

Abstract
The presence of refractory wake impairments in many individuals with severe sleep apnea led us to hypothesize that the hypoxia/reoxygenation events in sleep apnea permanently damage wake-active neurons. We now confirm that long-term exposure to hypoxia/reoxygenation in adult mice results in irreversible wake impairments. Functionality and injury were next assessed in major wake-active neural groups. Hypoxia/reoxygenation exposure for 8 weeks resulted in vacuolization in the perikarya and dendrites and markedly impaired c-fos activation response to enforced wakefulness in both noradrenergic locus ceruleus and dopaminergic ventral periaqueductal gray wake neurons. In contrast, cholinergic, histaminergic, orexinergic, and serotonergic wake neurons appeared unperturbed. Six month exposure to hypoxia/reoxygenation resulted in a 40% loss of catecholaminergic wake neurons. Having previously identified NADPH oxidase as a major contributor to wake impairments in hypoxia/reoxygenation, the role of NADPH oxidase in catecholaminergic vulnerability was next addressed. NADPH oxidase catalytic and cytosolic subunits were evident in catecholaminergic wake neurons, where hypoxia/reoxygenation resulted in translocation of p67phox to mitochondria, endoplasmic reticulum, and membranes. Treatment with a NADPH oxidase inhibitor, apocynin, throughout hypoxia/reoxygenation exposures conferred protection of catecholaminergic neurons. Collectively, these data show that select wake neurons, specifically the two catecholaminergic groups, can be rendered persistently impaired after long-term exposure to hypoxia/reoxygenation, modeling sleep apnea; wake impairments are irreversible; catecholaminergic neurons are lost; and neuronal NADPH oxidase contributes to this injury. It is anticipated that severe obstructive sleep apnea in humans destroys catecholaminergic wake neurons.
Keywords: sleep, apnea, oxidative injury, noradrenergic, dopaminergic, wake, NADPH oxidase
Introduction
Frequent, albeit brief, hypoxia/reoxygenation events across periods of sleep occur in at least 1% of the adult populations in developed countries; these individuals have undiagnosed or untreated sleep apnea (Young et al., 1993). When treated for obstructive sleep apnea, patients typically report less somnolence, but therapy does not fully reverse wake impairments in individuals with severe sleepiness at presentation (Bedard et al., 1991; Engleman et al., 1994; Meurice et al., 1997; Tiihonen and Partinen, 1998; Kingshott et al., 2000; Patel et al., 2003; Marshall et al., 2006). In support of irreversible wake impairments, neuroimaging studies of humans with sleep apnea have identified gray matter lesions (Macey et al., 2002; Morrell et al., 2003).
In animal models of sleep apnea oxygenation, repeated hypoxia/reoxygenation events result in wake impairments (Decker et al., 2003, 2005; Veasey et al., 2004a; Zhan et al., 2005a,b). The residual wake impairments, after hypoxia/reoxygenation exposure, are associated with oxidative injury (e.g., lipid peroxidation, nitration, and protein carbonylation) in several brain regions, including wake-active regions (Veasey et al., 2004a,b; Zhan et al., 2005a,b). Previously, we identified NADPH oxidase as a major contributor to both the oxidative injury and the wake impairments in the model of sleep apnea oxygenation. Specifically, mice with transgenic absence of a catalytic subunit of NADPH oxidase, Nox 2, and mice with pharmacological inhibition of Nox 2 catalysis throughout exposure to hypoxia/reoxygenation confer resistance to both wake impairments and oxidation (Zhan et al., 2005b). Therefore, NADPH oxidase may contribute to the vulnerability of select neurons to hypoxia/reoxygenation injury. Although, initially, brain NADPH oxidase was thought to be predominantly in microglia, NADPH oxidase subunits have been identified recently in subsets of neurons, including wake-active noradrenergic neurons in the locus ceruleus (LC) (Serrano et al., 2003; Wang et al., 2004; Zhan et al., 2005). Whether NADPH oxidase is present in other groups of wake neurons, and whether intraneuronal enzyme activation contributes significantly to the demise of wake-active neurons is not known. The present collection of studies demonstrates irreversible wake impairments in adult male mice after long-term intermittent hypoxia (LTIH) exposure. Wake impairments were associated with selective neuronal loss in the NADPH oxidase-containing dopaminergic ventral periaqueductal gray and noradrenergic locus ceruleus neurons. Other wake active groups appeared unperturbed. Remaining catecholaminergic wake neurons demonstrated vacuolization and activated caspase-3 within the dendrites and perikarya and with longer exposures, within the nuclei. In contrast, catecholaminergic groups A5 and A8 revealed neither NADPH oxidase nor caspase-3. It is anticipated that some individuals with severe obstructive sleep apnea have a similar neural injury, thereby raising the possibility of disease interactions with other neurodegenerative disorders, including Parkinson's disease.
Materials and Methods
Animals.
Male C57BL/6J (B6) male mice (The Jackson Laboratory, Bar Harbor, ME) were studied; mice were 2 months of age (young adult) at the onset of hypoxia/reoxygenation exposures and confirmed pathogen free at the time of behavioral and neuroanatomical studies. The methods and study protocols were approved in full by the Institutional Animal Care and Use Committee of the University of Pennsylvania and conformed with the revised National Institutes of Health Office of Laboratory Animal Welfare Policy. Food and water were provided ad libitum.
Long-term intermittent hypoxia protocol.
LTIH is an established model of sleep apnea oxygenation patterns (Gozal et al., 2001; Decker et al., 2003; Li et al., 2004; Row et al., 2004; Kheirandish et al., 2005; Zhan et al., 2005; Polotsky et al., 2006). The LTIH protocol details were published recently (Veasey et al., 2004a). Briefly, an automated nitrogen/oxygen profile system (Oxycycler model A84XOV; Biospherix, Redfield, NY) was used to produce brief reductions in ambient oxygen level from 21 to 10% for 5 s every 90 s for LTIH and from 21 to 20% for 5 s every 90 s for sham LTIH. The 21 to 10% ambient oxygen exposure results in fluctuations in oxyhemoglobin saturation from 94–98% to 76–84% lasting 2–7 s, modeling patterns observed in severe obstructive sleep apnea (Veasey et al., 2004a). Both LTIH and sham LTIH conditions were produced for 10 h of the lights-on period. Three durations of LTIH conditions were examined, as described below (2, 8, and 24 weeks). Humidity, ambient CO2, and environmental temperature were held constant within and across exposures.
Behavioral state recording and analysis.
Mice exposed to 8 weeks of LTIH have wake impairments persisting 2 weeks into recovery in normoxia conditions (Veasey et al., 2004a). To determine whether the wake impairments are reversible with a longer recovery, mice were randomized to either 8 weeks LTIH (n = 12) or sham LTIH (n = 12), and then both groups were allowed a 6 month recovery after IH exposures before examining sleep–wake behaviors. Surgical implantation of electrodes and electrophysiological recordings followed previously described methods (Veasey et al., 2000, 2004a). After 3d postoperative recovery, mice were connected to counter-weighted recording cables in individual cages, and 10 d after surgery (3 d after hook-up), sleep recordings were initiated. The ability to move freely within the entire cage and stand on hindlimbs to explore the top of the cage was confirmed in all mice studied. Baseline sleep was recorded for 5 d to ensure stable sleep–wake activities across days, where mice with >10% change across 5 d were excluded from analysis. Day 5 of stable baseline was used to analyze sleep–wake times. On recording day 6, a baseline murine multiple sleep latency test (MSLT) was performed (four nap opportunities between 2:00 and 4:00 P.M.) to measure wakefulness after sleep (Veasey et al., 2004c). The following day, enforced wakefulness by gentle handling (air puffs or introduction of murine nest building materials into the cage) was performed for 6 h, followed by a second MSLT (across the same clock hours as the baseline MSLT). The behavioral state acquisition and analysis program (ACQ/SSSSS 3.4) has been described previously (Benington et al., 1994; Veasey et al., 2000). The primary variables were total wake time/24 h across baseline conditions and mean sleep latency, before and after short-term sleep loss. As before, separate two-way ANOVAs were used to compare sleep times and sleep latencies in sham LTIH and LTIH mice (Veasey et al., 2004a,c).
Brain section procurement for immunohistochemistry.
Immunolabeling in wake-active brain regions was implemented to: (1) assess the c-fos response to enforced wakefulness in wake-active neurons to identify functional impairment within wake-implicated neural groups, (2) determine the extent of apoptosis/necrosis in wake-active groups, (3) perform cell count estimates of wake neurons, and (4) determine which wake-active neurons demonstrate intraneuronal NADPH oxidase. In all cases, brain tissue was procured after pentobarbital anesthesia followed by 4% paraformaldehyde transcardial perfusion. Whole brains were incubated in 4% paraformaldehyde and then cryopreserved before coronal sectioning (40 μm) from the medulla through the basal forebrain approximating murine coordinates 0.0 bregma to −6 mm bregma (Franklin and Paxinos, 1997). Sections were stored as 1:6 or 1:3 series in 0.1% sodium azide in PBS at 4°C before use.
Wake neuron identification.
Cholinergic, dopaminergic, histaminergic, noradrenergic, orexinergic, and serotonergic, neurons have all been implicated to play specific roles in wakefulness (Aston-Jones et al., 1991, 2001; Parmentier et al., 2002; Saper et al., 2005; Lu et al., 2006). Each implicated group was examined in the present study. Details of all primary antibodies, negative controls/antigen specificity, catalog number/source, dilutions for light microscopy, immunofluorescence, and electron microscopy where used are provided in the supplemental material (available at www.jneurosci.org). Cholinergic neurons in the basal forebrain (horizontal and vertical diagonal bands) and mesopontine groups, pedunculopontine tegmentum (PPT), and laterodorsal tegmentum (LDT) were labeled with goat anti-choline acetyltransferase (AB144P; Chemicon/Millipore, Billerica, MA). Noradrenergic locus ceruleus neurons were identified with sheep anti-dopamine β-hydroxylase (DBH) (PA3-925; ABR Affinity Bioreagents, Golden, CO) and mouse anti-tyrosine hydroxylase (TH; 22941; Immunostar, Hudson, WI). Anti-DBH was used to demarcate noradrenergic and dopaminergic mesopontine wake-active cell groups in mice, whereas anti-TH was used for double labeling studies (below) for its robust labeling of noradrenergic and dopaminergic axons and dendrites (Aston-Jones et al., 2004). Serotonergic neurons in the dorsal raphe nucleus were localized with a mouse monoclonal anti-5-hydroxytryptamine antibody (5-HT) (20079; Immunostar, Hudson, WI). Orexinergic and histaminergic neurons in the lateral hypothalamus were labeled with monoclonal mouse anti-Orexin A (Orex-A) (MAB763; R & D Systems, Minneapolis, MN), and a polyclonal rabbit anti-histidine decarboxylase (HDC; 03-16045; American Research Products, Belmont, MA), respectively. Secondary antibodies used were biotinylated donkey anti-goat, anti-mouse, anti-rabbit, or anti-sheep (Jackson ImmunoResearch, West Grove, PA) and labeled as below for specific studies.
c-fos response to wakefulness.
Mice exposed to 8 weeks of either LTIH (n = 4) or sham LTIH (n = 4) were examined for wake c-fos responses. As an index of neuronal activation in wakefulness, the percentage of neurons labeled with c-fos protein in the nucleus was assessed and compared across two conditions: unperturbed sleep–wake activity across 1:00–4:00 P.M. and enforced wakefulness across the same time period; both conditions were confirmed behaviorally and electrographically. For 2 weeks before experimentation, mice were handled daily for 30 min by the one individual observing sleep or enforcing wakefulness during testing. Three hours of enforced wakefulness was achieved on the day of experimentation by introducing nest building materials into the cages and by using similar gentle handling. At the end of experimentation, mice were perfused and brains were cryopreserved as above. Sections within the first well of the 1:6 series were doubled labeled in relevant regions with the above listed primary antibodies to identify wake neurons and with rabbit anti-fos (Ab-5, 1:2000; Calbiochem, La Jolla, CA). Noradrenergic, orexinergic, cholinergic, and dopaminergic wake neuron identifiers were labeled with Alexa Fluor 488 (green; Invitrogen, Carlsbad, CA), and c-fos was tagged with Alexa Fluor 594 (red). Because the primary antibody labeling histaminergic neurons (anti-HDC) was generated in the rabbit, c-fos for HDC labeled neurons was examined using DAB-nickel for c-fos labeling first, followed by rabbit anti-HDC and DAB labeling for both the ventral and caudal tuberomammillary nuclei, as previously delineated in the rat (Ko et al., 2003). In all cases, cells were deemed c-fos positive if c-fos labeling was evident in the nucleus. Two scorers, blinded to condition, counted numbers and c-fos labeling, and their scores were averaged. For each mouse, >150 neurons per region were examined. The mean percentage of c-fos labeled wake-implicated neurons was compared using two-way ANOVA (n = 5 mice per IH and behavioral state condition) with independent variables of behavioral state and LTIH condition. A Bonferroni's correction was performed for the number of wake groups analyzed (p < 0.01).
Cleaved caspase-3 immunohistochemistry.
In addition to demonstrating functional impairment with changes in wake c-fos responses, we sought to demonstrate neural injury, in part, by measuring activated (cleaved) caspase-3. Caspase-3 is a proteolytic enzyme that, when activated, destroys numerous critical cellular proteins; this enzyme is considered one of the key executioner proteins of apoptosis (Cohen, 1997; Springer, 2002; Rami, 2003). Thus, whereas cleaved caspase-3 is upregulated transiently in synaptic remodeling (Huesmann and Clayton, 2006), prolonged activation of caspase-3 is considered a marker of neural injury in models of ischemia, trauma, and neurodegeneration (Cohen, 1997; Springer, 2002; Rami, 2003). Additionally, cleaved caspase-3 has been identified in cortical neurons of animals exposed to LTIH (Xu et al., 2004) and thus may highlight injured wake-active neurons. The second and fifth 1:6 sets of sections from the above nonsleep-deprived mice (LTIH, n = 5; sham LTIH, n = 5) were used for double labeling of wake neuron identifier and cleaved caspase-3 (CC3) using polyclonal rabbit anti-cleaved caspase-3 primary antibody (1:500; Cell Signaling Technology, Danvers, MA). Specificity was confirmed with nonincubation with caspase-3 blocking peptide (Asp175; Cell Signaling Technology). Noradrenergic, cholinergic, dopaminergic, serotonergic, orexinergic, and histaminergic wake neurons were examined (Saper et al., 2005; Lu et al., 2006), using the above labeling techniques with one exception. Histaminergic neurons were labeled with guinea pig anti-HDC (265-1, 1:4000; Euro-Diagnostica, Arnhem, The Netherlands) and confirmed specific in the posterior hypothalamus with HDC−/− mice, as above. Antibodies were tagged with either Alexa Fluor 594 (donkey anti-rabbit for CC3, 1:400) or 488 (donkey anti-mouse for TH, orex, or HDC, 1:1000; Invitrogen). Wake neurons with visible nuclei were examined not only for nuclear CC3 but also for cytoplasmic CC3, as observed in hypoxic programmed necrosis in dopaminergic neurons (Niquet et al., 2003). Cytoplasmic and nuclear CC3 presence was determined separately using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD) to measure fluorescence intensity relative to background (adjacent non-TH region). For each group of wake-active neurons, the total number of nucleated wake neurons on all relevant sections were counted, and the percentage of these neurons with nuclear cleaved caspase-3 was then determined (Kerr et al., 1972); the relative fluorescent intensity of cytoplasmic-only cleaved caspase-3 was determined separately. Each parameter was compared using two-way ANOVA with independent variables of intermittent hypoxia condition and cell region, as above.
Neuronal counting.
In light of the impaired c-fos response at 8 weeks exposure, a second group of age-matched mice were exposed to 6 months of LTIH (n = 10) or sham LTIH (n = 10) to determine whether longer exposures result in loss of catecholaminergic wake neurons. For counts, 40 μm sections were placed in 1:3 series. Sections from the first series were stained with anti-tyrosine hydroxylase labeled with Vector SG substrate (SK-4700; Vector Laboratories, Burlingame, CA) and counterstained with Nissl, throughout the midbrain and pons, covering the ventral periaqueductal gray (VPAG) and LC, from −3.64 to −5.68 bregma (Franklin and Paxinos, 1997). Each immunostained section was assigned by its landmarks into one of the five groups: rostral VPAG (VPAG-r, −3.64 to −4.16 bregma), mid-VPAG (VPAG-m, −4.24 to −4.48), caudal VPAG (VPAG-c, −4.72 to −4.96), rostral LC (−5.20 to −5.34), and caudal LC (−5.34 to −5.68). Mean cell numbers were estimated using a Leitz DMRB microscope with motorized stage and StereoInvestigator version 6 (MicroBrightField, Williston, MA). Briefly, x,y boundaries of TH-immunoreactive neurons in the VPAG and locus ceruleus regions were traced for each section. The thickness of each section was then measured, and the z boundaries were designated to include all but 3 μm from the top and bottom of the section. Neither section thickness (LTIH, 10.1 μm; sham IH, 10.2 μm; t = 0.22; p = 0.82) nor nuclear vertical diameter (LTIH 1.2 μm ± 0.04; sham IH, 1.3 μm ± 0.02; t = 1.5; p = 0.14) varied with IH condition, thereby allowing us to use the Abercrombie (1946) correction factor to adjust cell count estimates for the relative thickness of the nucleus and the section. All full grids, each 100 × 100 μm, within the x,y demarcated regions were sampled, and TH-immunoreactive neurons were counted if the nucleus was in focus within the designated z-plane. Cell number estimates were averaged for each VPAG and LC region and then compared with two-way ANOVA for region and IH condition.
NADPH oxidase subunit immunohistochemistry.
Having identified NADPH oxidase as an important contributor to LTIH wake-active neuron oxidation and wake impairments (Zhan et al., 2005b), the presence of three NADPH oxidase subunits was looked for in cholinergic, dopaminergic, histaminergic, noradrenergic, orexinergic, and serotonergic wake neurons. Primary antibodies selected were anti-p47phox (rabbit, 1:500; 07-001, Upstate Biotechnology, Lake Placid, NY), anti-p67phox (rabbit, 1:500; 07-002, Upstate Biotechnology), and anti-gp91phox (mouse,1:500, clone 53; BD Biosciences, Franklin Lakes, NJ) and were used with the above wake neuron identifying antibodies and Alexa Fluor secondaries to label select groups of wake neurons and each NADPH oxidase subunit. Neurons were deemed positive for a given subunit if subunit immunoreactivity was observed within the perikarya across ± 3 μm z-plane views. Confirmation of the presence of each NADPH oxidase subunit in TH+ neurons was performed on the semithin sections prepared as described below for electron microscopy with DAB labeling of TH and silver-enhanced gold labeling of p67phox, p47phox, and gp91phox. In each animal, 100–200 neurons identified for each wake group were then assayed for subunit presence. The percentage of neurons was estimated for each mouse (Sham LTIH, n = 4; LTIH, n = 4) and analyzed with two-way ANOVA, Bonferroni's corrected for the three subunits.
Electron microscopy.
Immunoelectron microscopy was used for semiquantification of and subcellular localization of NADPH oxidase subunits in catecholaminergic neurons. Adult B6, mice were exposed to 2 weeks IH or sham IH. This short-term IH exposure was chosen to examine the early response to IH, before necrosis and other injuries. Mice were anesthetized as above, and brains were perfused with heparin and acrolein (3.8%) in 2% paraformaldehyde in PBS, pH 7.4, and postfixed as described in detail previously (Wang et al., 2004). Coronal sections (40 μm) were cut with a vibratome through the midbrain and pons, landmarked with an atlas (Franklin and Paxinos, 1997). Sections were treated with 1% NaBH4 in PBS and rinsed in PBS (Wang et al., 2004). Sections were next rinsed in Tris-buffered saline before incubation in bovine serum albumin to minimize nonspecific labeling. To double label NADPH oxidase subunit p67phox and TH, the above IHC primary antibodies were used. TH was detected with immunoperoxidase using DAB (Huang et al., 2003), and p67 was detected using 1ηm colloidal gold (Electron Microscopy Science, Fort Washington, PA) as described by Wang et al. (2004). Visualization of the gold was enhanced in silver solution (IntenSE M; GE Healthcare, Arlington Heights, IL) and postfixed (Wang et al., 2004). Precise location of TH-immunoreactive neurons in the LC or VPAG was performed as illustrated in Figure 1, using light microscopy imaging of the semithin (0.35 μm) section or low power thin section to identify neurons by configuration and adjacent tissue landmarks. Sequential ultrathin sections (70–80 ηm) were made using a Leica Ultracut E ultramicrotome (Deerfield, IL). Sections were collected onto mesh grids and counterstained with Reynold's lead citrate and uranyl acetate for ultrastructural analysis using a transmission electron microscope Phillips CM10 (Eindhoven, The Netherlands).
Figure 1.

Methodology for localization of noradrenergic locus ceruleus neurons for NADPH oxidase analysis. A, Immunoperoxidase (DAB; brown) labeling tyrosine hydroxylase neurons in a 50 μm section in the locus ceruleus of a mouse and silver-intensified gold labeling of NADPH oxidase subunit, p67phox. The arrow marks the same neuron in all three images. A blood vessel (BV) is observed ventral to the cell of interest, providing an additional landmark for unambiguous electron microscopy imaging of noradrenergic neuron. B, Low power (1200×) localizes the same cell (TH-S1) and clearly shows intraneuronal presence of p67phox. C, Higher power (3800×) of the same neurons (arrow) showing peroxidase labeling of TH. Although much p67phox in these LTIH-exposed TH neurons is dispersed throughout the cytoplasm, this typically cytosolic subunit is present on somata membranes, mitochondria (mit), lysozyme (Ly), and RER. GC, Golgi complex; Nuc, nucleus; TH-d, tyrosine hydroxylase-positive dendrite.
NADPH oxidase subunit p67 ultrastructural analysis was performed on mice exposed to IH (n = 3) and sham IH (n = 3) for 2 weeks, using methods of Haberstock-Debic et al. (2003) to analyze distribution of gold particle distribution in TH-positive soma (S), dendrites (Ds), and axon terminals (ATs) in the LC and VPAG. Specific variables measured were: (1) the number of gold particles in TH-positive S, D, and ATs, expressed as density (per cross sectional area); and (2) percentage of particles in each S, D, and AT on neuronal membrane, mitochondria, Golgi complex, rough endoplasmic reticulum (RER), and in cytoplasm or other subcellular location. Measures were compared across IH exposure condition using two-way ANOVA for subcellular region and brain region.
NADPH oxidase pharmacological inhibition.
To determine whether NADPH oxidase inhibition could prevent catecholaminergic neuronal demise, a third series of mice was implanted with osmotic pumps filled with NADPH oxidase inhibitor, apocynin (Sigma-Aldrich, St Louis, MO) concentrated to deliver subcutaneously 3 mg/kg/d drug (sham LTIM, n = 4; LTIH, n = 4; LTIH apocynin, n = 4; LTIH dimethyl sulfoxide/vehicle, n = 4) using a previously established dosing (Jacobson et al., 2003). The selected dose has been shown to prevent LTIH hypersomnolence in mice (Zhan et al., 2005b). Three days after pump implantation, mice were exposed to LTIH for a period of 8 weeks, exchanging pumps every 2 weeks. At the end of the LTIH exposure, mice were deeply anesthetized and perfused for morphology and CC3 immunohistochemistry studies as above.
Results
Wake impairments do not reverse in murine model of sleep apnea oxygenation
Hypoxia/reoxygenation exposure for 8 weeks was examined for recoverability of wake impairments by allowing a 6 month recovery and then examining sleep and sleep latency data. Adult male mice exposed to long-term intermittent hypoxia for 8 weeks were allowed to recover for 6 months in constant normoxia conditions and then compared with sham-treated mice (littermates) studied at the same age. All mice survived surgery. One mouse in the LTIH group had suboptimal EEG signals, and thus sample sizes for analysis were LTIH, n = 11 and sham LTIH, n = 12. Despite 6 months of recovery in normoxia, total wake times remained significantly lower in mice exposed to LTIH (120 min less wake time/24 h, relative to age-matched sham LTIH mice; t = 6.4; p < 0.01) (Fig. 2 A). Reduced wake time after LTIH resulted in increases in both non-rapid eye movement sleep and rapid eye movement (REM) sleep. To determine whether sleepiness, as measured by the multiple sleep latency test normalized with long-term recovery, baseline (unperturbed) mean sleep latencies were analyzed for LTIH mice allowed long-term recovery, and age-matched sham LTIH allowed the same 6 months recovery. The mean sleep latency after 24 weeks of recovery remained significantly lower in LTIH-exposed mice than in age-matched sham-treated mice (10.2 ± 0.6 vs 14.3 ± 1.2 min; t = 9.1; p < 0.001) (Fig. 2 B). Six hours of enforced wakefulness resulted in shortened sleep latencies in LTIH-exposed mice, relative to sham LTIH mice despite the long recovery. Thus, significant wake impairments remain after a 24 week recovery opportunity, and these effects are additive with sleep homeostatic effects on latency.
Figure 2.

Irreversibility of hypersomnolence and sleep propensity in mice after long-term exposure to hypoxia/reoxygenation, modeling sleep apnea. A, Despite a recovery opportunity of 6 months, total wake times in 24 h (dark hatched bar) remain significantly less than age-matched control (sham treated; light hatched bar). NREMS, Non-rapid eye movement sleep. B, Multiple sleep latency test values remain reduced across unperturbed sleep–wake cycles at the end of the rest period and also after 6 h of sleep deprivation for the same circadian time periods. The asterisks denote significant (p < 0.05) differences relative to sham hypoxia/reoxygenation-exposed mice.
Long-term hypoxia/reoxygenation impairs the immediate early gene response to wakefulness in catecholaminergic wake neurons
Enforced wakefulness for 1–3 h has been shown to induce a strong c-fos protein nuclear translocation response in orexinergic, cholinergic, histaminergic, dopaminergic, and select cholinergic wake-active groups and a weaker c-fos response in the locus ceruleus in the rat, but many of the c-fos responses have not been characterized in the mouse (Pompeiano et al., 1995; Cirelli et al., 1996; Ledoux et al., 1996; Greco et al., 2000; Estabrooke et al., 2001; Lu et al., 2006). As a first step, we determined in mice which wake-active groups show c-fos activation. In sham LTIH-exposed mice, neither dorsal raphe 5-HT nor basal forebrain/mesopontine cholinergic groups demonstrated increased nuclear c-fos expression with wakefulness, whereas c-fos activation in response to wakefulness was detected in nonserotonergic, cholinergic cells within the same nuclei/regions. For this reason, the wake neuronal groups included in this functional assay were the dopaminergic, histaminergic, and noradrenergic and orexinergic neurons. The effects of LTIH exposure on the c-fos response in wake-active groups studied are presented in Figure 3. Interobserver reliability for each wake neuronal group in each mouse was >85%. Overall, there were large suppressive effects of LTIH on wake c-fos responses in dopaminergic periaqueductal gray and noradrenergic locus ceruleus neurons (n = 4 mice per IH and behavioral state condition). In contrast, orexinergic and histaminergic neurons showed strong c-fos responses to waking, similar to wake responses in sham LTIH mice. Group data are summarized in Fig. 3 D. Although the wake c-fos response was not impaired in orexinergic neurons after LTIH, there was a reduction in the percentage of labeled c-fos neurons in mice allowed sleep, relative to sham LTIH mice allowed to sleep (Fig. 3 B). LTIH mice allowed spontaneous sleep slept more in the 3 h before deep anesthesia and brain procurement than sham LTIH mice allowed sleep (LTIH, 136 ± 10 vs sham LTIH 109 ± 4 min; p < 0.05). There was no effect of LTIH on histaminergic c-fos responses for either the unperturbed sleep or enforced wakefulness conditions. Thus, LTIH exposure manifested as impaired c-fos wakefulness responses in both catecholaminergic neural groups without disturbing c-fos wake responses in orexinergic and histaminergic neurons.
Figure 3.

Long-term intermittent hypoxia impairs the c-fos response to wakefulness in select groups of wake neurons. A, Immunofluorescence in the ventral periaqueductal gray of anti-tyrosine hydroxylase (green) and anti-c-fos (red) in sham LTIH control (top two panels) mice and mice exposed to 8 weeks of LTIH (bottom two panels) after 3 h of spontaneous sleep–wake activity (left) and after 3 h of enforced wakefulness (right). The arrows highlight several c-fos-positive nuclei in TH-labeled neurons. B, Immunofluorescence in the perifornicular region of the hypothalamus of anti-orexin A (green) and c-fos (red). Arrows delineate orexin-labeled neurons with c-fos immunoreactivity in nuclei. C, Immunolabeling in the ventral tuberomamillary nuclei of anti-histidine decarboxylase (DAB; brown) and c-fos (DAB/nickel; black). The arrows highlight double-labeled neurons. D, Percentage of neurons per region with nuclear c-fos labeling after 8 weeks exposure to IH or control (room air). Data are presented for mice allowed 3 h of spontaneous sleep activity before perfusion (sleep) and mice kept awake with enriched environment for 3 h before perfusion (wake). The asterisks and bars denote ANOVA (significance, p < 0.05; n = 5/group). Scale bars, 25 μm.
Long-term hypoxia/reoxygenation induces vacuolization in catecholaminergic wake-active neurons
We next examined neuronal demise, using morphologic parameters of injury (pruning of dendrites and vacuolization within axons, dendrites, and somata), and CC3 immunoreactivity in the five wake neural groups in mice exposed to LTIH or sham LTIH, both for 8 weeks (n = 4/IH condition). LTIH (8 weeks) did not result in appreciable nuclear CC3 (<2% of all TH neurons across the two groups, similar to sham LTIH TH neurons). After LTIH exposure for 24 weeks, nuclear anti-CC3 immunofluorescence was observed in 5–10% of TH+ wake neurons. In contrast, cytoplasmic CC3 was evident in the majority of catecholaminergic neurons at 8 weeks LTIH with minimal present in sham controls (Fig. 4). Cholinergic, histaminergic, serotonergic, and orexinergic neurons, in contrast, did not reveal CC3 immunoreactivity in nuclei or cytoplasm after either 8 or 24 weeks exposure to hypoxia/reoxygenation (Fig. 4). Nuclei were enlarged (>20 μm) in10–20% of neurons at LTIH 8 weeks, and vacuoles were identified in both the soma and proximal dendrites of LTIH mice (Fig. 5). In contrast, morphological effects were not evident in cholinergic, histaminergic, serotonergic, and orexinergic neurons from any of the mice exposed to LTIH.
Figure 4.

Long-term intermittent hypoxia increases cleaved caspase-3, and this effect is partially blocked by treatment across IH with apocynin, an inhibitor of the catalytic (Nox2) NADPH oxidase subunit. A, Immunofluorescence of anti-TH (green) and anti-CC3 (red) in the locus ceruleus across conditions of sham LTIH or LTIH for durations of 8 and 24 weeks (sham LTIH 8 weeks, sham LTIH 24 weeks, LTIH 8 weeks, LTIH 24 weeks). The arrows denote anti-TH and anti-CC3-labeled neurons. At 24 weeks, anti-CC3 was evident in nuclei of <10% of noradrenergic neurons. A similar effect of LTIH across duration was observed for the dopaminergic ventral periaqueductal gray neurons. The bottom two panels show perifornicular hypothalamus anti-orexin A neurons (green) and anti-CC3 (red) in the same mouse shown above for anti-TH neurons. B, Effects of vehicle control (veh) (dimethyl sulfoxide; top panel) and apocynin treatment (apo) throughout LTIH exposure. The arrows highlight anti-CC3 immunoreactivity in TH+ neurons in the locus ceruleus. Scale bar, 25 μm (for all images).C, Individual data for relative CC-3 immunofluorescence intensity.
Figure 5.
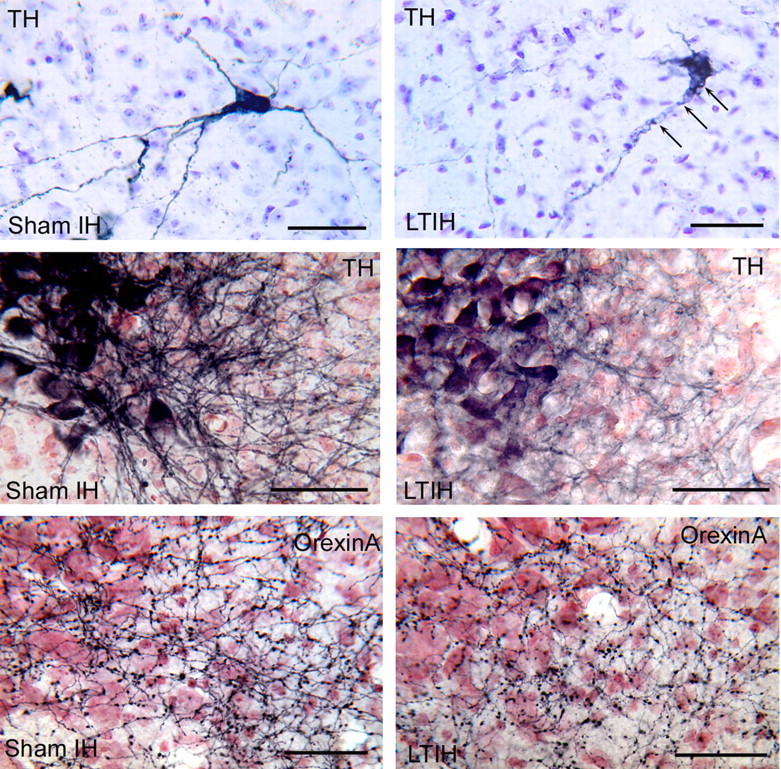
Effects of LTIH on cellular morphology of dopaminergic and noradrenergic wake neurons. The top two panels show ventrolateral periaqueductal gray neurons anti-TH-labeled Vector SG (blue) and Nissl background. LTIH-exposed neuron (top right panel) shows characteristic vacuolization in soma adjacent to dendrites and dendritic beading (arrows). The middle two panels compare dendrites of anti-TH-labeled neurons in locus ceruleus. The left panel shows prominent medial dendrites in sham LTIH mouse. In contrast, LTIH-exposed mouse shows loss of medial dendrites. The bottom panel shows adjacent region in the same mice, with preservation of orexinergic terminals into the dendritic region of the locus ceruleus. Scale bars, 50 μm.
NADPH oxidase subunits are present in select (catecholaminergic) wake-active neurons
In light of the previously described protection from LTIH wake impairments with NADPH oxidase inhibition (Zhan et al., 2005b), and the injury localized in catecholaminergic neurons, we hypothesized that LTIH upregulates NADPH oxidase in catecholaminergic neurons. Nonlocalization of NADPH oxidase in orexin, histaminergic, serotonergic, dopaminergic, and noradrenergic was determined for NADPH oxidase subunits: gp91phox, p47phox, and p67phox (n = 4 mice/IH condition). An exposure period of 2 weeks was selected to observe neurons before significant injury. None of the subunits were detected in orexinergic, serotonergic, or histaminergic neurons. There was trace p67phox and p47 phox in PPT cholinergic neurons and none evident in the magnocellular preoptic or LDT cholinergic neurons. In contrast, immunoreactivity was demonstrated for each subunit in the majority (>75%) of all dopaminergic and noradrenergic wake neurons of LTIH-exposed mice (Fig. 6), with predominant perinuclear envelope and proximal dendritic localizations. In summary, the catalytic subunit, gp91phox, and two of the cytosolic subunits are present in catecholaminergic wake neurons and are massively upregulated by early hypoxia/reoxygenation exposure. In summary, NADPH oxidase subunits are not evident in basal forebrain or LDT cholinergic neurons, orexinergic, serotonergic, or histaminergic neurons, but two cytosolic subunits of NADPH oxidase are present to a lesser degree in pedunculopontine cholinergic neurons.
Figure 6.

Immunolocalization of NADPH oxidase subunits in dopaminergic and noradrenergic wake neurons. Ventral periaqueductal gray neurons labeled for catalytic subunit (Nox2) with anti-gp91phox, silver enhanced gold and anti-TH immunoperoxidase labeled (DAB; brown) in 350 μm sections in sham LTIH, with subtle presence in dendrite (arrow) and in LTIH increased perinuclear and dendritic immunoreactivity (arrows). Locus ceruleus neurons with immunoreactivity for anti-p67phox in anti-TH labeled neurons in sham LTIH (left) and LTIH (right). The arrows highlight clustering of anti-p67phox labeling. Higher magnification shows intraneuronal labeling only (Figs. 1, 7). The bottom two panels show anti-p47phox immunoreactivity in TH-labeled locus ceruleus neurons. Scale bars, 25 μm.
The NADPH oxidase subunit p67phox response in noradrenergic neurons predicts injury to dendrites and the presence of cleaved caspase-3
To determine whether the indicators of injury (vacuolization and increased cytoplasmic cleaved caspase-3) responses to LTIH observed in the dopaminergic VPAG and noradrenergic LC were unique to these groups of catecholaminergic neurons, we examined the nearby A5, A7, and A8 noradrenergic neurons in the same mice analyzed above. A minority (<20% in all cases) of A7 neurons per section per mouse (n = 5/IH condition) showed trace p67phox and trace cleaved caspase-3 in response to 8 weeks LTIH. In contrast, none of the A5 or A8 neurons showed any trace of either NADPH oxidase p67phox or cleaved caspase-3. Vacuolization was not evident in any of the A5, A7, or A8 neurons in sham or LTIH mice (n = 5/IH condition). Thus, the presence of p67phox predicts prolonged activation of caspase-3, and this injury pattern is selective to the dopaminergic VPAG and noradrenergic LC neurons.
Subcellular density and distribution of wake neuron NADPH oxidase subunit p67phox in response to intermittent hypoxia
To estimate NADPH oxidase activation in neurons, we next determined whether the NADPH oxidase in VPAG and LC wake neurons translocates to organelles and/or membranes in response to IH (2 weeks exposure; n = 3/IH condition). As a measure of activation in TH neurons, we used immunoelectron microscopy to examine subcellular location shifts in NADPH oxidase subunits in response to IH. Previously, Western blots have been used to show membrane translocation, but here we sought to establish LTIH-induced translocation in the catecholaminergic neurons. In the locus ceruleus of mice exposed to IH, p67phox immunoreactivity was detected in the majority of dendrites (193 of 226) and somata (60 of 72) of TH-immunoreactive neurons. Few TH-positive axons were identified, but overall LTIH effects in the few axons paralleled changes in the dendrites and somata. Very little p67phox was evident in non-TH neurons and was rarely evident in glia in IH exposed mice. In sham IH mice, subunit p67 phox was identified in a smaller percentage of TH+ dendrites (166 of 310 dendrites) and in somata (22 of 65). A larger LTIH effect was observed regarding the density of p67phox immunoreactive particles (Fig. 7). Here, the density of p67phox particles in TH-positive dendrites and somata of IH-exposed mice increased twofold in dendrites (t = 7.1; p < 0.00001) and ninefold in somata (t = 5.6; p < 0.0001). Thus, LTIH increases the density of p67phox in the soma and dendrites of TH-positive locus ceruleus neurons. In addition to an overall increase in p67 immunoreactivity, a translocation of p67phox was observed to neuronal membranes and organelles (0.14 ± 0.02 particles/μm2 in sham IH and 0.34 ± 0.04; t = 4.6; p < 0.0001). IH-exposed mice showed increased gold particulate on endoplasmic reticulum, mitochondria, and Golgi (Fig. 7). The majority of p67phox immunoreactivity appeared to localize with ribosomes, as shown in several panels in Fig. 7.
Figure 7.

Ultrastructural localization of NADPH oxidase subunits in catecholaminergic wake neurons. Electron microscopic photomicrographs of noradrenergic locus ceruleus and dopaminergic VPAG neurons and dendrites labeled with anti-TH (stained with DAB) and cytosolic NADPH oxidase subunit, anti-p67phox (stained with silver-intensified gold). A, TH-immunoperoxidase-labeled LC neuron (TH-s) in sham LTIH-treated mouse with well-preserved cytoarchitecture, ER, mitochondria (mit), and Golgi complex (G). B, TH-immunoperoxidase-labeled LC neuron (TH-s) in LTIH-treated mouse with swollen mitochondria (mit), increased lysozymes (Ly), and clustered p67phox staining in ER around mitochondria (arrows) or on neuronal membrane. C–E, TH-immunoperoxidase-labeled dendrites (TH-d) in sham IH and LTIH-treated mice, with presence of silver-intensified gold staining of p67phox on mitochondria and membranes (D, E). F, In a mouse exposed to sham LTIH, well-preserved regions of RER with rare p67phox labeling were typical. G, In contrast, in LTIH-exposed mice, the RER showed, in regions of increased p67phox, a haziness of the ribosomes and membranes. The arrows highlight p67phox localization on RER. Scale bars: A, B, 1 μm; C–G, 500 ηm.
In the PAG, p67phox immunoreactive gold particles were observed in 58 of 62 TH-immunoreactive dendrites and 65 of 66 TH-soma in the mice exposed to IH. The majority of TH-positive axon terminals, identified by synaptic vesicles, also showed p67phox immunoreactive particles. In contrast, only a minority of dendrites, somata, and axons in the sham IH mice showed p67phox labeling. As in the LC, an increase in membrane and organelle p67phox labeling was significant with ninefold changes in both dendrites (t = 2.3; p < 0.05) and somata p67phox (t = 3.3; p < 0.01) (Fig. 7).
Inhibition of NADPH oxidase throughout LTIH exposure minimizes injury to catecholaminergic wake neurons
The effectiveness of apocynin, an NADPH oxidase inhibitor, in reducing catecholaminergic LTIH injury was tested by examining cleaved caspase-3 immunoreactivity in neurons across groups (sham LTIM, n = 4; LTIH, n = 4; LTIH apocynin, n = 4; LTIH vehicle, n = 4), where IH exposures were 8 weeks. Mid-VPAG and LC sections were matched (three/region/mouse) for semiquantification of catecholaminergic neuronal CC3 immunoreactivity. Vehicle (dimethyl sulfoxide) had no effect on caspase-3 immunoreactivity (Fig. 5). In contrast, apocynin-treated mice showed a significant reduction in CC3 immunoreactivity in catecholaminergic neurons but not full protection (Fig. 5). Overall, apocynin therapy across LTIH significantly reduced cleaved caspase-3 immunoreactivity in catecholaminergic wake neurons (Fig. 5).
LTIH induces early ultrastructural modifications in catecholaminergic neurons
Taking advantage of the electron micrographs obtained with 2 weeks exposure to hypoxia/reoxygenation, we next examined TH neuronal ultrastructure for sites of early injury. The most consistent ultrastructural modifications in mice exposed to LTIH were disruption of rough endoplasmic reticulum (a haziness or loss of distinction of both the ribosomes and the RER membranes) (Fig. 7 F,G) and mitochondria (increased variability in size with swelling of the intermembrane space) (Fig. 7 A,B). As a consequence of the latter finding, the cross-sectional diameter of mitochondria increased (sham IH, mean 210 ± 20 ηm; LTIH, mean 354 ± 62 ηm; t = 11; p < 0.001). In regions with minimal p67phox immunoreactive particulate, RER and mitochondria appeared preserved; however, in regions with more intense p67phox immunoreactivity, RER was difficult to delineate (Fig. 7 B,G), and mitochondria were enlarged. Thus, short-term exposure to intermittent hypoxia results in distortion of rough endoplasmic reticulum architecture and swelling of mitochondria.
LTIH results in neurodegeneration in the locus ceruleus and dopaminergic VPAG wake neurons
In light of the apoptosis present in noradrenergic and dopaminergic wake-active neurons at 8 weeks of LTIH, and minimal changes in other wake neural groups, we examined cell counts and CC-3 immunoreactivity at 6 months to better detect cell loss and to detect late-onset injury in noncatecholaminergic wake neurons. TH+ neurons and Nissl cell counts were performed throughout the PAG and locus ceruleus for both LTIH (n = 10) and sham LTIH (n = 10) mice on one set of every fourth section. Across the midbrain and pons, wake-active TH immunoreactive neurons with nuclei were reduced by 35–50% (Fig. 8). This achieved significance for both the VPAG dopaminergic neurons (t = 3.8; p < 0.01) and the locus ceruleus noradrenergic neurons (t = 7.2; p < 0.01). There was no change in nonlabeled neurons within the VPAG or LC to support downregulation of TH production in VPAG or LC neurons. Both sham and LTIH exposed mice showed some vacuolization in dopaminergic neurons (at 8 months of age); however, the percentage of TH-immunoreactive neurons with vacuolization was far greater in the LTIH-exposed mice. After 24 weeks of hypoxia/reoxygenation exposure, CC-3 immunoreactivity increased in both dopaminergic and noradrenergic wake neurons, yet remained absent in orexinergic and histaminergic groups. Thus, there is a select injury with neural loss to the noradrenergic and dopaminergic wake neural groups, without apparent effect on the noncatecholaminergic wake groups, even with considerable exposure time.
Figure 8.

Effects of 24 weeks LTIH on neuronal counts in catecholaminergic wake neural groups. Average cell count estimates per section of anti-TH-labeled neurons stained with vector SG per section in five matched regions across the complete dopaminergic VPAG and noradrenergic LC in sham LTIH (n = 10) and LTIH (n = 10). VPAG-r, Most rostral; VPAG-m, mid-VPAG; VPAG-c, caudal VPAG; LC-r, most rostral; LC, middle and caudal regions. Cell count ratios LTIH:sham LTIH (dark–light bars) are similar across the five regions. After Bonferroni's correcting for five brain regions, statistical significance in neuron number is present for four of the five dopaminergic and noradrenergic wake group regions (asterisks and bars represent p < 0.01).
Discussion
Long-term exposure to hypoxia/reoxygenation events, modeling sleep apnea oxygenation, induces irreversible and functionally significant injury in at least two wake-active neural groups: dopaminergic neurons in the ventral periaqueductal gray and noradrenergic neurons in the locus ceruleus. This injury, characterized as a 40% loss of wake neurons in each catecholaminergic nucleus, a 50% loss of dendrites and 70–90% reductions in c-fos protein nuclear translocation response to wakefulness across the two groups, is associated with irreversible wake impairments. In contrast to the multiple injury markers for catecholaminergic neural involvement, other monoaminergic, cholinergic, and orexinergic wake-active neural groups appear relatively spared from hypoxia/reoxygenation injury. Remaining catecholaminergic and noncatecholaminergic wake neurons fail to effectively compensate after this hypoxia/reoxygenation insult. We previously determined that both transgenic absence of functional NADPH oxidase and administration of NADPH oxidase inhibitor throughout hypoxia/reoxygenation fully prevent wake impairments in mice exposed to hypoxia/reoxygenation (Zhan et al., 2005b). We have now identified NADPH oxidase in the more vulnerable dopaminergic and noradrenergic neurons and provide evidence that hypoxia/reoxygenation events activate NADPH oxidase in these neurons by demonstrating translocation of a cytosolic subunit to membranes and organelles. In contrast, midbrain and pontine noradrenergic neurons without detectable NADPH oxidase show no evidence of LTIH injury. Additionally, inhibition of NADPH oxidase reduces, in part, injury to these vulnerable wake neurons in this model of sleep apnea.
Having identified persistent wake impairments 6 months after hypoxia/reoxygenation exposures, one primary objective for the present collection of studies was to identify the injured wake groups. Although noradrenergic locus ceruleus neurons are wake-active, it is unlikely that even a substantial lesion in this nucleus would result in wake impairments. Lesioning the noradrenergic innervation of the cortex with DSP-4 [N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine] does not reduce wake time/24 h (Cirelli and Tononi, 2004). In contrast, lesioning the dopaminergic ventral periaqueductal gray neurons with 6-hydroxydopamine results in reduced 24 h wake times similar to wake times observed in the present study (Lu et al., 2006). Although 6-hydroxydopamine injections result in a larger percentage reduction of dopaminergic neurons than LTIH, the loss of a c-fos response to wakefulness in the vast majority of remaining neurons suggests that the remaining neurons are functionally impaired.
Certainly, additional neural groups, including nonwake active neuronal populations, may be equally or even more severely injured in sleep apnea and in this model of sleep apnea oxygenation patterns. Although cell counts have not been performed in other brain regions to examine neurodegeneration, there is evidence of apoptosis in this model of sleep apnea oxygenation in the hippocampus and cortex (Goldbart et al., 2003; Xu et al., 2004; Kheirandish et al., 2005). The present study findings support exploration of neural injury in humans with severe obstructive sleep apnea, including the catecholaminergic wake neurons and other catecholaminergic groups.
Loss of noradrenergic locus ceruleus neurons has been identified as an early lesion in Alzheimer's disease, Parkinson's disease, and dementia with Lewy bodies (Zarow et al., 2003). Moreover, locus ceruleus injury has been shown to accelerate forebrain amyloid deposition and neural injury in a murine model of Alzheimer disease (Heneka et al., 2006; Kalinin et al., 2007). Having demonstrated in the present study that oxygenation patterns, as observed in sleep apnea, can significantly injure the locus ceruleus, it will be of interest to look for disease interactions between obstructive sleep apnea (present in >10% of elderly humans) and Alzheimer's disease. In Alzheimer's disease, locus ceruleus loss may be accompanied by compensatory changes, including dendritic and axonal sprouting (Szot et al., 2006). In the present model of hypoxia/reoxygenation injury, neuronal compensation is not evident, because dendrites are substantially reduced and many remaining dendrites show vacuolization.
The loss of noradrenergic dendrites in the locus ceruleus, without apparent injury to the orexinergic axonal projections in the same region, provides support that the injury is cell specific more than region specific. Injury to noradrenergic neurons in the locus ceruleus unveils the possibility of hypoxia/reoxygenation injury to other noradrenergic neurons, including premotor neurons. Catecholaminergic neurons in A1/C1, A5, A7, and to a lesser extent locus ceruleus, innervate upper airway motoneurons (Rukhadze and Kubin, 2007). Noradrenergic innervation of upper airway motoneurons contributes to activation of upper airway dilator motoneurons (Lai et al., 2001; Fenik et al., 2005; Chan et al., 2006; Rukhadze and Kubin, 2007). However, in our studies, at least the A5 and A7 groups appear relatively unperturbed by hypoxia/reoxygenation exposures. A careful look at all noradrenergic groups, including A1/C1, across longer exposures and in humans with severe sleep apnea should be initiated.
The susceptibility of dopaminergic neurons to oxidative neural injury is not surprising in light of the established susceptibility of dopaminergic neurons to oxidative injury, including NADPH oxidase injury, in models of Parkinson's disease (for review, see Przedborski and Ischiropoulos, 2005). The mechanisms by which Parkinson's disease imparts daytime wake impairments, however, on afflicted individuals are not known. The work in the present study supports the concept that obstructive sleep apnea in patients with Parkinson's disease might contribute to the hypersomnolence. Alternatively, these VPAG dopaminergic neurons may be injured by the same mechanisms underlying Parkinson's neuronal injury. The present findings of selective injury to the NADPH oxidase-containing dopaminergic and noradrenergic neurons supports the concept that neuronal NADPH oxidase activation by environmental or physiological perturbances may contribute to the vulnerability of these neurons in Parkinson's disease.
How NADPH oxidase activation translates into neural injury and demise are not fully understood. The primary function of NADPH oxidase is transfer of electrons across membranes (for review, see Bedard and Krause, 2007), and in neurons this enzyme may serve critical functions in signaling or plasticity (Tejada-Simon et al., 2005). However, the present findings of LTIH-induced NADPH oxidase subunit translocation to mitochondria, in association with mitochondrial swelling and disruption of endoplasmic reticulum, support the concept that NADPH oxidase activation in hypoxia/reoxygenation may extend beyond signaling to injury of mitochondria and endoplasmic reticulum. Both of these injuries are expected to further augment the oxidative burden. Having identified an important role for NADPH oxidase activation in the selective vulnerability of wake-active neurons, it will now be important to examine whether NADPH oxidase contributes to hypoxia/reoxygenation injury in other brain regions. This enzyme has been identified in subpopulations of neurons in many of the regions most susceptible to hypoxia/reoxygenation injury: cortex, striatum, amygdala, hippocampus, and thalamus (Serrano et al., 2003). Apocynin, the NADPH oxidase inhibitor used in the present studies to prevent injury to wake-active groups, has been shown to protect against hippocampal ischemic injury in rodents (Wang et al., 2006). Whether NADPH oxidase inhibition will prevent hippocampal injury and memory impairments should now be examined.
Although the present study demonstrates the importance of NADPH oxidase in the selective vulnerability of the catecholaminergic neurons to hypoxia/reoxygenation, the findings neither identify the mechanism by which LTIH activates NADPH oxidase nor provide information on the relative role of NADPH oxidase activation in LTIH injury to wake neurons. The LTIH model may result in independent injuries from hypoxia and reoxygenation, where NADPH oxidase may play a unique role in reoxygenation injury (Abramov et al., 2007). In the LTIH model, NADPH oxidase inhibition at doses sufficient to reduce carbonylation and a proinflammatory response, did not confer complete protection from vacuolization and cleaved caspase-3 immunoreactivity in catecholaminergic neurons. Thus, we anticipate the identification of NADPH oxidase independent mechanisms of injury to catecholaminergic neurons in both hypoxia/reoxygenation and sleep apnea. Although brief, the repeated hypoxia events are likely to burden the mitochondria. This is supported by mitochondrial swelling after brief (2 weeks) LTIH. Mitochondrial injury is expected to result in substantial production of reactive oxygen species (Sims and Anderson, 2002; Solenski et al., 2002; Schild and Reiser, 2005), where NADPH oxidase activation by LTIH would augment production of superoxide and minimize any protective reducing capacity. Thus, neuronal NADPH oxidase activation is expected to accentuate hypoxia/reoxygenation injury to neurons.
The present work has identified a significant vulnerability in catecholaminergic wake neurons to hypoxia/reoxygenation, modeling sleep apnea oxygenation. The injury includes neurodegeneration without functional compensation. This work also identifies an important source of oxidative injury in these neurons, NADPH oxidase, and suggests that hypoxia/reoxygenation results in early mitochondrial and endoplasmic reticulum perturbations. The work discovers unique groups of wake neurons most susceptible to hypoxia/reoxygenation and presents a contributing mechanism. Collectively, the findings support the potential for severe neural injury from long-term exposures to hypoxia/reoxygenation, as in obstructive sleep apnea.
Footnotes
This work was supported in part by National Institutes of Health Grants HL080492 and HL079555.
References
- Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Chiang C, Alexinsky T. Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog Brain Res. 1991;88:501–520. doi: 10.1016/s0079-6123(08)63830-3. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat Neurosci. 2001;4:732–738. doi: 10.1038/89522. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Zhu Y, Card JP. Numerous GABAergic afferents to locus ceruleus in the pericerulear dendritic zone: possible interneuronal pool. J Neurosci. 2004;24:2313–2321. doi: 10.1523/JNEUROSCI.5339-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bedard MA, Montplaisir J, Richer F, Malo J. Nocturnal hypoxemia as a determinant of vigilance impairment in sleep apnea syndrome. Chest. 1991;100:367–370. doi: 10.1378/chest.100.2.367. [DOI] [PubMed] [Google Scholar]
- Benington JH, Kodali SK, Heller HC. Scoring transitions to REM sleep in rats based on the EEG phenomena of pre-REM sleep: an improved analysis of sleep structure. Sleep. 1994;17:28–36. doi: 10.1093/sleep/17.1.28. [DOI] [PubMed] [Google Scholar]
- Chan E, Steenland HW, Liu H, Horner RL. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med. 2006;174:1264–1273. doi: 10.1164/rccm.200605-597OC. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Tononi G. Locus ceruleus control of state-dependent gene expression. J Neurosci. 2004;24:5410–5419. doi: 10.1523/JNEUROSCI.0949-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Pompeiano M, Tononi G. Neuronal gene expression in the waking state: a role for the locus coeruleus. Science. 1996;274:1211–1215. doi: 10.1126/science.274.5290.1211. [DOI] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;15(326):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker MJ, Hue GE, Caudle WM, Miller GW, Keating GL, Rye DB. Episodic neonatal hypoxia evokes executive dysfunction and regionally specific alterations in markers of dopamine signaling. Neuroscience. 2003;117:417–425. doi: 10.1016/s0306-4522(02)00805-9. [DOI] [PubMed] [Google Scholar]
- Decker MJ, Jones KA, Solomon IG, Keating GL, Rye DB. Reduced extracellular dopamine and increased responsiveness to novelty: neurochemical and behavioral sequelae of intermittent hypoxia. Sleep. 2005;28:169–176. doi: 10.1093/sleep/28.2.169. [DOI] [PubMed] [Google Scholar]
- Engleman HM, Martin SE, Deary IJ, Douglas NJ. Effect of continuous positive airway pressure treatment on daytime function in sleep apnoea/hypopnoea syndrome. Lancet. 1994;343:572–575. doi: 10.1016/s0140-6736(94)91522-9. [DOI] [PubMed] [Google Scholar]
- Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–1662. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenik VB, Davies RO, Kubin L. Noradrenergic, serotonergic and GABAergic antagonists injected together into the XII nucleus abolish the REM sleep-like depression of hypoglossal motoneuronal activity. J Sleep Res. 2005;14:419–429. doi: 10.1111/j.1365-2869.2005.00461.x. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Goldbart A, Row BW, Kheirandish L, Schurr A, Gozal E, Guo SZ, Payne RS, Cheng Z, Brittian KR, Gozal D. Intermittent hypoxic exposure during light phase induces changes in cAMP response element binding protein activity in the rat CA1 hippocampal region: water maze performance correlates. Neuroscience. 2003;122:585–590. doi: 10.1016/j.neuroscience.2003.08.054. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco MA, Lu J, Wagner D, Shiromani PJ. c-Fos expression in the cholinergic basal forebrain after enforced wakefulness and recovery sleep. NeuroReport. 2000;11:437–440. doi: 10.1097/00001756-200002280-00002. [DOI] [PubMed] [Google Scholar]
- Haberstock-Debic H, Wein M, Barrot M, Colago EE, Rahman Z, Neve RL, Pickel VM, Nestler EJ, von Zastrow M, Svingos AL. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J Neurosci. 2003;23:4324–4332. doi: 10.1523/JNEUROSCI.23-10-04324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci. 2006;26:1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Hara Y, Anrather J, Speth RC, Iadecola C, Pickel VM. Angiotensin II subtype 1A (AT1A) receptors in the rat sensory vagal complex: subcellular localization and association with endogenous angiotensin. Neuroscience. 2003;122:21–36. doi: 10.1016/s0306-4522(03)00606-7. [DOI] [PubMed] [Google Scholar]
- Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–1072. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinin S, Gavrilyuk V, Polak PE, Heneka MT, Feinstein DL. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28:1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirandish L, Gozal D, Pequignot JM, Pequignot J, Row BW. Intermittent hypoxia during development induces long-term alterations in spatial working memory, monoamines, and dendritic branching in rat frontal cortex. Pediatr Res. 2005;58:594–599. doi: 10.1203/01.pdr.0000176915.19287.e2. [DOI] [PubMed] [Google Scholar]
- Kingshott RN, Vennelle M, Hoy CJ, Engleman HM, Deary IJ, Douglas NJ. Predictors of improvements in daytime function outcomes with CPAP therapy. Am J Respir Crit Care Med. 2000;161:866–871. doi: 10.1164/ajrccm.161.3.9905053. [DOI] [PubMed] [Google Scholar]
- Ko EM, Estabrooke IV, McCarthy M, Scammell TE. Wake-related activity of tuberomammillary neurons in rats. Brain Res. 2003;992:220–226. doi: 10.1016/j.brainres.2003.08.044. [DOI] [PubMed] [Google Scholar]
- Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NADPH oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- Lai YY, Kodama T, Siegel JM. Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J Neurosci. 2001;21:7384–7391. doi: 10.1523/JNEUROSCI.21-18-07384.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux L, Sastre JP, Buda C, Luppi PH, Jouvet M. Alterations in c-fos expression after different experimental procedures of sleep deprivation in the cat. Brain Res. 1996;735:108–118. doi: 10.1016/0006-8993(96)00599-9. [DOI] [PubMed] [Google Scholar]
- Li RC, Row BW, Kheirandish L, Brittian KR, Gozal E, Guo SZ, Sachleben LR, Jr, Gozal D. Nitric oxide synthase and intermittent hypoxia-induced spatial learning deficits in the rat. Neurobiol Dis. 2004;17:44–53. doi: 10.1016/j.nbd.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of wake–active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey PM, Henderson LA, Macey KE, Alger JR, Frysinger RC, Woo MA, Harper RK, Yan-Go FL, Harper RM. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;166:1382–1387. doi: 10.1164/rccm.200201-050OC. [DOI] [PubMed] [Google Scholar]
- Marshall NS, Barnes M, Travier N, Campbell AJ, Pierce RJ, McEvoy RD, Neill AM, Gander PH. Continuous positive airway pressure reduces daytime sleepiness in mild to moderate obstructive sleep apnoea: a meta-analysis. Thorax. 2006;61:430–434. doi: 10.1136/thx.2005.050583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurice JC, Paquereau J, Neau JP, Caron F, Dore P, Ingrand P, Patte F. Long-term evolution of daytime somnolence in patients with sleep apnea/hypopnea syndrome treated by continuous positive airway pressure. Sleep. 1997;20:1162–1166. [PubMed] [Google Scholar]
- Morrell MJ, McRobbie DW, Quest RA, Cummin AR, Ghiassi R, Corfield DR. Changes in brain morphology associated with obstructive sleep apnea. Sleep Med. 2003;4:451–454. doi: 10.1016/s1389-9457(03)00159-x. [DOI] [PubMed] [Google Scholar]
- Niquet J, Baldwin RA, Allen SG, Fujikawa DG, Wasterlain CG. Hypoxic neuronal necrosis: protein synthesis-independent activation of a cell death program. Proc Natl Acad Sci USA. 2003;100:2825–2830. doi: 10.1073/pnas.0530113100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep–wake control. J Neurosci. 2002;22:7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SR, White DP, Malhotra A, Stanchina ML, Ayas NT. Continuous positive airway pressure therapy for treating sleepiness in a diverse population with obstructive sleep apnea: results of a meta-analysis. Arch Int Med. 2003;163:565–571. doi: 10.1001/archinte.163.5.565. [DOI] [PubMed] [Google Scholar]
- Pompeiano M, Cirelli C, Arrighi P, Tononi G. c-Fos expression during wakefulness and sleep. Neurophysiol Clin. 1995;25:329–341. doi: 10.1016/0987-7053(96)84906-9. [DOI] [PubMed] [Google Scholar]
- Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O'Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson's disease. Antioxid Redox Signal. 2005;7:685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- Rami A. Ischemic neuronal death in the rat hippocampus: the calpain-calpastatin-caspase hypothesis. Neurobiol Dis. 2003;13:75–88. doi: 10.1016/s0969-9961(03)00018-4. [DOI] [PubMed] [Google Scholar]
- Row BW, Kheirandish L, Li RC, Guo SZ, Brittian KR, Hardy M, Bazan NG, Gozal D. Platelet-activating factor receptor-deficient mice are protected from experimental sleep apnea-induced learning deficits. J Neurochem. 2004;89:189–196. doi: 10.1111/j.1471-4159.2004.02352.x. [DOI] [PubMed] [Google Scholar]
- Rukhadze I, Kubin L. Differential pontomedullary catecholaminergic projections to hypoglossal motor nucleus and viscerosensory nucleus of the solitary tract. J Chem Neuroanat. 2007;33:23–33. doi: 10.1016/j.jchemneu.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schild L, Reiser G. Oxidative stress is involved in the permeabilization of the inner membrane of brain mitochondria exposed to hypoxia/reoxygenation and low micromolar Ca2+ FEBS J. 2005;272:3593–3601. doi: 10.1111/j.1742-4658.2005.04781.x. [DOI] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988:193–198. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40:511–512. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- Solenski NJ, diPierro CG, Trimmer PA, Kwan AL, Helm GA. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke. 2002;33:816–824. doi: 10.1161/hs0302.104541. [DOI] [PubMed] [Google Scholar]
- Springer JE. Apoptotic cell death following traumatic injury to the central nervous system. J Biochem Mol Biol. 2002;35:94–105. doi: 10.5483/bmbrep.2002.35.1.094. [DOI] [PubMed] [Google Scholar]
- Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with Lewy bodies. J Neurosci. 2006;26:467–478. doi: 10.1523/JNEUROSCI.4265-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiihonen M, Partinen M. Polysomnography and maintenance of wakefulness test as predictors of CPAP effectiveness in obstructive sleep apnea. Electroencephalogr Clin Neurophysiol. 1998;107:383–386. doi: 10.1016/s0013-4694(98)00079-0. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Valladares O, Fenik P, Kapfhamer D, Sanford L, Benington J, Bucan M. An automated system for recording and analysis of sleep in mice. Sleep. 2000;23:1025–1040. [PubMed] [Google Scholar]
- Veasey SC, Davis C, Zhan G, Hsu YJ, Fenik P, Pratico D, Gow AJ. Intermittent hypoxia in mice: protracted hypersomnolence and basal forebrain redox alterations. Sleep. 2004a;27:194–201. doi: 10.1093/sleep/27.2.194. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Hsu Y-J, Thayer P, Fenik P. Murine multiple sleep latency test: phenotyping sleep propensity in mice. Sleep. 2004b;27:388–393. doi: 10.1093/sleep/27.3.388. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Zhan G, Fenik P, Pratico D. Long-term intermittent hypoxia: reduced excitatory hypoglossal nerve output. Am J Respir Crit Care Med. 2004c;170:665–672. doi: 10.1164/rccm.200403-261OC. [DOI] [PubMed] [Google Scholar]
- Wang G, Anrather J, Huang J, Speth RC, Pickel VM, Iadecola C. NADPH oxidase contributes to angiotensin II signaling in the nucleus tractus solitarius. J Neurosci. 2004;24:5516–5524. doi: 10.1523/JNEUROSCI.1176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Anrather J, Glass MJ, Tarsitano MJ, Zhou P, Pickel VM, Iadecola C. Nox2, Ca2+, and protein kinase C play a role in angiotensin II-induced free radical production in the nucleus tractus solitarius. Hypertension. 2006;48:482–489. doi: 10.1161/01.HYP.0000236647.55200.07. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. NEJM. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- Zhan G, Fenik P, Pratico D, Veasey SC. Inducible nitric oxide synthase in long-term intermittent hypoxia: hypersomnolence and brain injury. Am J Respir Crit Care Med. 2005a;171:1414–1420. doi: 10.1164/rccm.200411-1564OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan G, Serrano F, Hsu R, Kong L, Fenik P, Pratico D, Klann E, Veasey SC. NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med. 2005b;172:921–929. doi: 10.1164/rccm.200504-581OC. [DOI] [PMC free article] [PubMed] [Google Scholar]