

Abstract
In neuronal synapses, PDZ domains [postsynaptic density-95 (PSD-95)/Discs large/zona occludens-1] of PSD-95 proteins interact with C termini of NMDA receptor [NMDAR (NR)] subunits, linking them to downstream neurotoxic signaling molecules. Perturbing NMDAR/PSD-95 interactions with a Tat peptide comprising the nine C-terminal residues of the NR2B subunit (Tat-NR2B9c) reduces neurons' vulnerability to excitotoxicity and ischemia. However, NR subunit C termini may bind many of >240 cellular PDZs, any of which could mediate neurotoxic signaling independently of PSD-95. Here, we performed a proteomic and biochemical analysis of the interactions of all known human PDZs with synaptic signaling proteins including NR1, NR2A–NR2D, and neuronal nitric oxide synthase (nNOS). Tat-NR2B9c, whose interactions define PDZs involved in neurotoxic signaling, was also used. NR2A–NR2D subunits and Tat-NR2B9c had similar, highly specific, PDZ protein interactions, of which the strongest were with the PSD-95 family members (PSD-95, PSD-93, SAP97, and SAP102) and Tax interaction protein 1 (TIP1). The PSD-95 PDZ2 domain bound NR2A–NR2C subunits most strongly (EC50, ∼1 μm), and fusing the NR2B C terminus to Tat enhanced its affinity for PSD-95 PDZ2 by >100-fold (EC50, ∼7 nm). IC50 values for Tat-NR2B9c inhibiting NR2A–NR2C/PSD-95 interactions (∼1–10 μm) and nNOS/PSD-95 interactions (200 nm) confirmed the feasibility of such inhibition. To determine which of the PDZ interactions of Tat-NR2B9c mediate neuroprotection, one of PSD-95, PSD-93, SAP97, SAP102, TIP1, or nNOS expression was inhibited in cortical neurons exposed to NMDA toxicity. Only neurons lacking PSD-95 or nNOS but not PSD-93, SAP97, SAP102, or TIP1 exhibited reduced excitotoxic vulnerability. Thus, despite the ubiquitousness of PDZ domain-containing proteins, PSD-95 and nNOS above any other PDZ proteins are keys in effecting NMDAR-dependent excitotoxicity. Consequently, PSD-95 inhibition may constitute a highly specific strategy for treating excitotoxic disorders.
Keywords: PDZ domains, PSD-95, NMDA receptors, excitotoxicity, RNA interference, nitric oxide synthase
Introduction
At central excitatory synapses, ionotropic glutamate receptors are organized in multiprotein signaling complexes within the postsynaptic density (PSD) (Sheng, 2001). A prominent organizing protein is postsynaptic density-95 (PSD-95) (Kornau et al., 1995), which couples NMDA receptors [NMDARs (NRs)] to intracellular proteins and signaling enzymes (Brenman et al., 1996a; Sheng, 2001). NMDARs, in addition to signaling, have also long been known to mediate ischemic brain injury (Simon et al., 1984). However, blocking them is a failed strategy in stroke and can be deleterious to animals and humans (Fix et al., 1993; Morris et al., 1999; Davis et al., 2000). An alternative strategy is to block downstream excitotoxic signaling without blocking NMDARs or inhibiting excitatory neurotransmission. We accomplished this previously by targeting the NMDAR/PSD-95 complex. Initially, we suppressed PSD-95 expression in cortical neurons using antisense oligodeoxynucleotides. This reduced neuronal NMDAR-activated nitric oxide (NO) production and excitotoxicity without inhibiting NMDAR ionic currents and calcium signaling (Sattler et al., 1999). Because mutation or suppression of PSD-95 expression is therapeutically impractical, we next targeted the interactions of NMDAR NR2B subunits with PSD-95. Disrupting these reduced the excitotoxic vulnerability of neurons as previously observed in PSD-95-deficient cells. In addition, it rendered neurons resistant to focal cerebral ischemia in vivo (Aarts et al., 2002). The disruption of NR2B–PSD-95 interactions was achieved by the intracellular introduction of a peptide comprising the nine C-terminal residues of NR2B (KLSSIESDV; NR2B9c) (Kornau et al., 1995). NR2B9c was rendered cell permeant by fusing it to the cell-membrane transduction domain of the human immunodeficiency virus type 1 (HIV-1) Tat protein (YGRKKRRQRRR; Tat) (Schwarze et al., 1999) to obtain the 20 aa peptide Tat-NR2B9c.
However, the human genome contains hundreds of different PSD-95/discs large (DLG)/zona occludens-1 (ZO-1) (PDZ) domain-containing proteins (Giallourakis et al., 2006), and all NR subunit C termini possess a promiscuous type I PDZ interaction motif (T/SXV). Consequently NR subunits may bind many other cellular PDZ proteins, any of which could mediate neurotoxic signaling independently of PSD-95. Also, Tat-NR2B9c may mediate either neuroprotection or undesirable side effects by perturbing NR subunit–PDZ interactions other than with PSD-95.
To examine these possibilities, we cloned all publicly known human PDZ domains into expression vectors and produced them as recombinant glutathione S-transferase (GST):PDZ fusion proteins. These were then used in an ELISA-based competition assay to evaluate, among others, the interaction profiles of NMDAR NR1 and NR2A–NR2D subunits, neuronal nitric oxide synthase (nNOS), and Tat-NR2B9c. These analyses revealed all previously published and some new interactions of NMDARs, nNOS, and Tat-NR2B9c with PDZ domain-containing proteins. Then, to determine which of these interactions are involved in excitotoxic processes, we used RNA interference (RNAi) to suppress the expression of each key protein that interacts with Tat-NR2B9c, because this peptide's binding targets define a subset of interactions responsible for excitotoxicity in cortical neuronal cultures. The data provide an extensive database for >4100 interactions between synaptic proteins/receptors and cellular PDZ proteins among which are revealed novel interactions of key proteins involved in cellular signaling. The results also indicate a surprising specificity of neurotoxic signaling through PDZ proteins and provide the mechanism of action of a novel class of neuroprotectants.
Materials and Methods
Identification and cloning of PDZ domains.
A comprehensive set of human PDZ domains was determined using searchable databases (Table 1). In general, the protein-encoding sequences and gene identification numbers were obtained for the >470 human PDZ proteins in the SMART (http://smart.embl-heidelberg.de/; IPR001478) database and aligned using pairwise alignment algorithms of each individual domain to reveal a nonredundant set of ∼145 genes encoding proteins with PDZ domains (Schultz et al., 1998, 2000). Boundaries were aligned with other known PDZ domains to determine the location of the first β-sheet of the PDZ domain (Songyang et al., 1997) and confirmed using secondary structure prediction programs including Jpred and hydrophobicity analysis. Individual, unique PDZ domains were amplified from cDNA prepared from various human tissue sources using oligonucleotide primers (Integrated DNA Technologies, Coralville, IA) designed based on GenBank PDZ nucleic acid sequences with the addition of flanking restriction enzyme sites for in-frame cloning. Amplified PDZ DNA was purified from agarose gels using Sephaglas (GE Healthcare, Piscataway, NJ), digested with appropriate restriction enzymes, gel purified a second time, and ligated into the PGEX-3X vector (GE Healthcare) for inducible expression as a GST fusion protein. The restriction sites used most frequently were BamH1/EcoRI. In brief, the PDZ-encoding PCR fragment and digested pGEX-3X vector were ethanol precipitated and resuspended in 10 μl of standard ligation buffer. Ligation was performed for 4–10 h at 15°C using T4 DNA ligase (New England Biolabs, Beverly, MA). The plasmids were used to transform competent DH5α cells or BL21 λDE3 Escherichia coli bacterial strains for protein expression.
Table 1.
NMDA receptor subunit (10 μm native 20 aa C-terminal peptides) and nNOSd1 interactions
| PDZ domain construct | NR2A (Acc. NP_000824) | NR2B (Acc. NP_000825) | NR2C (Acc. NP_000826) | NR2D (Acc. NP_000827) | nNOSd1 |
|---|---|---|---|---|---|
| DLG1/SAP97d1 | |||||
| DLG1/SAP97d2 | Y | Y | Y | Y | Y |
| DLG2/PSD-93d2 | Y | Y | Y | Y | Y |
| NeDLG/SAP102d2 | Y | Y | Y | Y | |
| PSD-95d2 | Y | Y | Y | Y | Y |
| PSD-95d1 | Y | ||||
| INADLd8 | Y | ||||
| KIAA 0973 | Y | Y | Y | Y | |
| KIAA1634d1 | Y | Y | |||
| Lim mystique | Y | Y | |||
| Lim RIL 1 | Y | ||||
| MAGI-2d5 | Y | Y | Y | ||
| Outer membrane | Y | Y | Y | Y | |
| MAST2 | Y | Y | |||
| Syntrophin 1α | Y | Y | Y | Y | |
| Syntrophin 1β | Y | Y | Y | ||
| Syntrophin 1γ | |||||
| TIP1 | Y | Y | Y | Y | |
| TIP2 | Y | Y | Y | Y |
Acc., GenBank accession number; Y, yes (there is an interaction). PSD-95 family members are in bold lettering.
Protein expression and purification.
Detailed procedure of small-scale and large-scale fusion protein expression and purification are described in the GST gene fusion system handbook (second edition, revision 2; GE Healthcare). Protocol modifications are specified in this procedure. In brief, transformed bacteria were grown overnight in 50 ml of 2× YT media at 37°C in the presence of 100 μg/ml ampicillin. Then, 950 ml of prewarmed 2× YT media was inoculated with the 50 ml overnight culture and grown at 37°C to an OD600 of 0.6–0.8. At this point, the cultures were induced with 1 ml of 100 mm isopropyl-β-d-thiogalactopyranoside (100 μm final) for 1–3 h. Cells were harvested, resuspended in 10 ml of buffer A (50 mm dextrose, 50 mm Tris, pH 8.0, and 1 mm EDTA) and lysed in the presence of 8 mg/ml lysozyme for 10 min on ice. Lysis was stopped by the addition of 20 ml of buffer B (10 mm Tris, pH 8.0, 50 mm KCl, 1 mm EDTA, and 0.5% Tween 20). The mixture was then centrifuged to remove cellular debris, and the supernatant was collected. The supernatant was precleared by pouring through a 10 ml Sepharose CL-4B column and collected and incubated with 1 ml of glutathione Sepharose 4B (GE Healthcare) for 1–3 h at 4°C on a rocker or rotator table. The beads were poured into a column and washed five times with 20 ml of PBS. GST-PDZ protein was eluted with glutathione elution buffer (20 mm reduced glutathione in 100 mm Tris-HCl, pH 8, and 120 mm NaCl). The eluted protein concentration was checked by absorbance at 280 nm. The fractions containing the highest concentration of fusion protein were pooled, and purity was checked by SDS-PAGE. The pooled fractions were then stored with 35% glycerol at −80°C. Some fractions were dialyzed against PBS to reduce the glutathione content. Proteins were assessed for proper folding as reagents permitted, using binding assays with known or predicted protein or peptide ligands when available. Biotinylated peptides corresponding to known ligands were used in binding assays to confirm proper folding for >90% of the PDZ fusion proteins produced. Proteins that had low yields, were insoluble, or failed to bind known ligands were alternatively prepared using urea denaturation and renaturation. After the lysis step, buffer B containing 6 m urea and lacking detergent was added instead of buffer B. The suspension was then sonicated (ten 5 s bursts on ice) and centrifuged to remove insoluble material. The supernatant was dialyzed overnight against 10 L of PBS at 4°C. After dialysis, the lysate was placed in 50 ml conical tubes and cleared by centrifugation in a Sorvall (Newtown, CT) SA-600 at 12,000 rpm for 10 min. The supernatant was transferred to a fresh tube and incubated with glutathione Sepharose as above. Proteins not able to bind known ligands following either of these methods were recloned with different boundaries to increase solubility and proper folding.
ELISA-based PDZ domain interaction assays.
For each assay series, a 96-well plate (Nunc Maxisorp; ThermoFisherScientific, Rochester, NY) was coated overnight with anti-GST antibody in PBS (5 μg/ml; GE Healthcare). The plate was washed and then blocked with PBS containing 2% BSA. The blocking agent was removed, the plate was washed with PBS, and GST:PDZ fusion proteins (5 μg/ml in 2% BSA) were added to the appropriate wells and allowed to incubate 1–2 h at 4°C. Unbound fusion proteins were removed by washing three times with ice-cold PBS. Biotinylated test peptides (10 μm or less, as appropriate) were added and incubated for 10 min on ice followed by 20 min at room temperature. In some experiments, unlabeled peptide competitors were added in conjunction with the biotinylated peptides at this stage to test their effect on binding of the biotinylated peptides. In experiments aimed at EC50 determinations, test peptides were generally applied at concentrations ranging from 0.001 to 100 μm after titrating the amount of GST-PDZ protein on the plate to allow binding saturation below the absorbance limit of the microplate reader. Unbound peptide was removed by three washes with ice-cold PBS. Horseradish peroxidase (HRP) streptavidin (1:2000 dilution in 2% BSA; Zymed, South San Francisco CA) was added to each well and incubated on ice for 20 min. After five washes with PBS, TMB substrate was added (Dako, Milano, Italy). Plates were incubated at room temperature in the dark for 30 min, after which time 0.18 m H2SO4 was added to stop the reaction. Plates were read at 450 nm using a Molecular Dynamics (Sunnyvale, CA) plate reader. Raw data from the plate reader was imported and analyzed in SoftMax Pro (Molecular Dynamics). Interactions were deemed specific if they met the following criteria: (1) absorbance at 450 nm is >0.2; (2) signal is twice or more that of background (GST alone); and (3) SD between replicates is <20%. All experiments were replicated a minimum of three times. All test peptides were commercially synthesized and HPLC purified (Anaspec, San Jose, CA).
Primary murine cortical cultures.
Mixed cortical cell cultures containing both neurons and glia were prepared from embryonic day 16 CD1 Swiss mice as described previously (Sattler et al., 1998; Tymianski et al., 1998). In brief, cerebral cortices were extracted from embryos and incubated for 5 min in 0.05% trypsin-EDTA. Digested cortices were dissociated by trituration and plated on poly-l-ornithine-coated 24-well plates (Corning, Corning, NY) at 0.5–0.6 × 106 cells/well. All cell culture reagents were from Invitrogen (Burlington, Ontario, Canada). Plating medium consisted of minimum essential medium (MEM) supplemented with 10% heat-inactivated horse serum, 10% fetal bovine serum, 1% glutamine, and 25 mm glucose. After 4 d in vitro, growth of non-neuronal cells was halted by exposure to 10 μm FDU solution (5 μm uridine and 5 μm (+)-5-fluor-2′-deoxyuridine) for 48 h. Growth medium contained minimum essential media with 4.5 g/L glucose, 10% horse serum (Invitrogen), and 1× GlutaMAX (Invitrogen). Previous studies have shown that these cultures are >85% neurons (Sattler et al., 1997). Cultures were maintained at 37°C in a humidified 5% CO2 atmosphere until used at 12 d after plating.
NMDA toxicity assay.
All experiments were performed at 37°C. The cortical cultures were washed twice with HEPES solution [(in mm) 121 NaCl, 5 KCl, 10 HEPES acid, 7 HEPES sodium salt, 1 sodium-pyruvate, 1.8 CaCl2, 3 NaHCO3, 0.010 glycine, and 20 d-glucose, pH 7.4]. The HEPES solution also contained 2 μm nimodipine (Sigma-Aldrich, St. Louis, MO) and 10 μm CNQX (Sigma-Aldrich) to restrict the actions of applied NMDA to NMDARs by preventing the secondary activation of other pathways (Sattler et al., 1998). The cultures were then challenged with NMDA in HEPES solution and maintained at 37°C in a humidified chamber for 1 h. The NMDA challenge was terminated by a wash with fresh HEPES solution, followed by a wash in HEPES solution containing 10 μg/ml propidium iodide (PI; Invitrogen).
Quantification of in vitro cell death.
Cell death was determined by quantitative measurements of PI fluorescence using a multiwell plate fluorescence scanner (Fluoroskan Ascent FL; ThermoLab Systems, Burlington, ON) as described previously (Sattler et al., 1997, 1998; Tymianski et al., 1998). Baseline readings were taken before the NMDA challenge, and the plates were further incubated for 20 h at 37°C before obtaining final PI readings. The fraction of dead cells in each culture at a given time was calculated as follows: Fraction dead = (Ft − F0)/FNMDA, where Ft is PI fluorescence at time t, F0 is initial PI fluorescence at time 0, and FNMDA is background-subtracted PI fluorescence of identical cultures from the same dissection and plating, 20 h after a 60 min exposure to 1 mm NMDA at 37°C. Based on manual observations at the time of validation of this technique, this NMDA exposure routinely produced near-complete neuronal death in each culture but had no effect on surrounding glia (Bruno et al., 1994; David et al., 1996; Sattler et al., 1997). Adding Triton X-100 (0.1%) to cultures treated in this manner produced an additional 10–15% increase in PI fluorescence as a result of permeabilization of non-neuronal cell membranes, consistent with a 10–15% glial component in the cultures.
Reverse transcription-PCR.
Total RNA was isolated using TRIzol reagent (Invitrogen) as per the product insert, and reverse transcription and PCR were performed with 100 ng of total RNA. OneStep reverse transcription (RT)-PCR as described in the QIAGEN (Valencia, CA) OneStep PCR kit was used. The RNA template was combined with enzyme mix and 0.6 μm primers for 28 cycles (30 s at 95°C, 30 s at 60°C, and 60 s at 72°C). The primer sequences were designed based on GenBank sequences (accession number in parentheses) and are as follows: SAP97 (NM_007862), forward, 5′-TGGAGACATCCTCCATGTCA-3′; reverse, 5′-TGGAGACATCCTCCATGTCA-3′; PSD-93 (NM_0011807), forward, 5′-AGCCCTCAGTGACTCTCCAA-3′; reverse, 5′-TTGTTCCATAACCGTCGTCA-3′; SAP102 (NM_016747), forward, 5′-GACTTGCGGGAACAAATGAT-3′; reverse, 5′-TGTGCTTGCTGCAGTCTCTT-3′; PSD-95 (NM_007864), forward, 5′-CTGACCTCAGTGGGGAGCTA-3′; reverse, 5′-ACGGATGAAGATGGCGATAG-3′; nNOS (NM_008712), forward, 5′-CCATCAGCTCCTCTCCAGAC-3′; reverse, 5′-ATATGGCCTCCTTGCTCCTT-3′; Tax interaction protein 1 (TIP1; NM_029564), forward, 5′-CCGCCGTAGTGCAAAGAGTTGAAA-3′; reverse, 5′-TGACTGCTAAGACAGCATGGACTG-3′; and β-actin, forward, 5′-TGGAATCTTGTGGCATCCATGAACTGAATCTTGT-3′; reverse, 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′.
Small interfering RNA transfection.
Before transfection, cells were fed with fresh growth media. The initial transfection occurred on the seventh day in vitro (DIV). A presynthesized small interfering RNA (siRNA; Invitrogen) mix was prepared in DEPC water. The following mRNA target sequences were used in designing the siRNA: SAP97, 5′-AAACCCAAATCCATGGAAAAT-3′; PSD-93, 5′-CCCGATTGAAGACAGTGAAGTTCAA-3′; SAP102, 5′-GGCCAGACATACGAACAAGCAAATAA-3′; PSD-95, 5′-AAAGAGGCAGGTTCCATCGTT-3′ (Futai et al., 2007); nNOS, 5′-AAGAACAAGGGCGTCTTCAGA-3′; and TIP1, 5′-AAGTTGCGTCAAGGTGAGAAC-3′ (Kanamori et al., 2003).
The sequences were designed so that no other known genes exhibit sequence homology to these target sequences (GenBank search). For each transfection, 1.5 μl of Gene Silencer transfection reagent (Gene Therapy Systems, San Diego, CA) per 100 ng of siRNA was prepared in Opti-MEM solution (Invitrogen). The contents of the tubes containing Gene Silencer reagent and siRNA were mixed (i.e., total 100 μl reaction/tube), incubated at room temperature for 2–5 min, and then added to a well. For transfection controls, a nonsilencing (NS) siRNA was used (siRNA negative control 1; catalog number 4611; Ambion, Austin, TX). Transfection efficacy was determined using 40–60 pmol of a Cy3-conjugated NS siRNA (Ambion).
Protein harvest.
At 12 DIV, transfected neurons were harvested for protein. Cells were washed two times with cold PBS (Invitrogen). Thirty microliters of M-PER reagent (Pierce Biotechnology, Rockford, IL) per well were added, and cells were scraped off the surface. The suspension was triturated gently and placed in ice for 15 min with vortex treatments every few minutes throughout the incubation. Then tubes containing precipitated proteins were centrifuged at 4°C, 10,000 × g for 10 min.
Immunoblotting.
Protein samples were immunoblotted as described previously (Sattler et al., 2000) in 7% SDS gel. Sixty micrograms of lysates and 4× sample buffer (0.625 m Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, and 0.0125% bromophenol blue) were diluted to 1× and were loaded per well. The blotted proteins were probed with the following primary antibody dilutions: anti-SAP97 mouse monoclonal IgG1 (Stressgen, Victoria, British Columbia, Canada), 1:500; anti-PSD-93 rabbit Ig (Zymed), 1:250; anti-SAP102 mouse IgG1 (Stressgen), 1:250; anti-PSD-95 mouse IgG2a (Stressgen), 1:250; anti-nNOS mouse IgG2a (BD Biosciences Pharmingen, San Jose, CA), 1:2500; anti TIP1 monoclonal mouse IgG (Arbor Vita, Sunnyvale, CA), 1:250; and anti-β-actin mouse Ig (Sigma-Aldrich), 1:15,000. The membranes were incubated with secondary antibodies at a 1:5000 dilution. The HRP was conjugated to either a goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) or a goat anti-mouse IgG (Jackson ImmunoResearch Laboratories). Excess antibodies were washed away by Tris-buffered saline–Tween 20 (TBST) for three times at 10 min per washing, and then enhanced chemiluminescence labeling reagents (GE Healthcare) were used for Western blotting detection. The analysis of the bands was performed by Scion Image (Scion, Frederick, MD), in which each band's intensity was measured and subtracted from the background. The ratios of the bands of PSD-95 family members from both nonsilencing and specific siRNA-transfected knocked-down protein samples were calculated as follows: intensity of PSD-95 family member/intensity of respective β-actin band. Final calculations included another ratio: band ratio from knocked-down protein/band ratio from nonsilencing.
Immunocytochemistry.
Neuronal cultures grown on glass coverslips as above were treated with siRNAs targeting PSD-93, PSD-95, SAP97, or SAP102 or with nonsilencing siRNA as described. They were fixed and cryoprotected in 2.5% sucrose with 4% w/v paraformaldehyde in PBS at room temperature for 20 min. Immunostaining was performed as described previously (Sun et al., 2006). In brief, the coverslips were blocked using a 1% BSA, 3% goat serum, and 0.3% Triton X-100 in PBS solution for 90 min and labeled with mouse anti-NeuN antibody (1:100; Millipore, Billerica, MA) overnight at 4°C. The coverslips were subsequently washed, blocked briefly with the blocking solution, and incubated with goat anti-mouse FITC (1:100; Sigma-Aldrich) for 60 min at room temperature. Coverslips were mounted using ProLong Gold antifade reagent (Invitrogen). Images were viewed with a confocal laser-scanning microscope (see below). NeuN is a neuron-specific marker (Mullen et al., 1992).
DiI staining/assessment of cell morphology.
Neuronal morphology of neurons treated with the various siRNAs was examined by labeling cells with the phospholipid membrane tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (D-282; Invitrogen) as described previously (Sattler et al., 1998). In brief, coverslips were fixed in 4% w/v paraformaldehyde with 2.5% sucrose in PBS for 20 min at room temperature. DiI working solution (5 μg/ml in PBS) was diluted 1:100 in PBS from stock solution (0.5 mg/ml in absolute ethanol). Fixed cultures were incubated in DiI suspension for 90 min at room temperature, washed with PBS, and mounted using ProLong Gold antifade reagent (Invitrogen). Images were viewed with a confocal laser-scanning microscope (see below).
Confocal microscopy.
NeuN- and DiI-stained cultures were imaged with a laser-scanning confocal microscope (LSM 510 META; Zeiss, Thornwood, NY) using X25 and X63 lenses, and analyzed with a three-dimensional constructor (ImageJ software). Serial confocal images were taken at 0.5 μm intervals through the region of interest, and optical stacks of 10 images were produced for the figures as described previously (Sun et al., 2006).
Results
Proteomic analysis of the interactions of neuronal signaling proteins with human PDZ domains
Here we present a biochemical approach to the large-scale analysis of PDZ–ligand interactions. A comprehensive set of unique human PDZ domain-containing proteins was determined using searchable databases (see Materials and Methods) (partial list in supplemental Table S1a,b, available at www.jneurosci.org as supplemental material). All of the PDZ domains in these proteins were individually cloned into expression vectors and produced as properly folded GST-fusion recombinant proteins (see Materials and Methods) and plated on 96-well plates. They were then used in an ELISA-based assay (see Materials and Methods) to determine the interactions of each PDZ with a subset of C termini of neuronal signaling proteins, hereafter referred to as “PDZ ligands” or “PLs.” Each recombinant PDZ protein is a construct comprising at least one, but sometimes several, PDZ domains of a given parent protein. For example, in the case of PSD-95, we separately cloned and plated proteins consisting of GST fused to PSD-95d1, PSD-95d2, PSD-95d3, and also PSD-95d1,d3 and a single construct containing PSD-95d1, d2, and d3. Ideally, this ELISA-based assay should identify the specific PDZ domains bound by a given PL and concurrently be usable to assess both the relative affinity and selectivity of any identified interactions.
Because this project is focused on neurotoxic NMDAR signaling, the PLs chosen for this screen included the key NMDAR subunits NR1, NR2A, NR2B, NR2C, and NR2D. We also included Tat-NR2B9c, a peptide comprised of the 11-residue human HIV-1 Tat cell membrane protein transduction domain fused to the 9 C-terminal amino acids of the NMDA NR2B subunit. Because Tat-NR2B9c reduces the vulnerability of neurons by binding PDZ domains (Aarts et al., 2002), it was used here for identifying binding partners that represent important proteins in excitotoxic signaling. As a control for Tat-NR2B9c, we synthesized a peptide in which the 0 and −2 residues of the NR2B9c terminus are mutated to alanines (Tat-KLSSIEADA; termed TAT-NR2B9c-AA). The mutated PDZ-binding motif is not anticipated to bind PDZ proteins (Kornau et al., 1995; Christopherson et al., 1999) and is devoid of neuroprotective properties (Aarts et al., 2002).
The remaining PLs selected for this screen were for the purpose of validation and include a subset of proteins involved in diverse aspects of neuronal signaling, most of which have been previously shown to interact with PDZ proteins. Although these interactions are beyond the scope and focus of the current project, they serve as additional controls to demonstrate the assay's ability to demonstrate known PL–PDZ domain interactions. Unless otherwise stated, each PL was comprised of 19–20-mers recapitulating the C terminus of its parent protein. Because the affinity of PL–PDZ domain interactions is usually in the 1–10 μm range (Lim et al., 2002), the PLs were applied at a concentration of 10 μm in the initial screen unless otherwise indicated. This PL concentration was selected so as to minimize the probability of missing a relevant interaction on initial screening. Any interactions detected at these concentrations can then be titrated subsequently to determine the strength of the interaction. The results of this proteomic PDZ–PL interaction screen provide a database characterizing >6500 interactions of neuronal signaling PLs with all known human PDZs (∼4100 shown in Fig. 1). Potential limitations are reviewed in the Discussion.
Figure 1.
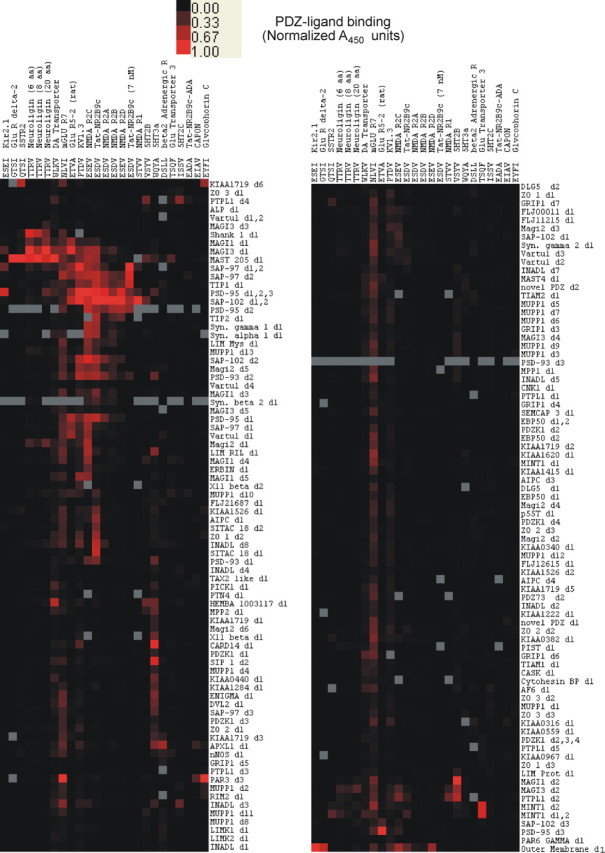
Clustered heat map of PDZ–ligand interactions. Columns represent peptides corresponding to PL C termini. Rows represent GST-PDZ fusion proteins containing the indicated human PDZ domains. The column headings provide the absolute C-terminal 4 aa of the PL peptides and the names of the proteins from which they are derived. The intensity of red color represents the observed colorimetric signal derived from four or more PDZ–PL binding experiments at 10 μm unless noted in the column header. For PLs with maximum binding signal >1 A450 unit, the data in that column were normalized to set the maximum observed binding to 1. The right half of the figure is a continuation of the heat map from the bottom of the left half. Gray boxes represent reactions that were not tested. PDZ domains labeled “d#” (e.g., “d2”) indicate the domain number as counted from the N terminus. When more than one domain is listed, the GST construct contains multiple domains. The results shown encompass a total of ∼4100 unique potential PDZ–PL interactions. All NR subunits and Tat-NR2B9c were tested against >240 unique PDZ domain constructs. For the purpose of clarity, rows of PDZ–PL interactions were not displayed if (1) the PDZ was tested against <22 of the PLs or (2) the PL did not significantly bind any of the PDZ domains bound by the NR2 subunits and Tat-NR2B9c (>2× background). To represent the binding profiles of PDZ proteins and PLs, hierarchical clustering with the centroid linkage method was performed (Schadt et al., 2001), using 1 − r as the distance metric, where r is the Pearson correlation coefficient, and the relative expression levels are displayed.
The efficient coclustering of NMDA receptor subunits with the core membrane-associated guanylate kinase (MAGUK) PDZs (Fig. 1) is one index of the specificity of the screening method. The NMDAR2 subunits are homologous in both C-terminal sequence and function and this is recapitulated in the figure. In addition, it is useful to note that many PDZ domains that bind class I PLs do not bind in a manner dictated solely by their three C-terminal residues, underscoring the importance of contributions from residues outside of the binding “core” (S/T-X-V/I/L) to specificity [compare NMDAR2 binding (SDV) to serotonin 5-HT2c (SSV) in Fig. 1]. This is also seen through examination of the C-terminal ligand sequences, in which hierarchical clustering by binding pattern appears to correlate with C-terminal sequence homology.
As one would expect, titration level of the ligand in the assay correlates with observed hits (compare Tat-NR2B9c at 10 μm, which is promiscuous, and Tat-NR2B9c at 7 nm, which is specific) (Fig. 1). As the ligand concentration is reduced, only the stronger interactions are maintained. Thus, this type of individual reaction screen appears appropriate to identify interactions over at least three or more orders of magnitude, but hits from this method must be titrated to determine a relative affinity.
To examine the effect of native sequence length on PDZ domain binding in vitro, three different constructs for neuroligin with similar total length but varying lengths of native C-terminal sequence were tested (neuroligin 20, TFAAGFNSTGLPHSTTRV; neuroligin 8, GGGGGSGGGGSGLPHSTTRV; neuroligin 6, GGGGGSGGGGSGGGHSTTRV). Although differences in relative binding strengths of individual interactions are observed, the three constructs all displayed similar binding patterns and were consecutive in the hierarchically clustered heat map. Thus, even the six C-terminal residues are sufficient to give an accurate interaction profile. When shorter total length peptides were used (from 10 down to 3 aa), more variance was observed in the outcome (data not shown). We believe this is attributable in part to differential solubility of the PL peptides but may be in part caused by steric hindrance of the biotin–HRP interaction when short peptides are bound to the PDZ domains.
Validation of the PDZ domain interaction assay: determination of established interactions of NR subunits and nNOS with PSD proteins
The validity of our proteomic screen was tested by focusing on the direct PDZ interactions of NMDAR subunits and of nNOS. All known PDZ interactions tested with these proteins were recapitulated. nNOS is a key signaling molecule downstream of NMDARs that is associated with MAGUKs and is implicated in normal and pathological neuronal function. The assay should reveal not only the known PDZ interactions of these proteins but also test predicted, and demonstrate any previously unknown, interactions. The NMDAR PLs were peptides corresponding to the C-terminal residues of the NMDAR NR1 and NR2A–NR2D subunits, and the nNOS PDZ was a fusion protein containing maltose-binding protein (MBP) fused to the first 120 residues of nNOS. This construct (hereafter termed MBP-nNOSd1) contains the PDZ domain and the internal β-finger domain that had been shown to bind PSD-95 domain 2 and syntrophin 1α (Brenman et al., 1996a).
NR1 splice isoforms NR1–NR3 and NR1–NR4 display a C-terminal PL sequence (-TVV) predicted to bind PDZ proteins (Kornau et al., 1995). A biotinylated 20 aa peptide corresponding to the 20 C-terminal residues of the human NMDA NR1 subunit (HPTDITGPLNLSDPSVSTVV; GenBank accession number, NP_000823) exhibited binding to five PDZ domain constructs (Figs. 1, 2A): DLG1/SAP97d1,d2, PSD-95d1,d2,d3, NeDLG/SAP102d1,d2, MAGI-3d2, and PTPL1d2.
Figure 2.

Representative PDZ-binding screens of the interactions between NR subunits assayed at a peptide concentration of 10 μm and >160 PDZ domain constructs. Each point on the x-axis represents a unique PDZ domain or multidomain construct. A450 indicates absorbance at 450 nm, with vertical bars indicating the average of four repetitions. A, PDZ interactions with the NR1 C terminus. B–E, PDZ interactions with the NR2A–NR2D C termini.
Three of the PDZ proteins that the NR1 C terminus binds (PSD-95, DLG1/SAP97, and NeDLG/SAP102) are MAGUKs that also bind NR2 subunits (described below). Whereas these interactions are established for NR2, they were less clearly determined for NR1 (Kornau et al., 1995) (but see Bassand et al., 1999). Our data confirm the feasibility of direct binding of these MAGUKs to NR1. Understanding the potential PDZ interactions and their relative in vitro affinities for individual domains may aid in the understanding of large multiprotein complexes such as the NMDA receptors.
The screen also detected previously described interactions of NR1 with membrane-associated guanylate kinase with inverted orientation (MAGI-3). This MAGUK localizes to epithelial cell tight junctions (Wu et al., 2000), may function as a transforming growth factor-α tail-binding protein (Franklin et al., 2005), and may be involved in the regulation of the JNK signaling pathway (Yao et al., 2004). It also interacts with RPTPβ (receptor tyrosine phosphatase β) and tyrosine-phosphorylated proteins (Adamsky et al., 2003). The NR1 PL was also shown to interact with PTPL1, a cytoplasmic protein tyrosine phosphatase expressed in multiple tissues including brain (Saras et al., 1994). PTPL1 has five PDZ domains and has previously been shown to interact with the C-terminal tail of Fas, a member of the tumor necrosis factor receptor family (Saras et al., 1997), and to participate in triggering apoptosis in human cancers (Bompard et al., 2002). These additional interactions of the NR1 subunit suggest additional pathways by which NMDARs may produce both physiological and pathological signals.
The array of PDZ proteins was also screened for interactions with peptides corresponding to the C termini of the human NR2A–NR2D subunits (Fig. 1), with representative screens provided in Figure 2B–E. When the PLs were assayed at 10 μm, we found a total of between nine and 20 high-affinity interactions (more than three times baseline relative binding at the test concentration) per NR2 subunit. There was significant overlap among the four different NR2 subunits in the PDZ proteins that they bound at the concentrations tested. Whereas the PSD-95 family members (PSD-95, SAP102, PSD-93, and SAP97) were all bound by each of NR2A–NR2D, some interactions were observed only for certain subunits at the concentrations tested (Table 1). These differences among subunit interactions could have functional consequences, accounting for the differential function or regulation of these subunits or complexes. Overall, the screen detected all of the NR2–PDZ interactions already reported in the literature, attesting to the robustness of the assay in generating protein–protein interaction data. In addition, several previously unanticipated interactions were detected (see Discussion).
Next, in addition to evaluating the PLs listed in supplemental Table S1b (available at www.jneurosci.org as supplemental material), we evaluated the interactions of the MBP-nNOSd1 construct with human PDZ proteins. Binding was assessed using a modification of the assay presented above, detecting binding through the use of an anti-MBP antibody (New England Biolabs) and an appropriate HRP-conjugated secondary antibody for detection (GE Healthcare). Only four PDZ domains were found to bind the internal β-finger of nNOS at high affinity: syntrophin 1α, PSD-95d2, DLG1d2/SAP97d2, and DLG2d2/PSD-93d2 (Table 1).
Determination of EC50 values for NR2:PDZ interactions
Our proteomic screen provides a database of >4100 interactions involving neuronal signaling receptors and PDZ proteins. In focusing on NMDARs, we next evaluated the affinities of binding for interactions of NR2 subunits with key target PDZs. Titrations were performed to determine EC50 values for seven of the PDZ proteins that bound NMDAR2 subunits with higher relative affinity (more than three times baseline relative binding at 10 μm; see Materials and Methods). As expected, the previously identified interactions with PSD-95d2, NeDLG/SAP102, and DLG2/PSD-93d2 appeared to have high relative affinities (Table 2). Our results are consistent with those of Husi et al. (2000), who isolated NMDA receptor multiprotein complexes through pulldown experiments using either an antibody against NMDAR1 or a peptide corresponding to the C terminus of NR2B and characterized their composition. Proteins coprecipitated or immunoprecipitated with the complex were identified using specific antibodies on Western blots and mass spectrometry. PSD-95, NeDLG/SAP102, and DLG2/PSD-93 were all identified as part of the complexes. In addition, the authors identified peptides in the complex corresponding to DLG1/SAP97 and the tight junction protein ZO-1, but at a lower frequency (based on the number of tryptic peptides identified from the complex belonging to these proteins). Based on the EC50 measurements in our studies, our data suggest that DLG1/SAP97 is likely to be a direct interaction, whereas ZO-1 may be indirect because the NR2 subunits do not bind it directly in our assay (Fig. 1). In addition, we observed higher relative affinities for DLG1d2 and TIP1. TIP1 was not identified by the mass spectrometry analysis of Husi et al. (2000), but the authors specifically excluded proteins <50 kDa from their analysis, and TIP1 is significantly smaller (∼14 kDa). Thus, our data are consistent with published work and demonstrate further the effectiveness of our assay in identifying relevant interactions. Based on the in vitro EC50 values for these interactions (Table 2), we have identified PSD-95 PDZ domain 2 and SAP102 as having the strongest PDZ domain interactions with NR2 subunits.
Table 2.
EC50s for PDZ:NR2 interactions (μm)
| PDZ construct | NR2A | NR2B | NR2C | TatNR2B9c |
|---|---|---|---|---|
| PSD-95d2 | 0.5 | 1.2 | 0.07 | 0.007 |
| NeDLG/SAP102d1,2 | 1.1 | 0.7 | 0.2 | |
| DLG2/PSD-93d2 | >11 | >5 | >1.4 | |
| DLG1/SAP97d1 | >9 | >5 | >1.4 | |
| TIP1 | >8 | >4 | 1.3 | |
| KIAA0973 | >15 | >2.7 | >0.5 | |
| TIP43 | >14 | >4 | ND |
″>″ indicates that a curve could be calculated but that binding saturation was not observed; thus, the true EC50 could be at or above the calculated value. ND, Not determined.
Interactions of PSD-95 with NR2 subunits and nNOS
Because PSD-95 is a key binding partner of NR2 subunits, we also evaluated in depth the interactions of each of NR2A–NR2D with each of the three PDZ domains of PSD-95. Titrations were performed in the ELISA PDZ-binding assay against GST-PDZ constructs comprising PSD-95d1, PSD-95d2, PSD-95d3, and PSD-95d1,d2,d3. Each of the 20 aa NR2A–NR2D C-terminal peptides was applied at concentrations ranging from 0.001 to 100 μm (Fig. 3A).
Figure 3.

Interactions of PSD-95 with NR2 subunits and nNOS. A, The indicated NR C-terminal peptides were titrated at concentrations ranging from 0.001 to 100 μm against the PSD-95 PDZ domain construct indicated on the x-axis. Numbers in parentheses indicate the PDZ domain of PSD-95 in the given construct. B, Titrations of the MBP-nNOSd1 or MBP alone against the indicated PSD-95 PDZ domain construct. Bars represent the mean ± SD of quadruplicate experiments.
There was significant similarity among the binding profiles of each of the NR2 subunits. Binding of NR2A–NR2D was strongest to constructs containing the PSD-95d2 PDZ, and each of NR2A–NR2C but not NR2D also bound PSD-95d1. None of the subunit C termini bound PSD-95d3. Notably, binding to the PSD-95d2 and to PSD-95d1,d2,d3 was similar, indicating that no significant additional effects on binding affinity were obtained from the larger construct.
Among the many binding partners of PSD-95 is nNOS (Stricker et al., 1997; Christopherson et al., 1999), the neuronal enzyme responsible for NO production. NO is a reactive nitrogen species that is a proposed downstream effector of excitotoxic neuronal death. Inhibition of nNOS using drugs such as l-NAME [N(G)-nitro-l-arginine methyl ester] or through gene knock-outs has demonstrated its contribution to excitotoxicity (Huang et al., 1994; Sattler et al., 1999). Suppressing the expression of PSD-95 or the addition of Tat-NR2B9c in primary cortical cultures reduces NO production, but without affecting nNOS function (Sattler et al., 1999; Aarts et al., 2002). Previous studies have shown that an internal PDZ ligand of nNOS can bind PDZ2 of PSD-95 but is unable to bind PSD-95 domain 1 (Jaffrey et al., 1998; Christopherson et al., 1999). Because these interactions may be key to excitotoxic mechanisms and the actions of Tat-NR2B9c, they were evaluated further. The MBP-nNOSd1 construct was titrated against GST-PDZ constructs comprising PSD-95d1,d3 and PSD-95d2,d3 (Fig. 3B). The data reveal that MBP-nNOSd1 only bound to the PSD-95d2 (2/3 indicates d2 of three domains) construct, but not to PSD-95d1 (1/3 indicates d1 of three domains), confirming that nNOS only interacts with the second PDZ domain of PSD-95. Because this is also the major domain bound by NR2 subunits, a higher-order complex must exist, because nNOS and NMDAR2 subunits cannot occupy the same PDZ-binding site concurrently.
Interactions and affinities of Tat-NR2B9c to its PDZ-binding targets
Because Tat-NR2B9c is neuroprotective against NMDA toxicity (Aarts et al., 2002), its interactions must define a subset of PDZs involved in excitotoxic vulnerability. Therefore, it was screened in the PDZ domain interaction assay. When tested at a peptide concentration of 10 μm, we observed significant binding to 44 PDZ domains (Fig. 1). However, testing at this concentration may greatly overestimate the number of PDZs bound by this peptide. Its C terminus recapitulates the 9 C-terminal amino acids of the NR2B subunit, whose strongest interactions are with PSD-95d2 and SAP102 (EC50, ∼1 μm) (Table 2). Therefore, a more pertinent screen for this peptide's relevant interactions would be at or near the EC50 of Tat-NR2B9c for PSD-95d2. To determine this, we titrated the peptide at concentrations ranging from 0.0001 to 100 μm against both PSD-95d1 and PSD-95d2. A representative experiment is provided (Fig. 4A), demonstrating that, like the NR2 subunits, Tat-NR2B9c binds PSD-95d1 and PSD-95d2, and that binding is not dependent on a combination of PDZs because it was not enhanced when Tat-NR2B9c was screened against PSD-95d1,d2,d3.
Figure 4.

Interactions and affinities of Tat-NR2B9c to its PDZ-binding targets. A, Titration of Tat-NR2B9c at concentrations ranging from 0.0001 to 100 μm against the indicated PSD-95 PDZ domain constructs. Bars represent the mean ± SD of quadruplicate experiments. B, Determination of the EC50 of Tat-NR2B9c for binding PSD-95d1 (left) and PSD-95d2 (right). Symbols represent the mean ± SD of duplicate experiments. C, Interaction profile of Tat-NR2B9c against the panels of PDZ domains displaying interactions over background (2 PDZ arrays of 4 shown). For each pair of panels, titration of 0.007 μm Tat-NR2B9c (top) and 10 μm Tat-NR2B9c-AA (bottom) are shown. Bar pairs represent two individual interactions on separate microtiter plates. Hits not consistent between plates are repeated (interactions are generally classified for a given peptide concentration as 2× background and <20% SD).
The EC50 of Tat-NR2B9c for PSD-95d2 was 0.0067 μm, representing a >100-fold higher affinity for this domain than for PSD-95d1 (0.67 μm) (Fig. 4B). This is strong evidence that PSD-95d2 is a primary target of this drug, which correlates with the observation that the PSD-95 multi-PDZ domain construct binds with similar affinity to PSD-95d2, but little added affinity is observed from the PSD-95d1 binding. Also, although similar in its nine C-terminal residues to the NR2B C terminus, the affinity of Tat-NR2B9c for PSD-95d2 was two orders of magnitude greater than that of the 20-residue NR2B peptide tested under the same conditions (Table 2). The mechanism by which an NR2B C terminus gains enhanced affinity to its target PDZ by combining it with the Tat PTD is currently unknown. Tat alone shows no binding to PSD-95 PDZ domains at this concentration (data not shown), but may potentially enhance the peptide's solubility and solution availability. It is also possible that when stabilized by NR2B9c peptide, the Tat moiety can provide stabilizing interactions that decrease the off rate for the interaction. In any case, the EC50 values for Tat-NR2B9c render it a practicable drug because of its high target affinity.
Next, we screened the interactions of Tat-NR2B9c in the PDZ domain interaction assay at 0.5 μm (similar to its EC50 for PSD-95d1) and 0.007 μm (at the EC50 for PSD-95d2). Lowering the peptide concentration from 10 to 0.5 μm and subsequently to 0.007 μm narrowed the set of higher-affinity interactions to five proteins (Table 3, Fig. 4C). Four of these were MAGUK family members: PSD-95, DLG1/SAP97, NeDLG/SAP102, and DLG2/PSD-93. The interactions of these four MAGUKs with NMDARs have already been established (Kornau et al., 1995; Kim et al., 1996; Muller et al., 1996; Christopherson et al., 1999). The fifth PDZ interacting strongly at 7 nm Tat-NR2B9c was TIP1, a protein originally identified on the basis of binding to the TAX oncoprotein from human T-cell leukemia virus that has subsequently been demonstrated to interact with β-catenin in oncogenic pathways (Kanamori et al., 2003). It is plausible that TIP1 could act as a tumor suppressor and that, when bound by a virus or Tat-NR2B9c, it allows derepression of oncogenic or anti-apoptotic pathways. The top five PDZ proteins bound by Tat-NR2B9c are all among the proteins identified above as interacting with at least NMDAR2A, NMDAR2B, and NMDAR2C (PSD-95d2, DLG1d2, DLG2d2, NeDLG, and TIP1). In addition, the additional PDZ interactions of Tat-NR2B9c when the concentration is raised to 0.05 μm are all represented in the list of PDZs binding to subsets of NMDAR2-binding PDZs (Table 1).
Table 3.
NR2B9c selectivity
| PDZ | 0.5 μ m | 0.007 μ m |
|---|---|---|
| Tip2 | Strong | — |
| PSD-95d1,2,3 | Strong | Strong |
| PSD-95d2 | Strong | Strong |
| DLG1d1,2 | Strong | Strong |
| TIP1 | Strong | Strong |
| DLG1d2 | Strong | Strong |
| Magi-1d3 | Strong | — |
| NeDLGd1,2 | Strong | Strong |
| Magi-2d5 | Strong | — |
| Synγ | Strong | — |
| DLG2d2 | Strong | Strong |
| MAST2 | Strong | — |
As a control, we also tested the interactions of 10 μm TatNR2B9c-AA in the PDZ interaction assay (see Materials and Methods). This peptide, which is not anticipated to bind PDZ proteins, and is devoid of neuroprotective properties (Aarts et al., 2002), had no binding partners in our screen (Fig. 1).
Inhibition of interactions by Tat-NR2B9c
The above demonstrates that at concentrations near the EC50 for PSD-95d2, Tat-NR2B9c has high-affinity interactions with only a few other PDZ proteins, and mainly with those with which NMDARs interact (Tables 1–3). Because this peptide binds PSD-95 with higher affinity than the NR subunits (Table 2), it may outcompete several of the native interactions. To investigate this further, we examined the inhibition by Tat-NR2B9c of the key NMDA receptor interactions identified in this project. As the NR2 C terminus also binds the same PDZ domain in PSD-95 as nNOS (Fig. 3B), we examined the ability of Tat-NR2B9c to inhibit the PSD-95/nNOS interaction as well.
The IC50 values for Tat-NR2B9c inhibiting the interaction between NR2A, NR2B, NR2C, and PSD-95 were determined by plating a constant amount of NMDA receptor subunit with PSD-95 protein that gives a subsaturated signal in the ELISA test and titrating the amount of Tat-NR2B9c added (Fig. 5A). The IC50 values for inhibiting NMDAR2A, NMDAR2B, and NMDAR2C binding to PSD-95 are 0.5 μm, ∼8 μm, and 0.75 μm, respectively (Table 4). A similar approach was taken to determine the IC50 value for Tat-NR2B9c inhibiting the interaction between PSD-95 and nNOS (Fig. 5B), and this revealed an IC50 of ∼0.2 μm (Table 4). Although there were similarities among the PDZs bound by the various NR subunits, there was significant variability in the IC50 values of Tat-NR2B9c in inhibiting the interactions between NR2A, NR2B, and NR2C and their various their target PDZs (Table 4).
Figure 5.

A, B, Determination of the IC50 values for Tat-NR2B9c inhibition of the interactions between NR2A, NR2B, and NR2C and PSD-95d2 (A) and the interaction between MBP-nNOSd1 and PSD-95d2 (B). Symbols indicate the mean ± SD of duplicate experiments. All interactions and inhibitions were titrated multiple independent times and showed consistent results.
Table 4.
IC50 of Tat-NR2B9c for NMDA receptor interactions
| PDZ ligand | PDZ protein | Tat-NR2B9c IC50 |
|---|---|---|
| NMDAR2A | PSD-95d2 | 0.5 μm |
| DLG1d2 | ∼10 μm | |
| NeDLG1,2 | ∼7 μm | |
| KIAA 0973 | ∼9 μm | |
| TIP-1 | ∼7 μm | |
| DLG2d2 | ∼3 μm | |
| NMDAR2B | PSD-95d2 | ∼8 μm |
| DLG1/SAP97d2 | >10 μm | |
| NeDLG/SAP102d1,d2 | ∼7 μm | |
| KIAA 0973 | >10 μm | |
| TIP-1 | ∼10 μm | |
| DLG2/PSD-93d2 | >10 μm | |
| NMDAR2C | PSD-95d2 | 0.75 μm |
| DLG1d2 | 0.5 μm | |
| NeDLG1,2 | ∼1.1 μm | |
| KIAA 0973 | ∼1.5 μm | |
| TIP-1 | 1 μm | |
| DLG2d2 | ∼1.2 μm | |
| nNOS internal | PSD-95d2 | 0.2 μm |
Suppression of Tat-NR2B9c-interacting proteins
The PDZs that Tat-NR2B9c binds constitute a subset of proteins involved in neurotoxic cascades. Consequently, we next evaluated the relative importance of these proteins in excitotoxicity. To this end, we suppressed the expression of one of PSD-95, PSD-93, SAP102, SAP97, TIP1, and nNOS in primary cultured cortical neurons that were subsequently exposed to an excitotoxic challenge with NMDA.
To suppress these PDZ domain-containing proteins, we used RNAi, a process of posttranscriptional gene silencing that inhibits, with high specificity, the expression of native genes in mammalian cells (Elbashir et al., 2001; Cullen, 2002). The transfection of siRNA duplexes into primary neurons was optimized previously using a nonsilencing FITC-conjugated siRNA to monitor its uptake into neurons (Aarts et al., 2003). Our approach (see Materials and Methods) resulted in >90% transfection efficacy as determined previously using cell counts (Aarts et al., 2003).
Here we transfected the cultures with 21 nt siRNA duplexes targeting our proteins of interest (see Materials and Methods). The effects were first optimized by examining the suppression of the target protein mRNA (Fig. 6A, left). Applying the siRNA at concentrations ranging from 40 to 60 pm achieved maximal suppression of mRNAs for the PSD-95 family members within ∼2 d, whereas maximal suppression of nNOS mRNA was observed on day 3 after transfection. Next, we examined the effects of the siRNA transfection on target protein levels at 5 d after transfection. Although mRNA suppression was sometimes near complete (Fig. 6A, left), we were only able to obtain partial (∼50%) suppression of the PSD-95 family member proteins and of nNOS as determined by densitometry (Fig. 6A, right, B).
Figure 6.

Suppression of PSD-95 family members and nNOS in primary murine neuron culture by RNAi. A, RT-PCR gels (left) and Western blots (right) of the indicated proteins. KD, Silencing siRNA. B, Quantitative summary of the degree of suppression of the indicated protein, harvested from siRNA-transfected cortical neurons, as assayed by immunoblotting. Each bar indicates the mean ± SEM of at least three separate experiments. *p < 0.05, differences from nonsilencing controls (each represented by the same gray bar). C, Assessment of the morphology of cultured cortical neurons treated with the indicated siRNAs. Cultures were stained with the neuron-specific marker NeuN (green) or with the lipophilic membrane tracer DiI (red) 5 d after siRNA transfection. Scale bar, 25 μm.
To examine the morphology and integrity of neurons treated with siRNAs targeting the PSD-95 family members, some cultures were stained with the neuron-specific marker NeuN or with the lipophilic membrane tracer DiI 5 d after siRNA transfection (see Materials and Methods). Knocking down PSD-93, PSD-95, SAP97, or SAP102 produced no differences in neuronal morphology as assessed by these techniques, compared with untransfected neurons or with neurons transfected with a control (nonsilencing; see Materials and Methods) siRNA (Fig. 6C). Moreover, neurons transfected by this method did not stain with PI, indicating good viability.
Because suppressing the expression of one protein in neurons could elicit compensatory changes in other protein levels, we also determined the levels of the PSD-95 family members and of nNOS protein for each instance in which one of these was suppressed. In cortical cultures in which we inhibited the expression of PSD-95, there were no detectable changes in the expression of PSD-93, SAP102, SAP97, and nNOS (Fig. 7A). This result is consistent with our past findings that suppressing PSD-95 expression using antisense oligodeoxynucleotides did not affect the levels of these proteins (Sattler et al., 1999). Also, inhibiting the expression of SAP102 or PSD-93 or SAP97 had no effect on the other PSD-95 family members or on nNOS (data not shown). However, suppressing the expression of nNOS protein also reduced PSD-95 expression (Fig. 7B), raising the possibility that there is interdependence in the expression of PSD-95 and of nNOS.
Figure 7.

A, B, Impact of siRNA suppression of PSD-95 (A) and nNOS (B) on other PSD-95 family proteins and nNOS. Cultures were treated with siRNA targeting PSD-95 (A) or nNOS (B), and the harvested protein was blotted with antibodies against the indicated proteins. Left, Representative immunoblots. Right, Quantitative analysis (mean ± SE) of three to six immunoblots per condition. *p < 0.05, **p <0.01, differences from NS siRNA controls.
The fifth PDZ protein that interacted with Tat-NR2B9c was TIP1 (Table 3, Fig. 4C). This was also suppressed in the cortical cultures by RNAi, as evidenced by RT-PCR detection of TIP1 mRNA and by immunoblotting for the protein (Fig. 8A, top and bottom, respectively). It was noted that TIP mRNA levels were substantially less in the cultured neurons compared with mRNAs of the PSD-95 family members (Fig. 8A, top). This may suggest that TIP1 is not strongly expressed in these cells. As in the case of the PSD-95 family members and nNOS, suppressing TIP1 produced no differences in neuronal morphology as assessed by staining with NeuN and DiI (Fig. 8B; compare with Fig. 6C, controls).
Figure 8.

A–D, Impact of suppressing the expression of TIP1 (A–C) and the PSD-95 family members or nNOS (D) on the excitotoxic vulnerability of cultured cortical neurons. A, Top, Lower levels of TIP1 mRNA compared with the PSD-95 family members in cultured cortical neurons. Bottom, Effectiveness of suppressing TIP1 by RNAi (TIP1 RNAi) compared with nonsilencing siRNA (NS RNAi). The remaining lanes are controls from untransfected cultures (Control Culture), mouse brain, and mouse kidney as indicated. B, Assessment of the morphology of cultured cortical neurons treated with siRNA targeted against TIP. Green, Immunostaining with NeuN antibody. Red, Staining with DiI. Scale bar, 50 μm. C, Effect of suppressing TIP1 protein on excitotoxic vulnerability. D, Effect of suppressing the indicated protein on vulnerability to NMDA excitotoxicity. Cultures were transfected with one of the following: NS siRNA or siRNA targeted against the indicated PSD-95 family member or nNOS. C, D, Cell death was assayed 20 h after a 1 h NMDA insult at the indicated concentration. *p < 0.05, differences in cell death compared with NS controls (ANOVA). Bars indicate the mean ± SE of three separate experiments.
Role of Tat-NR2B9c-interacting proteins in excitotoxicity
Suppression of PSD-95 using antisense oligodeoxynucleotides enhances the resilience of cultured cortical neurons to NMDA-mediated excitotoxicity, and this resilience paralleled a reduction in the production of nitric oxide by nNOS (Sattler et al., 1999). To examine the roles of all targets of Tat-NR2B9c in excitotoxicity, one of PSD-95, PSD-93, SAP102, SAP97, TIP1, or nNOS was suppressed by RNAi as above in the primary cortical cultures. These were then exposed to a 1 h challenge with NMDA (30–100 μm), and neuronal cell death was assessed at 24 h.
Suppressing the expression of TIP1 had no impact on the vulnerability of neurons to NMDA toxicity, suggesting that this protein is unlikely to participate directly in pathways that govern NMDA toxicity (Fig. 8C). However, suppressing the expression of PSD-95 or of nNOS each reduced the vulnerability of neurons to an excitotoxic NMDA challenge (Fig. 8D). This result confirms that the NMDAR–PSD-95–nNOS complex is important in mediating excitotoxic damage. Conversely, suppressing PSD-93, SAP102, or SAP97 in the cultures had no impact on the vulnerability of neurons to NMDA toxicity. This suggests that these proteins may play a lesser role in linking NMDA receptor activity to secondary neurotoxic signaling cascades. Given that we were unable to completely eliminate the expression of these proteins by RNAi (Fig. 6A), one cannot exclude the possibility that these proteins may still participate in excitotoxicity and that their expression must be suppressed to a greater level to perceive this impact. Alternatively, the relative lack of a role of these proteins in excitotoxicity may indicate a relatively lower expression level or less strategic cellular localization compared with PSD-95. Such reasoning may apply to PSD-93, which also binds nNOS (Brenman et al., 1996b), but whose contribution to linking NMDARs to nNOS may be exceeded by that of PSD-95. Consistent with our result, PSD-93 deficiency was not neuroprotective against hypoxia-ischemia in PSD-93 knock-out neonatal mice (Jiang et al., 2003). However, the reasons for this remain open to speculation as the intracellular interactions of each PSD-95 family member and the frequencies and strengths of these interactions are all quantitatively unknown.
Discussion
This project describes, validates, and uses a method for large-scale proteomic analysis of PDZ ligands with a comprehensive set of human PDZ domains. The validation provides a database examining >4100 interactions between synaptic signaling proteins and PDZ domain proteins (Fig. 1, Table 1). In focusing on NMDA receptors, the screen revealed all previously published interactions of NMDARs with PDZ proteins, as well as several unanticipated or underappreciated interactions of NR subunits (Fig. 2, Table 1). We were also able to examine the PDZ domain selectivity and the strength of key NR subunit interactions (Fig. 3, Table 2). Using Tat-NR2B9c as bait for important PDZ domain interactions that mediate excitotoxicity, its PDZ-binding partners, PDZ domain selectivities, and binding affinities were examined at relevant concentrations (Figs. 1, 4; Table 3). As a test of potential efficacy, we also determined this peptide's capacity to inhibit preformed NR2:PSD-95d2 interactions and nNOS:PSD-95d2 interactions (Fig. 5, Table 4). The interactions, PDZ domain selectivities, and affinities (Table 1, Fig. 3B) of nNOS, a key PDZ protein mediating excitotoxicity, were similarly determined. To test the participation of Tat-NR2B9c-binding PDZ proteins in excitotoxicity, they were suppressed in cortical neurons using RNAi (Figs. 6, 7). Surprisingly, of all the PDZ proteins examined, only PSD-95 and nNOS participated significantly in excitotoxic signaling (Fig. 8). Thus, despite the anticipated promiscuity of type I PDZ domain motifs of NMDA receptor subunits (Lim et al., 2002), excitotoxic signaling through PDZ domain proteins displays surprising, and unanticipated, specificity.
PDZ domain interaction assay
Signaling through PDZ domain scaffold, anchoring, and adaptor proteins is a common theme in cell functioning including epithelial polarity, organization of ion channels and photoreceptors, vesicular sorting, apoptosis, and synaptic communication (Pawson and Scott, 1997). Consequently, our assay could provide valuable insights into the organization and stoichiometry of multidomain PDZ protein complexes such as the Mint proteins (2 PDZs), MAGUKs (3 PDZs), MAGIs (5 PDZs), INAD-like (INADL) (8 PDZs), MUPP1 (multi-PDZ domain protein 1) (13 PDZs), and others. Such insights should lead to testable hypotheses on the biological functions of these interactions within the complex. Other uses might include the assessment of PDZ proteins and ligand complexes from biological samples or the evaluation of binding patterns of modulated PDZ ligand states (such as serine, threonine, or tyrosine phosphorylation) that may affect PDZ-binding selectivity (Su et al., 2004).
PDZ–PL interactions are also implicated in several disease pathologies, many of which may be amenable to therapeutic intervention. Examples include stroke (Sattler et al., 1999; Aarts et al., 2002), cancers (Kanamori et al., 2003; Hirai et al., 2004; Doorbar, 2006), infection (Excoffon et al., 2004; Xie et al., 2006), and disorders of lipid metabolism (Yesilaltay et al., 2005). Although we focused on relevant PDZ interactions of NMDAR subunits and of Tat-NR2B9c, the broader utility of this interaction assay as a drug discovery platform is evident. In such an instance, peptides recapitulating key PDZ ligands associated with important biological functions or diseases can be screened against the cloned and expressed human PDZ domain array to identify potentially important interactions. Then, as we have done here, biological experiments would be used to confirm interactions in cells and validate that disruption or modulation of the interaction is beneficial. After identification of therapeutically relevant interactions, peptide or small molecule drugs can be iteratively screened against these interactions to optimize drugs for bioavailability, stability, and specificity.
However, as this is an in vitro assay, it has certain limitations. Among these are that we have attempted to account for all splice variants of PDZ domains by isolating PDZs from different tissues types and searching various expressed sequence tag databases. However, we can never exclude the possibility that we have not identified all PDZs and/or splice variants. Also, this in vitro assay may have biases of its own. It could provide false positive results if a particular PDZ behaves differently when isolated away from its native protein and/or surrounding native interactions. A false negative might arise if native PDZs are modified (for example, phosphorylated). Nevertheless, our assay has robustly detected the known interactions of nNOS and NR2 subunits and, if anything, would tend to overestimate rather than underestimate any additional interactions of these proteins.
Novel interactions of NR subunits with PDZ proteins
In addition to uncovering previously published interactions of NMDARs and PDZ domain-containing proteins, the ELISA-based assay points at several previously unreported or underappreciated interactions that may mediate both physiological and pathological functions of glutamate receptors. Although beyond the scope of the current project, these interactions may merit additional future studies. For example, the assay picked up interactions of the NR2A subunit with INADL (Table 1). The INAD protein has been shown to interact via a PDZ domain with transient receptor potential (TRP) calcium channels that contribute to capacitative calcium entry into Drosophila photoreceptor cells, and may function as a TRP channel regulatory subunit (Shieh and Zhu, 1996). Human INAD-like (hINADl or INADL) protein contains 8 PDZ domains, is widely expressed in diverse tissues including the CNS, and is also concentrated in tight junctions of epithelial cells, whose integrity it may regulate (Lemmers et al., 2002). hINADl has significant sequence homology to the Drosophila INAD (Philipp and Flockerzi, 1997) and may therefore have similar signaling roles, such as regulating capacitative calcium entry into cells. The finding of a direct interaction of the NR2A PDZ C terminus with the eighth PDZ domain of hINADl suggests that this multi-PDZ protein may participate in postsynaptic protein organization, function, or both. Also interesting is that it provides a link between NMDARs and TRP channels, some of which have been implicated in CNS disorders, including ischemic neuronal death (Aarts et al., 2003).
Another set of PDZ proteins found to interact with NR2 subunits are LIM proteins (Table 1). In mammals, 10 genes are found that encode both a PDZ and one or several LIM domains. These genes are as follows: ALP, RIL (LIM-RIL), Elfin (CLP36), Mystique, Enigma (LMP-1), Enigma homolog (ENH), ZASP (Cypher, Oracle), LMO7, and the two LIM domain kinases (LIMK1 and LIMK2). All have been shown to associate with the actin cytoskeleton. The ALP and Enigma subfamily genes are together with LMO7 able to bind α-actinin via their PDZ domains. Important biological roles have been described for muscle and heart development (ZASP and ALP), bone morphogenesis (Enigma), and development of the nervous system and reproductive cells (LIM kinases). In addition, LMO7, Mystique, RIL, and the LIM kinases have all been linked to carcinogenesis and metastasis (Te Velthuis et al., 2007). LIM Mystique is an IGF-IR-regulated adapter protein located at the actin cytoskeleton that is necessary for the migratory capacity of epithelial cells (Loughran et al., 2005). In the nervous system, a role for RIL has been identified as directing the transport of GluR-A-containing AMPA receptors to and/or within dendritic spines, in an α-actinin/actin-dependent manner, thus promoting the synaptic accumulation of the receptors (Schulz et al., 2004).
As a final example, MAGI-2, also known as S-SCAM (synaptic scaffolding molecule), is a multi-PDZ domain scaffolding protein that interacts with several different ligands in brain, including PTEN (phosphatase and tensin homolog), dasm1 (dendrite arborization and synapse maturation 1), dendrin, axin, β- and δ-catenin, neuroligin, hyperpolarization-activated cation channels, β1-adrenergic receptors, and NMDA receptors (Hirao et al., 1998, 2000). MAGI-2 may also link complexes of AMPA receptors and their regulating proteins to other postsynaptic molecules and pathways, including NMDA receptors (Deng et al., 2006).
Implication for use of Tat-NR2B9c in vivo
Tat-NR2B9c is a putative drug for neuroprotection in stroke. It exhibits high-affinity binding to the second PDZ domain of PSD-95. This, and its ability to inhibit native protein–protein interactions, is in an appropriate range for its anticipated effects. In a previous publication, we demonstrated that the intravenous injection of Tat-NR2B9c into rats at a dose of 3 nmol/g reduced the vulnerability of neurons to focal cerebral ischemia (Aarts et al., 2002). In the most conservative estimate, assuming an even distribution of this peptide throughout the entire animal, this dose would translate to a tissue concentration of 3 μm. This conservative estimate of tissue concentration is in the same range of the IC50 values obtained for the ability of Tat-NR2B9c to disrupt NR–PSD-95 interactions, making it likely that the peptide was deliverable to tissues at effective concentrations. Notably, the NR2B–PSD-95d2 interaction is more difficult to disrupt than the nNOS/PSD-95 interaction (IC50, ∼8 μm and ∼0.2 μm for NR2B/PSD-95d2 and nNOS/PSD-95d2, respectively) (Table 4). Consequently, the most potent effect of Tat-NR2B9c may be via the prevention of nNOS binding to PSD-95d2 rather than of NMDAR2B binding to PSD-95 as originally anticipated (Aarts et al., 2002). In this scenario, even the conservative estimate of a 3 μm target tissue concentration is an order of magnitude above that required to uncouple nNOS from NMDARs.
Collectively, our results provide an extensive database of important PDZ–PL protein interactions and provide strong evidence for a surprising selectivity of NMDAR subunits in the PDZ proteins that they bind. This selectivity is based largely on differing affinities of the NR C terminus for different PDZ domain proteins. The results also provide a strong rationale for modulating native PDZ–PL interactions such as the NMDAR–PSD-95 interaction using exogenous peptides to target and disrupt a PDZ domain and, in the case of stroke, bode well for the rational use of drugs such as Tat-NR2B9c as neuroprotectants.
Footnotes
This work was supported by grants to M.T. from the Canadian Institutes of Health Research (MOP150807), the National Institutes of Health (NIH; NS048956), and the Canadian Stroke Network, and to P.S.L. from NIH Small Business Innovation Research Grant NS046954.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Adamsky K, Arnold K, Sabanay H, Peles E. Junctional protein MAGI-3 interacts with receptor tyrosine phosphatase beta (RPTP beta) and tyrosine-phosphorylated proteins. J Cell Sci. 2003;116:1279–1289. doi: 10.1242/jcs.00302. [DOI] [PubMed] [Google Scholar]
- Bassand P, Bernard A, Rafiki A, Gayet D, Khrestchatisky M. Differential interaction of the tSXV motifs of the NR1 and NR2A NMDA receptor subunits with PSD-95 and SAP97. Eur J Neurosci. 1999;11:2031–2043. doi: 10.1046/j.1460-9568.1999.00611.x. [DOI] [PubMed] [Google Scholar]
- Bompard G, Puech C, Prebois C, Vignon F, Freiss G. Protein-tyrosine phosphatase PTPL1/FAP-1 triggers apoptosis in human breast cancer cells. J Biol Chem. 2002;277:47861–47869. doi: 10.1074/jbc.M208950200. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996a;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci. 1996b;16:7407–7415. doi: 10.1523/JNEUROSCI.16-23-07407.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno VMG, Goldberg MP, Dugan LL, Giffard RG, Choi DW. Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen-glucose deprivation or excitatory amino acids. J Neurochem. 1994;63:1398–1406. doi: 10.1046/j.1471-4159.1994.63041398.x. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Cullen BR. RNA interference: antiviral defense and genetic tool. Nat Immunol. 2002;3:597–599. doi: 10.1038/ni0702-597. [DOI] [PubMed] [Google Scholar]
- David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SM, Lees KR, Albers GW, Diener HC, Markabi S, Karlsson G, Norris J. Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke. 2000;31:347–354. doi: 10.1161/01.str.31.2.347. [DOI] [PubMed] [Google Scholar]
- Deng F, Price MG, Davis CF, Mori M, Burgess DL. Stargazin and other transmembrane AMPA receptor regulating proteins interact with synaptic scaffolding protein MAGI-2 in brain. J Neurosci. 2006;26:7875–7884. doi: 10.1523/JNEUROSCI.1851-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin Sci (Lond) 2006;110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Excoffon KJ, Hruska-Hageman A, Klotz M, Traver GL, Zabner J. A role for the PDZ-binding domain of the coxsackie B virus and adenovirus receptor (CAR) in cell adhesion and growth. J Cell Sci. 2004;117:4401–4409. doi: 10.1242/jcs.01300. [DOI] [PubMed] [Google Scholar]
- Fix AS, Horn JW, Wightman KA, Johnson CA, Long GG, Storts RW, Farber N, Wozniak DF, Olney JW. Neuronal vacuolization and necrosis induced by the noncompetitive N-methyl-d-aspartate (NMDA) antagonist MK(+)801 (dizocilpine maleate): a light and electron microscopic evaluation of the rat retrosplenial cortex. Exp Neurol. 1993;123:204–215. doi: 10.1006/exnr.1993.1153. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Yoshiura K, Dempsey PJ, Bogatcheva G, Jeyakumar L, Meise KS, Pearsall RS, Threadgill D, Coffey RJ. Identification of MAGI-3 as a transforming growth factor-alpha tail binding protein. Exp Cell Res. 2005;303:457–470. doi: 10.1016/j.yexcr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95–neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giallourakis C, Cao Z, Green T, Wachtel H, Xie X, Lopez-Illasaca M, Daly M, Rioux J, Xavier R. A molecular-properties-based approach to understanding PDZ domain proteins and PDZ ligands. Genome Res. 2006;16:1056–1072. doi: 10.1101/gr.5285206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai A, Tada M, Furuuchi K, Ishikawa S, Makiyama K, Hamada J, Okada F, Kobayashi I, Fukuda H, Moriuchi T. Expression of AIE-75 PDZ-domain protein induces G2/M cell cycle arrest in human colorectal adenocarcinoma SW480 cells. Cancer Lett. 2004;211:209–218. doi: 10.1016/j.canlet.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof TC, Takai Y. A novel multiple PDZ domain-containing molecule interacting with N-methyl-d-aspartate receptors and neuronal cell adhesion proteins. J Biol Chem. 1998;273:21105–21110. doi: 10.1074/jbc.273.33.21105. [DOI] [PubMed] [Google Scholar]
- Hirao K, Hata Y, Yao I, Deguchi M, Kawabe H, Mizoguchi A, Takai Y. Three isoforms of synaptic scaffolding molecule and their characterization. Multimerization between the isoforms and their interaction with N-methyl-d-aspartate receptors and SAP90/PSD-95-associated protein. J Biol Chem. 2000;275:2966–2972. doi: 10.1074/jbc.275.4.2966. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang P, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snowman AM, Eliasson MJL, Cohen NA, Snyder SH. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interaction with PSD95. Neuron. 1998;20:115–124. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- Jiang X, Mu D, Sheldon RA, Glidden DV, Ferriero DM. Neonatal hypoxia-ischemia differentially upregulates MAGUKs and associated proteins in PSD-93-deficient mouse brain. Stroke. 2003;34:2958–2963. doi: 10.1161/01.STR.0000102560.78524.9D. [DOI] [PubMed] [Google Scholar]
- Kanamori M, Sandy P, Marzinotto S, Benetti R, Kai C, Hayashizaki Y, Schneider C, Suzuki H. The PDZ protein tax-interacting protein-1 inhibits beta-catenin transcriptional activity and growth of colorectal cancer cells. J Biol Chem. 2003;278:38758–38764. doi: 10.1074/jbc.M306324200. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17:103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Lemmers C, Medina E, Delgrossi MH, Michel D, Arsanto JP, Le BA. hINADl/PATJ, a homolog of discs lost, interacts with crumbs and localizes to tight junctions in human epithelial cells. J Biol Chem. 2002;277:25408–25415. doi: 10.1074/jbc.M202196200. [DOI] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- Loughran G, Healy NC, Kiely PA, Huigsloot M, Kedersha NL, O'Connor R. Mystique is a new insulin-like growth factor-I-regulated PDZ-LIM domain protein that promotes cell attachment and migration and suppresses Anchorage-independent growth. Mol Biol Cell. 2005;16:1811–1822. doi: 10.1091/mbc.E04-12-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF. Failure of the competitive N-methyl-d-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg. 1999;91:737–743. doi: 10.3171/jns.1999.91.5.0737. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Muller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Philipp S, Flockerzi V. Molecular characterization of a novel human PDZ domain protein with homology to INAD from Drosophila melanogaster. FEBS Lett. 1997;413:243–248. doi: 10.1016/s0014-5793(97)00877-6. [DOI] [PubMed] [Google Scholar]
- Saras J, Claesson-Welsh L, Heldin CH, Gonez LJ. Cloning and characterization of PTPL1, a protein tyrosine phosphatase with similarities to cytoskeletal-associated proteins. J Biol Chem. 1994;269:24082–24089. [PubMed] [Google Scholar]
- Saras J, Engstrom U, Gonez LJ, Heldin CH. Characterization of the interactions between PDZ domains of the protein-tyrosine phosphatase PTPL1 and the carboxyl-terminal tail of Fas. J Biol Chem. 1997;272:20979–20981. doi: 10.1074/jbc.272.34.20979. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M. Determination of the time-course and extent of neurotoxicity at defined temperatures in cultured neurons using a modified multi-well plate fluorescence scanner. J Cereb Blood Flow Metab. 1997;17:455–463. doi: 10.1097/00004647-199704000-00011. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Li C, Ellis B, Wong WH. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J Cell Biochem Suppl. 2001;37:120–125. doi: 10.1002/jcb.10073. [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TW, Nakagawa T, Licznerski P, Pawlak V, Kolleker A, Rozov A, Kim J, Dittgen T, Kohr G, Sheng M, Seeburg PH, Osten P. Actin/α-actinin-dependent transport of AMPA receptors in dendritic spines: role of the PDZ-LIM protein RIL. J Neurosci. 2004;24:8584–8594. doi: 10.1523/JNEUROSCI.2100-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse [see comments] Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Sheng M. Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci USA. 2001;98:7058–7061. doi: 10.1073/pnas.111146298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh BH, Zhu MY. Regulation of the TRP Ca2+ channel by INAD in Drosophila photoreceptors. Neuron. 1996;16:991–998. doi: 10.1016/s0896-6273(00)80122-1. [DOI] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Meldrum BS. Blockade of N-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- Stricker NL, Christopherson KS, Yi BA, Schatz PJ, Raab RW, Dawes G, Bassett DE, Jr, Bredt DS, Li M. PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences. Nat Biotechnol. 1997;15:336–342. doi: 10.1038/nbt0497-336. [DOI] [PubMed] [Google Scholar]
- Su Z, Xu P, Ni F. Single phosphorylation of Tyr304 in the cytoplasmic tail of ephrin B2 confers high-affinity and bifunctional binding to both the SH2 domain of Grb4 and the PDZ domain of the PDZ-RGS3 protein. Eur J Biochem. 2004;271:1725–1736. doi: 10.1111/j.1432-1033.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–2601. doi: 10.1152/jn.00970.2005. [DOI] [PubMed] [Google Scholar]
- Te Velthuis AJ, Isogai T, Gerrits L, Bagowski CP. Insights into the molecular evolution of the PDZ/LIM family and identification of a novel conserved protein motif. PLoS ONE. 2007;2:e189. doi: 10.1371/journal.pone.0000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Sattler R, Zabramski JM, Spetzler RF. A characterization of neuroprotection from excitotoxicity by moderate and profound hypothermia in cultured cortical neurons unmasks a temperature-insensitive component of glutamate neurotoxicity. J Cereb Blood Flow Metab. 1998;18:848–867. doi: 10.1097/00004647-199808000-00005. [DOI] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA. Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J Biol Chem. 2000;275:21477–21485. doi: 10.1074/jbc.M909741199. [DOI] [PubMed] [Google Scholar]
- Xie L, Yamamoto B, Haoudi A, Semmes OJ, Green PL. PDZ binding motif of HTLV-1 Tax promotes virus-mediated T-cell proliferation in vitro and persistence in vivo. Blood. 2006;107:1980–1988. doi: 10.1182/blood-2005-03-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Natsume Y, Noda T. MAGI-3 is involved in the regulation of the JNK signaling pathway as a scaffold protein for frizzled and Ltap. Oncogene. 2004;23:6023–6030. doi: 10.1038/sj.onc.1207817. [DOI] [PubMed] [Google Scholar]
- Yesilaltay A, Kocher O, Rigotti A, Krieger M. Regulation of SR-BI-mediated high-density lipoprotein metabolism by the tissue-specific adaptor protein PDZK1. Curr Opin Lipidol. 2005;16:147–152. doi: 10.1097/01.mol.0000162319.54795.e5. [DOI] [PubMed] [Google Scholar]