

Abstract
Gonadotropin-releasing hormone (GnRH) is released in a pulsatile manner that is dependent on circulating 17β-estradiol (E2) and glucose concentrations. However, the intrinsic conductances responsible for the episodic firing pattern underlying pulsatile release and the effects of E2 and glucose on these conductances are primarily unknown. Whole-cell recordings from mouse enhanced green fluorescent protein-GnRH neurons revealed that the KATP channel opener diazoxide induced an outward current that was antagonized by the sulfonylurea receptor 1 (SUR1) channel blocker tolbutamide. Single-cell reverse transcription (RT)-PCR revealed that the majority of GnRH neurons expressed Kir6.2 and SUR1 subunits, which correlated with the diazoxide/tolbutamide sensitivity. Also, a subpopulation of GnRH neurons expressed glucokinase mRNA, a marker for glucose sensitivity. Indeed, GnRH neurons decreased their firing in response to low glucose concentrations and metabolic inhibition. The maximum diazoxide-induced current was approximately twofold greater in E2-treated compared with oil-treated ovariectomized females. In current clamp, estrogen enhanced the diazoxide-induced hyperpolarization to a similar degree. However, based on quantitative RT-PCR, estrogen did not increase the expression of Kir6.2 or SUR1 transcripts in GnRH neurons. In the presence of ionotropic glutamate and GABAA receptor antagonists, tolbutamide depolarized and significantly increased the firing rate of GnRH neurons to a greater extent in E2-treated females. Finally, tolbutamide significantly increased GnRH secretion from the preoptic-mediobasal hypothalamus. Therefore, it appears that KATP channels and glucokinase are expressed in GnRH neurons, which renders them directly responsive to glucose. In addition, KATP channels are involved in modulating the excitability of GnRH neurons in an estrogen-sensitive manner that ultimately regulates peptide release.
Keywords: Kir6.2, SUR1, diazoxide, tolbutamide, glucokinase, glucose responsive, metabolic stress, GnRH secretion
Introduction
Control of the female reproductive cycle involves complex interactions between the gonads, pituitary, and brain. Gonadotropin-releasing hormone (GnRH), the primary neural signal involved in activation of luteinizing hormone (LH) release from the pituitary, is synthesized and secreted episodically by hypothalamic GnRH neurons into the hypothalamo-hypophysial portal system (Levine and Ramirez, 1980, 1982; Sarkar and Fink, 1980; Ching, 1982; Moenter et al., 1992). Estrogen appears to be the primary humoral signal responsible for the preovulatory LH and GnRH surges as well as negative feedback to suppress their secretion (Legan et al., 1975; Terasawa et al., 1979; Caraty et al., 1989; Terasawa, 1994). This can arise from estrogen modulation of synaptic inputs and/or intrinsic conductances that govern the excitability of GnRH neurons and hence the pulsatile release of the peptide (Leranth et al., 1985; Kalra, 1993; Herbison, 1998). Synchronized pulsatile activity may be intrinsic to GnRH neurons, because GT1–7 cells generate pulses of GnRH in the absence of exogenous input (Stojilkovic et al., 1990; Martínez de la Escalera et al., 1992; Wetsel et al., 1992). Also, acutely dissociated EGFP-GnRH neurons fire episodically in a relatively autonomous manner (Kuehl-Kovarik et al., 2002, 2005). The firing activity of these neurons is dependent on activation of an h-current (pacemaker current) and calcium currents to facilitate burst firing, calcium influx, and neurosecretion (Kelly and Wagner, 2002; Kato et al., 2003; Herbison, 2006). However, activation of these inward currents depends on the expression of K+ channels that hyperpolarize the cell membrane to activate (deinactivation) the responsible channels (McCormick and Pape, 1990; Erickson et al., 1993).
ATP-sensitive potassium (KATP) channels are potential mediators of the membrane hyperpolarization. These channels are a member (Kir6.x) of the inwardly rectifying K+ channel family (Reimann and Ashcroft, 1999). They are heteromultimeric complexes of sulfonylurea receptors (SURs; the regulatory subunit) and inwardly rectifying K+ channel (Kir6.x) subunits (Clement et al., 1997; Ashcroft and Gribble, 1998). Interestingly, KATP channels can couple membrane excitability to cellular metabolism by directly sensing and integrating intracellular changes in the concentration of nucleotides (Ashcroft and Gribble, 2000). The Kir6.2 plus SUR1 channel complex is activated by diazoxide and by metabolic inhibition and is blocked with high affinity by sulfonylureas such as glibenclamide and tolbutamide (Ashcroft and Gribble, 2000). Although sulfonylurea binding and electrophysiological studies have characterized neuronal KATP channels in a number of different brain areas, their role in the episodic firing and peptide release in GnRH neurons remains unexplored (Dunn-Meynell et al., 1998).
Because GnRH neurons are so critical for controlling the female reproductive cycle and GnRH secretion is sensitive to metabolic changes (Levine et al., 1995; Medina et al., 1998; Ohkura et al., 2000; Moenter et al., 2003; Herbison, 2006), we investigated whether GnRH neurons respond to activators of Kir6.2/SUR1 channels and whether the response is regulated by estrogen. We used a transgenic mouse model in which we could visualize EGFP-labeled GnRH neurons and measured the direct effects of the KATP channel opener diazoxide, the effects of in vivo estrogen treatment on this response, and the role of KATP channels in regulating neuronal firing. Finally, we determined whether these channels were sensitive to metabolic perturbations.
Materials and Methods
Animals and treatments.
All animal treatments described in this study are in accordance with institutional guidelines based on National Institutes of Health standards and were performed with institutional Animal Care and Use Committee approval at both the Oregon Health and Science University and Northwestern University. For the electrophysiology and single-cell reverse transcription-PCR (scRT-PCR) experiments, transgenic male and female mice expressing enhanced green fluorescent protein (EGFP) under the control of the GnRH promoter (EGFP-GnRH) were used in these studies (Suter et al., 2000). Animals were group-housed until surgery, at which time they were housed individually. All animals were maintained under controlled temperature and photoperiod (lights on at 6:00 A.M. and off at 6:00 P.M.) and given free access to food and water. Adult males were used intact. Adult females were ovariectomized (OVX) under isoflurane anesthesia, implanted with an oil or 17β-estradiol (E2) capsule (which yields plasma E2 levels of ∼30 pg/ml) for 4–7 d and killed at 10:00–11:00 A.M., at which time the uterus was removed and weighed. In addition, female mice used for the scPCR and real-time PCR were either intact or ovariectomized, implanted with oil or E2 capsules, and given a single injection of oil (100 μl) or estradiol benzoate (EB) (2 μg in 100 μl of oil, which yields plasma E2 levels of ∼75 pg/ml) 24 h before the experiment. The animals were killed at 10:00 A.M. during negative feedback (Christian et al., 2005). This supplemental estrogen treatment was used to increase mRNA expression, although it did not alter the E2-induced current.
For the measurements of GnRH release from preoptic-mediobasal hypothalamus, male Sprague Dawley rats (Charles River, Wilmington, MA) weighing 200–220 g were used. They were housed in temperature-controlled facilities under a 14/10 h light/dark illumination cycle (lights on, 5:00 A.M. to 7:00 P.M.) and provided standard laboratory rodent chow and water ad libitum.
Preparation of preoptic area-GnRH slices.
Intact male and OVX, oil- and estrogen-treated female mice, 6–7 weeks of age, were anesthetized with halothane and then decapitated. The brain was rapidly removed from the skull, and a block containing the diagonal band-preoptic area (DB-POA) was immediately dissected. The DB-POA block was submerged in cold (4°C) oxygenated (95%O2, 5% CO2) high-sucrose CSF containing the following (in mm): 208 sucrose, 2 KCl, 26 NaHCO3, 10 glucose, 1.25 NaH2PO4, 2 MgSO4, 1 MgCl2, 10 HEPES, pH 7.4. Coronal slices (200 μm) from the DB-POA were cut on a vibratome during which time (10 min) the slices were bathed in high-sucrose CSF at 4°C. The slices were then transferred to an auxiliary chamber where they were kept at room temperature (25°C) in artificial CSF (aCSF) consisting of the following (in mm): 124 NaCl, 5 KCl, 2.6 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, 10 HEPES, 10 glucose, pH 7.4, until recording (recovery for 2 h). A single slice at a time was transferred to the recording chamber, and was kept viable by continually perfusing with warm (35°C), oxygenated aCSF at 1.5 ml/min.
Visualized whole-cell patch recording using epifluorescence and infrared-differential interference contrast videomicroscopy.
Whole-cell patch recordings were made under a Zeiss (Jena, Germany) Axioskop FS out-fitted with epifluorescence (FITC filter set) and infrared-differential interference contrast (IR-DIC) video microscopy. The area containing GnRH neurons was initially identified under low power (5× objective) with UV illumination. Then, the GFP-tagged GnRH neurons were visualized through a 40× water immersion objective (Achroplan; Zeiss), and their position was marked on a monitor and compared carefully to its position under IR-DIC imaging. Patch pipettes (1.5 mm outer diameter borosilicate glass; A-M Systems, Seattle, WA) were pulled on a Brown/Flaming puller (model P-97; Sutter Instrument, Novato, CA) and were filled with the following solution (in mm): 128 potassium gluconate, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 2 ATP, 0.25 GTP (0.5% biocytin), adjusted to pH 7.3 with KOH, 295 mOsm. For recording miniature postsynaptic currents (mPSCs), the electrodes were filled with the following solution (in mm): 130 KCl, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 3 ATP, and 0.25 GTP. Pipette resistances were 2–6 MΩ when filled with the above pipette solutions. In whole-cell configuration, access resistance was 20–40 MΩ. In a subset experiment to measure the activation or inactivation parameters of ion channels, the access resistance was <20 MΩ and was 80% compensated. The input resistance was calculated by measuring the slope of the current–voltage relationship curve between −70 and −50 mV. The targeted GnRH neurons were visualized with IR-DIC for positioning the patch pipette, and standard whole-cell patch recording procedures and pharmacological testing were followed as described previously (Ibrahim et al., 2003). Voltage-clamp experiments were performed with an Axopatch 1D amplifier (2 kHz lowpass filter; Molecular Devices, Foster City, CA) under two conditions to measure the various currents. Steady-state current–voltage (I–V) plots were constructed with step command potentials from −120 to −50 mV with a step of 5 mV (holding potential, −60 mV) and duration of 1 s. In experiments to activate and measure the transient outward potassium current (IA), the above voltage protocol was followed by a 500 ms step to −50 or −60 mV. We found that IA was only significantly activated at −50 mV but not at −60 mV, so when we constructed the A-current inactivation curves, the currents activated at −60 mV were subtracted from those activated at −50 mV. Time-dependent KATP channel currents were measured at a holding potential of −60 mV. In a subset of experiments, loose-attached patch recording was used to detect the firing rate of GnRH neurons by recording action potential currents at a 0 mV holding potential (Nunemaker et al., 2003). Electrophysiological signals were digitized with Digidata 1200 or 1322A (Molecular Devices), and the data were analyzed using pClamp software (version 9.2; Molecular Devices).
Electrophysiological solutions/drugs.
Normal aCSF (in mm: 124 NaCl, 5 KCl, 2.6 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, 10 HEPES, 10 glucose) was used in most cases for electrophysiological recording. In whole-cell recordings, tetrodotoxin (TTX) was used to isolate the effect of presynaptic input. In loose-cell attached recordings, high Mg2+/low Ca2+ aCSF (in mm: 124 NaCl, 5 KCl, 2.6 NaH2PO4, 2 MgSO4, 2 MgCl2, 0.3 CaCl2, 26 NaHCO3, 10 HEPES, 0.1-5.0 glucose) and blockers for GABAA (100 μm picrotoxin) and ionic glutamate receptors (20 μm CNQX and 20 μm d-APV or 50 μm dl-APV) were used to isolate the effect from presynaptic input. In experiments where high K+ (15 mm KCl) was used to induce or increase firing of GnRH neurons, NaCl was replaced by KCl.
The outflow of 20 ml syringes with aCSF containing different drugs was controlled by a Gilson Mini-Plus Pump with a perfusion rate of 1.5 ml/min. The following ion channel blockers/activators were used (from Sigma, St. Louis, MO, unless otherwise noted): diazoxide (300 μm) (Ibrahim et al., 2003), tolbutamide (100 μm), MCC-134 (1-[4-(1H-imidazol-1-yl)benzoyl]-N-methyl-cyclobutanecarbothioamide; 300 μm; Mitsubishi Pharma, Osaka, Japan), CNQX (20 μm), dl-APV (50 μm), picrotoxin (100 μm), sodium azide (1 mm), and TTX (1 μm; Alomone Labs, Jerusalem, Israel).
Electrophysiology data analysis.
Comparisons between estrogen- and oil-treated groups were performed with an unpaired Student's t test. Changes in firing rate and mPSCs with glucose and metabolic perturbations were analyzed using a one-way ANOVA (with post hoc Bonferroni's paired analysis). Differences were considered significant if the probability of error was <5%.
Cell harvesting of dispersed GnRH neurons and scRT-PCR.
Two to three 300 μm diagonal band-POA slices were cut on a vibratome and placed in an auxiliary chamber containing oxygenated aCSF. The slices were allowed to recover for 1–2 h in the chamber before dispersion. A discrete region of the diagonal band-rostral POA was microdissected and incubated in 5–10 ml of aCSF (124 mm NaCl, 5 mm KCl, 2.6 mm NaH2PO4, 2 mm MgSO4, 2 mm CaCl2, 26 mm NaHCO3, 10 mm HEPES, 10 mm d-glucose, in DEPC-treated water, pH 7.3, 300 mOsm) containing 1 mg/ml protease for ∼17 min at 37°C. The tissue was then washed four times in low calcium aCSF (0.1 mm CaCl2) and two times in aCSF. The cells were isolated by trituration with flame-polished Pasteur pipettes. The cells were dispersed onto a 35 mm Petri dish and were visualized under a Nikon (Tokyo, Japan) inverted microscope equipped with fluorescence. The fluorescence cells or adjacent cells were patched and then harvested into the patch pipette by applying negative pressure. The contents of the pipette were expelled into a siliconized microcentrifuge tube containing 1 μl of 5× Colorless GoTaq Flexi buffer (Promega, Madison, WI), 15 U of RNAsin, 0.5 μl of 100 mm DTT, and DEPC-treated water in a 5 μl volume. In addition, two to three pools of five cells each were harvested from individual OVX oil- and E2-treated animals and expelled into siliconized microcentrifuge tubes as described above with the exception that the volume in each tube was increased to 8 μl. Each harvested cell or pool of cells was reverse transcribed as described previously (Ibrahim et al., 2003; Qiu et al., 2003). Briefly, the harvested cell solution and 25 ng of hypothalamic total RNA in 1 μl were denatured for 5 min at 65°C and then cooled on ice for 5 min. Single-stranded cDNA was synthesized from cellular RNA by adding 50 U of murine leukemia virus (MuLV) reverse transcriptase, 3 μl of 5× Colorless GoTaq Flexi buffer, 5 mm MgCl2, 0.2 μm dNTPs, 15 U of RNAsin, 10 mm DTT, and 100 ng of random hexamers in a total of 15 μl of DEPC-treated water for a final volume of 20 μl (25 μl for cell pools). Cells and tissue RNA used as negative controls were processed as described above but without MuLV RT. The reaction mixtures were incubated at 42°C for 60 min, denatured at 99°C for 5 min, and cooled on ice for 5 min.
Primers for the scPCR were designed using the Clone Manager software (Sci Ed Software, Cary, NC). The primers were as follows: mouse GnRH (239 nt product) accession number NM_008145, forward primer 21–40 nt, reverse primer 240–259 nt; mouse Kir6.2, which is intronless (262 nt product), accession number AF037313, forward primer 1302–1320 nt, reverse primer 1545–1563 nt; mouse SUR1 (227 nt product) accession number NM_011510, forward primer 3590–3610 nt, reverse primer 3795–3816 nt; mouse glucokinase (GK) (hexokinase IV; 388 nt product) accession number NM_010292, forward primer 165–183 nt, reverse primer 533–552 nt.
PCR was performed using 3 μl of cDNA template from each RT reaction in a 30 μl PCR volume containing the following: 6 μl of Colorless GoTaq Flexi buffer, 2–3 mm MgCl2 (concentration varies with each primer pair), 0.2 mm dNTPs, 2 U of GoTaq Flexi DNA polymerase (Promega), 0.22 μg of TaqStart Antibody (Clontech, Cambridge, UK), and 0.2 μm each of forward and reverse oligonucleotides. DNA polymerase and TaqStart Antibody were combined and incubated at room temperature for 5 min, and then the remainder of the reaction content was added to the tube. Fifty cycles of amplification were performed using an MJ Research (Watertown, MA) PTC-100 thermocycler in 0.5 ml of thin-walled PCR tubes according to one of the following protocols: 94°C, 2 min, 50 cycles of 94°C, 1 min; 58°C (Kir6.2), 60°C (GnRH), 61°C (SUR1, glucokinase), 1 min; 72°C, 1 min, with a final 72°C extension for 5 min. Ten microliters of the PCR products were visualized with ethidium bromide on a 2% agarose gel. In addition to the controls described above, harvested aCSF in the vicinity of the dispersed cells was used as a control in the RT-PCR.
Quantitative real-time PCR.
For SUR1, quantitative real-time PCR (qPCR) was performed using the Taqman Universal PCR mastermix (Applied Biosystems, Foster City, CA) with predesigned Taqman Gene Expression Assays (Applied Biosystems) (SUR1 assay ID Mm00803450_m1; β actin assay ID 4352341E) that include both primers and probes on the 7500 Fast Real-Time PCR System (Applied Biosystems). The assays were supplied with primers and probe concentrations of 900 and 250 nm, respectively. A multiplex qPCR contained 10 μl of 2× mastermix, 1 μl of 20× probe and primer for the target gene, 1 μl of 20× probe and primer for the control gene, 3 μl of cDNA and nuclease-free water to a 20 μl final volume. qPCR was performed on samples in duplicate under the following conditions: 95°C, 10 min; 45 cycles of amplification at 95°C, 15 s and 60°C, 1 min. For Kir6.2, qPCR was performed using the Power SybrGreen PCR mastermix (Applied Biosystems) method on the 7500 Fast Real-Time PCR System for increased sensitivity (the Taqman predesigned gene Expression Assay for Kir6.2 obtained from Applied Biosystems was less sensitive and did not consistently detect a product in GnRH neuronal pools). Primers were designed using the Clone Manager software. The primers were as follows: Kir6.2 (151 nt product) accession number AF037313, forward primer 1263–1282 nt, reverse primer 1394–1413 nt; β actin (63 nt product) accession number NM_007393, forward primer 849–867 nt, reverse primer 890–911 nt. The qPCR reaction for Kir6.2 or β actin contained 10 μl of 2× mastermix, 0.5 μm forward primer, 0.5 μm reverse primer, 2 μl of cDNA, and nuclease-free water to a 20 μl final volume. qPCR was performed on samples in duplicate under the following conditions: 95°C, 10 min; 45 cycles of amplification at 95°C, 15 s and 60°C, 1 min followed by a dissociation step for melting point analysis with 35 cycles of 95°C for 15 s, 60°C to 95°C in increments of 1°C for 1 min and 95°C for 15 s. Both Kir6.2 and SUR1 gene assays were tested to determine and compare the efficiencies of the target and control gene amplifications. Serially diluted cDNA (1:5, 1:10, 1:50, 1:100, 1:500, 1:2000) from mouse POA, each assayed in triplicates, was used to construct standard curves, and the efficiency was calculated according to the following formula: E = 10(−1/m) − 1, where m is the slope (Livak and Schmittgen, 2001; Pfaffl, 2001). Efficiencies were as follows: Kir6.2/β actin, 100/98%; SUR1/β actin, 100/97%. The Applied Biosystems Sequence Detection Software System (version 1.3) was used to generate a standard curve and to calculate the individual values for each sample.
Data analysis.
Eighty nine individual GnRH neurons were harvested from four intact mice and four OVX estradiol-treated mice. Because the number of neurons expressing Kir6.2, SUR1, or glucokinase did not differ between the two groups of animals, we combined the data from both groups of animals. Eight to 14 cells were harvested from each animal, and the mean number of cells expressing each transcript was determined for each animal and used for additional analysis of mean, SEM, and percentage expression. Similarly, the data from GnRH neuronal pools from OVX oil- and E2-treated mice were combined, because we found no difference in transcript expression between the two groups. Two to three pools of five cells each were harvested from each animal, and the average of neuronal pools expressing each transcript was determined and used for additional analysis of mean, SEM, and percentage expression.
For quantification of Kir6.2 and SUR1 expression differences between oil- and E2-treated animals, GnRH neuronal pools were analyzed using qPCR. Standard curves from diluted POA cDNA were included on each plate and used to calculate the relative mRNA expression (see Fig. 3A,D). Individual Kir6.2 and SUR1 values were then normalized to the endogenous control gene β actin, and the mean and SEM were calculated (see Fig. 3C,E). Statistical analysis was performed using the Student's two-tailed t test.
Figure 3.

qPCR analysis of Kir6.2 and SUR1 mRNA expression in GnRH neuronal cell pools. A–E, Kir6.2 expression was determined using the SYBR green method (A–C), and SUR1 expression was determined using the Taqman gene expression method (D, E). A, D, For both, cycle number is plotted against the normalized fluorescence intensity (Delta Rn) to visualize the PCR amplification. The cycle threshold (Ct) value (line with arrows) is the point in the amplification at which the sample values were calculated. A, D, POA cDNA serial dilutions and one representative GnRH neuronal pool (■-■) as well as the corresponding cycle number when the fluorescent signal was detected. B, The superimposed melting curves for Kir6.2 depict a single product. A, D, The standard curve regression line (inset) produced a slope of −3.3 for Kir6.2 and −3.2 for SUR1, which translates into similar efficiencies of 100%. C, E, Quantitative analysis of Kir6.2 and SUR1 mRNA expression in GnRH neuronal cell pools from oil- and E2-treated animals (mean ± SEM; n = 4 for each group).
Kir6.2 and SUR1 gene sequences were analyzed to look for estrogen response elements (EREs) using the Dragon Estrogen Response Element Finder, version 2 (Bajic et al., 2003). This program is a package for the specific discovery of estrogen response elements in DNA sequences. The consensus ERE is 5′-GGTCAnnnTGACC-3′, where n can be any nucleotide. To model the ERE, the program uses the Position Weight Matrix method in addition to the probability of pairing the half-sites by the transitional probabilities of the 3′ nucleotide of the 5′ half-site to the 5′ nucleotide of the 3′ half-site, ignoring spacer nucleotides. Details can be found on-line (http://sdmc.lit.org.sg/ERE-V2/index).
Kir6.2 (accession number AF037313) was found to have one ERE on the sense strand at the predicted position 1050 nt, GA-GGTCC-AGG-TGACC-AT. SUR1 (accession number NM_011510) was found to have one ERE on the sense strand at the predicted position 2752 nt, GT-GGTCT-TGG-TGACC-CA.
Superfusion of preoptic-mediobasal hypothalamus and measurement of GnRH.
Adult male rats were anesthetized with CO2 and quickly decapitated at 11:00 A.M. The preoptic-mediobasal hypothalamic tissue (POA-MBH) was rapidly dissected away from the brain and placed in a superfusion chamber. Three tissues per chamber were placed in the superfusion apparatus (Brandel 6000; Brandel, Gaithersburg, MD). The POA-MBH is defined rostrally by the anterior-most POA, laterally by the hypothalamic sulci, and caudally by the anterior extremity of the mammillary bodies, 1 mm dorsal to the surface of the brain. The superfusion medium, M199 (Invitrogen, Grand Island, NY), was supplemented with 1.25 g/L bacitracin and 2.2 g/L sodium bicarbonate and then equilibrated with 95%O2/5%CO2 before use. Throughout the experiments, the medium was continuously oxygenated with 95%O2/5%CO2. The superfusion flow rate of 80 μl/min (M199 medium) was maintained by Manostat Peristaltic Pump (Barnant Company, Barrington, IL). Concentrations of tolbutamide and diazoxide for the treatment were 100 and 200 μm, respectively. Tissues were superfused for 45 min as an equilibration period before collection of superfusates. Then, collection proceeded with media switches occurring at 45, 60, 120, 135, and 180 min. The four experimental groups consisted of tissues that were exposed to one of the following regimens of media in series: (1) B,B,B,B,K+; (2) B,B,D,B,K+; (3) B,B,T,B,K+; and (4) B,D,T&D,B,K+, where B is blank medium, T is tolbutamide, D is diazoxide, and K+ is 60 mm KCl solution. For each of the four groups, n = 4 chambers were superfused. The 60 mm KCl-containing medium (Invitrogen) was applied after drug challenges to test the viability of the tissues. Superfusate fractions were collected at 10 min intervals, boiled for 20 min, and stored at −20°C for subsequent GnRH radioimmunoassay.
Radioimmunoassay of GnRH was performed as described previously (Chappell and Levine, 2000) using the EL-14 GnRH antiserum (Ellinwood et al., 1985). The intra-assay and interassay coefficients of variance for the assays were 14.2 and 8.2%, respectively. Superfusion data were analyzed by two-way ANOVA with repeated measures and post hoc analysis with the Bonferroni's test.
Results
Identification of GnRH neurons and general characterization of membrane and ion channel properties
Whole-cell recordings were made from 92 female and 50 male GnRH-EGFP neurons from adult mice (DBA-C57BL/6J background) using visualized, whole-cell patch recording. We were confident that all of the cells that we targeted in the DB-POA were GnRH neurons based on our immunocytochemical staining, and scRT-PCR results that 99% of the neurons in this region expressing EGFP were identified as GnRH (Figs. 1A, 2A). For the electrophysiology analysis, only GnRH cells with gigaohm or better seals were included in this study. The mean input resistance was 1.1 ± 0.1 GΩ for female and 1.3 ± 0.1 GΩ for male mice. The mean resting membrane potential was −61.9 ± 1.3 mV for male and −63.3 ± 1.2 mV for female GnRH neurons at a 0 pA holding current. Moreover, all GnRH neurons showed a transient outward K+ current (IA) as described by DeFazio and Moenter (2002) (Fig. 1B). In addition, 50% of the neurons exhibited a time-dependent, hyperpolarization-activated, cation current (Ih) that was potently blocked by the selective h-channel antagonist ZD 7288 (Fig. 1C).
Figure 1.
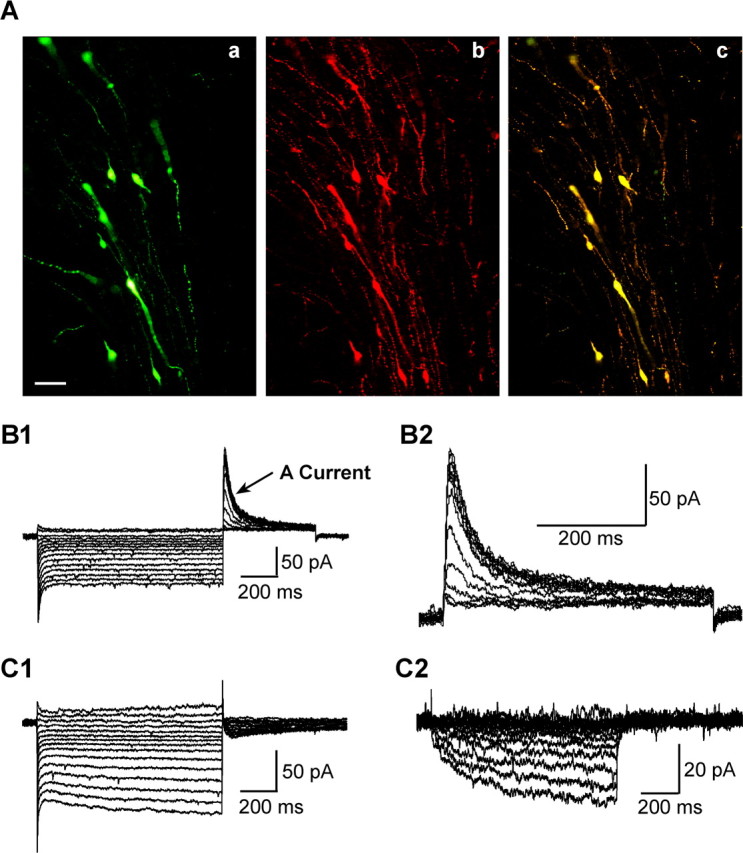
Characteristics of mouse GnRH neurons in hypothalamic slices. A, Identification of EGFP-labeled GnRH neurons by immunocytochemical staining for GnRH peptide. Aa, EGFP-GnRH neurons in a thin section (15 μm) of a coronal preoptic slice from a female mouse. Ab, The immunocytochemical staining of same section. Ac, Overlay of Aa and Ab. Scale bar, 25 μm. B, B1, Representative whole-cell voltage-clamp recordings showing that female mouse EGFP-GnRH neurons express transient outward potassium current (IA). Whole-cell currents were elicited by holding the cell at −60 mV and giving a series of 1 s prepulses ranging from −50 to −120 mV (in 5 mV increments) and then stepping back to −50 mV for 0.5 s to activate IA. B2, Expanded time scale of A-current activated at −50 mV after leak subtraction (see Materials and Methods). C, C1, A representative whole-cell voltage-clamp recording showing that female mouse EGFP-GnRH neurons express a hyperpolarization-activated cation current (Ih). Whole-cell currents were elicited by holding the cell at −60 mV and giving a series of 1 s prepulses ranging from −50 to −120 mV (in 5 mV increments) and measuring the slow activating steady-state current at the last 100 ms of the 1 s pulse. C2, ZD 7288 (50 μm)-sensitive component of the whole-cell currents shown in C1.
Figure 2.
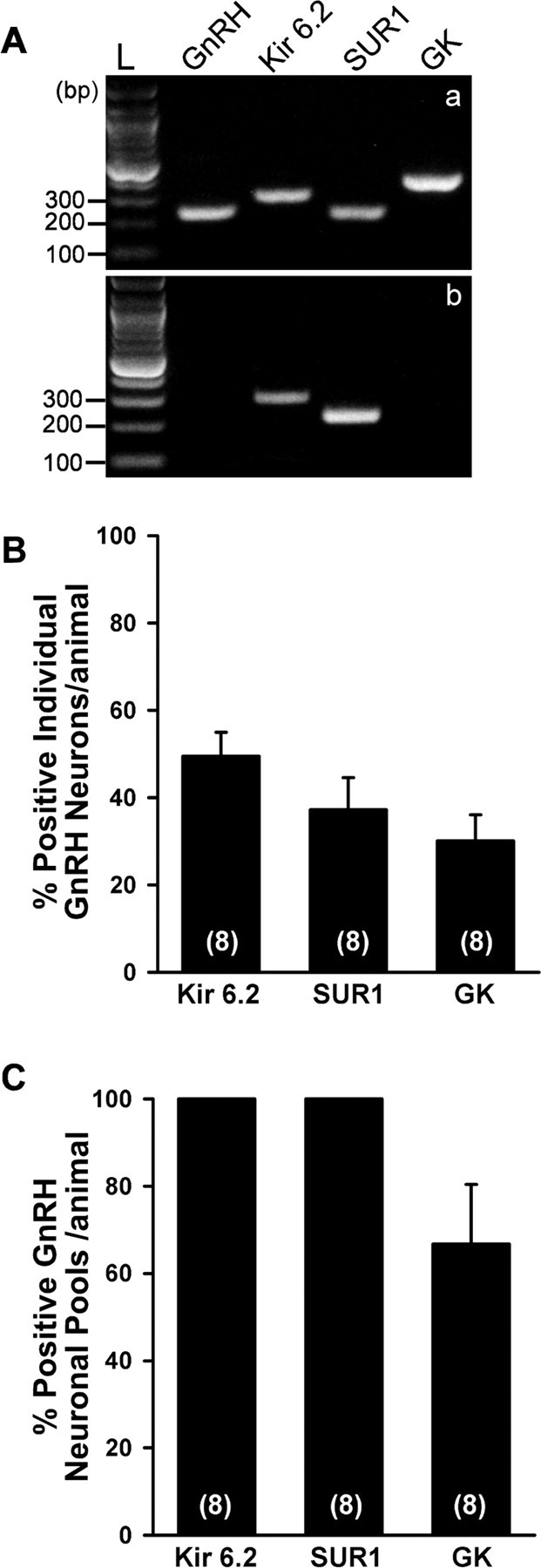
RT-PCR identification of KATP channel subunits and GK transcripts in GnRH neurons. A, A representative gel illustrating that harvested EGFP-GnRH neurons express mRNA for Kir6.2, SUR1, and GK (a). However, nonfluorescence (non-FL) cells (n = 12) expressed Kir6.2 (50%) and SUR1 (25%), but not GnRH or GK transcripts (b). In addition, the following controls were included: a tissue control and a cell without MuLV RT, aCSF from the dispersed cellular milieu, and water blank, all of which were negative after RT-PCR (data not shown). A base pair ladder (L) is given for determining the relative size of the transcripts. B, Quantitative analysis of Kir6.2, SUR1, and GK mRNA expression in individual GnRH neurons from eight animals, 8–14 cells per animal. Percentage expression was determined for each animal, and the mean ± SEM was calculated (n = 8). C, Quantitative analysis of Kir6.2, SUR1, and GK mRNA expression in GnRH neuronal pools from eight animals, two to three pools per animal. Percentage expression was determined for each animal, and the mean ± SEM was calculated (n = 8).
Expression of Kir6.2 and SUR1 transcripts in GnRH neurons
We used scRT-PCR to determine whether GnRH neurons express KATP channel transcripts and to ascertain the molecular composition of these channels in GnRH neurons (Fig. 2A). The analysis of 89 individual GnRH neurons from four intact and four E2-treated female mice revealed that 49.5 ± 5.5% of GnRH neurons express Kir6.2, 37.2 ± 7.3% express SUR1, and 24.7 ± 4.1% coexpress Kir6.2 and SUR1 transcripts (Fig. 2B). A similar profile of expression of Kir6.2 and SUR1 was measured in male GnRH neurons (data not shown). SUR2B transcripts were not detected in GnRH neurons (data not shown). GK (hexokinase IV) is thought to be the critical regulator of glucose-sensing in ventromedial hypothalamic neurons, and this high Km hexokinase is also present in pancreatic β cells (Dunn-Meynell et al., 2002). Using specific primers for glucokinase mRNA, we detected GK transcripts in GnRH neurons (30.1 ± 6.0%) but not in adjacent non-GnRH neurons (Fig. 2A).
Based on the whole-cell electrophysiology data in which the vast majority of GnRH neurons responded to selective Kir6.2/SUR1 openers and blockers (see below), we reasoned that the mRNA expression of these subunits must be below the level of detection in individual neurons. Therefore, we increased the template for the RT-PCR by pooling five individual GnRH neurons. Nine pools were harvested from four oil-treated animals and seven pools from four E2-treated animals. Indeed, by using these pools, we found that 100% of the neuronal pools expressed Kir6.2 transcripts and 100% expressed SUR1 transcripts. In addition, 66.7 ± 13.7% of the pooled cells expressed GK mRNA (Fig. 2C).
We used real-time PCR to quantify Kir6.2 and SUR1 mRNA expression in GnRH neuronal pools from oil- and E2-treated animals. As expected, the uterine weights of the E2-treated group (152 ± 10.5 mg) were significantly greater than the uterine weight of the oil-treated group (21 ± 1.4 mg), indicative of the circulating levels of estrogen (Christian et al., 2005). However, neither Kir6.2 nor SUR1 mRNA expression was altered with estrogen treatment (Fig. 3C,E).
Pharmacological identification of the subtypes of KATP channel expressed in GnRH neurons
To confirm the results of scRT-PCR, we measured whole-cell currents in GnRH neurons from intact female mice in response to the classical KATP channel opener diazoxide and the blocker tolbutamide. In the presence of 1 μm TTX and whole-cell voltage clamp, bath application of 300 μm diazoxide induced an outward current that was reversed by 100 μm tolbutamide (Fig. 4A) in all of the GnRH neurons examined. The maximum amplitude of the responses depended on the steroid state of the females (see below). The reversal potential (−80 mV) was close to the predicted Nernst equilibrium potential for potassium (EK+) (Fig. 4C). To further define the subtype of the SUR subunit, we compared the responses to MCC-134, a selective opener for SUR2A/2B but an inverse agonist for SUR1-containing KATP channels (Shindo et al., 2000), and diazoxide, a selective opener for SUR1 and SUR2B-containing KATP channels (SUR2A-containing KATP channels can only be activated by diazoxide in the presence of MgADP) (Babenko et al., 1998; D'hahan et al., 1999). Ninety-one percent of the cells tested (n = 11) showed a diazoxide (300 μm)-induced but not a MCC-134 (300 μm)-induced outward potassium current (Fig. 4B). Only one cell responded to the MCC-134 in addition to diazoxide response, but we did not measure any SUR2B transcripts in GnRH neurons (data not shown). Similar to females, bath application of 300 μm diazoxide induced an outward current in male GnRH neurons that was reversed by 100 μm tolbutamide (Fig. 5A) in all of the GnRH neurons examined (n = 6). The average current amplitude was 35.2 ± 5.3 pA, and the reversal potential was at the predicted EK+ (Fig. 5B). Therefore, similar to pancreatic β-cells, GnRH neurons express primarily Kir6.2/SUR1-type KATP channels based on both scRT-PCR and electrophysiological/pharmacological data.
Figure 4.
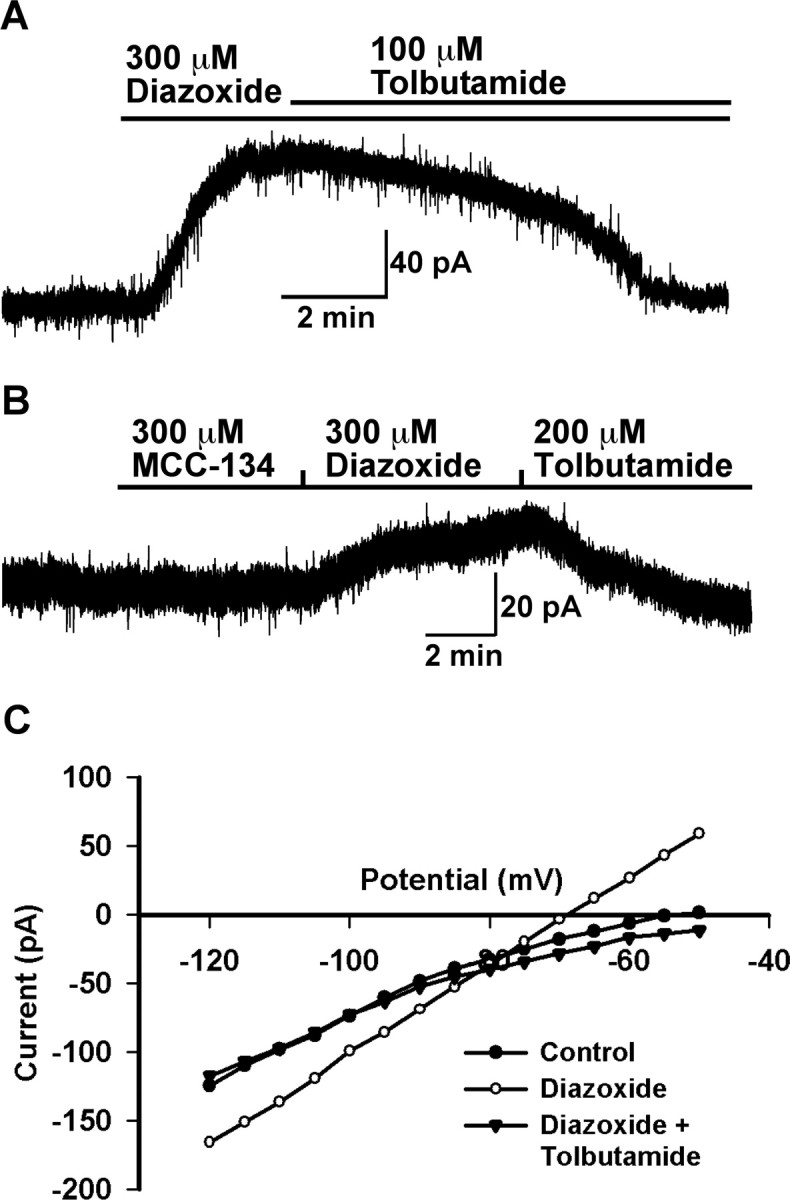
Pharmacological identification showed that female mice GnRH neurons mainly express SUR1-containing KATP channels. A, A representative recording showing that GnRH neurons express diazoxide- and tolbutamide-sensitive KATP channels. Diazoxide induced an outward current (76 pA) in a GnRH neuron, which was reversed by tolbutamide. Vhold = −60 mV. B, A representative recording showing that MCC-134 (300 μm), a selective SUR2-containing KATP channel opener, did not induce an outward current in a GnRH neuron, but diazoxide (300 μm) induced an outward current of 20 pA that was antagonized by tolbutamide (200 μm). Vhold = −60mV. C, Current–voltage plot taken from another cell at the time immediately before (control) and after application of diazoxide and tolbutamide. The diazoxide-induced outward current had a reversal potential of −80 mV, which is close to the predicted EK+. The voltage protocol consisted of 1 s steps every 5 mV from −50 to −120 mV. Vhold = −60 mV.
Figure 5.

Male GnRH neurons express diazoxide/tolbutamide-sensitive KATP current, and tolbutamide stimulates GnRH release from superfused POA-MBH tissues of adult male rats. A, Representative recording of diazoxide-induced outward current (30 pA) in a GnRH neuron that was reversed by tolbutamide. Vhold = −60 mV. B, Current–voltage plot taken from the recording shown in A at the time immediately before (Control) and after application of diazoxide. C, GnRH release rates (mean ± SEM) in successive 10 min superfusate collections, in which tissues were exposed to blank medium only, or diazoxide-containing medium at the times indicated. All tissues were exposed to isotonic medium containing 60 mm K+ after the drug challenges to confirm the viability of the preparations. D, GnRH release from tissues that were superfused with tolbutamide-containing medium, or tolbutamide and diazoxide-containing medium during the times indicated (* p < 0.05).
As further confirmation of the pharmacology and the role of KATP channels in modulating the excitability of GnRH neurons, we measured the effects of diazoxide and tolbutamide on the GnRH release from preoptic-hypothalamic blocks dissected from male rats. Under baseline conditions, GnRH was released from single preoptic-hypothalamic tissues at a steady rate of ∼50 pg/ml/10 min that was significantly augmented by perfusion with 60 mm KCl (Fig. 5C). Moreover, tolbutamide (100 μm) significantly increased the secretion of GnRH by twofold to threefold, and this was completely prevented by coperfusion of diazoxide (Fig. 5D). Perfusion with diazoxide alone resulted in GnRH release that tended to be lower than control values, but this was not a statistically significant effect (p > 0.05). The stimulated release by tolbutamide was equivalent to the KCl-stimulated release. Therefore, tolbutamide increases the excitability of GnRH neurons to the extent that it causes an augmentation of GnRH secretion.
GnRH neurons are responsive to changes in glucose concentrations
The major function of KATP channels as found in pancreatic β cells is to respond to changes in circulating levels of glucose. In the present study, we tested the response of GnRH neurons to low glucose concentrations (Fig. 6A,B) using loose-cell attached recording. Usually, the firing rate of GnRH neurons is low and varies over time (Nunemaker et al., 2001; Kuehl-Kovarik et al., 2002). Therefore, to obtain a relatively higher and stable firing rate in GnRH neurons, we perfused high potassium (15 mm) to increase the firing rate of GnRH neurons. In this case, high Mg2+/low Ca2+ CSF containing blockers for GABAA (100 μm picrotoxin) and ionic glutamate receptors (20 μm CNQX plus 20 μm d-APV) was used. In loose cell-attached recording, when the concentration of glucose decreased from 2.5 to 0.1 mm for 10–15 min, the firing rate of GnRH neurons decreased from 3.1 ± 0.5 to 0.6 ± 0.2 Hz. However, the firing rate returned to control levels (2.8 ± 0.7 Hz) when the glucose concentration was restored to 2.5 mm (Fig. 6). Five of nine GnRH neurons (56%) responded in such a manner. Hence, the glucose sensitivity of GnRH neurons appears to be similar to that of ventromedial and arcuate hypothalamic neurons (Song et al., 2001; Ibrahim et al., 2003; Wang et al., 2004).
Figure 6.
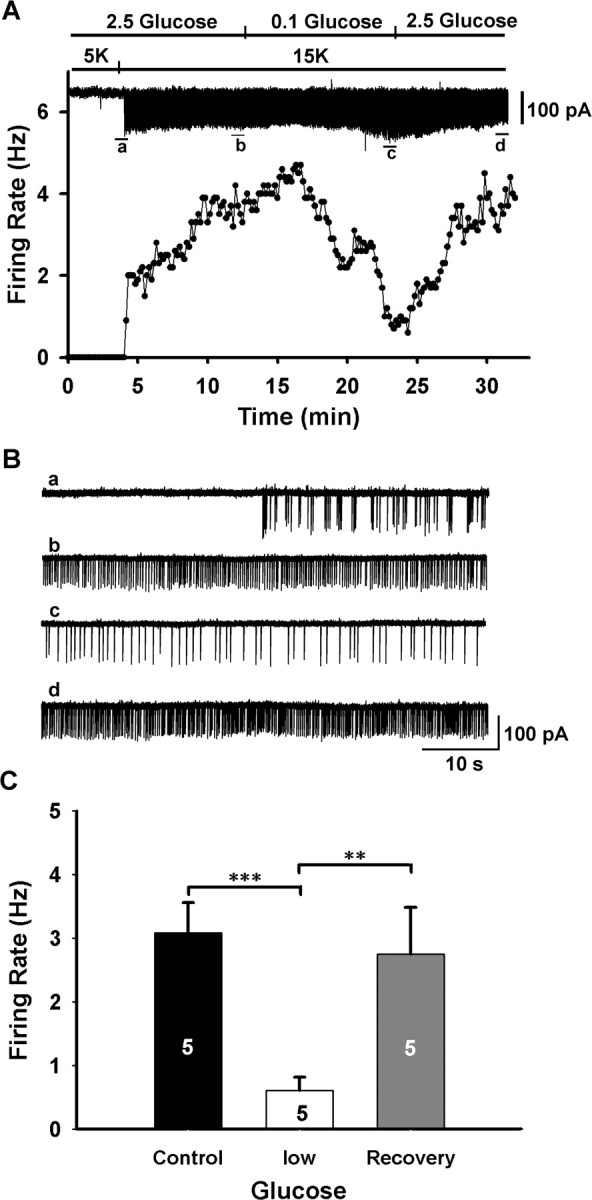
GnRH neurons are responsive to changes in glucose concentrations. A, Top, A representative loose cell-attached record showing the time course of the effect of low glucose (0.1 mm) on high K+ (15 mm) induced firing in a GnRH neuron. High Mg2+/low Ca2+ aCSF containing blockers for GABAA (100 μm picrotoxin) and ionic glutamate receptors (20 μm CNQX plus 20 μm d-APV) were used to isolate the cell from presynaptic input. Bottom, Firing frequency-time curve corresponding to the top record. B, Expanded recording traces indicated in the top panel of A. C, Summary of the effects of low glucose (0.1 mm) on the firing rate of GnRH neurons (n = 5 per group; **p < 0.01, *** p < 0.001).
To further verify that the major effects of altered glucose concentrations were mediated directly on GnRH neurons, we measured synaptic currents with altered glucose concentrations. Whole-cell recordings were done with the KCl internal solution (see Materials and Methods) in the presence of TTX, and the GnRH neurons were clamped at −60 mV. Under these conditions, both glutamatergic and GABAergic currents were measured as inward currents, mPSCs, which were analyzed off-line using Minianalysis (Synaptosoft, Decatur, GA). There was no change in mPSC frequency when glucose concentrations decreased from 2.5 to 0.1 mm, but there was a significant decrease (p < 0.01) in mPSC amplitude (from 27.5 ± 0.3 to 19.9 ± 1.2 pA; n = 4) that fully recovered (26.8 ± 1.4 pA) with a return to normal (2.5 mm) glucose concentrations. Therefore, the effects of altered glucose concentrations on mPSC amplitude but not frequency would indicate that the major effects of altered glucose concentrations are mediated postsynaptically on GnRH neurons, potentially through the shunting actions of an increase KATP channel conductance in these cells.
GnRH neurons are sensitive to metabolic perturbations
Because GnRH neurons were responsive to changes in glucose concentrations, we also wanted to ascertain the effects of metabolic inhibition on the excitability of these neurons. Therefore, we used loose cell-attached recordings to measure the effects of the metabolic inhibitor NaN3 on the firing rate of GnRH neurons. For this series of experiments, to obtain a relatively higher and stable firing rate in GnRH neurons, we perfused Kisspeptin (5 nm), a specific agonist that activates GnRH neurons (Han et al., 2005), to increase the firing rate. Once a stable firing rate was achieved, 1 mm NaN3 was applied to the cell and within ∼8 min, the firing rate was significantly inhibited (Fig. 7A). The inhibition by NaN3 was easily reversed within 4 min after washing out (Fig. 7A) or antagonism by tolbutamide (Fig. 7B). The results are summarized in Figure 7C, which shows that the mean firing rate in 1 mm NaN3 (0.08 ± 0.03 Hz; n = 4) was significantly lower than in control conditions (1.4 ± 0.2 Hz, n = 4, before metabolic inhibition; 1.7 ± 0.4 Hz, n = 4, after return to normal conditions). We also measured the effects of metabolic inhibition under whole-cell recording conditions with 3 mm ATP in the pipette (Fig. 7D), similar to what has been used in hippocampal CA1 neurons (Matsumoto et al., 2002). Application of 2 mm NaN3 caused hyperpolarization after 5–7 min that was reversed after washout. The averaged amplitude of hyperpolarization of GnRH neurons was −8.4 ± 1.1 mV (n = 7). Interestingly, metabolic inhibition produced the opposite response (i.e., depolarization) in hippocampal CA1 pyramidal neurons (data not shown), similar to what has been reported previously (Matsumoto et al., 2002). This indicates that metabolic inhibition hyperpolarizes GnRH neurons and that KATP channels may offer neuroprotection from metabolic insult, as has been suggested for other neuronal phenotypes (Liss and Roeper, 2001).
Figure 7.

GnRH neurons are sensitive to metabolic inhibition. A–C, Loose cell-attached patch recordings in the presence of 5 nm Kisspeptin-10, which was used to stimulate the firing activity. A, A representative recording showing that sodium azide (NaN3) strongly inhibited the kisspeptin induced firing of a GnRH neuron (top trace), and this inhibition was reversed after washout of sodium azide (bottom trace). B, The NaN3 inhibition was reversed by tolbutamide. C, Summary of the effects of sodium azide on the firing rate of GnRH neurons (n = 4; *p < 0.05, **p < 0.01). D, Whole-cell, voltage-clamp recording with 3 mm ATP in pipette showing that NaN3 strongly hyperpolarized a GnRH neuron by 7 mV, which reversed when returned to control conditions (washout). Vhold = −60 mV.
Estrogen increases KATP channel current
It is well documented that the neurosecretion of GnRH from preoptic GnRH neurons is regulated by estrogen feedback. However, the cellular mechanisms underlying feedback action of estrogen are primarily unexplored. Inward rectifying potassium channels are widely expressed in neurons and are hormonally regulated, in large part, through their coupling to metabotropic receptors. Because we established the expression of KATP channels in mouse GnRH neurons, we tested whether estrogen regulates the function of this inwardly rectifying K+ channel in GnRH neurons. Adult animals were ovariectomized and implanted with oil or E2 capsules and killed after 4–7 d. The uterine weight of the E2-treated group was 119.5 ± 6.9 mg and significantly greater (p < 0.001) than the uterine weight from the oil-treated group (18.3 ± 1.4 mg), indicative of the circulating levels of estrogen (Christian et al., 2005). GnRH neurons were voltage clamped at −60 mV in the whole-cell configuration, and diazoxide was applied to the cell through bath perfusion. As shown in Figure 8, diazoxide induced a whole-cell current in GnRH neurons from estrogen-treated animals (27.0 ± 3.4 pA; n = 14) that was twofold greater than the current from oil-treated animals (14.7 ± 1.5 pA; n = 11; p < 0.01). There was no difference in the whole-cell capacitance between the two groups (oil, 10.4 ± 0.6 pF vs estrogen, 10.6 ± 0.5 pF).
Figure 8.
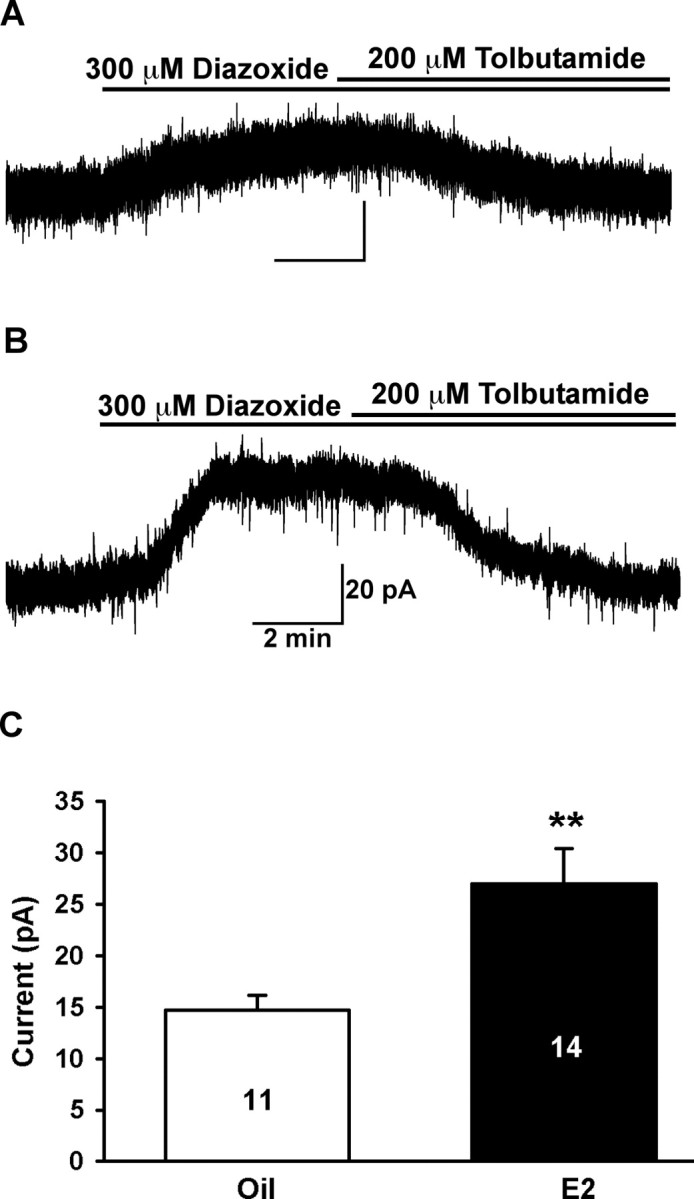
Estrogen enhanced the diazoxide-induced KATP channel currents in female GnRH neurons. Female mice were ovariectomized and implanted with oil or estrogen capsule for 4–7 d (see Materials and Methods). A, Whole-cell voltage-clamp recording in a GnRH neuron from an oil-treated female. Diazoxide (300 μm) induced an outward current of 16 pA that was antagonized by tolbutamide (200 μm). Vhold = −60 mV. B, Whole-cell voltage-clamp recording from a GnRH neuron from an estrogen-treated female. Diazoxide induced an outward current of 38 pA that was antagonized by tolbutamide. Vhold = −60 mV. C, Summary of the differences in the diazoxide-induced current in GnRH neurons between oil- and estrogen-treated females (**p < 0.01).
Estrogen enhances KATP- induced changes in membrane excitability
Because estrogen treatment increased the KATP channel conductance in GnRH neurons, we measured the contribution of KATP channels to the cell excitability. There was no difference in the mean resting membrane potential of GnRH neurons between estrogen- and oil-treated, OVX females (E2: −64.6 ± 1.7 mV, n = 8, vs oil: −62.4 ± 1.7 mV, n = 10). Although estrogen treatment per se did not affect the resting membrane potential, it did potentiate both the tolbutamide-induced depolarization (E2: 6.3 ± 0.5 mV, n = 5, vs oil: 3.8 ± 0.4 mV, n = 5; p < 0.01) and the diazoxide-induced hyperpolarization (E2: −11.1 ± 0.7 mV, n = 5, vs oil: −6.6 ± 1.1 mV, n = 5; p < 0.01) of GnRH neurons (Fig. 9A,B).
Figure 9.
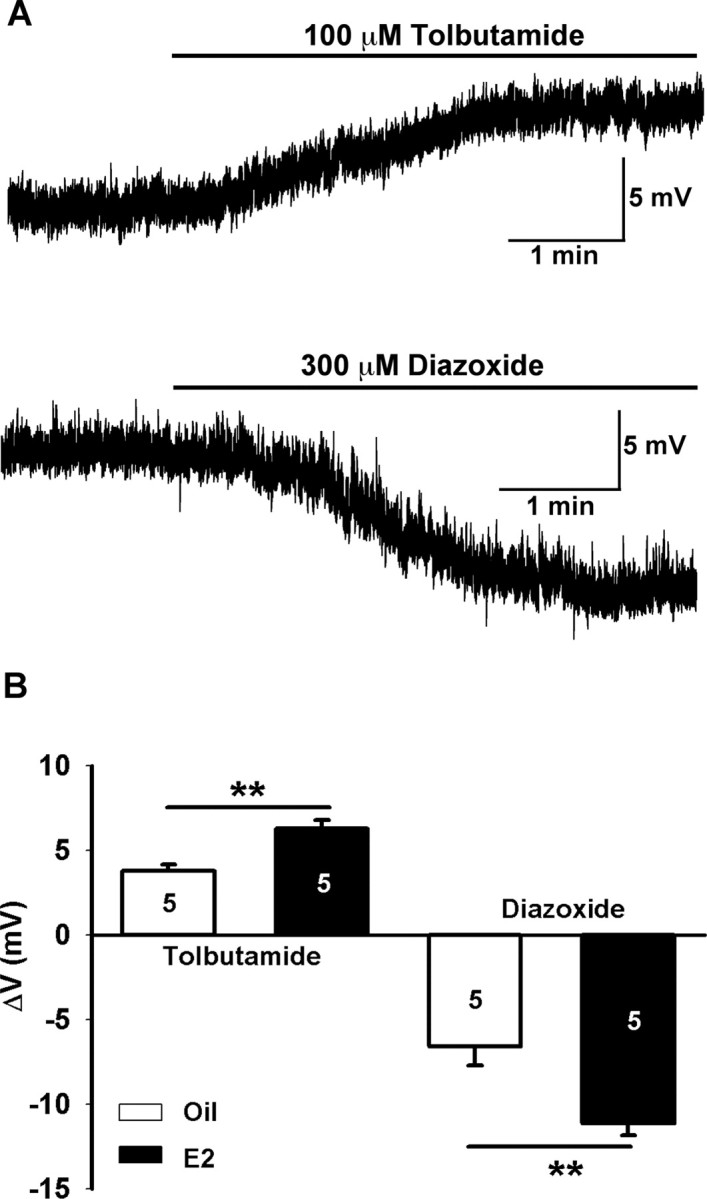
Tolbutamide differentially depolarizes and diazoxide hyperpolarizes GnRH neurons from oil- and estrogen-treated, ovariectomized female mice. A, Representative recordings from estrogen-treated female showing that tolbutamide induced a depolarization of 6 mV in one cell and diazoxide a hyperpolarization of 12 mV in another cell. B, Summary of the effects of tolbutamide and diazoxide on membrane potentials in GnRH neurons from oil- and estrogen-treated females (n = 5; **p < 0.01).
Furthermore, we measured the effects of tolbutamide on the spontaneous firing rate of female GnRH neurons in current-clamp recordings. It is generally believed that GABAergic and glutamatergic neurons comprise the major presynaptic input to GnRH neurons (Herbison, 2006). Thus, to prevent the indirect effect of tolbutamide on GnRH neurons through presynaptic neurons, we applied tolbutamide in the presence of ionotropic glutamate receptor blockers CNQX (20 μm) and AP-5 (50 μm) and GABAA receptor blocker picrotoxin (100 μm). In the absence of tolbutamide, the firing rate of E2-treated GnRH neurons was significantly lower than oil-treated GnRH neurons. Tolbutamide application increased the firing rate to a similar extent in both groups and induced continuous firing (Fig. 10A–C). However, when the increment in the firing rate was compared, tolbutamide increased the firing rate significantly more in estrogen-treated than in oil-treated, OVX females (E2: 4.6 ± 0.3 Hz, n = 5, vs oil: 3.1 ± 0.4 Hz, n = 5; p < 0.05)
Figure 10.

Tolbutamide differentially increased the firing rate of GnRH neurons from estrogen- and oil-treated, ovariectomized females. A, A representative recording from E2-treated female mouse showing spontaneous burst firing. Tolbutamide increased the firing rate and caused continuous firing. Resting membrane potential (RMP), −65 mV. B, Another representative recording from an oil-treated female showing a higher baseline firing rate. Tolbutamide also caused continuous firing. RMP, −63 mV. C, Summary of the effects of tolbutamide on the firing rate of GnRH neurons from oil- and E2-treated females. GnRH neurons from E2-treated females had significantly lower basal firing rates than GnRH neurons from oil-treated females (++p < 0.01; n = 5). Tolbutamide significantly increased the firing rate in both groups (oil, **p < 0.01, n = 5; E2, ***p < 0.001, n = 5). D, Tolbutamide increased the firing rate of GnRH neurons more in E2-treated versus oil-treated females (*p < 0.05).
Discussion
For the first time, we determined that GnRH neurons express Kir6.2/SUR1 KATP channel subunits that modulate the excitability of GnRH neurons in an estrogen-sensitive manner. In addition, the KATP channels were activated during metabolic stress, which would indicate that these channels play a neuroprotective role. Most importantly, a subset of the GnRH neurons expressed glucokinase, which rendered them glucose responsive. Therefore, it appears that GnRH neurons can process multiple neural and humoral inputs to generate the appropriate neurosecretory output during different metabolic states, which is vital for control of the reproductive cycle.
GnRH neurons predominately express Kir6.2/SUR1 KATP channel subunits
Both male and female mice GnRH neurons express functional KATP channels, specifically Kir6.2 and SUR1 subunits. Greater than 90% of GnRH neurons responded to the KATP channel opener diazoxide with an outward current, which exhibited weak rectification and was blocked by tolbutamide. The scRT-PCR data substantiated our electrophysiological findings that GnRH neurons express Kir6.2 and SUR1 subunit transcripts. Thus, the combined electrophysiology, pharmacology, and molecular biology techniques have clearly identified functional KATP channels in GnRH neurons. Finally, in an in vitro hypothalamic thick slice preparation, we were able to measure GnRH release that was significantly augmented by bath perfusion of tolbutamide, an effect that was completely blocked by coperfusion with diazoxide, providing additional evidence for the critical contribution of KATP channels to GnRH neuronal excitability.
Hypothalamic pro-opiomelanocortin (POMC) neurons express the same compliment of KATP channel subunits, which renders them glucose responsive (Ibrahim et al., 2003). In general, KATP channels couple membrane excitability to cellular metabolism by directly sensing and integrating intracellular concentration changes of nucleotides such as ATP and ADP (Reimann and Ashcroft, 1999). Sulfonylurea binding and electrophysiological studies have characterized KATP channels with different properties in a variety of cell types, including many CNS neurons (Babenko et al., 1998). The Kir6.2 and SUR1 subunits are widely distributed in rat brain and are present in neurons expressing tyrosine hydroxylase, NPY, and glutamic acid decarboxylase (GAD65) (Dunn-Meynell et al., 1998; Miki et al., 2001; Ibrahim et al., 2003). Previous studies have shown that GABAergic and dopamine neurons in the substantia nigra and ventral tegmental dopamine neurons also express both subunits (Liss et al., 1999, 2005; Liss and Roeper, 2001). In cells that express Kir6.2 and SUR1 channel subunits, diazoxide and metabolic inhibition open the channel, and the sulfonylurea tolbutamide closes the channel.
GnRH neurons respond to changes in glucose concentrations and metabolic inhibition
The reproductive activity of mammals is linked to metabolic state (for review, see Wade and Jones, 2004). In fact, there appears to be a positive correlation between circulating glucose levels and pulsatile LH release (Medina et al., 1998; Ohkura et al., 2000). In addition, reproductive behavior is affected by nutritional status, which is dependent on the expression of CNS GnRH (type II) receptors at least in shrews (Temple et al., 2003; Kauffman and Rissman, 2004). However, it is believed that metabolic signals are transmitted to GnRH neurons via the area postrema and the mediobasal hypothalamus (Wade and Jones, 2004; The ESHRE Capri Workshop Group, 2006). Mediobasal hypothalamic POMC and NPY neurons project directly onto GnRH to modulate GnRH neuronal activity through μ-opioid and NPY5 receptors, respectively (Lagrange et al., 1995; Campbell et al., 2001; Zheng et al., 2005). However, until now, there has been little evidence that GnRH neurons respond directly to metabolic factors such as glucose. Notably, the anatomical location of the GnRH neurons in the organum vasculosum of the lamina terminalis area, a site of a leaky blood–brain barrier, permits direct access of metabolic factors to these neurons. Indeed, we found that high concentrations of glucose excited GnRH neurons and low glucose and metabolic inhibition uniformly inhibited the firing activity of GnRH neurons. Therefore, GnRH neurons can be classified “glucose-responsive” neurons based on the original definition (Oomura et al., 1969). Interestingly, 20 years ago, it was shown that ventromedial nucleus (VMH) neurons accelerate their firing activity through a decrease in a K+ conductance in response to increasing glucose concentrations (Minami et al., 1986). In GnRH neurons, there is a direct correlation between firing rate and the glucose sensitivity of GnRH neurons, which is similar to that of POMC neurons (Ibrahim et al., 2003). Therefore, similar to other hypothalamic neurons, glucose responsiveness in GnRH neurons can be linked to KATP channel activity (Minami et al., 1986; Muroya et al., 1999; Song et al., 2001; Ibrahim et al., 2003; Wang et al., 2004; Song and Routh, 2005).
In addition to providing a mechanism for monitoring circulating glucose concentrations, KATP channels are an avenue for the actions of leptin, insulin, and free fatty acids in the brain (Spanswick et al., 1997, 2000; Lam et al., 2005). For example, KATP channels transduce, at least in part, the effects of leptin and insulin in POMC neurons (Plum et al., 2006). This would indicate that the KATP channels play a more global role in controlling energy homeostasis. We found that GnRH neurons responded to metabolic inhibition with a pronounced hyperpolarization and a decrease in firing activity, which was antagonized by tolbutamide. Hence, in states of low energy availability, GnRH neuronal activity would be inhibited, which would correlate with decreased reproductive states. Indeed, the LH pulse frequency in ewes is correlated with central glucose levels (Ohkura et al., 2000). Moreover, in the extreme situation of starvation, the reproductive cycle in humans is abrogated (The ESHRE Capri Workshop Group, 2006).
It is believed that one requirement for cells to be glucose responsive is that they express glucokinase, a high Km hexokinase (IV), which is found in pancreatic β cells and VMH neurons (Dunn-Meynell et al., 2002; Kang et al., 2006). Indeed, the Levin laboratory has shown that transfection of VMH neurons with small interfering glucokinase RNA abrogated virtually all of the glucose responsiveness of these neurons (Kang et al., 2006). We measured glucokinase mRNA in 65% of the pooled GnRH neurons that corresponded to the percentage of GnRH neurons that were glucose responsive. In addition, a recent report has shown that glucokinase (partial) knock-out mice exhibit reduced fertility (reduced liter sizes) consistent with the role of GnRH neurons as glucose-responsive neurons (Yang et al., 2007).
Estrogen upregulates KATP channel function in GnRH neurons
Perhaps unique to GnRH neurons, estrogen increased the diazoxide-induced outward (KATP channel) current by approximately twofold. We investigated whether this change could affect the function of GnRH neurons, because it is known that these K+ channels play a critical role in modulating excitability (Reimann and Ashcroft, 1999). Under whole-cell recording conditions with 2 mm ATP in the pipette solution, there was a small (−2 mV) increase in the resting membrane potential of GnRH neurons from estrogen-treated animals. However, application of tolbutamide produced a significant depolarization (∼4 mV) from oil-treated animals and an even greater depolarization (∼6 mV) of GnRH neurons from estrogen-treated animals. Likewise, the KATP channel opener diazoxide hyperpolarized GnRH neurons from the estrogen-treated females to a greater extent (∼11 mV) than GnRH neurons from oil-treated animals (∼7 mV). This increased activity during the estrogen-negative feedback state (Christian et al., 2005) does not appear to be the result of increased expression of the channel based on our quantitative PCR results (Fig. 3). Therefore, it is more likely that there is a change in signaling molecules that impinge on the channel. For example, changes in PIP2 levels can significantly affect the channel kinetics (Shyng and Nichols, 1998). Indeed, we found that activation of serotonin 5HT2C receptors inhibit GIRK (G-protein-gated inwardly rectifying K+ channel) channel activity in POMC neurons through direct hydrolysis of PIP2 by phospholipase C (Qiu et al., 2007). Therefore, E2 via an mER receptor that is Gq coupled (Qiu et al., 2003) may have a similar effect in GnRH neurons. In contrast, the effects of estrogen may be indirect via altered synaptic input (Rønnekleiv and Kelly, 2005; Herbison, 2006). Regardless of the mode and mechanism of action, estrogen increases KATP channel activity in GnRH neurons, which would significantly affect the overall excitability of these vital neurosecretory neurons.
As a functional assay of overall excitability, we measured the effects of tolbutamide on the spontaneous firing rate of GnRH neurons with glutamate and GABA ionotropic receptors blocked. Under baseline conditions, GnRH neurons fired in an episodic or bursting mode, and tolbutamide uniformly converted GnRH neurons to a continuous firing mode. However, the net increase in tolbutamide-induced firing was significantly greater in GnRH neurons from estrogen-treated animals. Therefore, it appears that KATP channels govern the firing activity of GnRH neurons in an estrogen-dependent manner. Because K+ channels are important regulatory targets of hormone and neurotransmitter release (Yamada et al., 1998), it will be of interest to determine whether KATP channels are also regulated by other signaling molecules in GnRH neurons.
In summary, we found that GnRH neurons predominately express Kir6.2/SUR1 KATP channel subunits as well as glucokinase mRNA that render them sensitive to glucose, metabolic inhibition, and estrogen. Therefore, these GnRH neurons are uniquely poised to integrate neural sensory input, metabolic changes, and hormonal fluctuations to serve as the hypothalamic command neurons to control reproduction in the mammal.
Footnotes
This work was supported by United States Public Health Service Grants NS 38809, NS 43330, DK 68098, and HD 20677. We thank Dr. Suzanne Moenter (University of Virginia, Charlottesville, VA) for providing the transgenic EGFP-GnRH mice and Elizabeth A. Rick for her excellent technical assistance. We also recognize Dr. Asami Umeda (Mitsubishi Pharma, Osaka, Japan) for his generous gift of MCC 134.
References
- Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. New windows on the mechanism of action of KATP channel openers. Trends Pharmacol Sci. 2000;21:439–445. doi: 10.1016/s0165-6147(00)01563-7. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Aguilar-Bryan L, Bryan J. A view of SUR/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Ffrench-Mullen JMH, Cowley MA, Smith MS, Grove KL. Hypothalamic circuitry of neuropeptide Y regulation of neuroendocrine function and food intake via the Y5 receptor subtype. Neuroendocrinology. 2001;74:106–119. doi: 10.1159/000054676. [DOI] [PubMed] [Google Scholar]
- Caraty A, Locatelli A, Martin GB. Biphasic response in the secretion of gonadotrophin-releasing hormone in ovariectomized ewes injected with oestradiol. J Endocrinol. 1989;123:375–382. doi: 10.1677/joe.0.1230375. [DOI] [PubMed] [Google Scholar]
- Chappell PE, Levine JE. Stimulation of gonadotropin-releasing hormone surges by estrogen. I. Role of hypothalamic progesterone receptors. Endocrinology. 2000;141:1477–1485. doi: 10.1210/endo.141.4.7428. [DOI] [PubMed] [Google Scholar]
- Ching M. Correlative surges of LHRH, LH and FSH in pituitary stalk plasma and systemic plasma of rat during proestrus. Neuroendocrinology. 1982;34:279–285. doi: 10.1159/000123313. [DOI] [PubMed] [Google Scholar]
- Christian CA, Mobley JL, Moenter SM. Diurnal and estradiol-dependent changes in gonadotropin-releasing hormone neuron firing activity. Proc Natl Acad Sci USA. 2005;102:15682–15687. doi: 10.1073/pnas.0504270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Paten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Moenter SM. Estradiol feedback alters potassium currents and firing properties of gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16:2255–2265. doi: 10.1210/me.2002-0155. [DOI] [PubMed] [Google Scholar]
- D'hahan N, Moreau C, Prost A-L, Jacquet H, Alekseev AE, Terzic A, Vivaudou M. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci USA. 1999;96:12162–12167. doi: 10.1073/pnas.96.21.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res. 1998;814:41–54. doi: 10.1016/s0006-8993(98)00956-1. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. doi: 10.2337/diabetes.51.7.2056. [DOI] [PubMed] [Google Scholar]
- Ellinwood WE, Rønnekleiv OK, Kelly MJ, Resko JA. A new antiserum with conformational specificity for LHRH: usefulness for radioimmunoassay and immunocytochemistry. Peptides. 1985;6:45–52. doi: 10.1016/0196-9781(85)90075-0. [DOI] [PubMed] [Google Scholar]
- Erickson KR, Rønnekleiv OK, Kelly MJ. Role of a T-type calcium current in supporting a depolarizing potential, damped oscillations, and phasic firing in vasopressinergic guinea pig supraoptic neurons. Neuroendocrinology. 1993;57:789–800. doi: 10.1159/000126438. [DOI] [PubMed] [Google Scholar]
- Han S-K, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci. 2005;25:11349–11356. doi: 10.1523/JNEUROSCI.3328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE. Multimodal influence of estrogen upon gonadotropin-releasing hormone neurons. Endocr Rev. 1998;19:302–330. doi: 10.1210/edrv.19.3.0332. [DOI] [PubMed] [Google Scholar]
- Herbison AE. Physiology of the gonadotropin-releasing hormone neuronal network. In: Neill JD, editor. Knobil and Neill's physiology of reproduction, Ed 3. Dunedin, New Zealand: Elsevier; 2006. pp. 1–68. [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose-responsive and express K-ATP channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- Kalra SP. Mandatory neuropeptide-steroid signaling for the preovulatory luteinizing hormone-releasing hormone discharge. Endocr Rev. 1993;14:507–538. doi: 10.1210/edrv-14-5-507. [DOI] [PubMed] [Google Scholar]
- Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, Eikis J, Zhang BB, Levin BE. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes. 2006;55:412–420. doi: 10.2337/diabetes.55.02.06.db05-1229. [DOI] [PubMed] [Google Scholar]
- Kato M, Ui-Tei K, Watanabe M, Sakuma Y. Characterization of voltage-gated calcium currents in gonadotropin-releasing hormone neurons tagged with green fluorescent protein in rats. Endocrinology. 2003;144:5118–5125. doi: 10.1210/en.2003-0213. [DOI] [PubMed] [Google Scholar]
- Kauffman AS, Rissman EF. A critical role for the evolutionarily conserved gonadotropin-releasing hormone II: mediation of energy status and female sexual behavior. Endocrinology. 2004;145:3639–3646. doi: 10.1210/en.2004-0148. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Wagner EJ. GnRH neurons and episodic bursting activity. Trends Endocrinol Metab. 2002;13:409–410. doi: 10.1016/s1043-2760(02)00698-7. [DOI] [PubMed] [Google Scholar]
- Kuehl-Kovarik MC, Pouliot WA, Halterman GL, Handa RJ, Dudek FE, Partin KM. Episodic bursting activity and response to excitatory amino acids in acutely dissociated gonadotropin-releasing hormone neurons genetically targeted with green fluorescent protein. J Neurosci. 2002;22:2313–2322. doi: 10.1523/JNEUROSCI.22-06-02313.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl-Kovarik MC, Pouliot WA, Partin KM, Handa RJ, Dudek FE. Spike-dependent depolarizing afterpotentials contribute to endogenous bursting in gonadotropin releasing hormone neurons. Neuroscience. 2005;134:295–300. doi: 10.1016/j.neuroscience.2005.03.047. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ. Estradiol-17β and μ-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback? Endocrinology. 1995;136:2341–2344. doi: 10.1210/endo.136.5.7720682. [DOI] [PubMed] [Google Scholar]
- Lam TKT, Pocai A, Guiterrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, Schwartz GJ, Rossetti L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- Legan SJ, Coon GA, Karsch FJ. Role of estrogen as initiator of daily LH surges in the ovariectomized rat. Endocrinology. 1975;96:50–56. doi: 10.1210/endo-96-1-50. [DOI] [PubMed] [Google Scholar]
- Leranth C, MacLusky NJ, Sakamoto H, Shanabrough M, Naftolin F. Glutamic acid decarboxylase-containing axons synapse on LHRH neurons in the rat medial preoptic area. Neuroendo. 1985;40:536–539. doi: 10.1159/000124127. [DOI] [PubMed] [Google Scholar]
- Levine JE, Ramirez VD. In vitro release of luteinizing hormone-releasing hormone estimated with push-pull cannulae from the mediobasal hypothalami of ovariectomized, steroid-primed rats. Endocrinology. 1980;107:1782–1790. doi: 10.1210/endo-107-6-1782. [DOI] [PubMed] [Google Scholar]
- Levine JE, Ramirez VD. Luteinizing hormone-releasing hormone release during the rat estrous cycle and after ovariectomy, as estimated with push-pull cannulae. Endocrinology. 1982;111:1439–1448. doi: 10.1210/endo-111-5-1439. [DOI] [PubMed] [Google Scholar]
- Levine JE, Chappell P, Besecke LM, Bauer-Dantoin AC, Wolfe AM, Porkka-Heiskanen T, Urban JH. Amplitude and frequency modulation of pulsatile luteinizing hormone-releasing hormone release. Cell Mol Neurobiol. 1995;15:117–140. doi: 10.1007/BF02069562. [DOI] [PubMed] [Google Scholar]
- Liss B, Roeper J. A role for neuronal K-ATP channels in metabolic control of the seizure gate. Trends Pharmacol Sci. 2001;22:599–601. doi: 10.1016/s0165-6147(00)01861-7. [DOI] [PubMed] [Google Scholar]
- Liss B, Bruns R, Roeper J. Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. EMBO J. 1999;18:833–846. doi: 10.1093/emboj/18.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Haeckel O, Wildmann J, Miki T, Seino S, Roeper J. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat Neurosci. 2005;8:1742–1751. doi: 10.1038/nn1570. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martínez de la Escalera G, Choi ALH, Weiner RI. Generation and synchronization of gonadotropin-releasing hormone (GnRH) pulses: intrinsic properties of the GT1–1 GnRH neuronal cell line. Proc Natl Acad Sci USA. 1992;89:1852–1855. doi: 10.1073/pnas.89.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Komiyama S, Akaike N. Pre- and postsynaptic ATP-sensitive potassium channels during metabolic inhibition of rat hippocampal CA1 neurons. J Physiol (Lond) 2002;541:511–520. doi: 10.1113/jphysiol.2002.018267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Pape H-C. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol (Lond) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina CL, Nagatani S, Darling TA, Bucholtz DC, Tsukamura H, Maeda K, Foster DL. Glucose availability modulates the timing of the luteinizing hormone surge in the ewe. J Neuroendocrinol. 1998;10:785–792. doi: 10.1046/j.1365-2826.1998.00264.x. [DOI] [PubMed] [Google Scholar]
- Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- Minami T, Oomura Y, Sugimori M. Electrophysiological properties and glucose responsiveness of guinea-pig ventromedial hypothalamic neurones in vitro. J Physiol (Lond) 1986;380:127–143. doi: 10.1113/jphysiol.1986.sp016276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moenter SM, Brand RC, Karsch FJ. Dynamics of gonadotropin-releasing hormone (GnRH) secretion during the GnRH surge: insights into the mechanism of GnRH surge induction. Endocrinology. 1992;130:2978–2984. doi: 10.1210/endo.130.5.1572305. [DOI] [PubMed] [Google Scholar]
- Moenter SM, DeFazio RA, Straume M, Nunemaker CS. Steroid regulation of GnRH neurons. Ann NY Acad Sci. 2003;1007:143–152. doi: 10.1196/annals.1286.014. [DOI] [PubMed] [Google Scholar]
- Muroya S, Yada T, Shioda S, Takigawa M. Glucose-sensitive neurons in the rat arcuate nucleus contain neuropeptide Y. Neurosci Lett. 1999;264:113–116. doi: 10.1016/s0304-3940(99)00185-8. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Geusz ME, Herzog ED, Pitts GR, Moenter SM. Long-term recordings of networks of immortalized GnRH neurons reveal episodic patterns of electrical activity. J Neurophysiol. 2001;86:86–93. doi: 10.1152/jn.2001.86.1.86. [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, DeFazio RA, Moenter SM. A targeted extracellular approach for recording long-term firing patterns of excitable cells: a practical guide. Biol Proced Online. 2003;5:53–62. doi: 10.1251/bpo46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura S, Tanaka T, Nagatani S, Bucholtz DC, Tsukamura H, Maeda K-I, Foster DL. Central, but not peripheral, glucose-sensing mechanisms mediate glucoprivic suppression of pulsatile luteinizing hormone secretion in the sheep. Endocrinology. 2000;141:4472–4480. doi: 10.1210/endo.141.12.7853. [DOI] [PubMed] [Google Scholar]
- Oomura Y, Ono T, Ooyama H, Wayner MJ. Glucose and osmosensitive neurones of the rat hypothalamus. Nature. 1969;222:282–284. doi: 10.1038/222282a0. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Münzberg H, Shanabrough M, Burdakov D, Rother E, Janoschek R, Alber J, Belgardt BF, Koch L, Seibler J, Schwenk F, Fekete C, Suzuki A, Mak TW, Krone W, Horvath TL, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest. 2006;116:1886–1901. doi: 10.1172/JCI27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Rønnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Xue C, Bosch MA, Murphy JG, Fan W, Rønnekleiv OK, Kelly MJ. Serotonin 5HT2C receptor signaling in hypothalamic POMC neurons: role in energy homeostasis in females. Mol Pharmacol. 2007 doi: 10.1124/mol.107.038083. in press. [DOI] [PubMed] [Google Scholar]
- Reimann F, Ashcroft FM. Inwardly rectifying potassium channels. Curr Opin Cell Biol. 1999;11:503–508. doi: 10.1016/S0955-0674(99)80073-8. [DOI] [PubMed] [Google Scholar]
- Rønnekleiv OK, Kelly MJ. Diversity of ovarian steroid signaling in the hypothalamus. Front Neuroendocrinol. 2005;26:65–84. doi: 10.1016/j.yfrne.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Sarkar DK, Fink G. Luteinizing hormone releasing factor in pituitary stalk plasma from long-term ovariectomized rats: effects of steroids. J Endocrinol. 1980;86:511–524. doi: 10.1677/joe.0.0860511. [DOI] [PubMed] [Google Scholar]
- Shindo T, Katayama Y, Horio Y, Kurachi Y. MCC-134, a novel vascular relaxing agent, is an inverse agonist for the pancreatic-type ATP-sensitive K+ channel. J Pharmacol Exp Ther. 2000;292:131–135. [PubMed] [Google Scholar]
- Shyng S-L, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of K-ATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2005;54:15–22. doi: 10.2337/diabetes.54.1.15. [DOI] [PubMed] [Google Scholar]
- Song ZT, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2001;50:2673–2681. doi: 10.2337/diabetes.50.12.2673. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford MLJ. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- Stojilkovic S, Iida T, Virmani MA, Izumi S-I, Rojas E, Catt KJ. Dependence of hormone secretion on activation-inactivation kinetics of voltage-sensitive Ca2+ channels in pituitary gonadotrophs. Proc Natl Acad Sci USA. 1990;87:8855–8859. doi: 10.1073/pnas.87.22.8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter KJ, Song WJ, Sampson TL, Wuarin J-P, Saunders JT, Dudek FE, Moenter SM. Genetic targeting of green fluorscent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology. 2000;141:412–419. doi: 10.1210/endo.141.1.7279. [DOI] [PubMed] [Google Scholar]
- Temple JL, Millar RP, Rissman EF. An evolutionarily conserved form of gonadotropin-releasing hormone coordinates energy and reproductive behavior. Endocrinology. 2003;144:13–19. doi: 10.1210/en.2002-220883. [DOI] [PubMed] [Google Scholar]
- Terasawa E. Steroid modulation of pulsatile LHRH release in the rhesus monkey. Horm Behav. 1994;28:406–416. doi: 10.1006/hbeh.1994.1037. [DOI] [PubMed] [Google Scholar]
- Terasawa E, Rodriguez JS, Bridson WE, Wiegand SJ. Factors influencing the positive feedback action of estrogen upon luteinizing hormone surge in the ovariectomized guinea pig. Endocrinology. 1979;104:680–686. doi: 10.1210/endo-104-3-680. [DOI] [PubMed] [Google Scholar]
- The ESHRE Capri Workshop Group. Nutrition and reproduction in women. Hum Reprod Update. 2006;12:193–207. doi: 10.1093/humupd/dmk003. [DOI] [PubMed] [Google Scholar]
- Wade GN, Jones JE. Neuroendocrinology of nutritional infertility. Am J Physiol Regul Intergr Comp Physiol. 2004;287:R1277–R1296. doi: 10.1152/ajpregu.00475.2004. [DOI] [PubMed] [Google Scholar]
- Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes. 2004;53:1959–1965. doi: 10.2337/diabetes.53.8.1959. [DOI] [PubMed] [Google Scholar]
- Wetsel WC, Valença MM, Merchenthaler I, Liposits Z, López FJ, Weiner RI, Mellon PL, Negro-Vilar A. Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc Natl Acad Sci USA. 1992;89:4149–4153. doi: 10.1073/pnas.89.9.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Inanobe A, Kurachi Y. G protein regulation of potassium ion channels. Pharmacol Rev. 1998;50:723–757. [PubMed] [Google Scholar]
- Yang X-J, Mastaitis J, Mizuno T, Mobbs CV. Glucokinase regulates reproductive function, glucocorticoid secretion, food intake, and hypothalamic gene expression. Endocrinology. 2007;148:1928–1932. doi: 10.1210/en.2006-1312. [DOI] [PubMed] [Google Scholar]
- Zheng SX, Bosch MA, Rønnekleiv OK. Mu-opioid receptor mRNA expression in identified hypothalamic neurons. J Comp Neurol. 2005;487:332–344. doi: 10.1002/cne.20557. [DOI] [PubMed] [Google Scholar]