ΔK280 and P301L, are very similar to wild-type tau with respect to diffusion and dwell time on MT. This means that the mutations in the tau gene do not affect MT interactions as such but operate on other levels, possibly isoform distribution, aggregation, phosphorylation, or degradation.
Regarding the mechanism of long-range transport of tau, according to the work of Utton et al. (2005), we expected to observe tau particles moving along axonal MTs in a rapid stop-and-go manner, consistent with the accepted view of slow axonal transport (Wang et al., 2000). We indeed observed inhomogeneous tau accumulations with typical sizes in the 1–10 μm range. However, we believe that this is not the normal physiological mechanism but rather a sign of degenerating axons, akin to incipient axonal swellings. The reasons are that (1) tau particles occur only at high levels of tau expression and (2) more importantly, they originate not in the cell body or proximal axon, but in the distal regions of axons. It is known that tau induces defects in axonal traffic because it interferes with motor–microtubule interactions (Mandelkow et al., 2004). This causes gradual axonal degeneration, starting at the extremities and accompanied by the accumulations of proteins, vesicles, and organelles. We consider the tau particles to be a sign of this process. In contrast, the physiological tau carriers (MT fragments) are barely visible (see above); they represent a small fraction and originate at the cell body or proximal axon.
Given the rapid diffusion of tau, one may ask why tau is confined to axons in differentiated neurons or, conversely, why the appearance in the somatodendritic compartment is a sign of a disease state. The sorting mechanism for tau and other MAPs becomes effective only toward the end of the fetal stage, concomitant with the expression of adult tau isoforms and a reduction of phosphorylation level (Drubin and Kirschner, 1986; Garcia and Cleveland, 2001). Several mechanisms of axonal sorting have been envisaged, e.g., preferential routing of tau mRNA to the axon (Hirokawa et al., 1996; Aronov et al., 2002), preferential stabilization in the axon (e.g., by MT interactions and/or phosphorylation), and preferential loss of MT binding and degradation in the dendrites (Kanai and Hirokawa, 1995). In fact, when tau protein is microinjected, it rapidly diffuses into both axons and dendrites, but subsequently it persists only in axons (Hirokawa et al., 1996). Our data show that a fraction of tau moves actively on MT fragments. This takes place even under conditions in which tau also fills cell bodies and dendrites, similar to the distribution in AD. The anterograde traffic of MT fragments is sustained by the motor protein dynein (50%) and actin filaments (50%) (He et al., 2005). This feature, combined with the rapid diffusion, would explain why tau transport works even if the kinesin-dependent transport of vesicles and organelles is already inhibited. In this view, the initial distribution of expressed CFP-tau can mostly be accounted for by diffusion, whereas the long-range distribution requires an active transport component. Conversely, the pathological somatodendritic distribution of tau in AD would not be caused by missorting in the strict sense but rather to a higher effective diffusion rate of hyperphosphorylated tau because it is largely detached from microtubules, combined with a failure of tau degradation.
Footnotes
This work was supported in part by the Deutsche Forschungsgemeinschaft. We thank N. Gehrke and F. Baltruschat for cloning and adenovirus production, Dr. J. Biernat for providing many tau constructs, and Dr. G. Steinberg (Max Planck Institute, Marburg, Germany) for providing a 3x-eGFP plasmid.
References
- Ahmad FJ, Baas PW. Microtubules released from the neuronal centrosome are transported into the axon. J Cell Sci. 1995;108:2761–2769. doi: 10.1242/jcs.108.8.2761. [DOI] [PubMed] [Google Scholar]
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Aronov S, Aranda G, Behar L, Ginzburg I. Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J Cell Sci. 2002;115:3817–3827. doi: 10.1242/jcs.00058. [DOI] [PubMed] [Google Scholar]
- Baas PW, Buster DW. Slow axonal transport and the genesis of neuronal morphology. J Neurobiol. 2004;58:3–17. doi: 10.1002/neu.10281. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Developments in neuronal cell culture. Nature. 1988;336:185–186. doi: 10.1038/336185a0. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- Bulinski JC, Odde DJ, Howell BJ, Salmon TD, Waterman-Storer CM. Rapid dynamics of the microtubule binding of ensconsin in vivo. J Cell Sci. 2001;114:3885–3897. doi: 10.1242/jcs.114.21.3885. [DOI] [PubMed] [Google Scholar]
- Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Svitkina TM, Borisy GG, Popov SV. Speckle microscopic evaluation of microtubule transport in growing nerve processes. Nat Cell Biol. 1999;1:399–403. doi: 10.1038/15629. [DOI] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Cowan NJ, Hirokawa N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature. 1992;360:674–677. doi: 10.1038/360674a0. [DOI] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia ML, Cleveland DW. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol. 2001;13:41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Funakoshi T, Sato-Harada R, Kanai Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J Cell Biol. 1996;132:667–679. doi: 10.1083/jcb.132.4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton W. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hirokawa N. Sorting mechanisms of tau and MAP2 in neurons: suppressed axonal transit of MAP2 and locally regulated microtubule binding. Neuron. 1995;14:421–432. doi: 10.1016/0896-6273(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Finch EA. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J Neurosci. 1987;7:3142–3153. doi: 10.1523/JNEUROSCI.07-10-03142.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lim SS, Sammak PJ, Borisy GG. Progressive and spatially differentiated stability of microtubules in developing neuronal cells. J Cell Biol. 1989;109:253–263. doi: 10.1083/jcb.109.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Kosik KS. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol Biol Cell. 2001;12:171–184. doi: 10.1091/mbc.12.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Shakiryanova D, Vardya I, Popov SV. Quantitative analysis of microtubule transport in growing nerve processes. Curr Biol. 2004;14:725–730. doi: 10.1016/j.cub.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol. 2004;167:99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercken M, Fischer I, Kosik KS, Nixon RA. Three distinct axonal transport rates for tau, tubulin, and other microtubule-associated proteins: evidence for dynamic interactions of tau with microtubules in vivo. J Neurosci. 1995;15:8259–8267. doi: 10.1523/JNEUROSCI.15-12-08259.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Direct evidence for coherent low velocity axonal transport of mitochondria. J Cell Biol. 2006;173:373–381. doi: 10.1083/jcb.200510097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted JB, Stemple DL, Saxton W, Neighbors B, McIntosh JR. Cell cycle-dependent changes in the dynamics of MAP 2 and MAP 4 in cultured cells. J Cell Biol. 1989;109:211–223. doi: 10.1083/jcb.109.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Coffee P, Smith G, Liem RK, Brady ST, Black MM. Neurofilaments are transported rapidly but intermittently in axons: implications for slow axonal transport. J Neurosci. 2000;20:6849–6861. doi: 10.1523/JNEUROSCI.20-18-06849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabry J, O'Connor TP, Kirschner MW. Axonal transport of tubulin in Ti1 pioneer neurons in situ. Neuron. 1995;14:1247–1256. doi: 10.1016/0896-6273(95)90271-6. [DOI] [PubMed] [Google Scholar]
- Samsonov A, Yu JZ, Rasenick M, Popov SV. Tau interaction with microtubules in vivo. J Cell Sci. 2004;117:6129–6141. doi: 10.1242/jcs.01531. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, III, Mumby MC, Bloom GS. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem. 1999;274:25490–25498. doi: 10.1074/jbc.274.36.25490. [DOI] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;1739:280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112:2355–2367. doi: 10.1242/jcs.112.14.2355. [DOI] [PubMed] [Google Scholar]
- Utton MA, Connell J, Asuni AA, van Slegtenhorst M, Hutton M, de Silva R, Lees AJ, Miller CC, Anderton BH. The slow axonal transport of the microtubule-associated protein tau and the transport rates of different isoforms and mutants in cultured neurons. J Neurosci. 2002;22:6394–6400. doi: 10.1523/JNEUROSCI.22-15-06394.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utton MA, Noble WJ, Hill JE, Anderton BH, Hanger DP. Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J Cell Sci. 2005;118:4645–4654. doi: 10.1242/jcs.02558. [DOI] [PubMed] [Google Scholar]
- Vershinin M, Carter B, Razafsky D, King SJ, Gross SP. Multiple-motor based transport and its regulation by Tau. Proc Natl Acad Sci USA. 2006;104:87–92. doi: 10.1073/pnas.0607919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brown A. Rapid movement of microtubules in axons. Curr Biol. 2002;12:1496–1501. doi: 10.1016/s0960-9822(02)01078-3. [DOI] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RK, Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2:137–141. doi: 10.1038/35004008. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Desai A, Bulinski JC, Salmon ED. Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr Biol. 1998;8:1227–1230. doi: 10.1016/s0960-9822(07)00515-5. [DOI] [PubMed] [Google Scholar]