

Abstract
We generated a mouse line harboring an autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE) mutation: the α4 nicotinic receptor S248F knock-in strain. In this mouse, modest nicotine doses (1–2 mg/kg) elicit a novel behavior termed the dystonic arousal complex (DAC). The DAC includes stereotypical head movements, body jerking, and forelimb dystonia; these behaviors resemble some core features of ADNFLE. A marked Straub tail is an additional component of the DAC. Similar to attacks in ADNFLE, the DAC can be partially suppressed by the sodium channel blocker carbamazepine or by pre-exposure to a very low dose of nicotine (0.1 mg/kg). The DAC is centrally mediated, genetically highly penetrant, and, surprisingly, not associated with overt ictal electrical activity as assessed by (1) epidural or frontal lobe depth-electrode electroencephalography or (2) hippocampal c-fos-regulated gene expression. Heterozygous knock-in mice are partially protected from nicotine-induced seizures. The noncompetitive antagonist mecamylamine does not suppress the DAC, although it suppresses high-dose nicotine-induced wild-type-like seizures. Experiments on agonist-induced 86Rb+ and neurotransmitter efflux from synaptosomes and on α4S248Fβ2 receptors expressed in oocytes confirm that the S248F mutation confers resistance to mecamylamine blockade. Genetic background, gender, and mutant gene expression levels modulate expression of the DAC phenotype in mice. The S248F mouse thus appears to provide a model for the paroxysmal dystonic element of ADNFLE semiology. Our model complements what is seen in other ADNFLE animal models. Together, these mice cover the spectrum of behavioral and electrographic events seen in the human condition.
Keywords: nicotine, epilepsy, ADNFLE, dystonia, mecamylamine, synaptosome
Introduction
Nocturnal frontal lobe epilepsy (NFLE) is characterized by clusters of stereotypic episodes of arousal from sleep associated with dystonic neck, limb, and trunk movements that occur during stages 2–4 of non-rapid eye movement (REM) sleep (Montagna, 1992; Plazzi et al., 1995; Provini et al., 1999; Provini et al., 2000). Less commonly, there are prolonged ballistic limb and trunk movements, resulting in injury (Scheffer et al., 1995; Combi et al., 2004). Autosomal-dominant NFLE (ADNFLE) (Scheffer et al., 1995), a familial subtype of NFLE, was initially associated with a missense (S248F) mutation in the α4 nicotinic acetylcholine receptor subunit (α4-nAChR) gene (Steinlein et al., 1995) at position 6′ in the M2 transmembrane region. Subsequently, in other ADNFLE pedigrees, additional mutations were discovered at the α4 M2–10′ (Steinlein et al., 1997; Hirose et al., 1999), M2–17′ (Steinlein et al., 1997; Hirose et al., 1999), and β2 subunit M2–22′ positions and one at the β2 M3 position that contacts M2 in most structural models (De Fusco et al., 2000; Phillips et al., 2001; Bertrand et al., 2005). A mutation in α2 (Aridon et al., 2006) was identified in a pedigree with atypical nocturnal epilepsy.
Electrophysiological analyses of heterologously expressed ADNFLE alleles suggest that ADNFLE mutant receptors display a gain of function (Bertrand et al., 2002; Rodrigues-Pinguet et al., 2003, 2005). Two mouse strains with extreme gain-of-function mutations in α4 at the M2–9′ position (not linked to ADNFLE) were generated. Adult 9′ knock-in mice do not display spontaneous seizures (Fonck et al., 2003, 2005) but are hypersensitive to many effects of nicotine. The reported L9′A mutant low-dose nicotine-induced “behavioral seizures” are not associated with epileptiform spike-and-wave electroencephalographic (EEG) changes (Fonck et al., 2005); in this respect, the L9′A nicotine-induced behavior resembles most ADNFLE attacks (Combi et al., 2004).
Two ADNFLE knock-in strains have been described: the Chrna4S252F (same as our S248F M2–6′ mutation) and the α4 in-frame insertion of a leucine at M2–17′ (Klaassen et al., 2006). Both lines display spontaneous seizures of two types: brief periods of behavioral arrest and extended attacks of wild running associated with falls. In contrast to typical ADNFLE seizures, which display epileptiform electroencephalograms in only one-third of patients (Combi et al., 2004), these mice display frequent spontaneous seizures associated with high-voltage EEG spikes throughout the day. However, ADNFLE includes a spectrum of clinical manifestations. Although brief dystonic “extrapyramidal” semiology is a signature of ADNFLE (Scheffer et al., 1995; Combi et al., 2004; Bertrand et al., 2005), occasional patients display violent, prolonged hyperkinetic activity (Scheffer et al., 1995). Thus, the Klaassen lines could represent a more severe model of ADNFLE.
Here we report that α4 S248F knock-in mice respond to modest nicotine injections with a novel behavior that closely resembles the dystonic head and limb semiology of ADNFLE attacks (Scheffer et al., 1995; Combi et al., 2004). Consonant with the majority of ADNFLE attacks in humans (Combi et al., 2004), the dystonic paroxysms in S248F mice are not accompanied by overt epileptiform changes.
Materials and Methods
Materials.
[3H]Dopamine (40–60 Ci/mmol), [3H]GABA (35 Ci/mmol), 86Rb+, and [125I]epibatidine were obtained from PerkinElmer (Boston, MA). All reagent-grade salts and the following chemicals were products of Sigma-Aldrich (St. Louis, MO): acetylcholine iodide, ascorbic acid, atropine sulfate, bovine serum albumin, carboxymethoxylamine (aminooxyacetic acid), cytisine, diisophosphofluorophosphate, glucose, NNC-711, nomifensine, pargyline, and tetrodotoxin. HEPES was from Roche Diagnostics (Indianapolis, IN), and sucrose was from Fisher Chemical (Fairlawn, NJ). Mecamylamine used in synaptosome assays was a gift from Merck, Sharp and Dohme Research Lab (Rahway, NJ). Mecamylamine, carbamazepine, 2-hydroxypropyl-β-cyclodextrin, and nicotine used in behavioral studies were obtained from Sigma (St. Louis, MO); nicotine doses stated refer to free base. Chlorisondamine diiodide was obtained from Tocris (Ellisville, MO). Chloral hydrate was obtained from Crown Scientific (Melbourne, Australia). Mecamylamine used in the oocyte study was obtained from Sigma.
Animals.
All procedures involving the use of live animals conformed to the Australian National Health and Medical Research Council code of practice. The guidelines of the National Institutes of Health (NIH) were followed for all animal procedures at the University of Colorado and the California Institute of Technology.
Generation of S248F knock-in mice.
An ∼4000 bp fragment containing exon 5 of the α4-nAChR was excised from the L9S′ targeting construct (Labarca et al., 2001) using an SpeI restriction site and cloned into pBlueScript (Stratagene, La Jolla, CA). Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) to correct the original L9S′ mutation and introduce the S248F mutation. Primers used for mutagenesis reaction were a 40 bp sense 5′-AGGTCACGCTGTGCATCTTTGTGCTGCTTTCTCTCACCGT-3′ and 40 bp antisense 5′-ACGGTGAGAGAAAGCAGCACAAAGATGCACAGCGTGACCT-3′. The ∼4000 bp SpeI fragment containing the S248F mutation was recloned into the targeting construct backbone. The final targeting construct was a 15.2 kb plasmid containing the S248F carrying exon 5 with a LoxP-flanked neomycin phosphotransferase-resistance cassette cloned 163 bp downstream of exon 5. The diphtheria toxin-A chain gene is cloned adjacent to the 5′-flanking homologous limb for negative selection against random insertion. The construct was linearized with NotI and electroporated into a 129/Sv line of embryonic stem (ES) cells (W9.5). Homologous recombinant events were identified by an EcoRI/BamHI digest Southern blot using an ∼1000 bp flanking probe complementary to a genomic DNA sequence lying upstream of the 5′ homologous flank. The wild-type (WT) allele was identified as a 9.7 kb BamHI–BamHI fragment, whereas the recombinant allele was a 7.7 kb BamHI–EcoRI fragment (see Fig. 1A). Recombinant ES clones identified by Southern blotting and confirmed to have the S248F knock-in mutation by direct sequencing were injected into C57BL/6 blastocysts for generation of chimeric mice. Chimeras that were consistently germline transmitting were mated with CD1 background adenovirus EIIa promoter-driven Cre recombinase-expressing homozygous transgenic mice (Lakso et al., 1996) so as to excise the neomycin selection cassette. The presence of the mutation at the RNA level was verified by extracting total-brain RNA, synthesizing cDNA using the First-Strand cDNA synthesis kit (Invitrogen, San Diego, CA) and sequencing amplicons generated by PCR. Heterozygous CD1/129Sv background mice were crossed with each other to generate homozygous mice or were mated with C57BL/6 mice to generate mice on a C57BL/6 genetic background. High-percentage chimeras were also mated with pure 129Sv background WT mice (Howard Florey Institute) to generate neomycin intact heterozygous 129Sv background mice, and these were used in crosses with EIIa/Cre activator lines when chimeras were no longer generating progeny. PCR using sequence-specific primers that were based on the sequence of the original L9S′ plasmid (Labarca et al., 2001) spanning the residual LoxP site were used to identify transgenic progeny. Forward primers with the sequence 5′-GAGGGTACATAGTGTAGTCT-3′ and a backward primer with the sequence 5′-AAACAGCCTGAACACT-TCAA-3′ were used.
Figure 1.

Generation of S248F ADNFLE mouse. A, Gene-targeting strategy. The targeting construct contained exon 5 of the mouse α4-nAChR subunit gene with the S248F mutation and a LoxP-flanked neomycin phosphotransferase cassette located 163 bp downstream of this exon. Homologous recombination results in the insertion of a novel EcoRI site. A flanking probe complementary to DNA downstream of a BamHI site was used to confirm correct targeting of a BamHI–EcoRI genomic digest where a WT allele generates a 9.7 kb BamHI–BamHI band, whereas a targeted allele gives a 7.7 kb BamHI–EcoRI band. DTA, Diphtheria toxin-A chain. B, Random insert ES cell clones contained only the WT 9.7 kb fragments (lanes 1, 2), whereas the correctly targeted ES clones contained the WT 9.7 kb and the targeted 7.7 kb bands (lanes 3, 4). C, High-percentage chimeras containing the S248F mutation with a LoxP-flanked neomycin gene were mated with homozygous adenovirus EIIa/Cre recombinase mice to splice out the neomycin cassette and allow full expression of the targeted α4-nAChR allele. D, The presence of the mutation was confirmed by sequencing analysis of amplicons synthesized from total brain cDNA. WT mice showed the original serine codon (TCG), whereas heterozygous and homozygous mice displayed the introduced phenylalanine codon (TTT) in a gene dose-dependent manner. The result of sequencing of WT, homozygous (HT), and homozygous (HM) brain-derived amplicons.
EEG monitoring of mice.
Mice were anesthetized with chloral hydrate (4.0%, 0.01 ml/g). Four electrodes were fitted onto a plastic cap to occupy the following position on a mouse skull: front right and left, 1.5 mm anterior to bregma and 1.5 mm lateral to the midline; posterior right and left, 3 mm posterior to bregma and 1.5 mm lateral to the midline (Franklin and Paxinos, 1997). The cap and electrodes were secured to the mouse skull using dental cement. The mice were allowed to recover from the procedure for at least 2 weeks before EEG recordings commenced. The potential difference (voltage) between two frontal electrodes was recorded as the anterior EEG channel, whereas the difference between the two posterior electrodes was recorded as the posterior EEG channel. A Molecular Devices Digi-Data programmable signal conditioner and CyberAmp 380 were connected to a Pentium 4 personal computer (PC) to record EEG data. Both the anterior and posterior channels were sampled at 1 kHz with bandpass filtering at 1–80 Hz. Mice were monitored for baseline changes and for evidence of spontaneous electrographic seizures both during extended daytime and overnight as described below. Five minutes of baseline electroencephalograms were recorded before nicotine was administered intraperitoneally. All postinjection behavior was monitored with a camera attached to the PC. Electroencephalogram and video scorers were blinded to the genotype of the mice.
Combined depth and epidural electrode EEG studies.
Simultaneous epidural and depth-electrode video–electroencephalogram recordings were conducted on eight adult female heterozygous CD1/129 genetic background S248F knock-in mice (24–29 g), and each animal was monitored between the hours of 10:00 A.M. and 5:00 P.M. for at least 10 h over a 2 d period. Mice were anesthetized by intraperitoneal injection with a mixture of ketamine (60 mg/kg; Parnell Laboratories, Alexandra, New South Wales, Australia) and xylazine (10 mg/kg; Troy Laboratories, Smithfield, New South Wales, Australia), and their temperature was monitored and maintained using a heated blanket (Frederick Haer Company, Bowdoinham, ME). Mice were placed in a mouse-adapted stereotaxic frame (MyNeuroLab, St. Louis, MO) in a flat skull position. Monopolar electrodes made of insulated silver wire (0.125 mm in diameter) were implanted in the left ventral orbital cortex (VO). The VO is located 2.1 mm anterior to bregma, 0.9 mm lateral to midline, and 2.7 mm dorsoventral as described previously (Franklin and Paxinos, 1997). The VO was chosen for depth-electrode placement because this part of the mouse brain (together with the nearby medial orbital and cingulate cortex) represents a relatively cholinesterase-rich region of the mouse cortex (Franklin and Paxinos, 1997) corresponding to the human homologs of ictal focus (mesial and orbital frontal cortex) as identified by video–electroencephalogram monitoring, interictal positron emission tomography, and focal hyperperfusion on ictal single photon emission computerized tomography studies in ADNFLE (Hayman et al., 1997). The animals were also equipped with monopolar epidural electrodes made of insulated stainless steel wire (0.051 mm in diameter) with insulation removed at the tip, positioned over the right somatosensory cortex (0.9 mm posterior to bregma and 2.5 mm lateral to midline) and the left and right visual cortex (4.5 mm caudal to bregma and 2.5 mm lateral to midline) (Franklin and Paxinos, 1997). Each electrode was connected to an electrode interface board (Qube3, Melbourne, Australia), which was attached to the skull and cemented in place using dental cement. The animals were allowed to recover, and all recordings were made from animals between 3 and 10 days after surgery. EEG activities were recorded with a computer-based digital acquisition system (Chart version 5.4.2; ADInstruments, Sydney, Australia) with concurrent video recording for behavioral analysis (Quicktime Capture Module version 1.2.2). All animals were placed in a glass cage and allowed ad libitum access to food and water. EEG signals were referenced to signals from a ground screw implanted in the bone over the frontal association cortex. EEG recording from each animal was monitored for several hours between 9:00 A.M. and 4:00 P.M. immediately before injection of nicotine. Animals were then given injections of nicotine (2 mg/kg, i.p.) followed by pentylenetetrazole (PTZ) (70 mg/kg, s.c.) to ensure that all electrodes were adequate for recording seizure activity. After completion of the recording experiments, animals were given injections of a lethal dose of Nembutal (100 mg/kg, i.p.) and perfused with 4% paraformaldehyde (PFA). Brains were removed and equilibrated in sucrose, and 50 μm sections were cut on a cryostat. Histological analysis was performed after neutral red staining to verify the location of the VO depth-electrode.
Epidural electrodes only were used in electroencephalograms undertaken on 10th backcrossed C57BL/6 S248F knock-in and WT mice. For these experiments, monopolar epidural electrodes made of insulated stainless steel wire (0.051 mm in diameter) with insulation removed at the tip were positioned over the left and right somatosensory cortex (0.9 mm posterior to bregma and 2.5 mm lateral to midline) and the left and right visual cortex (4.5 mm caudal to bregma and 2.5 mm lateral to midline) (Franklin and Paxinos, 1997).
Behavioral studies.
All drugs used in this study for intraperitoneal injections were made up in 0.9% saline and were injected in a volume ratio of 100 μl/13 g body weight. For the seizure study, drug-naive male and female heterozygous and WT mice between 11–17 weeks of age, from three different genetic backgrounds [mice derived by mating germline-transmitting chimeras with CD1 congenic EIIa/Cre homozygous mice (CD1/129Sv), mice derived from crossing CD1/129Sv heterozygous mice once with C57BL/6 mice (BL6 N1), and mice backcrossed five times on a C57BL/6 background (BL6 N5)], and α4−/− knock-out mice and littermate control mice (both of hybrid 129SvJ/C57BL/6 background) (Ross et al., 2000) were used in these experiments. All experiments were performed between 9:00 A.M. and 1:00 P.M. Each animal received a single intraperitoneal injection of 2, 6, or 8 mg/kg body weight nicotine immediately before being placed in a glass aquarium, and their seizure-like behavior was scored using a modified behavioral scale (Franceschini et al., 2002): 0, no identifiable seizure-like activity as defined by the presence of tremors, wild running, tonic–clonic seizure activity, or death; 1, head and/or body oscillatory tremors; 2, wild running; 3, loss of righting response and clonic seizure; 4, death after a clonic seizure. These behavioral end points were chosen for inclusion in our seizure scale because they were either correlated with unequivocal cortical EEG spike-and-wave activity or were noted to give rise to hippocampal correlates of seizure activity on c-fos-regulated gene expression studies (data not shown). All experiments were videotaped to allow post hoc evaluation. In all cases, experimenters were blind to genotype and drug dose. Fifteen male and 15 female heterozygous mice and the same number of WT mice were used for each of the three doses of nicotine used in each of the three genetic backgrounds tested.
For the mecamylamine experiments, heterozygous BL6 N1 female mice 14–16 weeks of age were given intraperitoneal injections of either 0.5 mg/kg mecamylamine (n = 9) or saline (n = 6). Ten minutes later, mice received 8 mg/kg nicotine intraperitoneally and were placed in a glass aquarium, and their behavior was monitored and recorded.
For the carbamazepine study, BL6 N1 female heterozygous mice (n = 35) were administered carbamazepine intraperitoneally at 0, 40, or 60 mg/kg. Carbamazepine was dissolved in 45% w/v 2-hydroxypropyl-β-cyclodextrin. Twenty minutes after drug administration, locomotion was scored using a simple quadrant entry paradigm. Briefly, the floor of the glass-walled observation arena (36 × 20 × 20 cm) was evenly divided into six quadrants. The number of quadrants the mouse entered with all four limbs over a 2 min period was scored by an experimenter who was blind to drug dose. Immediately after completion of locomotion scoring, the mouse was administered 1 mg/kg nicotine intraperitoneally and returned to the arena for behavioral scoring. The scoring period lasted 5 min. All experiments were videotaped to allow post hoc analysis. Two weeks later, animals were randomly assigned to different doses of carbamazepine, and the experiment repeated using a nicotine challenge (2 mg/kg, i.p.).
Fos–Tau–LacZ study.
Individually housed WT and S248F knock-in mice heterozygous for the S248F mutation and Fos–Tau–LacZ (FTL) transgene [BL6 N1 mice (both male and female)] were given intraperitoneal injections with saline and 2 or 6 mg/kg nicotine. Two hours after drug administration, animals were deeply anesthetized using Lethabarb (0.35 mg/g) and perfused with 50 ml of ice-cold PBS, followed by 50 ml of 2% PFA and 0.005% glutaraldehyde. Whole brains were removed and stored overnight in 30% sucrose in PBS. The following day, 40 μm coronal sections were cut using a cryostat and collected in cryoprotectant. Sections were rinsed in PBS and incubated overnight at 37°C in 5 mm potassium ferricyanide, 5 mm MgCl2, and 0.4 mg/ml X-gal (Wilson et al., 2002). The reaction was stopped by the addition of 0.5% PFA in PBS, and the sections were washed and mounted in 0.5% gelatin. Sections representative of 2.2 mm posterior to bregma (Paxinos and Franklin, 2001) were identified, and images were obtained using Pictureframe software. The number of LacZ-positive neurons in the CA1, dentate gyrus, and pyramidal cell layer of the hippocampus were determined using ImageJ software.
Intracerebroventricular injections.
Female heterozygous CD1/129Sv and BL6 N1 hybrid genetic background mice were anesthetized with 5% chloral hydrate in saline and secured in a stereotaxic apparatus, and the skull was leveled between bregma and lambda. A 23 gauge stainless steel guide cannula was implanted into the lateral ventricle, 0.5 mm posterior to bregma, 1 mm lateral of the midline, and 1.2 mm below the surface of the skull (Franklin and Paxinos, 1997). The cannula was secured to the skull using dental acrylic and plugged with a stainless steel obturator. Mice were allowed 1 week to recover from surgery. A prefilled 30 gauge injector was inserted into the cannula, and the mouse was placed in the test arena. One minute later, 17.5 μg of nicotine (at a concentration of 3.5 μg/μl) was administered over 1 min and behavior was scored. The injector remained in position throughout the experiment. Chlorisondamine is a sympathetic cholinergic ganglion blocker that does not readily cross the blood–brain barrier (Clarke, 1984). Chlorisondamine (1 mg/kg in 0.2 ml) was administered intraperitoneally 30 min before intraperitoneal nicotine (2 mg/kg) challenge. Chlorisondamine (25 nmol/mouse in 5 μl) was then administered by intracerebroventricular injection 30 min before nicotine (2 mg/kg, i.p.) to determine whether the dystonic arousal complex (DAC) was mediated by central nicotinic receptors.
[125I]Epibatidine autoradiography.
The procedure was based on a previous study (Pradhan et al., 2002). Briefly, brain sections from mice were thawed to room temperature and preincubated for 10 min at room temperature in buffer containing (in mm) 50 Tris, 120 NaCl, 5 KCl, 1 MgCl2, and 2.5 CaCl2, pH 7.0. The slides were then incubated for 2 h at room temperature in buffer with 15 pm [125I]epibatidine (2200 Ci/mmol). Nonspecific binding was defined by the addition of 300 μm nicotine bitartrate. Sections were then rinsed in ice-cold buffer (twice for 5 min) and distilled water (2 s). Dried sections were apposed to x-ray film (X-Omat AR; Kodak, Rochester, NY) together with [14C] standards for 12 h before developing. The developed films were subsequently quantified using the Scion (Frederick, MD) Image system (PC version 1.4 of NIH Image), by comparing the optical densities of autoradiographic images with those of corresponding standard microscales. After autoradiographic films were developed, the brain slices were stained with 0.1% thionin, cleared, and coverslipped. Mouse brain nuclei were subsequently defined with reference to a mouse stereotaxic atlas (Franklin and Paxinos, 1997). The density of binding to individual brain nuclei is expressed as dpm/mm2. In every experimental condition, total binding was measured from four consecutive sections per region and nonspecific binding from two sections per region from each individual mouse for each region analyzed. Unless indicated otherwise, data are reported as the mean ± SEM. The statistical program SigmaStat (Jandel Scientific, San Rafael, CA) was used throughout. One-way ANOVA followed by the Dunnett's test or a Kruskal–Wallis ANOVA by ranks followed by the Dunn's test was performed for the comparison of radioligand binding parameters between heterozygous or homozygous mice and WT counterparts. A significance level of p < 0.05 was used throughout.
[125I]Epibatidine binding to membrane fragments.
A previously described method (Whiteaker et al., 2000b) was used to prepare membranes from four brain regions: cortex, hippocampus, striatum, and thalamus from three genotypes (wild type, heterozygous, and homozygous) of S248F knock-in mice. Saturation binding experiments were performed as described previously (Whiteaker et al., 2000b). In separate experiments, inhibition of [125I]epibatidine binding was determined using 200 pm [125I]epibatidine with 0, 50, and 150 nm cytisine. Data were calculated using a two-site inhibition model.
Synaptosome preparation.
Mice were killed by cervical dislocation, brains were removed and placed on an ice-cold platform, and cortex, hippocampus, striatum, and thalamus were dissected. Each region was individually homogenized by hand in ice-cold sucrose (0.32 m) buffered to pH 7.5 with HEPES (5 mm) using a glass-Teflon tissue grinder. The homogenates were divided into appropriate aliquots to enable assaying for several functions from each preparation and centrifuged at 20,000 × g for 20 min. The supernatants were removed, and the P1 pellets were resuspended in the appropriate buffer for uptake of radioactive tracer.
Uptake of radioactive tracers.
Previously described methods were used for 86Rb+ uptake (Marks et al., 1999), for [3H]GABA uptake (Lu et al., 1998), and for tritiated dopamine ([3H]DA) uptake (Salminen et al., 2004). All synaptosomes were treated with diisopropyl fluorophosphate (10 μm) during the last 5 min of the uptake procedure to inactivate acetylcholinesterase.
Neurotransmitter release and 86Rb+ efflux assays.
After uptake of radioactive neurotransmitter tracer, synaptosomes were aliquotted onto filters on a perfusion apparatus and perfused for 10 min with buffer (128 mm NaCl, 2.4 mm KCl, 3.2 mm CaCl2, 1.2 mm KH2PO4, 1.2 mm MgSO4, 25 mm HEPES, pH 7.5, 10 mm glucose, 0.1% bovine serum albumin, and 1 μm atropine) containing the relevant reuptake inhibitor (100 nm NNC-711 for GABA or 1 μm nomifensine for DA). ACh solutions were made fresh daily. For experiments using α-conotoxin MII (αCtxMII) to inhibit the α6*-nAChR, synaptosomes were exposed to αCtxMII (50 nm) for the last 5 min of the 10 min buffer perfusion and immediately preceding the exposure to ACh. The kinetics of recovery from inhibition by αCtxMII is sufficiently slow that this inhibitor was not included with ACh (Salminen et al., 2004). In experiments with mecamylamine, the indicated concentration was added to the αCtxMII-containing buffer, and synaptosomes were perfused for 5 min with both antagonists before exposure to agonist with the same concentration of mecamylamine. For [3H]GABA release, a 16 s exposure to ACh or K+ was used; for [3H]DA, the ACh or K+ exposure was for 20 s. For both assays, perfusion speed was 0.7 ml/min, and 10 s fractions were collected into 96-well plates using a Gilson (Middleton, WI) FC204 fraction collector. Optiphase “Super-Mix” scintillation mixture was added, and radioactivity was determined using a 1450 MicroBeta Trilux scintillation counter (instrument and mixture from PerkinElmer–Wallac, Turku, Finland). Instrument efficiency was 40%.
86Rb+ efflux was determined by perfusion of synaptosomes with buffer (135 mm NaCl, 5 mm CsCl, 1.5 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 25 mm HEPES hemisodium, 20 mm glucose, 50 nm tetrodotoxin, 1 μm atropine, and 0.1% bovine serum albumin, pH 7.5) at 2.5 ml/min (Marks et al., 1999). Efflux was monitored continuously for 3 min per sample by pumping effluent through a Cherenkov cell in a β-RAM Radioactivity HPLC Detector (IN/US Systems, Tampa, FL). A 5 s exposure to agonist was used. For mecamylamine treatment, the indicated concentration was added to the buffer and also included with agonist.
Xenopus oocyte electrophysiology.
Rat WT and mutant α4β2-nAChRs were expressed in Xenopus oocytes as described previously (Rodrigues-Pinguet et al., 2003). Surgical excision of the oocytes from Xenopus was performed according to NIH and California Institute of Technology guidelines. Isolated oocytes were injected with 10 ng of either WT α4 or α4 (S252F, which aligns with mouse S248F) and 15 ng of β2 cRNA. After injection, the oocytes were incubated in a modified Barth's solution containing 96 mm NaCl, 5 mm HEPES, 2.5 mm sodium pyruvate, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 2.5 μg/ml gentamicin (Sigma), and 5% horse serum, pH 7.4 (Irvine Scientific, Santa Ana, CA) for 3–6 d before recording. To measure the ACh responses of the expressed receptors, the oocytes were voltage clamped at −50 mV with 3 m KCl-filled microelectrodes; continually superfused with a nominally Ca2+-free saline containing (mm) 98 NaCl, 1 MgCl2, and 5 HEPES, pH 7.4, at 20–23°C; and bath applied with 2 μm ACh. In the mecamylamine experiments, mecamylamine was applied 2 min before, during, and for 2 min after the second ACh application. The responses were filtered at 20 Hz and digitized at 100 Hz using a PC equipped with pCLAMP 9.2 software (Molecular Devices). One-way ANOVA and the Dunnett's test were used to test for significant differences between the peak responses.
Results
S248F knock-in mice express the mutant allele
A targeting construct used to generate the α4 L9S′ mouse (Labarca et al., 2001) was modified to correct the original L9S′ mutation and introduce the S248F mutation at the 6′ position in the putative M2 region of the α4-nAChR subunit gene. The targeting vector contained the exon 5 portion of the α4-nAChR subunit carrying the S248F (TCG→TTT) mutation. The linearized construct (Fig. 1A) was electroporated into the W9.5 ES cell line, and recombinant clones were identified by Southern blotting (Fig. 1B, lanes 3 and 4). High-percentage agouti coat color germline-transmitting chimeric mice were mated with CD1 background adenovirus EIIa/Cre homozygous transgenic mice (Lakso et al., 1996) to splice out the neomycin phosphotransferase selection cassette, thereby ensuring full expression of the mutated allele in heterozygous mice (Fig. 1C). This breeding strategy ensured that heterozygous and agouti WT control mice had an identical genetic background (i.e., 50% 129/Sv and 50% CD1, and both had a single copy of the EIIa/Cre transgene). Home-cage behavior, weight (data not shown), and fertility were normal in both heterozygous and homozygous mice. Sequencing of whole-brain-derived cDNA amplicons confirmed the presence of the S248F mutation at the mRNA level (Fig. 1D).
The S248F knock-in mutation has minor effects on receptor binding properties
Quantitative ligand autoradiography with [125I]epibatidine [a ligand for α4β2*-nAChRs (Gerzanich et al., 1995; Davila-Garcia et al., 1997)], demonstrated similar binding levels in most brain regions examined in male WT and heterozygous mice. Homozygous mice showed higher than WT binding in a few regions: fasciculus retroflexus, posterior hypothalamus, and dorsolateral geniculate (Fig. 2A).
Figure 2.

[125I]Epibatidine binding in S248F knock-in mice. A, Quantitative autoradiography of [125I]epibatidine binding to mouse brain. WT (n = 10), heterozygous (HT; n = 9), and homozygous (HM; n = 9) mice were assessed, and binding data were plotted as percentage of wild type (mean ± SEM) for comparative purposes. *p < 0.05 vs WT mice; #p < 0.05 vs heterozygous mice. B, [125I]Epibatidine saturation binding to membranes. Binding data (means ± SEM) are shown (n = 4 for each genotype) using cortex and thalamus membranes and eight concentrations of [125I]epibatidine (4–350 pm). Data shown are specific binding (total binding minus nonspecific binding determined by the addition of 100 μm cytisine). Hippocampus and striatum membrane binding had patterns similar to cortex (data not shown). Binding was reduced in the thalamus of HT and HM S248F mice. *p < 0.05, significant differences from wild type. C, Total specific binding of [125I]epibatidine to membranes prepared from four brain regions. Each bar represents specific binding (mean ± SEM) from five mice of each genotype, using 200 pm [125I]epibatidine. No significant differences were found. D, E, Cytisine-sensitive and cytisine-resistant [125I]epibatidine binding. Total specific binding (shown in C) as well as binding in the presence of cytisine (50 and 150 nm) were measured, and the data were fitted to a two-site model to determine the amounts of binding sensitive and resistant to cytisine (Whiteaker et al., 2000b). EPI, [125I]Epibatidine; CPu, caudate putamen; CTX, cortex; fr, fasciculus retroflexus; MHb, medial habenular; PH, posterior hypothalamus; TH, thalamus; Tu, tubercle; SNc, substantia nigra pars compacta; DLG, dorsolateral geniculus; VTA, ventral tegmental area; IPn, interpeduncular nucleus; SC, superior colliculus; ST, striatum.
In a separate experiment, total [125I]epibatidine binding (200 pm) in membrane preparations from four brain regions was assessed (Fig. 2C). No significant differences were detected between genotypes. To further assess nAChR density, saturation binding of [125I]epibatidine was performed in membrane preparations from four regions of WT, heterozygous, and homozygous S248F mice. In three regions [cortex (Fig. 2B), hippocampus, and striatum (data not shown)], there were no differences between genotypes. A small decrease was seen in both heterozygous and homozygous mice compared with wild type in thalamic membranes (Fig. 2B, bottom).
To isolate different populations of nAChR subtypes, cytisine-sensitive and cytisine-resistant [125I]epibatidine binding were determined in membrane preparations from four regions (Fig. 2D,E). Cytisine-sensitive sites represent mainly the α4β2*-nAChRs, whereas cytisine-resistant subtypes include a mixed population of nAChRs containing α3, α6, β2, β3, and β4 subunits (Whiteaker et al., 2000b; Marks et al., 2006). No significant differences were observed between genotypes for either cytisine-sensitive or cytisine-resistant [125I]epibatidine binding. Overall, the binding data indicate that the S248F mutation confers little change in receptor numbers in most regions, although there are rather small increases or decreases in a few regions.
S248F knock-in mice display altered synaptosomal nAChRs function
The function of nAChRs was assessed in synaptosomal preparations using several assays. ACh-stimulated [3H]DA release from striatal synaptosomes (Fig. 3A), 86Rb+ efflux from thalamic synaptosomes (Fig. 3B), and [3H]GABA release from hippocampal synaptosomes (Fig. 3C) were measured from WT, heterozygous, and homozygous mice. Dopamine release is mediated by two classes of nAChRs (Salminen et al., 2004): those resistant to inhibition by αCtxMII (Fig. 3A, top), which are α4β2*-nAChRs, and those sensitive to αCtxMII inhibition (Fig. 3A, bottom), which are α6β2*-nAChR subtypes. The results show that dopamine release mediated by the αCtxMII-resistant subtypes is affected by the S248F mutation, whereas release mediated by the αCtxMII-sensitive subtypes does not differ with genotype. 86Rb+ efflux is mediated by two populations of nAChRs, a low- and a high-affinity form, both thought to be α4β2* subtypes (Marks et al., 2007). In addition, [3H]GABA release appears to be mediated by α4β2*-nAChRs (Whiteaker et al., 2000a).
Figure 3.
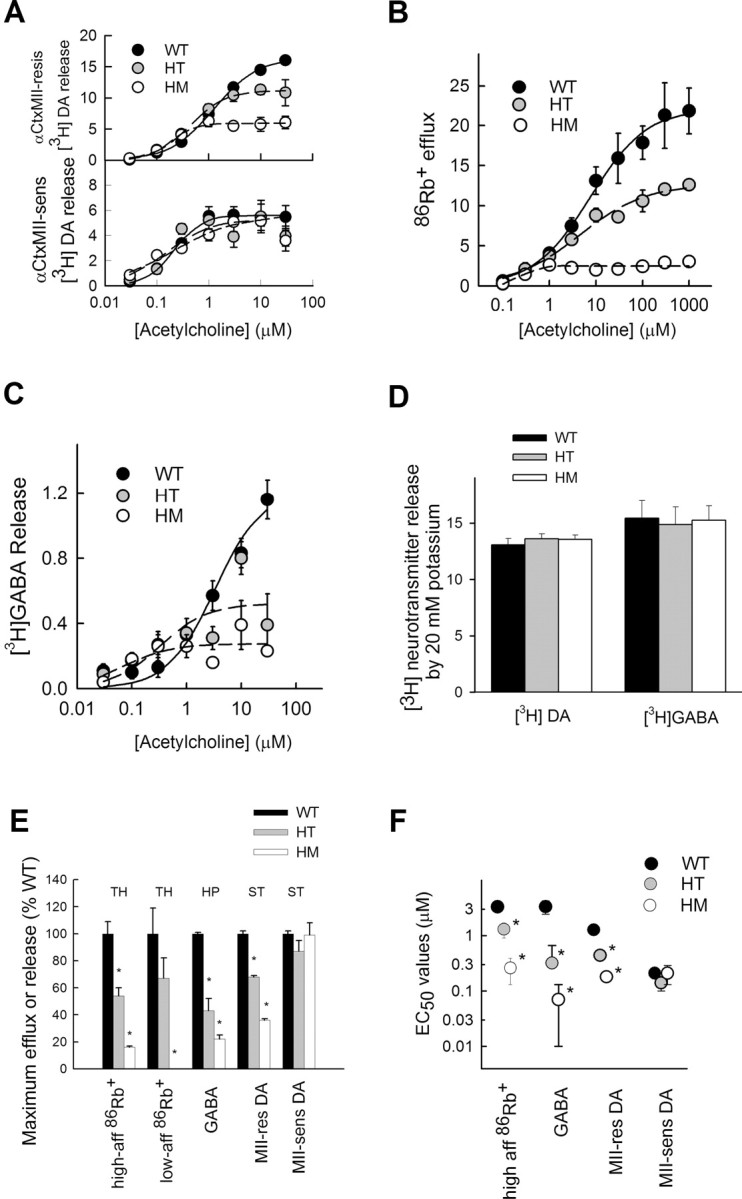
Synaptosomal function in S248F knock-in mice. A, [3H]DA release from striatal synaptosomes. Release was stimulated by a 20 s exposure to the indicated concentrations of ACh. Data shown are mean ± SEM for five mice of each genotype. In the top graph, synaptosomes were exposed to αCtxMII (50 nm) for 5 min before ACh exposure to determine release mediated by nAChR subtypes resistant to that treatment. In the bottom graph, the amount of release resistant to αCtxMII was subtracted from release by ACh without exposure to αCtxMII. This difference represents release mediated by nAChR subtypes sensitive to αCtxMII. Release is in units (counts per minute normalized to baseline counts per minute). B, 86Rb+ efflux from thalamic synaptosomes. Efflux of 86Rb+ was stimulated by a 5 s exposure to ACh. Data shown are means ± SEM for five mice of each genotype. Efflux from cortical synaptosomes showed the same pattern (data not shown). C, [3H]GABA release from hippocampal synaptosomes. [3H]GABA was stimulated by a 16 s exposure to the indicated concentrations of ACh. Data shown are means ± SEM for five mice of each genotype. Release from cortical synaptosomes showed the same pattern (data not shown). D, Comparison of neurotransmitter release stimulated by 20 mm K+. Data shown are means ± SEM for five mice of each genotype. E, Comparison of maximal effect of ACh for various measures of synaptosomal function. Data were compared by one-way ANOVA with Duncan's post hoc test. Significant differences from wild type are shown (*p < 0.05). F, Comparison of EC50 values for ACh for various assays of synaptosomal function. Data were compared by one-way ANOVA with Duncan's post hoc test. Significant differences from wild type are shown (*p < 0.05). HT, Heterozygous; HM, homozygous; TH, thalamus; HP, hippocampus; ST, striatum.
The patterns of change are similar for all three functions mediated by α4β2*-nAChRs, including αCtxMII-resistant dopamine release, high- and low-affinity 86Rb+ efflux, and [3H]GABA release (Fig. 3A–C). At low agonist concentrations, the S248F knock-in mice have nearly normal nAChR function, and at high agonist concentrations, the S248F mice have decreased responses. Figure 3, E and F, present dose–response parameters abstracted for those measures mediated by the α4β2* subtypes of nAChRs functions. Figure 3E shows that the S248F mutation results in significantly decreased α4β2* maximal function. This mutation results in a significantly higher-affinity nAChR as seen in the shifts in EC50 values for ACh (Fig. 3F) for both the heterozygous and homozygous S248F knock-in mice. The function of the αCtxMII-sensitive subtypes, assessed both by maximal activity and by EC50 value, are unchanged by this mutation. The loss of function seen with high agonist concentrations is similar to what has been reported previously (Kuryatov et al., 1997, 2005) and was postulated to result from a change in stoichiometry of subunit assembly (Kuryatov et al., 2005).
S248F knock-in mice do not have spontaneous seizures
In an initial set of experiments, EEG recordings with video monitoring of mutant mice [heterozygous (n = 8) and homozygous (n = 5)] and WT littermates (n = 3) undertaken over a 12–16 h overnight period and for periods of 4–6 h during the daylight hours were normal. In particular, mutant mice displayed no behavior suggestive of spontaneous seizures and no electrographic abnormalities (data not shown). The same EEG paradigm has been shown to detect behavior and electrographic signs of PTZ-induced seizures (McColl et al., 2003) and spontaneous seizures in another mutant line (Gantois et al., 2007). Initial studies on S248F mice were performed on mice of two different genetic backgrounds including the CD1/129Sv (n = 5 heterozygous, n = 5 homozygous, and n = 3 WT littermates) and CD1/129Sv backcrossed four generations (N4) into the C57BL/6 strain (called BL6 N4; n = 3 heterozygous). In a second set of experiments, epidural electroencephalograms (using the second epidural recording paradigm described above) with video monitoring were recorded for 10 h during daylight hours from a total of 11 mice backcrossed 10 generations on a C57BL/6 genetic background (BL6 N10; two WT and eight heterozygous mice and one homozygous mouse). Electrographic spike-and-slow wave seizures were never seen.
In summary, a total of 22 S248F knock-in mice were monitored with epidural electrodes, and no epileptiform spike-and-wave events were detected. Spontaneous behavioral seizures were never seen in this cohort or in a separate cohort of 40 heterozygous and 21 homozygous mice on a BL6 N10 background observed during routine laboratory handling. Furthermore, spontaneous motor seizures were never observed during the day-to-day handling of 160 homozygous and >1000 heterozygous mice observed over a 2 year period by staff trained in the recognition of mouse seizures. In conclusion, S248F mutant mice do not appear to have behavioral or electrographic spontaneous seizures.
S248F knock-in mice display the novel nicotine-induced DAC
When we injected heterozygous mice of the CD1/129Sv background with 2 mg/kg nicotine, we noted three distinct stereotypic behaviors collectively termed the DAC (see S248F DAC1 video, available at www.jneurosci.org as supplemental material). The first component, termed saccadic behavior, involves aroused, jerky, exploration-like darting of the head in random directions accompanied by whole-body movements in the same direction. This continues for 9 ± 1 s (mean ± SEM; n = 24) and is then followed by the second stereotypical phase, termed the forelimb dystonia phase. In this phase, the trunk is extended forward, as if the mouse is stretching: natural, upright supported posture is lost, and the animal becomes prone (Fig. 4A). In extreme cases, extended and supinated forelimbs cross the midline in a dystonic posture (Fig. 4B). The arms supinate and forelimbs flex at the elbows, wrists, and digits, so that eventually the paws clasp together below the chin (Fig. 4B). Although the second phase occurs in males and females, forelimb crossing and digital clasping is more common and prominent in female mice. In the third component of the DAC, the tail occupies the Straub configuration, bent 180° from its natural position (Fig. 4A). The Straub tail begins simultaneously with trunk extension and forelimb dystonia. Occasionally, prominent flexion–extension head and body jerking is seen as the tail collapses. The three phases of the DAC last 36 ± 3 s (mean ± SEM), at which normal upright posture and natural exploratory behavior is re-established. Balance is preserved during the DAC. This can be appreciated by examining the S248F DAC2 video (available at www.jneurosci.org as supplemental material) in which the mouse manages to remain upright and not fall off the platform despite its hindquarters hanging over the edge and a hand-clap noise startle. Both DAC videos provided show digital spreading, which also accompanies the forelimb dystonic posturing. Please note that upper-limb dystonic posturing with digital spreading was reported by Scheffer et al. (1995).
Figure 4.

DAC. A, A heterozygous mutant mouse on a CD1/129Sv genetic background that received an injection of 2 mg/kg nicotine displays the 180° Straub tail (black arrow). B, An example of marked forelimb dystonia, part of the DAC, in a S248F knock-in mouse that received an injection of 2 mg/kg nicotine shows midline crossing of the forelimbs culminating in the clasping of digits (black arrow). C, A WT mouse that received an injection of 2 mg/kg nicotine displays no behaviors attributable to the DAC. An incomplete Straub tail, as exemplified by this mouse, is rarely beyond 90° to the horizontal plane, and the mouse shows normal upright forelimb posturing.
A system of scoring the Straub tail and forelimb dystonia components of the DAC was developed to quantify the magnitude of the DAC response. Mice were scored from 0 to 4 for Straub tail depending on the angle that the long axis of the tail makes with the horizontal plane (0, no Straub tail observed; 1, a rigid tail between 0 and 45°; 2, tail raised between 45 and 90°; 3, tail raised between 90 and 135°; 4, tail raised beyond 135°) and from 0 to 3 for the degree of forelimb dystonia [0, no dystonia observed; 1, isolated forward stretching of the forelimbs; 2, stretching plus supination (rotation of the forelimbs along a long axis in a direction away from the midline); 3, midline crossing or folding of stretched and supinated forelimbs].
The DAC is a centrally mediated phenotype modified by gender, genetic background, and S248F expression levels
The DAC scoring system was applied to quantify the response of heterozygous mice (n = 23 males and n = 23 females on the 129/CD1 background) that received injections of 2 mg/kg nicotine intraperitoneally. One hundred percent of female heterozygous mice displayed all three component behaviors of the DAC (saccadic behavior, Straub tail score of ≥3, and forelimb dystonia) compared with only 75% of heterozygous males (z test, p = 0.033). Compared with males, female heterozygous mice achieved significantly higher scores for Straub tail (females, 3.7 ± 0.1; males, 3.0 ± 0.2; Mann–Whitney rank sum test, p = 0.004) and forelimb dystonia (females, 2.2 ± 0.1; males, 1.5 ± 0.1; Mann–Whitney rank sum test, p = 0.002). Saccadic behavior and forelimb dystonia was never seen in WT CD1/129Sv background mice (n = 23 females and n = 24 males), but WT mice did display a low-grade Straub tail. A Straub tail was seen in 96% of WT females compared with only 64% of males (z test, p = 0.019). Female WT mice scored significantly higher than males for Straub tail (Mann–Whitney rank sum test, p < 0.001). Overall, the WT Straub tail response was less than for the Straub tail scored as part of the DAC response in heterozygous mice (average score: male WT mice, 0.8 ± 0.1 vs male heterozygous mice, 3.0 ± 1.8; p < 0.001; female WT mice, 1.6 ± 1.2 vs female heterozygous mice, 3.7 ± 0.1; Mann–Whitney rank sum test, p < 0.001). Hence, a Straub tail score of ≥3 was considered the threshold for defining a DAC-like Straub tail. The same experiment was undertaken in heterozygous mice on a (BL6 N1) background (n = 36 males and n = 36 females) given injections of 2 mg/kg nicotine intraperitoneally. Ninety-four percent of female heterozygous mice displayed all three component behaviors of the DAC (saccadic behavior, Straub tail of ≥3, and forelimb dystonia) compared with only 89% of heterozygous males. Female heterozygous mice achieved significantly higher scores for forelimb dystonia (females, 2.5 ± 0.1; males, 1.5 ± 0.1; Mann–Whitney rank sum test, p < 0.01) on this background although there was no difference observed in scores for Straub tail in this background. These data suggest that the DAC has a greater penetrance in females. For this reason, experiments undertaken to investigate the DAC were mostly undertaken on female mice.
The latency to the DAC was surprisingly brief, 24 ± 3 s (mean ± SEM), raising the possibility that it was triggered by activation of peripheral rather than central nAChR receptors. The DAC could be reproduced by direct intracerebroventricular injection of nicotine (17.5 μg; n = 8). Moreover, the DAC was completely abolished by previous intracerebroventricular (25 nmol/mouse in 5 μl; n = 7), but not peripheral (1 mg/kg in 0.2 ml; n = 2), administration of chlorisondamine, a nicotinic receptor antagonist that does not readily cross the blood–brain barrier (Clarke, 1984).
Although the DAC was consistently observed in mice with various genetic backgrounds including CD1/129Sv and BL6 N1, the phenotype was subtly modified in BL6 N1 mice and substantially modified in mutant mice backcrossed five generations into the C57BL/6 background (BL6 N5). BL6 N1 mice typically continued to roam after onset of the DAC. In BL6 N5 background heterozygous mutant mice, the Straub tail was still present to the same degree, but the forelimbs were rarely totally out-stretched, supinated, or crossed with associated digital clasping; rather, mice repeatedly dragged their forelimbs under their body and across the midline as they walked. In the BL6 N5 background, saccadic behavior was markedly reduced [percentage of mice displaying saccadic behavior after 2 mg/kg nicotine: CD1/129Sv, 87% (n = 30); BL6 N1, 100% (n = 30); BL6 N5, 33% (n = 30); BL6 N5 mice are significantly different from other groups; Kruskal–Wallis test followed by post hoc Tukey's test, p < 0.05], but mice moved forward in jerk-like steps, and forelimb dystonia was subtle and manifest as forelimb supination with clenched fists and rhythmic forelimb retraction and extension during accelerated jerky ambulation. The accentuated Straub tail response was maintained in this genetic background. A 2 mg/kg dose of nicotine often induced body tremors in the BL6 N5 mice, although this also occurred in mice with WT α4 receptors (Table 1). The attenuated DAC in heterozygous BL6 N5 mice (loss of saccadic behavior and modification of dystonia) seemed to result from response competition related to locomotor hyperactivity and body tremors.
Table 1.
Nicotine-induced seizure dose responses
| Background | Genotype | Nicotine | Score 0 | Score 1 | Score 2 | Score 3 | Score 4 | Score 3 + 4 |
|---|---|---|---|---|---|---|---|---|
| CD1/129Sv | WT | 2 | 83.33 (25) | 13.33 (4) | 3.33 (1) | 0.00 (0) | 0.00 (0) | 0 (0) |
| 6 | 3.33 (1) | 16.67 (5) | 33.33 (10) | 43.33 (13) | 3.33 (1) | 47 (14) | ||
| 8 | 0.00 (0) | 3.33 (1) | 13.33 (4) | 50.00 (15) | 33.33 (10) | 83 (25) | ||
| Heterozygous | 2 | 96.67 (29) | 0.00 (0) | 3.33 (1) | 0.00 (0) | 0.00 (0) | 0 (0) | |
| 6 | 16.67 (5) | 3.33 (1) | 33.33 (10) | 40.00 (12) | 6.67 (2) | 47 (14) | ||
| 8 | 3.33 (1) | 6.67 (2) | 30.00 (9) | 46.67 (14) | 13.33 (4) | 60 (18) | ||
| BL6 N1 | WT | 2 | 83.33 (25) | 16.67 (5) | 0.00 (0) | 0.00 (0) | 0.00 (0) | 0 (0) |
| 6 | 0.00 (0) | 6.67 (2) | 33.33 (10) | 40.00 (12) | 20.00 (6) | 60 (18) | ||
| 8 | 6.67 (2) | 6.67 (2) | 13.33 (4) | 20.00 (6) | 53.33 (16) | 73 (22) | ||
| Heterozygous | 2 | 96.67 (29) | 3.33 (1) | 0.00 (0) | 0.00 (0) | 0.00 (0) | 0 (0) | |
| 6 | 20.00 (6) | 13.33 (4) | 23.33 (7) | 16.67 (5) | 26.67 (8) | 43 (13) | ||
| 8 | 10.00 (3) | 0.00 (0) | 16.67 (5) | 30.00 (9) | 43.33 (13) | 73 (22) | ||
| BL6 N5 | WT | 2 | 53.33 (16) | 40.00 (12) | 0.00 (0) | 6.67 (2) | 0.00 (0) | 6 (20) |
| 6 | 0.00 (0) | 0.00 (0) | 0.00 (0) | 46.67 (14) | 53.33 (16) | 100 (30) | ||
| 8 | 0.00 (0) | 0.00 (0) | 0.00 (0) | 36.67 (11) | 63.33 (19) | 100 (30) | ||
| Heterozygous | 2 | 66.67 (20) | 26.67 (8) | 3.33 (1) | 3.33 (1) | 0.00 (0) | 3 (1) | |
| 6 | 10.00 (3) | 13.33 (4) | 0.00 (0) | 56.67 (17) | 20.00 (6) | 76 (23) | ||
| 8 | 0.00 (0) | 0.00 (0) | 0.00 (0) | 53.33 (16) | 46.67 (14) | 100 (30) | ||
| 129SvJ/C57/B6 | WT | 2 | 93.33 (28) | 6.67 (2) | 0.00 (0) | 0.00 (0) | 0.00 (0) | 0 (0) |
| 6 | 20.00 (6) | 40.00 (12) | 10.00 (3) | 23.33 (7) | 6.67 (2) | 30 (9) | ||
| 8 | 6.67 (2) | 16.67 (5) | 3.33 (1) | 40.00 (12) | 33.33 (10) | 73 (22) | ||
| Homozygous | 2 | 96.67 (29) | 3.33 (1) | 0.00 (0) | 0.00 (0) | 0.00 (0) | 0 (0) | |
| 6 | 23.33 (7) | 6.67 (2) | 16.67 (5) | 33.33 (10) | 20.00 (6) | 53 (16) | ||
| 8 | 13.33 (4) | 0.00 (0) | 3.33 (1) | 40.00 (12) | 43.33 (13) | 83 (25) |
A nicotine seizure dose–response relationship was determined for WT and heterozygous mice in several genetic backgrounds (CD1/129Sv, BL6 N1, and BL6 N5). Mice were scored for body tremors (score 1), wild running (score 2), clonic seizures (score 3), and death associated with clonic seizures (score 4). Thirty mice were used for each dose of nicotine tested for each genotype and genetic background. The highest score achieved was recorded for each mouse. The data represent the percentage of mice obtaining a specific score and the number of 30 (in parentheses) for each test condition. The seizure score was analyzed using ordinal logistic regression as described. After testing for interactions, a main-effects model was fitted. This showed that the main or average effect of genotype was highly statistically significant (p < 0.001); WT mice had a worse seizure profile than heterozygous mice in all genetic backgrounds. The effect of nicotine dose was also statistically significant, with higher doses leading to worse outcomes (p < 0.001). There was also a statistically significant difference of background overall (p < 0.001), with CD1/129Sv being the least affected and C57B6 N5 being the most severely affected. In contrast, there was no significant difference between the nicotine seizure profile of homozygous α4 knock-out and littermate control WT mice.
Neomycin cassette intact CD1/129Sv background heterozygous mice were also available for testing. These mice were generated from matings between neomycin intact pure 129Sv background heterozygous knock-in mice and homozygous adenovirus EIIa/Cre mice in which recombination failed entirely [a phenomenon seen previously with this line of Cre activator mice (Drago et al., 1998)]. In this experiment, mice were tested only once. Twenty-eight of 29 mice examined displayed saccadic behavior and/or Straub tail but not forelimb dystonia when given 2 mg/kg nicotine. One of 29 mice displayed forelimb dystonia with this dose of nicotine. This compares with 18 of 30 neomycin cassette-deleted CD1/129Sv mice that displayed forelimb dystonia in this study (z test, p < 0.001). At higher doses of nicotine, the forelimb dystonia reappeared (6 mg/kg, 12 of 29 mice; 8 mg/kg 11 of 30 mice), although the degree of dystonia was less (average dystonia score at 8 mg/kg nicotine: CD1/129Sv, 2.2 ± 0.2; neomycin intact CD1/129Sv, 0.7 ± 0.2; Mann–Whitney rank sum test, p < 0.001). The observation that gene expression is reduced by the presence of the neomycin cassette at this gene locus (Fonck et al., 2003) suggests that the most ephemeral component of the DAC is forelimb dystonia and that the complete phenotype requires full expression of at least one mutant allele.
The DAC was not seen in WT mice of several genetic backgrounds (CD1/129Sv, BL6 N1, 129SvJ/C57BL/6, BL6 N5) at a range of nicotine doses (2, 4, and 8 mg/kg; n = 120 for each nicotine dose amounting to a total of 360 WT mice examined). Other than in the BL6 N5 background, locomotor activity was reduced in WT mice given low doses of nicotine (2 mg/kg). The mice remained upright but were immobile and sat in a resting position (Fig. 4C). The tail elevation displayed by WT mice rarely exceeded 90° above the horizontal plane (Fig. 4C), and the onset was 104 ± 12.8 s (mean ± SEM; n = 90) after nicotine injection. This incomplete Straub tail in WT mice was usually observed with convulsive doses of nicotine (6–10 mg/kg) and was typically followed by a tonic–clonic seizure.
The DAC is not associated with epileptiform changes on epidural or depth-electrode electroencephalogram or with changes in hippocampal c-fos-regulated gene expression
Epidural electroencephalograms recorded in mice with a CD1/129Sv and BL6 N4 genetic background during the DAC were normal (Fig. 5A), in contrast to tonic–clonic seizure events at high nicotine doses that showed prominent paroxysmal ictal activity (Fig. 5B). Mutant mice given injections of convulsive doses of nicotine (6–10 mg/kg) showed, in sequence, the DAC associated with a normal electroencephalogram followed by a clonic seizure associated temporally with classic ictal electrographic spike-and-wave activity. The early onset low-dose nicotine-induced behavioral seizures previously reported in adult L9′A mice (Fonck et al., 2005) were described in both heterozygous and homozygous mice. They were always preceded by a Straub tail, followed by loss of the righting response and ending with forelimb clonus. These behavioral seizures were not associated with spike-and-wave EEG changes. When adult L9′A mice (Fonck et al., 2005) were given injections of high doses of nicotine, two behaviors were observed: early-onset behavioral seizures as described above and late-onset classic tonic–clonic seizures. Only the latter were associated with epileptiform EEG changes. There are therefore clear parallels between what has been previously reported (Fonck et al., 2005) and the EEG-negative DAC described in our study.
Figure 5.

Electroencephalograms. A, Traces from epidural electroencephalograms during nicotine-induced DAC do not differ from normal background electroencephalograms. Ai shows a trace from a male WT mouse (CD1/129Sv genetic background) 21 weeks of age before and after 2 mg/kg nicotine injection. The open arrowhead indicates the 29 s time point after nicotine injection. Aii shows a trace from a male heterozygous (HT) mouse (CD1/129Sv genetic background) 21 weeks of age before and after 2 mg/kg nicotine injection. The onset of the DAC (at 29 s after nicotine injection) is indicated by the arrowhead above the trace, and the offset of the DAC is indicated by the arrowhead below the trace. B, Electroencephalograms during nicotine-induced tonic–clonic seizures display large paroxysmal discharges with an ictal spike/wave configuration. The trace shows an electroencephalogram from a heterozygous female CD1/129Sv genetic background mouse 21 weeks of age that received an injection of nicotine. The paroxysmal spiking activity starts 120 s after injection of 10 mg/kg nicotine. C, D, Recordings from the VO (Ci, Di) and epidural electroencephalograms (Cii, Dii) from a female CD1/129Sv genetic background heterozygous mouse 12 weeks of age after injection of nicotine (2 mg/kg) and PTZ (70 mg/kg). In Ci and Cii, the onset of the DAC is indicated by the arrowhead above the trace, and the offset of the DAC is indicated by the arrowhead below the trace. The period of animal handling and nicotine injection is indicated by the horizontal bar above the beginning of the trace (C), and the period of animal handling and injection of PTZ is indicated by the horizontal bar (D). In this experiment, PTZ was injected 30 min after nicotine injection. E, An expanded section of the recording in D marked by the asterisk. After nicotine injection, no activity was evident in either the VO electrode or the epidural EEG electrodes during the DAC. In contrast, after PTZ injection, spikes were evident on both the VO and epidural traces associated with myoclonic activity.
In additional experiments, depth-electrode and epidural long-term EEG recording with video monitoring was undertaken on eight female CD1/129Sv background adult heterozygous S248F mice after injection of nicotine (2 mg/kg, i.p.) or PTZ (50–70 mg/kg, s.c.). No spontaneous electrographic seizures were observed during a minimum of 10 h of cumulative baseline daytime recording undertaken before nicotine injections in any of the eight heterozygous mice. After injection of nicotine, a typical DAC was observed in all mice. During these events, no abnormal EEG events or changes in neuronal activity in the VO were observed (Fig. 5C). These experiments were then terminated by a positive control manipulation, injection of a convulsive dose of PTZ (Fig. 5D,E). PTZ typically elicited brief myoclonic jerks, progressing to clonic seizures, and a hindlimb extension tonic maximal seizure. There was a corresponding increase in neuronal activity in the depth-electrode and polyspikes in the surface EEG recordings during PTZ-induced myoclonic jerks and clonic seizure (Fig. 5D,E).
Mutant S248F knock-in mice on a BL6 N10 genetic background were also examined using a standard epidural recording paradigm (the second epidural paradigm described in Materials and Methods). Two WT, one homozygous, and six heterozygous mice (all on a BL6 N10 genetic background) were given injections of 2 mg/kg nicotine and monitored by simultaneous video and EEG recording. All knock-in S248F BL6 N10 mice had a modified DAC as seen typically in this genetic background characterized by side-to-side head movements, jerky ambulation and paroxysmal forelimb extension/supination, and a Straub tail response. Epileptiform activity was not observed during EEG monitoring of the DAC.
In summary, we monitored a total of 30 S248F knock-in mice for at least 10 h each and did not detect electrographic seizures. Twenty-eight of these were also given injections of nicotine, and all showed the DAC, but in the absence of signature epileptiform changes on the electroencephalogram. All nicotine injections were undertaken between 9:00 A.M. and 6:00 P.M.
To determine the parts of the brain activated during the DAC, the S248F mutants were bred with c-fos promoter-driven Tau–LacZ reporter mice (Wilson et al., 2002). Mice were given injections of a DAC-inducing dose of nicotine (2 mg/kg) or of a convulsive dose of nicotine (6 mg/kg). A total of 33 mice were used for this study. Background brain LacZ levels were determined by examining six saline-injected heterozygous S248F/FTL mice and six saline-injected FTL single transgenic mice. Four mice heterozygous for both the S248F mutation and the FTL transgene and six FTL single transgenic mice were given injections of 2 mg/kg nicotine. Six mice heterozygous for both the S248F mutation and the FTL transgene and five FTL single positive mice were given injections of 6 mg/kg nicotine. As expected, LacZ staining was prominent in the hippocampal dentate gyrus in all mice displaying tonic–clonic seizures after administration of 6 mg/kg nicotine (three of six heterozygous S248F mice and one in five FTL single transgenic mice), indicating strong activation in this region (Fig. 6C). LacZ staining in the hippocampus (Fig. 6A,B) and other brain regions (data not shown) was at background levels (i.e., the same as saline-injected mice) in FTL single transgenic mice, in all S248F/FTL double transgenic mice displaying only the DAC in response to low-dose nicotine challenge, and in all FTL single and S248F/FTL double-transgenic mice that displayed no clonic seizure or tremors after administration of high-dose nicotine. Increased hippocampal dentate gyrus c-fos-regulated gene expression was also sometimes seen after body tremors alone (two of six S248F heterozygous mice given injections of 6 mg/kg nicotine). In summary, a total of five heterozygous mice had a DAC alone (four after 2 mg/kg and one in response to a 6 mg/kg injection), and yet no change in LacZ expression was detected in the dentate gyrus or elsewhere in the hippocampus.
Figure 6.

c-fos-regulated gene expression in the hippocampus of phenotypic WT and FTL/S248F double-transgenic mice treated with nicotine. A, An FTL transgenic mouse that received an injection of 2 mg/kg nicotine shows no prominent LacZ staining in the hippocampus. An identical picture is seen in FTL/S248F double-transgenic mice that received an injection of saline (not shown). B, C, A FTL/S248F double-transgenic mutant mouse displaying the DAC with 2 mg/kg nicotine shows no LacZ staining in the hippocampus (B), whereas both FTL-positive WT (shown) and FTL/S248F double-transgenic (not shown) mice show prominent LacZ staining in the dendate gyrus of the hippocampus after sustaining a tonic–clonic seizure at a nicotine dose of 6 mg/kg (C). Scale bars, 250 μm.
The cohort of mice used in the study showed significant background cortical LacZ activity. Therefore, unlike the situation in the hippocampus, it was not possible to exclude DAC-associated activation of the neocortex in this experiment. There was no increased signal-to-noise ratio in the basal ganglia in mice that displayed the DAC.
Heterozygous S248F knock-in mice are relatively protected from nicotine-induced seizures
A nicotine seizure dose–response relationship was determined for WT and heterozygous mice in several genetic backgrounds (CD1/129Sv, BL6 N1, and BL6 N5) (Table 1). Mice were scored for body tremors, wild running, clonic seizures, and death associated with clonic seizures. Forelimb dystonia and Straub tail were not scored on the seizure scale because we had no direct evidence from extensive EEG analysis and c-fos gene-regulated activation studies to confirm that these behaviors were seizure phenomena. The seizure score was analyzed using ordinal logistic regression (McCullagh, 1980), with background (CD1/129Sv, BL6 N1, and BL6 N5), genotype (wild type/heterozygous S248F knock-in), and nicotine dose (2, 6, and 8 mg/kg) as three explanatory variables. This model assumes only that the outcome variable, seizure score is ordinal in nature. This analysis showed statistically significant effects of each of these three variables. The data are consistent with the same underlying genotype difference across genetic backgrounds and nicotine doses. The interaction between background and genotype was not statistically significant (p = 0.42). After testing for interactions, a main effects model was fitted. This showed that the main or average effect of genotype was highly statistically significant (p < 0.001); WT mice had more severe seizures than heterozygous mice in all genetic backgrounds. The effect of nicotine dose was also statistically significant in the expected direction (higher doses leading to more severe seizures; p < 0.001). There was also a statistically significant difference of background overall (p < 0.001). CD1/129Sv was the least affected, BL6 N1 was more severe, and BL6 N5 was the most severely affected. The pairwise differences had the following p values: CD1/129Sv versus BL6 N1, p = 0.04; CD1/129Sv versus BL6 N5, p < 0.001; BL6 N1 versus BL6 N5, p < 0.001.
The latency to seizure onset (defined as the point at which the mouse lost its righting reflex) was independent of genotype but did vary significantly with nicotine dose and genetic background, with mice of the BL6 N5 background having more rapid-onset seizures than CD1/129Sv background mice as well as more severe seizures. At 6 mg/kg, latency for BL6 N5 mice was 92 ± 2.7 s and CD1/129Sv had a latency of 128 ± 5.1 s, and at 8 mg/kg, BL6 N5 had a latency of 84 ± 2.1 s and CD1/129Sv had a latency of 106 ± 3.5 s (p < 0.001). Heterozygous S248F mice of all genetic backgrounds examined displayed the DAC when given injections of nicotine (2, 6, or 8 mg/kg). The DAC occurred before seizure onset in cases in which convulsive doses of nicotine (i.e., 6 or 8 mg/kg) were used.
The DAC is not the result of complete loss of function of the α4-nAChR and is mecamylamine resistant
Ninety α4-nAChR knock-out (α4−/−) mice (Ross et al., 2000) (45 males and 45 females) and the same number of littermate control mice on a hybrid 129SvJ/C57BL/6 genetic background were given injections of 2, 6, and 8 mg/kg nicotine. The DAC was not seen in the α4−/− mice at any nicotine dose. This is an important observation given the results of our synaptosome assay showing that at high agonist doses, the mutant receptor showed impaired function. Ordinal logistic regression (McCullagh, 1980) was also used to analyze the knock-out data (Table 1); here there was only one background, so the two explanatory variables were genotype and nicotine dose. The effect of nicotine dose was statistically significant in the expected direction (higher doses leading to worse outcomes; p < 0.001). The difference between the genotypes was not statistically significant (p = 0.11); the direction of the observed effect was that the homozygous knock-out mice did worse than the WT mice. Collectively, these data suggest (1) that the DAC is not the result of complete loss of function of the α4-nAChR subunit and (2) that nicotine-induced seizures in WT mice are not α4-nAChR subunit mediated, because loss of this receptor subunit did not diminish nicotine-induced seizures.
Pretreatment of heterozygous S248F BL6 N1 background mice (n = 9) with the nicotinic channel antagonist mecamylamine (0.5 mg/kg, i.p.) 10 min before administration of 8 mg/kg nicotine did not disrupt the DAC in any manner, although it did completely inhibit all seizure-like behavior (tremors, wild running, clonic seizures, and death associated with clonic seizures were not seen). In contrast, mice administered nicotine after saline pretreatment (n = 6) had a typical spectrum of seizures seen with this dose of nicotine. Two of the six mice experienced tremors and wild running (maximum score of 2) and three of six mice died in the setting of clonic seizures (maximum score of 4) (Fisher's exact test, p = 0.002).
The anticonvulsant sodium channel blocker carbamazepine blocks the dystonic component of the DAC
The anticonvulsant sodium channel blocker carbamazepine is effective in the treatment of ADNFLE (Provini et al., 2000). We administered S248F heterozygous mice carbamazepine (40–60 mg/kg, i.p.) before challenge with nicotine (1–2 mg/kg, i.p.) and monitored the DAC. Carbamazepine at 40 mg/kg had no significant effect on ambulation, but at 60 mg/kg, it reduced ambulatory scores to 25% of baseline (data not shown). Mutant mice had a dose-dependent attenuation of nicotine-induced forelimb dystonia (Fig. 7) (z test, 0 mg/kg vs 60 mg/kg, p = 0.006), but nicotine-induced saccadic behavior and Straub tail responses were present at both doses of carbamazepine tested (Fig. 7). Carbamazepine (60 mg/kg) completely ablated forelimb dystonia produced in response to 1 mg/kg nicotine (Fig. 7). These data confirm that the dystonic component of the DAC was reduced by a drug that produces a therapeutic effect in human ADNFLE. Although mice were partially sedated at 60 mg/kg carbamazepine, other studies had shown that even extreme sedation does not prevent the emergence of the DAC (data not shown).
Figure 7.

Carbamazepine inhibits the dystonic component of the DAC. A–D, The percentage of mice that displayed saccadic behavior (A), Straub tail (B), forelimb dystonia (C), and the complete phenotype (D) after intraperitoneal administration of carbamazepine (CBZ) at 0 mg/kg (black bar), 40 mg/kg (gray bar), or 60 mg/kg (white bar), followed 22 min later with either 1 or 2 mg/kg nicotine intraperitoneally. *p = 0.006, significantly different from 0 mg/kg CBZ at the same nicotine dose. Data are expressed as a percentage of control mice that were treated with vehicle. n = 7–9 mice per group.
The DAC is abolished by preinjection of a small dose of nicotine
Although we believe that the S248F mouse represents a good model for aspects of ADNFLE, a possible objection is that nicotine injections elicit ADNFLE-like behaviors (i.e., DAC) in S248F mice but nicotine prevents and/or diminishes events in ADNFLE patients. As in all studies of nicotinic receptors, however, one must note that nicotine either activates or desensitizes receptors depending on the concentration or dose administered. There are at least two possible mechanisms. First, at relatively low nicotine doses, ADNFLE-like behaviors may be suppressed because nicotinic receptors are inactivated by desensitization. Second, higher nicotine doses may trigger behavioral responses such as the DAC by widespread nicotinic receptor activation. To date, two reports have shown the therapeutic benefit of nicotine in ADNFLE patients: Willoughby et al. (2003) reported an almost complete elimination of seizures in an ADNFLE patient after treatment with a nicotine patch (this therapeutic benefit continues in this patient; J. Willoughby, personal communication), and the study by Brodtkorb and Picard (2006) showed a significant inverse correlation between tobacco use and nocturnal seizure frequency. In both studies, the authors argue that nicotine acts by desensitizing nicotinic receptors. It would be reasonable to postulate that chronic nicotine exposure during the waking period may lead to receptor desensitization through stages 2–4 of sleep at night, when most ADNFLE events occur. In addition, both behavioral actions of nicotine were demonstrated in a knock-in mouse strain engineered to express hypersensitive α4 nicotinic receptors (L9′A) (Fonck et al., 2005): nicotine either initiated behavioral seizures (at 2 mg/kg) or prevented them (at 0.1 mg/kg, 10 min before 2 mg/kg).
We performed similar experiments on heterozygous S248F mice (n = 9, three male and six female mice on a CD1/129 background). A dose of nicotine (0.1 mg/kg) too small to give the full DAC was able to block the DAC when mice were given injections of 2 mg/kg nicotine 10 min later (none of nine mice displayed the complete DAC). In contrast, saline preinjection had no effect on the DAC in the same cohort of heterozygous mice (eight of nine mice displayed the full DAC) (z-test, p < 0.001) tested 1 week later. These data are consistent with the published clinical studies that suggest that chronic low-dose nicotine is effective in treating ADNFLE by a mechanism of receptor desensitization.
The S248F mutation confers mecamylamine resistance on α4β2-containing nAChRs
Previous studies show that 6′ mutations make muscle nAChRs resistant to channel block by QX-222 (Leonard et al., 1988). Mecamylamine is a noncompetitive α4β2-nAChR antagonist (Papke et al., 2001) and probably acts as a channel blocker (Giniatullin et al., 2000). To determine whether the failure of mecamylamine to block the DAC phenotype was caused by mecamylamine resistance of the mutated S248F receptors, the effects of mecamylamine were studied on the ACh response of rat α4β2-nAChRs expressed in oocytes. The rat gene α4(S252F) mutation (which aligns with the mouse α4 S248F mutation at the M2 6′ position) conferred mecamylamine resistance on rat α4β2-nAChRs expressed in oocytes (Fig. 8). We used an ACh concentration (2 μm) near the previously reported rat WT and α4(S252F)β2 EC50 for ACh (Kuryatov et al., 1997; Figl et al., 1998) and a mecamylamine concentration (0.5 μm) near the range of IC50 values previously reported for human α4β2-nAChRs (Papke et al., 2001). As reported previously (Weiland et al., 1996; Kuryatov et al., 1997; Bertrand et al., 1998; Figl et al., 1998), the α4(S252F) mutation enhanced agonist-induced desensitization. Successive applications of 2 μm ACh at 3 min intervals had no significant effect on the peak WT response but reduced the peak mutant response significantly (p < 0.05) (Fig. 8A). To compensate for this effect, we applied ACh alone to the oocytes three times and measured the ratios of the second- and third-peak ACh responses to that of the first in a separate series of experiments. We then divided the corresponding responses in the mecamylamine experiments by these ratios. As reported previously (Giniatullin et al., 2000), mecamylamine persistently inhibited the WT response. However, it produced no reduction in the mutant response beyond that produced by cumulative agonist-induced desensitization alone. The peak WT response in 0.5 μm mecamylamine was 66 ± 5% (n = 5 oocytes) less than the control and remained 51 ± 4% (n = 5) less after a 3 min wash with saline (p < 0.05) (Fig. 8B). In contrast, after correcting for agonist-induced cumulative desensitization, the mutant responses in 0.5 μm mecamylamine and after a 3 min wash were not significantly different from the control response (Fig. 8B). Thus, α4(S252F)β2-nAChRs expressed in oocytes were resistant to mecamylamine block.
Figure 8.

The S248F mutant receptors are resistant to mecamylamine block. A, The current traces are voltage-clamp ACh responses from oocytes expressing rat α4β2 WT receptors or α4(S252F)β2 mutant receptors. The horizontal bars above the traces show the timing of the 2 μm ACh applications, at 3 min intervals. Three brief (10–30 s) ACh applications to the WT receptors produced no significant change in the peak response (top left). In contrast, the same protocol resulted in significant cumulative desensitization of the peak mutant response (top right). Mecamylamine (0.5 μm) persistently inhibited the peak WT response but produced no further reduction in the mutant response beyond that by produced by ACh alone (bottom right). In the two lower sets of traces, mecamylamine was present throughout the entire middle trace (second ACh application). B, The normalized peak WT and mutant responses in 0.5 μm mecamylamine and after a 3 min wash with control saline corrected for cumulative desensitization. After correction, mecamylamine had no significant effect on the mutant response. The asterisks denote a significant difference (p < 0.05) from control. C, Effect of mecamylamine on dopamine release from striatal synaptosomes of WT, heterozygous (HT), and homozygous (HM) S248F mice. Synaptosomes were perfused for 5 min with the indicated concentration of mecamylamine plus αCtxMII (50 nm) and subsequently exposed to ACh (3 μm for 20 s) containing the indicated concentration of mecamylamine. Data are expressed as percentage of response without mecamylamine. Control responses were as follows: WT mice (n = 9), 15.6 ± 1.0 U; HT mice (n = 5), 10.8 ± 1.9 U; HM mice (n = 11), 7.8 ± 1.6 U. D, Effect of mecamylamine on 86Rb+ efflux from thalamic synaptosomes. The indicated concentration of mecamylamine was present for 2 min before and also during the exposure to ACh (30 μm for 5 s). Data are expressed as percentage of control response without mecamylamine. Control responses (n = 5 mice per group) were as follows: WT mice, 20.6 ± 1.5 U; HT mice, 15.0 ± 1.2 U; HM mice, 4.9 ± 0.9 U. E, Effect of mecamylamine on 86Rb+ efflux from cortical synaptosomes. The indicated concentration of mecamylamine was present for 2 min before and also during the exposure to ACh (30 μm for 5 s). Data are expressed as percentage of control response without mecamylamine. Control responses (n = 5 mice per group) were as follows: WT mice, 6.6 ± 0.6 U; HT mice, 2.7 ± 0.4 U; HM mice, 1.9 ± 0.2 U.
The activity of synaptosomal preparations were also investigated to determine whether this resistance also occurs in heterozygous or homozygous S248F knock-in mice using two assays shown above to be affected by the S248F mutation. Dopamine release mediated by αCtxMII-resistant nAChRs and stimulated by ACh (3 μm) was measured using striatal synaptosomes from WT, heterozygous, and homozygous mice. Synaptosomes from S248F mice were perfused for 5 min with αCtxMII (50 nm) and mecamylamine (0–100 μm) before exposure to ACh (3 μm for 20 s) (Fig. 8C). Both the heterozygous and homozygous mice are resistant to the effects of mecamylamine for this activity. The 86Rb+ efflux properties of the S248F-nAChR were investigated using synaptosomes from thalamus (Fig. 8D) and cortex (Fig. 8E). For both regions, synaptosomes from WT mice were blocked by considerably lower concentrations of mecamylamine than those from either the heterozygous or homozygous S248F mice. Similar results were obtained for the dopamine release and the 86Rb+ efflux assays using nicotine as agonist (data not shown).
The concentration of mecamylamine achieved in a mouse by the 0.5 mg/kg intraperitoneal dose is estimated to be ∼1 μm from an elimination time course for [3H]mecamylamine in mouse brain (Collins et al., 1994). This concentration does not block the S248F-nAChR in the in vitro experiments above. In conclusion, the in vitro experiments confirm that a behavior, such as the DAC, mediated by the S248F-nAChRs would be resistant to this level of the nAChR blocker mecamylamine in vivo.
Discussion
Acute nicotine administration reveals a highly penetrant and robust phenotype in S248F knock-in mice not previously described in other models of ADNFLE. The DAC recapitulates two behaviors seen in ADNFLE patients. In typical ADNFLE attacks, sleep is interrupted by sudden elevation of the head and trunk and dystonic posturing of the upper limbs (Scheffer et al., 1995; Zucconi et al., 1997; Oldani et al., 1998; Provini et al., 1999, 2000). The human video accompanying this report (Human ADNFLE video, available at www.jneurosci.org as supplemental material) shows an example of an ADNFLE attack. The patient wakes from sleep in a startle, sits upright, and looks forward. He turns his head to the left and right and at times stares straight ahead. The patient extends the arms forward in an asymmetrical dystonic limb posture as the head-turning movements continue. The attack is brief, and the patient communicates at the end of the attack. Dystonic upper-limb posturing and head and body thrashing are described in detail in other patients in the literature (Scheffer et al., 1995; Bertrand et al., 2005). The stereotypical jerky exploratory-like darting that we observed in our S248F mice involves head and trunk movements analogous to those in ADNFLE attacks. The stereotypical forelimb dystonia is probably the most convincing correlate with the dystonic semiology of ADNFLE: both mutant mice and humans display dystonic twisting of the limbs, which may be tonic and sustained (as in CD1/129Sv background S248F mice) or rhythmic (as in BL6 N5 background S248F mice). The attenuation of the dystonic component of the DAC by carbamazepine suggests that the DAC represents the murine equivalent of dystonic-predominant ADNFLE attacks, because this drug effectively reduces the frequency of NFLE attacks by ∼68% (Provini et al., 1999). In accordance with some clinical studies (Willoughby et al., 2003; Brodtkorb and Picard, 2006) showing a benefit of chronic low-dose nicotine in reducing ADNFLE severity, and the inhibition of nicotine-induced behavioral seizures in L9′A mice (Fonck et al., 2005), we were also able to block the DAC by pretreatment with a low dose of nicotine (0.1 mg/kg). The failure to detect an electrographic correlate for the DAC was consistent with the absence of ictal EEG activity in most ADNFLE attacks (Combi et al., 2004).
Surprisingly, heterozygous mice had less severe nicotine-induced seizures than WT mice of the same genetic background. Indeed, α4−/− mice had a normal nicotine-induced seizure profile, confirming other reports (Damaj et al., 1999; Dobelis et al., 2003) that nicotine-induced seizures in WT mice are not mediated through the α4-nAChR subunit; studies have implicated α3, β4, and α5 subunits (Franceschini et al., 2002; Kedmi et al., 2004). Finally, mecamylamine achieved complete abrogation of nicotine-induced major motor seizures without inhibiting the DAC, suggesting that the two processes may be mechanistically different. Interestingly, this mecamylamine resistance is also manifest in the agonist-induced currents for the S248F mutation expressed in oocytes, as well as in agonist-induced ion flux and transmitter release for synaptosomes from S248F mice.
A spectrum of severity in ADNFLE mouse models
Several other ADNFLE-related models have been reported. There are two mouse strains with extreme gain-of-function mutations in α4 at the M2–9′ position, which have not been linked to ADNFLE. Although initially no spontaneous seizures were observed in the L9′ mouse lines (Fonck et al., 2003, 2005), more recently, we have identified spontaneous seizures (Fonck and Lester, 2006) in a few L9′A mice. These prolonged tonic–clonic seizures associated with cortical c-fos activation seen only in young mice (age, 12–29 d) sometimes result in death. Severe motor seizures restricted to a narrow postnatal time window would be atypical for ADNFLE. Adult L9′A mice (Fonck et al., 2005) were reported to display low-dose nicotine-induced behavioral seizures not associated with spike-and-wave EEG changes.
Two other α4 subunit ADNFLE knock-in strains have been independently generated and studied: the Chrna4S252F (the same mutation as reported here) and the α4 +L264 lines (Klaassen et al., 2006). Both lines have spontaneous seizures of two types: brief periods of behavioral arrest and prolonged hyperkinetic motor seizures resembling bursts of wild running seen with convulsive doses of nicotine (Franceschini et al., 2002). Spontaneous motor seizures occur commonly in both lines and are associated with high-voltage spiking activity on subdural EEG recordings. Despite the frequency and severity of motor seizures, premature death is not reported. The finding of electrographic seizures in this model is consistent with reported electrographic seizures occurring in ADNFLE (Scheffer et al., 1995; Hayman et al., 1997; Phillips et al., 1998; Provini et al., 2000; Steinlein et al., 2000). It should be noted that although ADNFLE is a disease of non-REM sleep, the spontaneous seizures and EEG changes noted in the Klaassen lines were not correlated to any sleep/wake cycle.
A β2 subunit V287L (M2–22′ position) ADNFLE mutant mouse strain has also been reported briefly (Xu et al., 2006). The few spontaneous seizures (<2% of animals) have not yet been systematically characterized.
The reported ADNFLE-related mice display a spectrum of behavioral phenotypes that reflect the range of motor phenotypes previously reported for ADNFLE (Scheffer et al., 1995). The human phenotypes include thrashing/hyperkinetic activity in the minority (16 of 47) of cases and tonic stiffening in the majority (36 of 47) of cases, 23 of which also had superimposed clonic jerking. Our BL6 N5 S248F knock-in mice display rhythmic forelimb dystonic posturing resembling the ADNFLE attacks described in this clinical study.
In summary, the most severe spontaneous seizures are noted in a small proportion of recently born juvenile L9′A mice. The Klaassen α4 strains display frequent spontaneous electrographic major motor seizures. Infrequent as yet uncharacterized spontaneous seizure-like behaviors are seen in β2 subunit V287L mice, whereas our S248F mice and the adult L9′A mice display no such events. All strains reported to date display modest to low-dose nicotine-induced behaviors that differ from what is seen in WT mice. Importantly, these nicotine-induced ADNFLE like-behaviors are EEG negative in the adult L9′A, in our S248F mice, and in the β2 strain (C. Fonck, B. N. Cohen, J. Xu, H. A. Lester, and S. F. Heinemann, unpublished data). The behavioral effects of modest to low-dose nicotine in the Klaassen α4 strains were described in a seizure scale, but associated EEG findings were not reported.
Epilepsy or movement disorder?
If an epileptic seizure is defined as a transient occurrence of signs and/or symptoms because of abnormal excessive or synchronous neuronal activity in the brain (Fisher et al., 2005), then there are only two possible explanations for failure to identify an electrographic correlate to the DAC. First, perhaps the EEG paradigms used in this study, and in previous studies that identified EEG negative behavioral seizures, were not appropriately sensitive or were not directed to the correct site in the brain. A systematic unbiased and dense cortical and depth-electrode survey throughout the entire mouse brain may identify such a discrete focus and address this caveat. This view is supported by observations in human ADNFLE that single individuals may show a continuum of paroxysmal events, beginning with arousals that lack distinctive epileptiform features and evolving into dystonic attacks and more violent events including frank tonic–clonic seizures with overt epileptiform activity suggesting a common epileptic mechanism (Scheffer et al., 1995; Oldani et al., 1998; Provini et al., 1999, 2000).
Second, perhaps the DAC is not an epileptic phenomenon but rather represents a paroxysmal movement disorder. A number of studies have reported the coexistence of epilepsy and movement disorders in the same individuals or in different individuals within a family (Guerrini et al., 1999, 2002; Guerrini, 2001). In a recent report, a mutation was identified in a calcium-sensitive potassium channel in childhood-onset paroxysmal nonkinesigenic dyskinesia with absence epilepsy associated with rare tonic–clonic seizures and generalized spike-wave complexes on electroencephalogram (Du et al., 2005). Of the 13 cases described, 7 had paroxysmal dyskinesia alone, 5 had paroxysmal dyskinesia and seizures, and 1 had only seizures. The clinical phenotype of a genetically defined channelopathy can therefore be either epilepsy or a paroxysmal movement disorder.
The influence of modifier genes/genetic background may also explain the difference in the phenotype seen between our S248F mice and the Klaassen lines. Two notable differences include the CD1 background in our S248F mice and use of the J1 ES line by Klaassen et al. (2006) versus our use of W9.5 cells. The potential confounding effect of modifier genes on phenotype was underscored by a report on sodium channel 1α heterozygous knock-out mice that model severe myoclonic epilepsy in infancy. The mice showed a dramatically different phenotype depending on genetic background: frequent spontaneous seizures in a C57BL/6 background and no seizures in a 129/SvJ background (Yu et al., 2006). The late appearance and low penetrance of the dramatic juvenile spontaneous seizures in L9′A mice in a line established some years beforehand also argues for potential modifier gene influence although an epigenetic effect is not excluded.
Conclusions
Our S248F line appears to predominantly model dystonic aspects of ADNFLE semiology. Exploiting this line to identify channel blockers (possibly related structurally to mecamylamine) that selectively block S248F mutant channel function would be useful in treating carbamazepine-resistant cases of ADNFLE.
Footnotes
This work was supported by National Health and Medical Research Council (NHMRC) Program Grant 236805 and National Institutes of Health Grant MH53631 to J.M.M.; National Institute on Drug Abuse Grants DA003194, DA012242, and DA015663; National Institute of Neurological Disorders and Stroke Grants NS43800 and NS046464; and Science Foundation Ireland Grant 02-1N1B227. J.D. is an NHMRC Practitioner fellow. We thank Vincenzo Ferreri, Jim Massalas, Keith Buxton, and Yvette Wilson for technical assistance and Dr. Rachael Nally and Associate Prof. Ian Gordon for advice with statistical analysis. We are grateful to Dr. Jim Boulter for providing genomic DNA clones used to build the original L9S′ targeting construct. We are grateful to Prof. Ingrid Scheffer for critical input and discussion on clinical aspects of ADNFLE.
References
- Aridon P, Marini C, Di Resta C, Brilli E, De Fusco M, Politi F, Parrini E, Manfredi I, Pisano T, Pruna D, Curia G, Cianchetti C, Pasqualetti M, Becchetti A, Guerrini R, Casari G. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. 2006;79:342–350. doi: 10.1086/506459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand D, Picard F, Le Hellard S, Weiland S, Favre I, Phillips H, Bertrand S, Berkovic SF, Malafosse A, Mulley J. How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia. 2002;43(Suppl 5):112–122. doi: 10.1046/j.1528-1157.43.s.5.16.x. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Elmslie F, Hughes E, Trounce J, Sander T, Bertrand S, Steinlein OK. The CHRNB2 mutation I312M is associated with epilepsy and distinct memory deficits. Neurobiol Dis. 2005;20:799–804. doi: 10.1016/j.nbd.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Weiland S, Berkovic SF, Steinlein OK, Bertrand D. Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol. 1998;125:751–760. doi: 10.1038/sj.bjp.0702154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodtkorb E, Picard F. Tobacco habits modulate autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy Behav. 2006;9:515–520. doi: 10.1016/j.yebeh.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Clarke PB. Chronic central nicotinic blockade after a single administration of the bisquaternary ganglion-blocking drug chlorisondamine. Br J Pharmacol. 1984;83:527–535. doi: 10.1111/j.1476-5381.1984.tb16517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Luo Y, Selvaag S, Marks MJ. Sensitivity to nicotine and brain nicotinic receptors are altered by chronic nicotine and mecamylamine infusion. J Pharmacol Exp Ther. 1994;271:125–133. [PubMed] [Google Scholar]
- Combi R, Dalpra L, Tenchini ML, Ferini-Strambi L. Autosomal dominant nocturnal frontal lobe epilepsy—a critical overview. J Neurol. 2004;251:923–934. doi: 10.1007/s00415-004-0541-x. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Glassco W, Dukat M, Martin BR. Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther. 1999;291:1284–1291. [PubMed] [Google Scholar]
- Davila-Garcia MI, Musachio JL, Perry DC, Xiao Y, Horti A, London ED, Dannals RF, Kellar KJ. [125I]IPH, an epibatidine analog, binds with high affinity to neuronal nicotinic cholinergic receptors. J Pharmacol Exp Ther. 1997;282:445–451. [PubMed] [Google Scholar]
- De Fusco M, Becchetti A, Patrignani A, Annesi G, Gambardella A, Quattrone A, Ballabio A, Wanke E, Casari G. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26:275–276. doi: 10.1038/81566. [DOI] [PubMed] [Google Scholar]
- Dobelis P, Hutton S, Lu Y, Collins AC. GABAergic systems modulate nicotinic receptor-mediated seizures in mice. J Pharmacol Exp Ther. 2003;306:1159–1166. doi: 10.1124/jpet.103.053066. [DOI] [PubMed] [Google Scholar]
- Drago J, Padungchaichot P, Wong JY, Lawrence AJ, McManus JF, Sumarsono SH, Natoli AL, Lakso M, Wreford N, Westphal H, Kola I, Finkelstein DI. Targeted expression of a toxin gene to D1 dopamine receptor neurons by cre-mediated site-specific recombination. J Neurosci. 1998;18:9845–9857. doi: 10.1523/JNEUROSCI.18-23-09845.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- Figl A, Viseshakul N, Shafaee N, Forsayeth J, Cohen BN. Two mutations linked to nocturnal frontal lobe epilepsy cause use-dependent potentiation of the nicotinic ACh response. J Physiol (Lond) 1998;513:655–670. doi: 10.1111/j.1469-7793.1998.655ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J., Jr Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- Fonck C, Nashmi R, Deshpande P, Damaj MI, Marks MJ, Riedel A, Schwarz J, Collins AC, Labarca C, Lester HA. Increased sensitivity to agonist-induced seizures, Straub tail, and hippocampal theta rhythm in knock-in mice carrying hypersensitive α4 nicotinic receptors. J Neurosci. 2003;23:2582–2590. doi: 10.1523/JNEUROSCI.23-07-02582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonck C, Cohen BN, Nashmi R, Whiteaker P, Wagenaar DA, Rodrigues-Pinguet N, Deshpande P, McKinney S, Kwoh S, Munoz J, Labarca C, Collins AC, Marks MJ, Lester HA. Novel seizure phenotype and sleep disruptions in knock-in mice with hypersensitive α4* nicotinic receptors. J Neurosci. 2005;25:11396–11411. doi: 10.1523/JNEUROSCI.3597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonck CI, Lester HA. Spontaneous seizures and nicotine-induced c-fos in knock-in mice with hypersensitive α4* nicotinic receptors. Society for Neuroscience. 2006 Abstracts 326.316/C384. [Google Scholar]
- Franceschini D, Paylor R, Broide R, Salas R, Bassetto L, Gotti C, De Biasi M. Absence of alpha7-containing neuronal nicotinic acetylcholine receptors does not prevent nicotine-induced seizures. Brain Res Mol Brain Res. 2002;98:29–40. doi: 10.1016/s0169-328x(01)00309-6. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Gantois I, Fang K, Jiang L, Babovic D, Lawrence AJ, Ferreri V, Teper Y, Jupp B, Ziebell J, Morganti-Kossmann CM, O'Brien TJ, Nally R, Schütz G, Waddington J, Egan G, Drago J. Ablation of D1 dopamine receptor-expressing cells generates mice with seizures, hindlimb dystonia, hyperactivity and impaired oral behavior. Proc Natl Acad Sci USA. 2007;104:4182–4187. doi: 10.1073/pnas.0611625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 1995;48:774–782. [PubMed] [Google Scholar]
- Giniatullin RA, Sokolova EM, Di Angelantonio S, Skorinkin A, Talantova MV, Nistri A. Rapid relief of block by mecamylamine of neuronal nicotinic acetylcholine receptors of rat chromaffin cells in vitro: an electrophysiological and modeling study. Mol Pharmacol. 2000;58:778–787. doi: 10.1124/mol.58.4.778. [DOI] [PubMed] [Google Scholar]
- Guerrini R. Idiopathic epilepsy and paroxysmal dyskinesia. Epilepsia. 2001;42(Suppl 3):36–41. doi: 10.1046/j.1528-1157.2001.042suppl.3036.x. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Bonanni P, Nardocci N, Parmeggiani L, Piccirilli M, De Fusco M, Aridon P, Ballabio A, Carrozzo R, Casari G. Autosomal recessive rolandic epilepsy with paroxysmal exercise-induced dystonia and writer's cramp: delineation of the syndrome and gene mapping to chromosome 16p12-11.2. Ann Neurol. 1999;45:344–352. doi: 10.1002/1531-8249(199903)45:3<344::aid-ana10>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Sanchez-Carpintero R, Deonna T, Santucci M, Bhatia KP, Moreno T, Parmeggiani L, Bernardina BD. Early-onset absence epilepsy and paroxysmal dyskinesia. Epilepsia. 2002;43:1224–1229. doi: 10.1046/j.1528-1157.2002.13802.x. [DOI] [PubMed] [Google Scholar]
- Hayman M, Scheffer IE, Chinvarun Y, Berlangieri SU, Berkovic SF. Autosomal dominant nocturnal frontal lobe epilepsy: demonstration of focal frontal onset and intrafamilial variation. Neurology. 1997;49:969–975. doi: 10.1212/wnl.49.4.969. [DOI] [PubMed] [Google Scholar]
- Hirose S, Iwata H, Akiyoshi H, Kobayashi K, Ito M, Wada K, Kaneko S, Mitsudome A. A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 1999;53:1749–1753. doi: 10.1212/wnl.53.8.1749. [DOI] [PubMed] [Google Scholar]
- Kedmi M, Beaudet AL, Orr-Urtreger A. Mice lacking neuronal nicotinic acetylcholine receptor beta4-subunit and mice lacking both alpha5- and beta4-subunits are highly resistant to nicotine-induced seizures. Physiol Genomics. 2004;17:221–229. doi: 10.1152/physiolgenomics.00202.2003. [DOI] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci USA. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. J Neurosci. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–1851. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, Nashmi R, Kofuji P, Dang H, Shi W, Fidan M, Khakh BS, Chen Z, Bowers BJ, Boulter J, Wehner JM, Lester HA. Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci USA. 2001;98:2786–2791. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard RJ, Labarca CG, Charnet P, Davidson N, Lester HA. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 1988;242:1578–1581. doi: 10.1126/science.2462281. [DOI] [PubMed] [Google Scholar]
- Lu Y, Grady S, Marks MJ, Picciotto M, Changeux JP, Collins AC. Pharmacological characterization of nicotinic receptor-stimulated GABA release from mouse brain synaptosomes. J Pharmacol Exp Ther. 1998;287:648–657. [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Calcaterra J, Stitzel JA, Bullock AE, Grady SR, Picciotto MR, Changeux JP, Collins AC. Two pharmacologically distinct components of nicotinic receptor-mediated rubidium efflux in mouse brain require the beta2 subunit. J Pharmacol Exp Ther. 1999;289:1090–1103. [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Collins AC. Deletion of the alpha7, beta2, or beta4 nicotinic receptor subunit genes identifies highly expressed subtypes with relatively low affinity for [3H]epibatidine. Mol Pharmacol. 2006;70:947–959. doi: 10.1124/mol.106.025338. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Meinerz NM, Drago J, Collins AC. Gene targeting demonstrates that a4 nicotinic acetylcholine receptor subunits contribute to expression of diverse [3H]epibatidine binding sites and components of biphasic 86Rb+ efflux with high and low sensitivity to stimulation by acetylcholine. Neuropharmacology. 2007;53:390–405. doi: 10.1016/j.neuropharm.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl CD, Horne MK, Finkelstein DI, Wong JY, Berkovic SF, Drago J. Electroencephalographic characterisation of pentylenetetrazole-induced seizures in mice lacking the alpha 4 subunit of the neuronal nicotinic receptor. Neuropharmacology. 2003;44:234–243. doi: 10.1016/s0028-3908(02)00369-6. [DOI] [PubMed] [Google Scholar]
- McCullagh P. Regression models for ordinal data (with discussion) J R Stat Soc B. 1980;42:109–142. [Google Scholar]
- Montagna P. Nocturnal paroxysmal dystonia and nocturnal wandering. Neurology. 1992;42:61–67. [PubMed] [Google Scholar]
- Oldani A, Zucconi M, Asselta R, Modugno M, Bonati MT, Dalpra L, Malcovati M, Tenchini ML, Smirne S, Ferini-Strambi L. Autosomal dominant nocturnal frontal lobe epilepsy. A video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain. 1998;121:205–223. doi: 10.1093/brain/121.2.205. [DOI] [PubMed] [Google Scholar]
- Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. San Diego: Academic; 2001. The mouse brain in stereotaxic coordinates, Ed 2. [Google Scholar]
- Phillips HA, Scheffer IE, Crossland KM, Bhatia KP, Fish DR, Marsden CD, Howell SJ, Stephenson JB, Tolmie J, Plazzi G, Eeg-Olofsson O, Singh R, Lopes-Cendes I, Andermann E, Andermann F, Berkovic SF, Mulley JC. Autosomal dominant nocturnal frontal-lobe epilepsy: genetic heterogeneity and evidence for a second locus at 15q24. Am J Hum Genet. 1998;63:1108–1116. doi: 10.1086/302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HA, Favre I, Kirkpatrick M, Zuberi SM, Goudie D, Heron SE, Scheffer IE, Sutherland GR, Berkovic SF, Bertrand D, Mulley JC. CHRNB2 is the second acetylcholine receptor subunit associated with autosomal dominant nocturnal frontal lobe epilepsy. Am J Hum Genet. 2001;68:225–231. doi: 10.1086/316946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plazzi G, Tinuper P, Montagna P, Provini F, Lugaresi E. Epileptic nocturnal wanderings. Sleep. 1995;18:749–756. doi: 10.1093/sleep/18.9.749. [DOI] [PubMed] [Google Scholar]
- Pradhan AA, Cumming P, Clarke PB. [125I]Epibatidine-labelled nicotinic receptors in the extended striatum and cerebral cortex: lack of association with serotonergic afferents. Brain Res. 2002;954:227–236. doi: 10.1016/s0006-8993(02)03340-1. [DOI] [PubMed] [Google Scholar]
- Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122:1017–1031. doi: 10.1093/brain/122.6.1017. [DOI] [PubMed] [Google Scholar]
- Provini F, Plazzi G, Montagna P, Lugaresi E. The wide clinical spectrum of nocturnal frontal lobe epilepsy. Sleep Med Rev. 2000;4:375–386. doi: 10.1053/smrv.2000.0109. [DOI] [PubMed] [Google Scholar]
- Rodrigues-Pinguet N, Jia L, Li M, Figl A, Klaassen A, Truong A, Lester HA, Cohen BN. Five ADNFLE mutations reduce the Ca2+ dependence of the mammalian alpha4beta2 acetylcholine response. J Physiol (Lond) 2003;550:11–26. doi: 10.1113/jphysiol.2003.036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues-Pinguet NO, Pinguet TJ, Figl A, Lester HA, Cohen BN. Mutations linked to autosomal dominant nocturnal frontal lobe epilepsy affect allosteric Ca2+ activation of the α4β2 nicotinic acetylcholine receptor. Mol Pharmacol. 2005;68:487–501. doi: 10.1124/mol.105.011155. [DOI] [PubMed] [Google Scholar]
- Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, Drago J. Phenotypic characterization of an α4 neuronal nicotinic acetylcholine receptor subunit knock-out mouse. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, Andermann F, Desbiens R, Keene D, Cendes F, Manson JI, Constantinou JE, McIntosh A, Berkovic SF. Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain. 1995;118:61–73. doi: 10.1093/brain/118.1.61. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Magnusson A, Stoodt J, Bertrand S, Weiland S, Berkovic SF, Nakken KO, Propping P, Bertrand D. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet. 1997;6:943–947. doi: 10.1093/hmg/6.6.943. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Stoodt J, Mulley J, Berkovic S, Scheffer IE, Brodtkorb E. Independent occurrence of the CHRNA4 Ser248Phe mutation in a Norwegian family with nocturnal frontal lobe epilepsy. Epilepsia. 2000;41:529–535. doi: 10.1111/j.1528-1157.2000.tb00205.x. [DOI] [PubMed] [Google Scholar]
- Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett. 1996;398:91–96. doi: 10.1016/s0014-5793(96)01215-x. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Marks MJ, Grady SR, Lu Y, Picciotto MR, Changeux JP, Collins AC. Pharmacological and null mutation approaches reveal nicotinic receptor diversity. Eur J Pharmacol. 2000a;393:123–135. doi: 10.1016/s0014-2999(00)00052-2. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol. 2000b;131:729–739. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby JO, Pope KJ, Eaton V. Nicotine as an antiepileptic agent in ADNFLE: an N-of-one study. Epilepsia. 2003;44:1238–1240. doi: 10.1046/j.1528-1157.2003.58102.x-i1. [DOI] [PubMed] [Google Scholar]
- Wilson Y, Nag N, Davern P, Oldfield BJ, McKinley MJ, Greferath U, Murphy M. Visualization of functionally activated circuitry in the brain. Proc Natl Acad Sci USA. 2002;99:3252–3257. doi: 10.1073/pnas.042701199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ferraro NV, Zhu Y, Fonck C, Deshpande P, Marks MJ, Collins AC, Lester AC, Heinemann SF. Increased sensitivity to nicotine-induced seizures in β2 V287L knock-in mice. Soc Neurosci. 2006 Abstr 36:326.314/C382. [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zucconi M, Oldani A, Ferini-Strambi L, Bizzozero D, Smirne S. Nocturnal paroxysmal arousals with motor behaviors during sleep: frontal lobe epilepsy or parasomnia? J Clin Neurophysiol. 1997;14:513–522. doi: 10.1097/00004691-199711000-00008. [DOI] [PubMed] [Google Scholar]