

Abstract
Postsynaptic morphogenesis of dendrites is essential for the establishment of neural connectivity in the brain, but the mechanisms that govern postsynaptic dendritic differentiation remain poorly understood. Sumoylation of the transcription factor myocyte enhancer factor 2A (MEF2A) promotes the differentiation of postsynaptic granule neuron dendritic claws in the cerebellar cortex. Here, we identify the protein PIASx as a MEF2 SUMO E3 ligase that represses MEF2-dependent transcription in neurons. Gain-of-function and genetic knockdown experiments in rat cerebellar slices and in the postnatal cerebellum in vivo reveal that PIASx drives the differentiation of granule neuron dendritic claws in the cerebellar cortex. MEF2A knockdown suppresses PIASx-induced dendritic claw differentiation, and expression of sumoylated MEF2A reverses PIASx knockdown-induced loss of dendritic claws. These findings define the PIASx-MEF2 sumoylation signaling link as a key mechanism that orchestrates postsynaptic dendritic claw morphogenesis in the cerebellar cortex and suggest novel functions for SUMO E3 ligases in brain development and plasticity.
Keywords: sumoylation, transcription factor, signal transduction, dendrite, cerebellar cortex, granule neurons
Introduction
Postsynaptic differentiation of dendrites is a fundamental process in synapse formation. During brain development, dendrite morphogenesis begins with the specification and growth of dendrites from the neuronal cell body, followed by a phase of maturation comprised of dendritic remodeling and the differentiation of postsynaptic specializations at sites of synaptic contact with other neurons (Cline, 2001; Luo, 2002; Jan and Jan, 2003). Disruption of postsynaptic dendritic morphology is thought to play a key role in the pathogenesis of several neurologic and psychiatric disorders, including mental retardation and neurodegenerative diseases (Lin et al., 1997; Irwin et al., 2000; Kaufmann and Moser, 2000; Fiala et al., 2002; Spires and Hyman, 2004). Therefore, defining the pathways that regulate postsynaptic dendritic morphogenesis is essential for a better understanding of both brain development and disease.
Granule neurons in the rodent cerebellar cortex provide an excellent model system for investigating the mechanisms that underlie neuronal morphogenesis and connectivity. After neurogenesis and migration, granule neurons settle in the internal granule layer (IGL) of the cerebellar cortex (Hatten and Heintz, 1995; Altman and Bayer, 1997). Here, they issue several dendrites, which are later pruned. The remaining few dendrites form on their ends dendritic cups that surround mossy fiber terminals and Golgi neuron axons. As these connections mature, the smooth surface of granule neuron dendritic cups becomes serrated and undulating, transforming into claw-like structures on which mossy fiber terminals and Golgi neuron axons synapse (Palay and Chan-Palay, 1974; Hamori and Somogyi, 1983; Ramon y Cajal, 1995; Altman and Bayer, 1997). The postsynaptic protein postsynaptic density-95 (PSD-95) is enriched in dendritic claws compared with dendrite shafts of granule neurons in the cerebellar cortex (Shalizi et al., 2006). In addition, the presynaptic marker synaptophysin is found within the cleft or apposed to dendritic claws (Shalizi et al., 2006). Dendritic claws in granule neurons thus represent specialized dendritic structures, analogous to the smaller dendritic spines in principal neurons that house synapses with afferent neurons (Hamori and Somogyi, 1983; Altman and Bayer, 1997; Ethell and Pasquale, 2005; Lippman and Dunaevsky, 2005; Shalizi et al., 2006; Tada and Sheng, 2006). The formation of dendritic claws in granule neurons constitutes a critical step of postsynaptic dendritic differentiation that is necessary for normal synapse development with mossy fibers and Golgi neuron axons in the cerebellar cortex.
We found that the transcription factor myocyte enhancer factor 2A (MEF2A) plays a critical role in postsynaptic dendritic morphogenesis in the brain (Shalizi et al., 2006). Genetic knockdown of MEF2A by RNA interference (RNAi) in rat cerebellar slices and in the developing postnatal rat cerebellum in vivo revealed an essential function for MEF2A in granule neuron dendritic claw differentiation (Shalizi et al., 2006). Importantly, a small ubiquitin-related modifier (SUMO)-modified form of MEF2A that acts as a transcriptional repressor induces postsynaptic dendritic differentiation (Shalizi et al., 2006). These findings raise the major question of the identity of the enzyme that stimulates MEF2A sumoylation and thereby drives postsynaptic dendritic morphogenesis.
Sumoylation, the covalent linkage of a small ubiquitin-related modifier to the ε-amine of lysine residues in target proteins, requires the activities of three sets of enzymes (Johnson, 2004). SUMO is first attached to the bipartite SUMO-activating enzyme Aos1/Uba2 (E1) in an ATP-dependent manner (Gong et al., 1999), followed by transfer of SUMO to the SUMO-conjugating enzyme Ubc9 (E2) (Desterro et al., 1997; Gong et al., 1997). In turn, Ubc9 catalyzes the transfer of SUMO to a substrate protein, a reaction that is facilitated by a SUMO E3 ligase (Melchior et al., 2003; Gill, 2004). The PIAS [protein inhibitors of activated STAT (signal transducer and activator of transcription)] proteins form the largest family of SUMO E3 ligases (Schmidt and Muller, 2003; Shuai and Liu, 2005). These proteins were originally isolated based on their ability to inhibit STAT proteins, hence the name protein inhibitors of activated STAT (Chung et al., 1997; Liu et al., 1998). The PIAS proteins were subsequently found to encode SUMO E3 ligase activity (Jackson, 2001; Schmidt and Muller, 2003; Shuai and Liu, 2005). The biological functions of the PIAS SUMO ligases in the nervous system remained unknown.
In this study, we defined a novel function for PIASx as a MEF2A SUMO E3 ligase that promotes dendritic claw differentiation in the cerebellar cortex. By controlling MEF2A sumoylation and consequent postsynaptic dendritic differentiation in the cerebellar cortex, PIASx may play a pivotal role in the establishment of neuronal connectivity.
Materials and Methods
Plasmids and antibodies.
The pBJ5-FLAG-HDAC4 expression plasmid was a gift from Dr. Stuart Schreiber (Harvard University, Cambridge, MA). The pCDNA3-HA-calcineurin A* was generated by cloning the cDNA encoding constitutively active calcineurin A into pCDNA3. The MEF2A and yeast galactose response gene 4-MEF2A (GAL4-MEF2A) expression plasmids, including MEF2A and GAL4-MEF2A sumoylation mutants, pCDNA3-HA-SUMO1, and luciferase and renilla reporter constructs, have been described previously (Shalizi et al., 2006).
The PIASx RNAi plasmids were generated by cloning the following oligonucleotides into pBS/U6 or pBS/U6-cmv-GFP, where the italics indicates the targeted sequence of PIASx: piasx1 5′-AACAGAAGCGCCCTGGACGCTTCAAGCTTGCGTCCAGGGCGCTTCTGTTCTTTTTG-3′; piasx2 5′-GGGTTCTCATGTATCAGCCATACAAGCTTTGGCTGATACATGAGAACCCCTTTTTG-3′. The RNAi-resistant PIASx-Res construct was generated by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) according to the protocol of the manufacturer and incorporated the following silent mutations indicated by lowercase letters: 5′-GTg CTa ATG TAc CAa-3′.
The PIASx antibodies used are described previously (Moilanen et al., 1999; Arora et al., 2003). The FLAG monoclonal antibody was purchased from Sigma (St. Louis, MO). The hemagglutinin (HA) polyclonal, MEF2 polyclonal, and GAL4 monoclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The green fluorescent protein (GFP) polyclonal antibody was purchased from Invitrogen (Eugene, OR). The extracellular signal-regulated kinase 1/2 (ERK1/2) antibody was purchased from Promega (Madison, WI). The MEF2A-pS408 polyclonal antibody was described previously (Shalizi et al., 2006).
Cell culture and transfections.
Cultures of primary granule neurons were isolated from postnatal day 6 (P6) Long–Evans rats as described previously (Shalizi et al., 2003). Granule neurons were maintained in full medium [basal medium, Eagle (BME) plus 10% calf serum (Hyclone, Logan, UT), 1 mm each penicillin, streptomycin, and l-glutamine, and 25 mm KCl]. Granule neurons were transfected in DMEM by DNA-calcium phosphate precipitation as described previously (Konishi et al., 2002).
Human embryonic kidney 293T (HEK293T) cells were maintained in DMEM supplemented with 10% calf serum and 1 mm each of penicillin, streptomycin, and l-glutamine. 293T cells were transfected by DNA-calcium phosphate precipitation as described previously (Shalizi et al., 2006). Medium was replaced 24 h after transfection, and cells were harvested 48 h after transfection for in vivo sumoylation assays, coimmunoprecipitation studies, or luciferase-reporter assays and 72–96 h after transfection for RNAi studies.
Protein immunoprecipitation and sumoylation assays.
In vivo sumoylation assays were performed as described previously (Shalizi et al., 2006). Briefly, HEK293T cells cotransfected with expression plasmids for full-length MEF2A or GAL4-MEF2A, HA-SUMO1, and other proteins as indicated were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mm NaCl, 10 mm Na2HPO4, pH 7.2, 2 mm EDTA, 50 mm NaF, 1 mm NaVO4, 1% NP-40, 0.1% SDS, 0.75% sodium deoxycholate, 1 mm PMSF, 10 mm N-ethylmaleimide, 10 μg/ml aprotinin) and precleared with protein A-Sepharose beads. Five percent of this starting material was retained for detection of input proteins, and the remainder was subjected to immunoprecipitation overnight at 4°C. For experiments using full-length MEF2A, a MEF2 polyclonal antibody (Santa Cruz Biotechnology) was used together with protein A-Sepharose beads, and for experiments using GAL4-MEF2A, an agarose-conjugated GAL4 monoclonal antibody (Santa Cruz Biotechnology) was used. Immune complexes were washed five times with RIPA buffer at 4°C and resuspended in Laemli's buffer. Immune complexes and input samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies.
Coimmunoprecipitation experiments were performed as described previously (Becker and Bonni, 2006) with minor modifications. Briefly, HEK293T cells cotransfected with expression plasmids for FLAG-PIASx and MEF2A-WT or MEF2A-S408A were lysed in coimmunoprecipitation buffer (150 mm NaCl, 50 mm Tris HCl, pH 7.5, 1 mm EDTA, 50 mm NaF, 1 mm NaVO4, 1% NP-40, 1 mm PMSF, 10 mm N-ethylmaleimide, 10 μg/ml aprotinin), and precleared with protein G-Sepharose beads. Five percent of this starting material was retained for detection of input proteins, and the remainder was subjected to immunoprecipitation with anti-FLAG antibodies for 4 h at 4°C. Immune complexes were bound to protein G-Sepharose beads for 1 h at 4°C, washed twice with coimmunoprecipitation buffer, once with PBS (pH 7.4), and resuspended in Laemli's buffer. Immune complexes and input samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies.
Luciferase assays.
Luciferase assays were performed as described previously (Shalizi et al., 2006) with minor modifications. Granule neurons were transfected with the reporter constructs pNur77-luc or pNur77mut-luc and renilla luciferase-thymidine kinase promoter (pRL-TK) and the indicated hairpin RNA (hpRNA) expression plasmids, and an expression construct for Bcl-XL. Granule neurons were switched from full medium to fresh BME supplemented with 5% calf serum (Hyclone) 72 h after transfection and incubated overnight. HEK293T cells maintained as described were cotransfected with p5G4luc or pMEF2x3luc and pRL-TK reporter constructs and the indicated expression plasmids by DNA-calcium phosphate precipitation. Fresh growth media were added within 24 h of transfection. Cells were lysed 48 h after transfection. In both neurons and HEK293T cells, firefly- and renilla-luciferase activities were determined using a dual-luciferase assay kit (Promega) according to the instructions of the manufacturer.
Reverse transcription-PCR.
RNA was prepared from HEK293T cells or granule neurons using TRIzol (Invitrogen) according to the instructions of the manufacturer. Purified RNA was subjected to reverse transcription (RT)-PCR using the SuperScript II one-step RT-PCR system (Invitrogen) according to the protocol of the manufacturer. Amplification conditions were as follows: cDNA synthesis for 30 min at 55°C followed by 1 min at 95°C and 25 [glyceraldehyde-3-phosphate dehydrogenase (GAPDH)] or 30 (PIASx) cycles of amplification at 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min, with a final extension at 72°C for 5 min. PCR products were separated by agarose gel electrophoresis in 1× Tris-acetate-EDTA. Primers for GAPDH have been described previously (Shalizi et al., 2003). Primers for PIASx were sense 5′-CCTTTGCCTGGCTATGCACC-3′ and antisense 5′-CAGGACAAATCCAGGTGGGC-3′.
Cerebellar slices.
Slices were prepared and processed as described previously (Gaudilliere et al., 2004; Shalizi et al., 2006). Briefly, cerebella from P9 or P10 rats were dissected in HHGN (2.5 mm HEPES, 35 mm glucose, 4 mm NaHCO3 diluted in Cellgro HBSS) and cut into 400 μm sagittal slices using a tissue chopper (McIllwain), transferred to a porous membrane (Millicell-CM Low Height Culture Plate Insert) that allows for an air-media interface, and maintained in MEM supplemented with 25% horse serum, 2.5% 10× Cellgro HBSS, 1% Invitrogen Penicillin-Streptomycin-Glutamine, 12.5 mm HEPES, and 22 mm glucose. At 4 d in vitro (DIV), slices were transfected using biolistics (Helios Gene Gun; Bio-Rad, Hercules, CA) as described previously (Gaudilliere et al., 2004; Shalizi et al., 2006). At 8 DIV, slices were fixed in 4% paraformaldehyde, then blocked and permeabilized in 0.4% Triton X-100 in 10% goat serum/PBS. Slices were incubated with a rabbit GFP antibody (Invitrogen A6455) at 1:500 in 1% goat serum, 0.05% BSA, 0.025% sodium azide, 0.4% Triton X-100 in PBS overnight at 4°C, and then with goat anti-rabbit secondary antibody conjugated with Cy2 or Cy3 (GE Healthcare, Arlington Heights, IL) at 1:500 for 2 h at room temperature. Nuclei were stained with the DNA dye bisbenzimide (Hoechst 33258).
Microscopy was performed as described previously (Gaudilliere et al., 2004; Shalizi et al., 2006). For confocal imaging of cerebellar slices, Z series (0.5 μm) of images of transfected granule neurons were obtained at 60× magnification on a Nikon (Tokyo, Japan) TE2000-U spinning disc confocal microscope. Two-dimensional reconstruction of Z-series images was then performed using a maximum brightness projection algorithm (Volocity imaging software). Images of transfected granule neurons were analyzed using SPOT software for dendritic length and number of primary and secondary dendrites as described previously (Gaudilliere et al., 2004; Shalizi et al., 2006; Ramos et al., 2007).
In vivo electroporation in the postnatal cerebellum.
Rat pups (P3) were subjected to in vivo electroporation as described previously (Konishi et al., 2004; Stegmuller et al., 2006) and analyzed by immunohistochemical analysis at P12 as described previously (Shalizi et al., 2006).
Cerebellar and cortical lysates.
The cerebellum and cerebral cortex were dissected from P6, P10, and P14 rat pups in HHGN. After isolation, these structures were transferred to lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 1 mm DTT, 50 mm NaF, 1 mm sodium orthovanadate, 3 μg/ml aprotinin, 1 μg/ml leupeptin, 2 μg/ml pepstatin) and homogenized using a Brinkmann Polytron homogenizer (Kinematica, Newark, NJ). After 10 min, the homogenates were spun down at 14,000 rpm, and supernatant was collected.
Results
PIASx is a MEF2A SUMO E3 ligase
The finding that sumoylation of MEF2A plays a key role in postsynaptic dendritic morphogenesis raises the fundamental issue of identifying the SUMO E3 ligase that stimulates MEF2A sumoylation and thereby promotes postsynaptic dendritic differentiation. The PIAS proteins comprise the largest family of SUMO E3 ligases (Shuai and Liu, 2005; Sharrocks, 2006). Although the PIAS proteins are expressed in the nervous system (Wu et al., 1997; Garcia-Dominguez et al., 2006), their functions in the nervous system remain primarily unknown.
Using a candidate approach, we asked whether a PIAS protein might promote SUMO modification of MEF2A in a sumoylation assay in cells. We coexpressed MEF2A and HA-tagged SUMO alone or together with members of the PIAS family of proteins (PIAS1, PIAS3, PIASxα, PIASxβ, PIASy) in HEK293T cells. Because class IIa histone deacetylases (HDACs) interact with MEF2 proteins and are associated with SUMO E3 ligase activity (Gregoire and Yang, 2005; Zhao et al., 2005), we also tested the ability of HDAC4 to stimulate MEF2A sumoylation. Lysates of transfected cells were subjected to immunoprecipitation of MEF2A followed by immunoblotting with an antibody to HA to detect SUMO-modified MEF2A. Among the different SUMO E3 ligases, only PIASxα and PIASxβ efficiently increased the level of SUMO-modified MEF2A (Fig. 1 A). PIASxα and PIASxβ, which represent the products of spliced mRNAs encoded by the same gene (Shuai and Liu, 2005), promoted MEF2A sumoylation to a similar extent. For the sake of clarity in the remainder of the study, we refer to the two isoforms collectively as PIASx.
Figure 1.

PIASx is a SUMO E3 ligase for MEF2A. A, HEK293T cells were cotransfected with expression plasmids encoding GAL4-MEF2A, HA-SUMO1, and the indicated FLAG-tagged candidate SUMO E3 ligase. After 48 h, cells were lysed and subjected to immunoprecipitation with a GAL4 antibody. Input and immunoprecipitated proteins were immunoblotted with the indicated antibodies. B, HEK293T cells were transfected with full-length MEF2A, HA-SUMO1, and increasing concentrations of distinct PIAS proteins. After 48 h, cells were lysed and subjected to immunoprecipitation with a MEF2 antibody, followed by immunoblotting with the indicated antibodies. PIASx, but not other PIAS proteins or HDAC4 (class IIa HDAC), enhanced MEF2A sumoylation. C, PIASx inhibits MEF2-dependent transcription. HEK293T cells were transfected with expression plasmids encoding MEF2A, a MEF2-responsive luciferase reporter gene, a renilla-luciferase reporter under control of the TK promoter (pRL-TK) as transfection control, and increasing concentrations of PIASx. After 48 h, cells were lysed, and the relative activity of firefly luciferase normalized to renilla-luciferase was determined. Data are shown as percentage of activity relative to cells transfected with MEF2A alone. PIASx significantly reduced MEF2A-dependent transcription (*p < 0.005; ANOVA; n = 3). D, PIASx sumoylates MEF2A on Lysine 403. Cells were transfected with expression plasmids encoding wild-type (WT) or Lys403Arg (K403R) GAL4-MEF2A and HA-SUMO1 with or without PIASx and analyzed as in A. In addition, immunoblot of total lysates with the ERK1/2 antibody served as loading control. PIASx enhanced sumoylation of WT but not K403R MEF2A. E, PIASx interacts with MEF2A independently of Ser408 phosphorylation. Cells were transfected with expression plasmids encoding WT or Ser408Ala (S408A) MEF2A with or without FLAG-PIASx. After 48 h, cells were lysed and subjected to immunoprecipitation with a FLAG antibody, followed by immunoblotting with a MEF2A or FLAG antibody. Both WT and S408A MEF2A interacted with FLAG-PIASx. F, PIASx induces MEF2A sumoylation in a Ser408 phosphorylation-dependent manner. Cells were cotransfected with expression plasmids encoding WT or S408A GAL4-MEF2A and HA-SUMO1 with or without PIASx and analyzed as in A. In addition, immunoblot of total lysates with the ERK1/2 antibody served as loading control. PIASx enhanced sumoylation of WT but not S408A MEF2A. G, Activated calcineurin reduces PIASx enhancement of MEF2A sumoylation. Cells were cotransfected with expression plasmids encoding GAL4-MEF2A and HA-SUMO1 with or without constitutively active calcineurin (CnA*) and with or without FLAG-PIASx, and analyzed as in A. Expression of constitutively active calcineurin inhibited PIASx-induced MEF2A sumoylation. Ctrl, Control; WB, Western blot.
We compared the potency of the different PIAS proteins in promoting the sumoylation of MEF2A by transfecting increasing amounts of PIAS1, PIAS3, PIASx, and PIASy in cells (Fig. 1 B). In these experiments, PIASx most robustly induced MEF2A sumoylation (Fig. 1 B). In contrast, expression of PIAS proteins other than PIASx often reduced the amount of SUMO-modified MEF2A (Fig. 1 A,B). In coimmunoprecipitation experiments, PIASx, PIAS1, PIAS3, and PIASy interacted with MEF2A (Fig. S1, available at www.jneurosci.org as supplemental material). These results are consistent with the possibility that PIAS proteins other than PIASx may have titrated critical cofactors of the machinery necessary for MEF2A sumoylation.
Sumoylation of MEF2A represses MEF2-dependent transcription (Shalizi et al., 2006). We therefore tested the effect of PIASx expression on MEF2-dependent transcription in cells. We transfected 293T cells with a luciferase reporter gene controlled by MEF2 response elements (MRE-luciferase) together with a PIASx expression plasmid or its control vector. In these experiments, PIASx potently inhibited MRE-luciferase reporter gene expression (Fig. 1 C). Thus, consistent with its ability to stimulate MEF2A sumoylation, PIASx represses MEF2-dependent transcription.
The sumoylation of MEF2A occurs on Lysine 403, which is part of a conserved peptide motif within the MEF2 repressor domain (Shalizi et al., 2006). Importantly, efficient sumoylation of MEF2A is dependent on the phosphorylation of MEF2A at the nearby site of Serine 408 (Shalizi et al., 2006). We asked whether PIASx induces sumoylation of MEF2A at Lysine 403 and whether the PIASx-induced MEF2A sumoylation is controlled by Serine 408 phosphorylation. In assays of sumoylation, whereas PIASx stimulated the sumoylation of wild-type MEF2A, PIASx failed to trigger the sumoylation of a MEF2A mutant in which Lysine 403 was replaced with arginine (MEF2AK403R) (Fig. 1 D). In other experiments, we found that although PIASx interacted with a MEF2A mutant in which Serine 408 was replaced with alanine (MEF2AS408A) as efficiently as with wild-type MEF2A (Fig. 1 E), PIASx failed to induce the robust sumoylation of MEF2AS408A (Fig. 1 F). The ability of PIASx to induce the sumoylation of wild-type MEF2A was also significantly reduced after coexpression of the activated form of the phosphatase calcineurin (Fig. 1 G), which induces the dephosphorylation of MEF2A at Serine 408 (Shalizi et al., 2006). These results suggest that PIASx induces the sumoylation of MEF2A at Lysine 403 in a Serine 408 phosphorylation-dependent manner. Collectively, based on distinct lines of evidence, we conclude that PIASx represents a bona-fide MEF2A SUMO E3 ligase.
PIASx promotes dendritic claw differentiation in the cerebellar cortex
The identification of PIASx as a MEF2A SUMO E3 ligase led us to investigate the function of PIASx in neurons. We first characterized the expression of PIASx in granule neurons in the cerebellum. PIASx mRNA and protein were detected in primary cerebellar granule neurons by RT-PCR and immunoblotting analyses (Fig. 2 A,B). Immunohistochemical analysis of the developing rat cerebellar cortex revealed expression of PIASx in both Purkinje neurons and granule neurons within the IGL (Fig. 2 C,D). PIASx expression was present in the cerebellar cortex in rat pups during the first and second weeks postnatally (Fig. 2 E). This pattern of expression overlaps with that of MEF2A (Shalizi et al., 2006). Because PIASx acts as a MEF2A SUMO E3 ligase, the overlapping pattern of PIASx and MEF2A expression in the cerebellar cortex suggested that PIASx might regulate MEF2A function in neurons.
Figure 2.

PIASx is expressed in the cerebellar cortex. A, PIASx mRNA is expressed in cerebellar granule neurons. RNA was extracted from HEK293T cells (293T) transfected with the PIASx expression plasmid or control plasmid as well as primary granule neurons from P6 rat pups cultured for 5 d (P6 plus 5 DIV) and subjected to RT-PCR with primers specific for PIASx or GAPDH as an internal control. B, PIASx protein is expressed in cerebellar granule neurons. Lysates of granule neurons (P6 plus 5 DIV; 30 μg of lysate), HEK293T cells transfected with the PIASx expression plasmid (5 μg of lysate), were immunoblotted with a PIASx antibody. The asterisk indicates a nonspecific band in HEK293T cell lysates. In granule neurons, the PIASx antibody recognizes two distinct isoforms of PIASx (PIASxα and the larger PIASxβ) indicated by two arrowheads. In HEK293T cells, the antibody recognizes overexpressed PIASxα and several bands that run with much higher mobility that represent posttranslationally modified PIASxα. C, PIASx is expressed in the cerebellar cortex. Left, Immunostaining of cryosectioned P8 cerebella with a rabbit antibody to PIASx (Moilanen et al., 1999) reveals PIASx immunoreactivity in cells within the IGL (arrowhead) and in Purkinje cells (arrow). Right, Nuclear stain with the DNA dye bisbenzimide (Hoechst) showing layers of the cerebellar cortex. Scale bar, 100 μm. D, PIASx is expressed in Purkinje neurons and IGL neurons. Immunohistochemistry of P8 cerebella with the PIASx antibody (I, IV, VI) as in C together with a mouse monoclonal antibody to calbindin (II) or the neuron-specific tubulin type III (Tuj1) (V, VI). Hoechst is shown in III. The top panels (I–III) show that PIASx is expressed in calbindin-positive Purkinje neurons. Arrowheads in VI indicate a few examples of IGL granule neurons double-labeled with Tuj1 and PIASx. Scale bar: top, 100 μm; bottom, 50 μm. E, PIASx is expressed in the rat cerebellum in the first and second weeks postnatally. Lysates of the cerebellum of animals with the indicated age were immunoblotted with the PIASx or the ERK1/2 antibody, the latter serving as control for loading. Ctrl, Control; EGL, external granule layer; 293T, HEK293T.
To determine the role of endogenous PIASx in the control of MEF2-regulated transcription in neurons, we used a plasmid-based method of RNAi to acutely knockdown PIASx (Gaudilliere et al., 2002). We constructed plasmids that encode hpRNAs targeting two distinct regions of PIASx (U6/piasx1 and U6/piasx2). Expression of each PIASx hairpin RNA but not a control-scrambled hairpin RNA induced the efficient knockdown of PIASx protein in cells (Fig. 3 A). PIASx RNAi induced the specific knockdown of PIASx but not the related protein PIAS1 (Fig. 3 A). In addition, PIASx RNAi reduced endogenous PIASx immunoreactivity in primary granule neurons stained with the antibody used in Figure 2 C (Fig. S2, available at www.jneurosci.org as supplemental material). Finally, PIASx RNAi reduced efficiently the expression of both PIASxα and PIASxβ in cells (Fig. S3, available at www.jneurosci.org as supplemental material). Collectively, these experiments indicate that PIASx RNAi reduces the expression of PIASx in cells and primary neurons.
Figure 3.

PIASx represses MEF2-dependent transcription in cerebellar granule neurons. A, PIASx RNAi induces specific knockdown of PIASx in cells. HEK293T cells were cotransfected with FLAG-tagged PIASx or PIAS1 expression plasmids, together with the control RNAi plasmid (U6/bs), PIASx RNAi plasmids encoding hpRNAs targeting distinct regions of PIASx mRNA (U6/piasx1 and U6/piasx2, respectively), or a control RNAi plasmid encoding scrambled hairpin RNAs (U6/scri), and FLAG-14-3-3 expression plasmid as a transfection control. After 72 h, cells were lysed and subjected to immunoblotting for FLAG-tagged proteins. Expression of PIASx hpRNAs, but not the scrambled hpRNAs, specifically reduced PIASx expression but not PIAS1 or 14-3-3 expression. B, PIASx represses the Nur77 promoter in primary cerebellar granule neurons. Granule neurons (P6 plus 4 DIV) were cotransfected with a luciferase reporter gene controlled by the Nur77 promoter (pNur77-luc), the PIASx RNAi or control RNAi plasmids, and pRL-TK as a control for transfection efficiency. After 72 h, cells were transferred to medium containing 5 mm KCl, and firefly and renilla luciferase activities were determined 24 h later. Expression of both piasx1 and piasx2 hpRNAs, but not the control scrambled hpRNA or empty RNAi vector, significantly enhanced Nur77 promoter activity (*p < 0.001; ANOVA; n = 4). C, MEF2 binding is necessary for PIASx-repression of the Nur77 promoter in neurons. Granule neurons (P6 plus 4 DIV) were transfected with wild-type (WT) or MEF2 binding site mutant (MREmut) pNur77 luc, the PIASx or control RNAi plasmids, and pRL-TK, and analyzed as in B. Data are normalized as percentage of activation by PIASx hpRNAs relative to the control RNAi plasmid. PIASx hpRNAs significantly enhanced the activity of WT but not MREmut pNur77-luc reporter gene (*p < 0.002; ANOVA; n = 3).
We transfected primary rat cerebellar granule neurons with the U6/piasx1, U6/piasx2, or control U6 RNAi plasmid together with a luciferase reporter gene controlled by the Nur77 promoter containing two MREs. Nur77 is a direct repressed target gene of sumoylated MEF2A in granule neurons, the repression of which promotes postsynaptic dendritic differentiation in the cerebellar cortex (Shalizi et al., 2006). We found that knockdown of PIASx, using either the U6/piasx1 or U6/piasx2 RNAi plasmid, significantly increased the level of Nur77 promoter-mediated transcription in granule neurons (Fig. 3 B). Expression of the control-scrambled hairpin RNA had no effect on the level of the Nur77 promoter (Fig. 3 B). In other experiments, we found that PIASx RNAi failed to induce the expression of a luciferase reporter gene driven by a Nur77 promoter-containing mutant MREs (Fig. 3 C). These results demonstrate that PIASx knockdown derepresses Nur77 promoter-mediated transcription in an MRE-dependent manner. Together, our experiments support the conclusion that PIASx represses MEF2-dependent transcription in primary neurons.
We next determined the role of PIASx in neuronal morphogenesis. We found recently that sumoylation of MEF2A promotes the differentiation of dendritic claws in granule neurons of the developing cerebellum (Shalizi et al., 2006). To determine PIASx function in dendritic morphogenesis in the cerebellar cortex, we induced PIASx knockdown in rat cerebellar slices (Fig. 4 A). Using a biolistics approach, we transfected cerebellar slices prepared from P10 rat pups with the U6/piasx1, U6/piasx2, or control U6 plasmid that also encoded GFP bicistronically. The U6/piasx-cmvGFP RNAi constructs were confirmed to induce the knockdown of PIASx (Fig. 4 B). Four days later, cerebellar slices were subjected to immunohistochemistry with an antibody to GFP to visualize transfected neurons within the cerebellar cortex. Granule neurons in the IGL were found with their typical small cell body and associated parallel fiber axons (Fig. 4 C, top panels). As expected, the dendrites of control U6-transfected neurons often harbored dendritic claws (Fig. 4 C, top panels). Dendritic claws were identified on the basis of classic descriptions as dendritic structures that are present at the end of dendrites, having cuplike or sicklelike appearance with inner serrated or undulating surfaces (Palay and Chan-Palay, 1974; Hamori and Somogyi, 1983; Ramon y Cajal, 1995; Altman and Bayer, 1997; Shalizi et al., 2006). Dendritic claws visualized in cerebellar slices are enriched with postsynaptic protein PSD-95 puncta, indicating that dendritic claws represent sites of postsynaptic differentiation (Shalizi et al., 2006).
Figure 4.
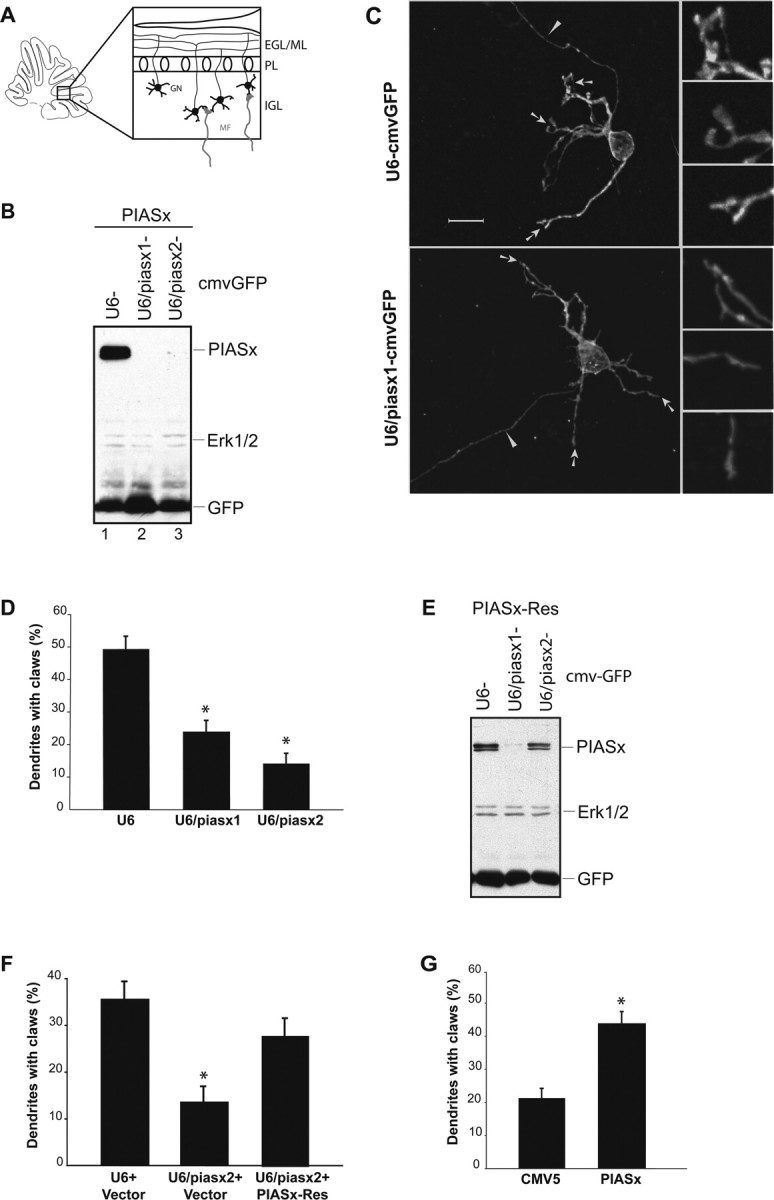
The SUMO E3 ligase PIASx promotes granule neuron dendritic claw differentiation in cerebellar slices. A, Schematic of a sagittal section through the developing cerebellar cortex. Granule neurons (GN) migrate from the external granule layer (EGL) and reside in the IGL, where they generate dendrites and dendritic claws on which mossy fibers (MF) and Golgi neuron axons form synapses. Parallel fiber axons of granule neurons extend in the molecular layer (ML) where they contact dendrites of Purkinje neurons, the cell bodies of which are in the Purkinje cell layer (PL). B, The U6/piasx1-cmvGFP and U6/piasx2-cmvGFP RNAi plasmids encoding two distinct hairpin RNAs to PIASx and expressing GFP bicistronically trigger knockdown of PIASx. Lysates of HEK293T cells transfected with each of the two RNAi plasmids or the control U6-cmvGFP RNAi plasmid together with a plasmid encoding FLAG-PIASx were immunoblotted with the FLAG, ERK1/2, and GFP antibodies. C, PIASx knockdown impairs the differentiation of granule neuron dendritic claws. Rat cerebellar slices were prepared from P10 rat pups, transfected 4 d later with the U6/piasx1-cmvGFP, U6/piasx2-cmvGFP, or control U6-cmvGFP RNAi plasmid, and subjected to immunohistochemistry with the GFP antibody. Representative confocal images of transfected GFP-positive granule neurons from the piasx1 hpRNA-expressing or control RNAi plasmid are shown. Arrows and arrowheads indicate dendritic claws and axons, respectively. Insets show higher-magnification views of ends of dendrites. Granule neurons in control-transfected slices have robust dendritic claws. In contrast, PIASx knockdown granule neurons have tapered dendritic ends. Scale bar, 5 μm. D, Quantification of the effect of PIASx knockdown on dendritic claw differentiation in rat cerebellar slices. The percentage of primary dendrites per neuron that harbor dendritic claws was measured in transfected granule neurons in the IGL in a blinded manner. The number of dendritic claws was significantly reduced in PIASx knockdown granule neurons induced by both PIASx hpRNAs, compared with granule neurons transfected with the control U6 RNAi plasmid (*p < 0.0001; ANOVA; total neurons measured, n = 106). E, Expression of an RNAi-resistant form of PIASx: PIASx-Res is encoded by cDNA designed to be resistant to piasx2 hairpin RNAs and encodes wild-type PIASx protein. Lysates of HEK293T cells transfected with an expression plasmid encoding FLAG-PIASx-Res and U6/piasx1-cmvGFP or U6/piasx2-cmvGFP or the control RNAi plasmid were immunoblotted with the FLAG, ERK1/2, and GFP antibodies. PIASx-Res is knocked down by piasx1 but not piasx2 hairpin RNAs. F, Cerebellar slices (P10) were transfected with the control U6-cmvGFP or U6/piasx2-cmvGFP RNAi plasmid together with the PIASx-Res expression plasmid or its control vector and analyzed as in C and D. The number of dendritic claws was significantly reduced in slices transfected with PIASx knockdown but not in PIASx-Res-expressing neurons in the presence of PIASx knockdown (*p < 0.005; ANOVA; total neurons measured, n = 118). G, Expression of PIASx promotes dendritic claw differentiation. Cerebellar slices prepared from P9 rat pups were transfected 4 d later with the PIASx expression plasmid or its control vector together with a GFP expression plasmid and analyzed as in C and D. The number of dendritic claws was significantly increased in PIASx-expressing granule neurons when compared with control-transfected neurons (*p < 0.0001; t test; total number of neurons measured, n = 63).
Strikingly, PIASx knockdown neurons had significantly fewer dendritic claws than control U6-transfected neurons, and dendrites of PIASx knockdown neurons typically displayed tapered ends (Fig. 4 C, bottom panels). There was a 50 and 70% reduction in the number of dendritic claws in cerebellar slices transfected with the piasx1 and piasx2 RNAi plasmids, respectively (Fig. 4 D). The PIASx knockdown-induced dendritic claw phenotype was not caused by impaired dendritic growth or branching, because PIASx RNAi did not lead to a reduction in dendritic length or the number of branches in cerebellar slices (Fig. S4, available at www.jneurosci.org as supplemental material). Together, these results suggest that PIASx plays a critical role in the differentiation of granule neuron dendritic claws.
To rule out the possibility that the PIASx knockdown-induced dendritic claw phenotype was the result of activation of the RNAi machinery per se, we performed a rescue experiment. We constructed an expression plasmid encoding wild-type PIASx protein using cDNA designed to be resistant to piasx2 hpRNAs (PIASx-Res). Although expression of piasx1 hpRNAs robustly induced knockdown of PIASx-Res, piasx2 hpRNAs failed to effectively trigger knockdown of PIASx-Res (Fig. 4 E). In cerebellar slices, whereas PIASx RNAi led to a significant reduction of dendritic claw number in granule neurons, expression of PIASx-Res reversed the piasx2 RNAi-induced dendritic phenotype, restoring the number of claws to nearly 80% of control U6-transfected cerebellar slices (Fig. 4 F). These results indicate that the PIASx RNAi-induced dendritic claw phenotype is the result of specific knockdown of PIASx rather than off-target effects of RNAi.
Having identified a requirement for endogenous PIASx in the differentiation of the postsynaptic dendritic claws in granule neurons, we next tested the effect of increasing the levels of PIASx above its endogenous levels on dendritic claw differentiation. We transfected cerebellar slices with the PIASx expression plasmid or its control vector together with a GFP expression plasmid. In these experiments, we used slices prepared from P9 rat pups instead of P10 to have a lower baseline of dendritic claw number in control-transfected slices. We found that expression of PIASx robustly increased the number of dendritic claws in IGL granule neurons (Fig. 4 G), suggesting that PIASx stimulates dendritic claw differentiation. Thus, on the basis of both loss-of-function knockdown experiments and gain-of-function experiments, we conclude that PIASx plays a key role in dendritic claw morphogenesis in the cerebellar cortex.
To establish the importance of PIASx in granule neuron dendritic development in vivo, we induced the knockdown of PIASx in the postnatal rat cerebellum using electroporation-mediated gene transfer (Fig. 5 A). We injected the U6/piasx1-cmvGFP or control U6-cmvGFP RNAi plasmid into the cerebellar cortex of P3 rat pups and examined the morphology of IGL granule neurons 9 d later in these animals at P12 (Fig. 5 A). In control-transfected cerebella, granule neurons had well-defined dendritic claws at the ends of dendrites (Fig. 5 A,B). In contrast, in animals transfected with the PIASx RNAi plasmid granule neurons displayed tapered or bulbous dendritic ends (Fig. 5 B). Quantitative analyses revealed that PIASx knockdown led to a significant reduction in the number of dendritic claws (Fig. 5 C). These results show that endogenous PIASx plays a critical role in the differentiation of dendritic claws in vivo in the postnatal rat cerebellar cortex.
Figure 5.

PIASx is required for dendritic claw morphogenesis in the postnatal cerebellum in vivo. A, P3 rat pups were injected and electroporated with the U6-cmvGFP plasmid into the cerebellum (Cb). Animals were killed 9 d later, and sections of U6-cmvGFP-electroporated cerebellum were prepared and subjected to immunofluorescence with the GFP antibody. Representative images of granule neurons with cell bodies that reside in the IGL of a cerebellar section are shown at 20× (top; scale bar, 60 μm) and 60× (bottom; scale bar, 10 μm). Arrowheads and arrows, respectively, indicate parallel fiber axons and dendritic claws of transfected granule neurons. B, Representative images of the tips of dendrites of granule neurons in sections of animals (P12) that were injected at P3 with the control U6-cmvGFP or U6/piasx1-cmvGFP RNAi plasmid in vivo. Arrows indicate dendritic claws or tapered dendritic ends. Granule neuron dendrites in control-transfected animals have robust dendritic claws. In contrast, dendrites in PIASx knockdown animals have tapered or bulbous ends. C, Quantification of dendritic claws of experiments performed in B reveals that the number of granule neuron dendritic claws is significantly reduced in PIASx knockdown pups compared with control-transfected pups (p < 0.001; t test; total number of neurons measured, n = 80).
PIASx drives dendritic claw differentiation via MEF2 sumoylation
We next determined whether the novel function of PIASx in dendritic claw differentiation is mediated via sumoylation of MEF2A. We first asked whether the gain-of-function effect of PIASx-induced dendritic claw differentiation requires the presence of MEF2A protein. Knockdown of MEF2A on its own as expected dramatically reduced the number of dendritic claws in rat cerebellar slices (Fig. 6 A) (Shalizi et al., 2006), an effect that has been demonstrated to be secondary to loss of the sumoylated transcriptional repressor form of MEF2A (Shalizi et al., 2006). We found that knockdown of MEF2A completely suppressed the ability of PIASx overexpression to increase the number of dendritic claws in cerebellar slices (Fig. 6 A). These results are consistent with the conclusion that MEF2A acts downstream of PIASx in dendritic claw differentiation.
Figure 6.

PIASx promotes dendritic claw differentiation via MEF2 sumoylation. A, Cerebellar slices were transfected with the PIASx expression plasmid or its control vector together with the U6/mef2a RNAi plasmid that bicistronically encoded GFP (U6/mef2a-cmvGFP) or its control U6-cmvGFP plasmid. Slices were then analyzed as in Figure 4, C and D. Representative images of dendritic ends of transfected GFP-positive neurons are shown in the right panels. Arrows point to dendritic claws or tapered ends. Scale bar, 5 μm. The left panel shows quantitation of these experiments. Knockdown of MEF2A as well as knockdown of MEF2A together with PIASx expression significantly reduced the number of dendritic claws compared with PIASx expression alone or to control vector-transfected neurons (p < 0.05; ANOVA; total number of neurons measured, n = 139). B, Cerebellar slices were transfected with the U6/piasx2-cmvGFP RNAi plasmid together with an expression plasmid encoding wild-type MEF2A, a MEF2A-SUMO fusion protein, or the control vector, and analyzed as in Figure 4, C and D. Representative images of dendritic ends of transfected GFP-positive neurons are shown in the right panels. Arrows point to dendritic claws or tapered ends. Scale bar, 5 μm. The left panel shows quantitation of these experiments. Expression of ME2A-SUMO, but not MEF2A, significantly increased the number of dendritic claws in granule neurons in the background of PIASx RNAi (p < 0.0001; ANOVA; total number of neurons measured, n = 93). C, Cerebellar slices were transfected with the U6/piasx2-cmvGFP RNAi plasmid together with an expression plasmid encoding a dominant interfering form of Nur77 (DN Nur77), wild-type Nur77 (WT Nur77), or the control expression vector, and analyzed as in Figure 4, C and D. Expression of DN Nur77, but not WT Nur77, significantly increased the number of dendritic claws in granule neurons in the background of PIASx RNAi (p < 0.001; ANOVA; total number of neurons measured, n = 104). D, A model of the PIASx-MEF2 sumoylation pathway in the control of postsynaptic dendritic claw differentiation in the cerebellar cortex.
We next tested the effect of expression a MEF2A-SUMO fusion protein on the dendritic claw phenotype induced by PIASx knockdown. The MEF2A-SUMO fusion protein mimics the effect of SUMO that is covalently linked to MEF2A on the native lysine and thus acts as a transcriptional repressor that promotes postsynaptic dendritic claw differentiation (Shalizi et al., 2006). We found that expression of MEF2A-SUMO, but not MEF2A, robustly increased the number of dendritic claws in the background of PIASx RNAi (Fig. 6 B). Thus, sumoylated MEF2A suppresses the PIASx knockdown-induced dendritic claw phenotype.
Sumoylated MEF2A drives dendritic claw differentiation via repression of the orphan nuclear receptor Nur77 (Shalizi et al., 2006). We therefore tested the effect of expression of a dominant interfering form of Nur77 (DN Nur77) on dendritic claw differentiation in the background of PIASx RNAi in rat cerebellar slices. We found that expression of DN Nur77, but not the wild-type Nur77 (WT Nur77), significantly increased the number of dendritic claws in the background of PIASx RNAi (Fig. 6 C). Thus, Nur77 inhibition mimicked the ability of sumoylated MEF2A to suppress the PIASx knockdown-induced dendritic claw phenotype. Collectively, our results support the conclusion that by acting as a MEF2A SUMO E3 ligase, PIASx promotes the morphogenesis of granule neuron dendritic claws in the cerebellar cortex (Fig. 6 D).
Discussion
In this study, we uncovered a novel function for PIASx as a MEF2 SUMO E3 ligase that promotes dendritic claw differentiation in the cerebellar cortex. The major findings in our study are: (1) among the PIAS family of proteins, only PIASx stimulates the robust sumoylation of MEF2A and thereby represses MEF2-dependent transcription; (2) PIASx induces MEF2A sumoylation at the key regulatory site of Lysine 403 in a Serine 408 phosphorylation-dependent manner; (3) PIASx overexpression and inhibition studies in rat cerebellar slices and in vivo in the postnatal cerebellum demonstrate a function for PIASx in the differentiation of granule neuron dendritic claws in the cerebellar cortex; and (4) expression of sumoylated MEF2A or inhibition of the sumoylated MEF2A-repressed target gene Nur77 restores the appearance of dendritic claws in the background of PIASx knockdown, suggesting that PIASx increases dendritic claw number via MEF2 sumoylation.
Elucidation of a function for PIASx in postsynaptic dendritic morphogenesis in the cerebellar cortex in this study represents, to our knowledge, the first report of a biological role for a SUMO E3 ligase in the mammalian brain. In future studies, it will be interesting to consider the role of other enzymes that alter MEF2 sumoylation (Johnson, 2004). In particular, it will be important to identify the SUMO proteases that inhibit the sumoylation of MEF2A at Lysine 403 and thereby regulate dendritic claw differentiation. Although we focused on regulation of dendritic claw differentiation in the cerebellar cortex by the PIASx-MEF2A sumoylation pathway, MEF2 proteins, including MEF2A, MEF2C, and MEF2D, are widely expressed in the developing brain (Lyons et al., 1995). In addition to MEF2A regulation of postsynaptic differentiation in the cerebellar cortex, MEF2A and MEF2D have also been implicated in the control of synapse number in hippocampal neurons (Flavell et al., 2006; Shalizi et al., 2006). All MEF2 proteins except MEF2B are covalently conjugated with SUMO at a key regulatory site corresponding to Lysine 403 in MEF2A (Gregoire et al., 2006; Hietakangas et al., 2006; Shalizi et al., 2006). It is conceivable that PIASx may also act as a SUMO E3 ligase in the sumoylation of other MEF2 proteins. It will be important to determine whether PIASx, by sumoylating MEF2A or other MEF2 proteins, plays a role in postsynaptic dendritic development in diverse regions of the brain. In addition to its expression in the developing cerebellar cortex, PIASx is expressed elsewhere in the brain, including the cerebral cortex and hippocampus (Fig. S5, available at www.jneurosci.org as supplemental material) (www.brainatlas.org). Therefore, it will be interesting to determine whether the PIASx-MEF2 signaling link might play a role in the refinement of postsynaptic dendritic morphology and synaptic plasticity.
An important question for future studies is how the PIASx-MEF2 sumoylation pathway promotes the differentiation of the postsynaptic dendritic claws in the cerebellar cortex. Two nonmutually exclusive possibilities are that this pathway stimulates the formation of dendritic claws or that this pathway stabilizes dendritic claws. Addressing this question will require time-lapse analyses of dendrite development in the cerebellar cortex. Another important question is how PIASx function is regulated in neurons. PIASx-induced sumoylation of MEF2A at Lysine 403 is dependent on the phosphorylation of MEF2A at the nearby site of Serine 408 (Shalizi et al., 2006). The Serine 408 phosphorylation does not appear to recruit PIASx, because MEF2A interacts with PIASx regardless of the Serine 408 phosphorylation status. Thus, phosphorylation may render the Lysine 403 peptide a better substrate for the PIASx-induced sumoylation. Structural studies using the MEF2A peptides together with the sumoylation enzyme machinery should provide insights into how the Serine 408 phosphorylation facilitates PIASx-induced sumoylation of MEF2A at Lysine 403. In addition to modifications of the MEF2A peptide, it will be important to consider whether PIASx itself might be subject to posttranslational modifications that in turn affect its ability to promote MEF2A sumoylation.
Because the MEF2 transcription factors have diverse biological roles in development and homeostasis (McKinsey et al., 2002; Shalizi and Bonni, 2005), identification of the PIASx-MEF2 signaling connection suggests that PIASx may control diverse biological responses. Within the brain, beyond the control of dendritic morphogenesis, it will be interesting to determine whether PIASx-induced MEF2 sumoylation impacts on the function of MEF2 in neuronal survival (Mao et al., 1999; Gaudilliere et al., 2002; Okamoto et al., 2002; Shalizi et al., 2003; Heidenreich and Linseman, 2004). Outside the brain, PIASx may also regulate the established functions of MEF2 in muscle differentiation and muscle fiber type switching (Wu et al., 2001).
Although the biochemical reactions of sumoylation and its consequences on transcription factors are increasingly understood (Johnson, 2004), major gaps in the biological functions of protein sumoylation remain. The brain offers an excellent system for elucidation of the biological functions of protein sumoylation. Our findings raise the prospect that SUMO E3 ligases may have critical roles in nervous system development and function. Recent evidence suggests that sumoylation may contribute to the pathogenesis of neurodegenerative diseases (Steffan et al., 2004). Therefore, a better understanding of protein sumoylation in neurons will serve to enhance our understanding of both brain development and disorders of the nervous system.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS41021 (A.B.), an NIH training grant to A.S., and a Charles A. King Trust/Bank of America/Co-Trustee postdoctoral fellowship to J.S. We thank Stuart Schreiber and Anjana Rao for providing plasmids, Jorma Palvimo for providing an antibody to PIASx, and Paola Merlo and other members of the Bonni laboratory for helpful discussions and reading this manuscript.
References
- Altman J, Bayer S. Development of the cerebellar system: in relation to its to its evolution, structure, and functions. New York: CRC; 1997. The third stage of cerebellar development: maturation of the cerebellar system; pp. 323–536. [Google Scholar]
- Arora T, Liu B, He H, Kim J, Murphy TL, Murphy KM, Modlin RL, Shuai K. PIASx is a transcriptional co-repressor of signal transducer and activator of transcription 4. J Biol Chem. 2003;278:21327–21330. doi: 10.1074/jbc.C300119200. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Pin1 mediates neural-specific activation of the mitochondrial apoptotic machinery. Neuron. 2006;49:655–662. doi: 10.1016/j.neuron.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- Cline HT. Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol. 2001;11:118–126. doi: 10.1016/s0959-4388(00)00182-3. [DOI] [PubMed] [Google Scholar]
- Desterro JM, Thomson J, Hay RT. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 1997;417:297–300. doi: 10.1016/s0014-5793(97)01305-7. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Garcia-Dominguez M, Gilardi-Hebenstreit P, Charnay P. PIASxbeta acts as an activator of Hoxb1 and is antagonized by Krox20 during hindbrain segmentation. EMBO J. 2006;25:2432–2442. doi: 10.1038/sj.emboj.7601122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem. 2002;277:46442–46446. doi: 10.1074/jbc.M206653200. [DOI] [PubMed] [Google Scholar]
- Gaudilliere B, Konishi Y, de la Iglesia N, Yao G, Bonni A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–241. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- Gong L, Kamitani T, Fujise K, Caskey LS, Yeh ET. Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J Biol Chem. 1997;272:28198–28201. doi: 10.1074/jbc.272.45.28198. [DOI] [PubMed] [Google Scholar]
- Gong L, Li B, Millas S, Yeh ET. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. 1999;448:185–189. doi: 10.1016/s0014-5793(99)00367-1. [DOI] [PubMed] [Google Scholar]
- Gregoire S, Yang XJ. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol Cell Biol. 2005;25:2273–2287. doi: 10.1128/MCB.25.6.2273-2287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire S, Tremblay AM, Xiao L, Yang Q, Ma K, Nie J, Mao Z, Wu Z, Giguere V, Yang XJ. Control of MEF2 transcriptional activity by coordinated phosphorylation and sumoylation. J Biol Chem. 2006;281:4423–4433. doi: 10.1074/jbc.M509471200. [DOI] [PubMed] [Google Scholar]
- Hamori J, Somogyi J. Differentiation of cerebellar mossy fiber synapses in the rat: a quantitative electron microscope study. J Comp Neurol. 1983;220:365–377. doi: 10.1002/cne.902200402. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Linseman DA. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol. 2004;29:155–166. doi: 10.1385/MN:29:2:155. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci USA. 2006;103:45–50. doi: 10.1073/pnas.0503698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Jackson PK. A new RING for SUMO: wrestling transcriptional responses into nuclear bodies with PIAS family E3 SUMO ligases. Genes Dev. 2001;15:3053–3058. doi: 10.1101/gad.955501. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. The control of dendrite development. Neuron. 2003;40:229–242. doi: 10.1016/s0896-6273(03)00631-7. [DOI] [PubMed] [Google Scholar]
- Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- Lin RC, Matesic DF, Connor JA. The role of dendritic dysfunction in neurodegeneration. Ann NY Acad Sci. 1997;825:134–145. doi: 10.1111/j.1749-6632.1997.tb48424.x. [DOI] [PubMed] [Google Scholar]
- Lippman J, Dunaevsky A. Dendritic spine morphogenesis and plasticity. J Neurobiol. 2005;64:47–57. doi: 10.1002/neu.20149. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc Natl Acad Sci USA. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Lyons GE, Micales BK, Schwarz J, Martin JF, Olson EN. Expression of mef2 genes in the mouse central nervous system suggests a role in neuronal maturation. J Neurosci. 1995;15:5727–5738. doi: 10.1523/JNEUROSCI.15-08-05727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- Melchior F, Schergaut M, Pichler A. SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem Sci. 2003;28:612–618. doi: 10.1016/j.tibs.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Moilanen AM, Karvonen U, Poukka H, Yan W, Toppari J, Janne OA, Palvimo JJ. A testis-specific androgen receptor coregulator that belongs to a novel family of nuclear proteins. J Biol Chem. 1999;274:3700–3704. doi: 10.1074/jbc.274.6.3700. [DOI] [PubMed] [Google Scholar]
- Okamoto S, Li Z, Ju C, Scholzke MN, Mathews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2002;99:3974–3979. doi: 10.1073/pnas.022036399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palay S, Chan-Palay V. New York: Springer; 1974. Cerebellar cortex: cytology and organization. [Google Scholar]
- Ramon y Cajal S. The cerebellum. In: Swanson N, Swanson L, editors. Histology of the nervous system. New York: Oxford UP; 1995. pp. 3–86. [Google Scholar]
- Ramos B, Gaudilliere B, Bonni A, Gill G. Transcription factor Sp4 regulates dendritic patterning during cerebellar maturation. Proc Natl Acad Sci USA. 2007;104:9882–9887. doi: 10.1073/pnas.0701946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Muller S. PIAS/SUMO: new partners in transcriptional regulation. Cell Mol Life Sci. 2003;60:2561–2574. doi: 10.1007/s00018-003-3129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, Gaudilliere B, Donovan N, Han J, Konishi Y, Bonni A. Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci. 2003;23:7326–7336. doi: 10.1523/JNEUROSCI.23-19-07326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Gaudilliere B, Yuan Z, Shirogane T, Stegmuller J, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic dendritic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- Shalizi AK, Bonni A. Brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol. 2005;69:239–266. doi: 10.1016/S0070-2153(05)69009-6. [DOI] [PubMed] [Google Scholar]
- Sharrocks AD. PIAS proteins and transcriptional regulation–more than just SUMO E3 ligases? Genes Dev. 2006;20:754–758. doi: 10.1101/gad.1421006. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004;15:267–278. doi: 10.1515/revneuro.2004.15.4.267. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Stegmuller J, Konishi Y, Huynh MA, Yuan Z, Dibacco S, Bonni A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Wu H, Rothermel B, Kanatous S, Rosenberg P, Naya FJ, Shelton JM, Hutcheson KA, DiMaio JM, Olson EN, Bassel-Duby R, Williams RS. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001;20:6414–6423. doi: 10.1093/emboj/20.22.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wu H, Ma L, Sangiorgi F, Wu N, Bell JR, Lyons GE, Maxson R. Miz1, a novel zinc finger transcription factor that interacts with Msx2 and enhances its affinity for DNA. Mech Dev. 1997;65:3–17. doi: 10.1016/s0925-4773(97)00032-4. [DOI] [PubMed] [Google Scholar]
- Zhao X, Sternsdorf T, Bolger TA, Evans RM, Yao TP. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol. 2005;25:8456–8464. doi: 10.1128/MCB.25.19.8456-8464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]