

Abstract
N-terminal acylation of the α-subunits of heterotrimeric G-proteins is believed to play a major role in regulating the cellular localization and signaling of G-proteins, but physiological evidence has been lacking. To examine the functional significance of N-acylation of a well understood G-protein α-subunit, transducin (Gαt), we generated transgenic mice that expressed a mutant Gαt lacking N-terminal acylation sequence (GαtG2A). Rods expressing GαtG2A showed a severe defect in transducin cellular localization. In contrast to native Gαt, which resides in the outer segments of dark-adapted rods, GαtG2A was found predominantly in the inner compartments of the photoreceptor cells. Remarkably, transgenic rods with the outer segments containing GαtG2A at 5–6% of the Gαt levels in wild-type rods showed only a sixfold reduction in sensitivity and a threefold decrease in the amplification constant. The much smaller than predicted reduction may reflect an increase in the lateral diffusion of transducin and an increased activation rate by photoexcited rhodopsin or more efficient activation of cGMP phosphodiesterase 6 by GαtG2A; alternatively, nonlinear relationships between concentration and the activation rate of transducin also potentially contribute to the mismatch between the amplification constant and quantitative expression analysis of GαtG2A rods. Furthermore, the G2A mutation reduced the GTPase activity of transducin and resulted in two to three times slower than normal recovery of flash responses of transgenic rods, indicating the role of Gαt membrane tethering for its efficient inactivation by the regulator of G-protein signaling 9 GTPase-activating protein complex. Thus, N-acylation is critical for correct compartmentalization of transducin and controls the rate of its deactivation.
Keywords: phototransduction, retina, transducin, N-acylation, G-proteins, vision
Introduction
Heterotrimeric G-proteins mediate the transduction of a variety of extracellular signals that activate G-protein-coupled receptors at the plasma membrane (Bourne, 1997; Pierce et al., 2002; Cabrera-Vera et al., 2003). The vertebrate rod phototransduction cascade represents one of the best model systems to study the mechanisms of G-protein signaling. Rod transducin (Gt) couples photoexcitation of rhodopsin (R) to the activation of cGMP phosphodiesterase 6 (PDE6). The GTP-bound α-subunit of Gt (GαtGTP) binds to the γ-subunit of PDE6 (Pγ), activating the enzyme. The GαtGTP/PDE6 complex is deactivated when bound GTP is hydrolyzed as a result of intrinsic GTPase activity of Gαt controlled by the GTPase-activating protein (GAP) complex, regulator of G-protein signaling 9-1 (RGS9-1)/Gβ5/RGS9 anchor protein (R9AP) (Burns and Baylor, 2001; Burns and Arshavsky, 2005).
Lipid modifications of G-proteins are thought to play a major role in the regulation of G-protein cellular localization and signaling (Wedegaertner et al., 1995; Higgins and Casey, 1996; Chen and Manning, 2001). Most G-protein γ-subunits (Gγs) are modified with a thioether-linked isoprenoid geranylgeranyl (C20) attached to Cys residues within the C-terminal “CAAX” box (Escriba et al., 2006). In contrast, the rod-specific Gγ1 carries the farnesyl moiety (C15), which facilitates light-dependent translocation of Gt from the rod outer segment (ROS) to the inner compartments, thereby contributing to light adaptation (Kassai et al., 2005). Gα-subunits are typically modified with an amide-linked fatty acid myristate (C14:0) at the extreme N-terminal Gly residue and/or with a thioester-linked palmitate (C16:0) at Cys residues near the N termini (Wedegaertner et al., 1995; Chen and Manning, 2001). Uniquely, rod Gαt is heterogeneously fatty N-acylated with C12:0, C14:0, C14:1, and C14:2 moieties (Neubert et al., 1992). The lower hydrophobicity of the Gαt acyl residues also assists light-dependent redistribution of transducin between the outer segment (OS) and inner regions of rods (Lobanova et al., 2007).
Although the heterogeneity of Gαt lipid modification may influence certain aspects of phototransduction and light adaptation (Neubert and Hurley, 1998; Lobanova et al., 2007), the significance of the lipid modification itself is expected to be much more profound. The N-terminal acylation may be required for proper transport and localization of transducin to the OS. On the disc membrane, the Gαt lipid anchor may control the lateral diffusion and collisional coupling of Gαt with photoexcited rhodopsin (R*). Furthermore, the Gαt lipid is potentially critical for PDE6 activation and inactivation. The activated GαtGTP/PDE6 complex is membrane bound and stabilized by the Gαt and PDE6 lipid modifications (Catty et al., 1992; Clerc and Bennett, 1992). The ability of the GAP complex to rapidly inactivate the GαtGTP/PDE6 complex depends on the membrane attachment of the GAP complex via the anchor protein, R9AP (Lishko et al., 2002, Hu et al., 2003).
Despite growing data supporting the roles of N-acylation of Gα subunits for localization and transduction efficiency of G-proteins, functional evidence for the importance in cell signaling is lacking. To probe the functional significance of the N-acylation of Gαt in intact cells, we have produced and examined transgenic mice expressing an acylation-deficient mutant, GαtG2A, in rod photoreceptors.
Materials and Methods
Generation of GαtG2A transgenic mice.
A pBRH Gαt transgenic construct contained the mouse Gαt genomic sequence of ∼5.5 kb (Raport et al., 1989) flanked by the 4.4 kb mouse rod opsin promoter fragment (Lem et al., 1991) and polyadenylation signal (Fig. 1A). The transgene sequence was mutated previously to introduce the Glu-Glu (EE) monoclonal antibody epitope to assess expression levels of mutant transducins in mouse retinas (Kerov et al., 2005; Moussaif et al., 2006). The G2A substitution was created using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA). A DNA fragment of 9 kb was released from the pBRH-GαtEEG2A plasmid by restriction with NotI and gel purified. The DNA fragment was microinjected into mouse embryos at the transgenic core facility at the University of Iowa. Transgenic mice were identified by PCR of mouse tail DNA with a pair of primers chosen to amplify a 300 bp fragment surrounding the junction between the Gαt and polyadenylation signal sequences. Six potential GαtG2A transgenic mouse founders were identified and mated to C57BL/6 mice. The founder line with the highest expression level of GαtG2A was selected for breeding with Gαt knock-out mice (Gαt−/−) to produce GαtG2A/Gαt+/− transgenic mice hemizygous for native Gαt allele. GαtG2A/Gαt+/− mice were further mated with Gαt−/− mice to move the transgene into the knock-out (Gαt−/−) background. GαtG2A/Gαt−/− mice were identified by the absence of PCR product using primers designed to amplify a DNA sequence only from the wild-type Gαt allele. All experimental procedures involving the use of mice were performed in accordance with the National Institutes of Health (NIH) guidelines and the protocol approved by the University of Iowa Animal Care and Use Committee.
Figure 1.

A, Transgenic construct used for the generation of mutant GαtG2A mice. B, Expression levels of G2A mutant in GαtG2A/Gαt+/− and GαtG2A/Gαt−/− mice. Retinas from transgenic and control (Gαt+/+) mice were homogenized and subjected to SDS-PAGE. Sample loads are in micrograms of retinal protein (ret. pr.). Immunoblot (IB) analysis was performed with anti-rod Gαt and anti-EE antibodies. C, Quantification of protein levels from the anti-EE antibody immunoblot in B. The relative level of GαtG2A in GαtG2A/Gαt−/− mice is 1.5-fold higher than in GαtG2A/Gαt+/− mice (calculated as the ratio of the linear fit slopes). D, Immunoblot analyses of key phototransduction proteins. Samples contained 2.5, 5, and 10 μg of retinal homogenate from 4-month-old GαtG2A/Gαt−/− or control WT mice. The antibodies are described in Materials and Methods. E, Quantification of the relative protein levels from the immunoblot in D. Integrated densities for each protein band were plotted as a function of retinal homogenate load to determine the linear fit slopes and the ratios of the slopes for WT and mutant mice. Arr, Arrestin.
Immunoblot analysis and quantification of transgene expression.
Total mouse retinal homogenates were obtained by solubilization of two retinas in 100 μl of 10% SDS-Na using brief sonication. Protein concentrations were determined using the DC protein assay (Bio-Rad, Hercules, CA) and bovine serum albumin dissolved in 10% SDS-Na as the standard. Typically, the total protein content of a homogenate obtained from a single mouse retina was ∼500 μg. Samples of retinal homogenates were subjected to SDS-PAGE in 12% gels, transferred onto nitrocellulose membranes BA 85 (Schleicher and Schuell, Keene, NH), and probed with the following primary antibodies (1:1000 dilution): anti-rod Gαt (K-20), anti-Gβ1 (M-14), and anti-Gγ1 (P-19) (Santa Cruz Biotechnology, Santa Cruz, CA); EE monoclonal (Covance, Princeton, NJ); monoclonal anti-rhodopsin 1D4 (National Cell Culture Center, Minneapolis, MN); anti-PDE6αβ MOE antibodies (Cytosignal, Irvine, CA); anti-arrestin (PA1–731; ABR, Golden, CO), and anti-RGS9 antibodies (Elmira Biologicals, Iowa City, IA). According to the manufacturer, the epitope of anti-rod Gαt antibody K-20 is located within residues 80–120 of human Gαt. Thus, the affinity of this antibody for Gαt should not be affected by the G2A or EE mutations. The antibody–antigen complexes were detected using secondary anti-rabbit, anti-mouse, or anti-sheep antibodies (1:10,000) conjugated to horseradish peroxidase (Santa Cruz Biotechnology) and ECL reagent (GE Healthcare, Piscataway, NJ). Nitrocellulose membranes were exposed to film, and integrated densities of scanned individual bands were measured with Image J 1.33u software.
Immunohistochemistry.
For dark adaptation, mice were kept in the dark for at least 12 h. All dark procedures were performed under infrared illumination using night vision goggles. Eyeballs were enucleated, and the corneas were perforated with a 21 gauge needle, fixed in PBS with 4% formaldehyde for 1 h, and cut in half. The cornea and lens were removed, and the eyecups were submersed in a 30% sucrose solution in PBS for 5 h at 4°C. The eyecups were then embedded in tissue freezing medium (Triangle Biomedical Sciences, Durham, NC) and frozen on dry ice. Radial sectioning (10 μm) of the retina was performed using a cryomicrotome HM 505E. Retinal cryosections were air dried and kept at −80°C. The sections were warmed up to room temperature and incubated in PBS/0.1% Triton X-100 for 30 min. Next, 2% goat serum and 5% BSA in PBS was added, and incubation continued for 30 min. Sections were then incubated with rabbit anti-rod Gαt antibody (K-20; 1:2000; Santa Cruz Biotechnology) and a monoclonal anti-EE antibody (1:2000; Covance) for 3 h. After a 1 h incubation with goat anti-rabbit AlexaFluor 555 or goat anti-mouse AlexaFluor 488 secondary antibodies (1:1000; Invitrogen, Carlsbad, CA), the sections were visualized using a Zeiss (Thornwood, NY) LSM 510 confocal microscope. To evaluate mouse retina morphology, eyeballs were fixed in 2.5% gluteraldehyde in 0.1 m cacodylate buffer for 2 h at 22°C, cut in half to remove the cornea and lens, dehydrated through a series of acetones, and embedded in Epon. Radial sections (1 μm) were stained with toluidine blue and photographed through a Nikon (Tokyo, Japan) Optiphot microscope using a Spot camera (Diagnostic Instruments, Sterling Heights, MI).
Biochemical analysis of transducin function in mouse ROS membranes.
Mouse ROS membranes were prepared using a procedure that allows isolation of osmotically intact and sealed ROS, thereby guarding against loss of OS proteins (Tsang et al., 1998). Rhodopsin concentrations were determined using the difference in absorbance at 500 nm before and after photobleaching of ROS preparations solubilized in 1% Ammonix LO. To measure light-dependent binding of GTPγS, ROS (10 μl) from dark-adapted GαtG2A/Gαt−/− or control Gαt+/+ mice containing 10 μm rhodopsin were mixed with 5 μl of 10 μm [35S]GTPγS under infrared illumination. After 10 s of incubation, the amount of bound GTPγS was determined in a 6 μl aliquot by the nitrocellulose filter binding assay. The rest of the sample was bleached to determine the GTPγS binding in the light.
The single-turnover GTPase measurements were initiated by mixing photobleached ROS (12 μl) containing 10 μm rhodopsin with 3 μl of 0.1 μm [γ-32P]GTP. The time course of 32Pi formation was determined using the activated charcoal procedure after the reactions were stopped with perchloric acid. The GTPase rate constants were calculated by fitting the experimental data to an exponential function:
where k is a rate constant for GTP hydrolysis.
To measure PDE6 activation by transducin, photobleached ROS (3 μm R) were incubated with 3 μm cGMP for 3 min to stabilize the PDE6 basal activity (Tsang et al., 1998). The reactions were started by the addition of 4 mm [3H]cGMP with or without 10 μm GTPγS. To activate PDE by limited trypsinolysis, ROS samples were incubated with tosyl phenylalanyl chloromethyl ketone-treated trypsin (50 μg/ml) for 4 min on ice followed by incubation with soybean trypsin inhibitor (250 μg/ml) for 5 min at 25°C. cGMP hydrolysis was terminated after 30 s by heating the samples for 2 min at 100°C. After cooling, 0.1 U of bacterial alkaline phosphatase (Sigma-Aldrich, St. Louis, MO) was added to each sample followed by incubation for 20 min at 37°C. Nonhydrolyzed cGMP was removed with AG1-X2 anion exchange resin (Bio-Rad), and [8-3H]guanosine was counted in a liquid scintillation counter.
To assess pertussis toxin-catalyzed Gβ1γ1-dependent ADP ribosylation of Gαt, dark-isolated or bleached wild-type (WT) ROS (0.6 μm R) and GαtG2A/Gαt−/− ROS membranes (6 μm R) were incubated with 3 μg/ml of pertussis toxin (preactivated by 15 min incubation at 30°C with 100 mm dithiothreitol and 0.2% SDS) and 5 μm [32P]nicotinamide adenine dinucleotide ([32P]NAD) for 1 h at 25°C in the presence or absence of 30 μm AlCl3 and 10 mm NaF. The reactions were stopped by the addition of SDS-sample buffer and analyzed by SDS-PAGE in 12% gels and autoradiography.
Suction electrode recordings and analysis.
Mice were cared for and handled following an approved protocol from the Animal Care and Use Committee of the University of California, Davis and in compliance with NIH guidelines for the care and use of experimental animals. Before an experiment, animals were dark adapted overnight for a minimum of 12 h. Animals were anesthetized and killed under infrared light, and the retinas were dissected and stored on ice in L-15 solution with 10 mm glucose and 0.1 mg/ml bovine serum albumin (Sigma-Aldrich). For recording, a small piece of retina was chopped into small pieces in a chamber containing the L-15 solution, and the chopped tissue was then placed in a recording chamber perfused with bicarbonate solution, pH 7.4, supplemented with 10 mm glucose held at 35–37°C. Individual rods were visualized under infrared light using a CCD camera (Stanford Photonics, Palo Alto, CA). The outer segment of an individual rod was gently drawn into the tip of a capillary pipette containing HEPES solution, pH 7.4. The membrane currents were recorded by a current-to-voltage converter (Axopatch 1B; Molecular Devices, Union City, CA) and low-pass filtered (eight-pole Bessel; Frequency Devices, Haverhill, MA) using 30 Hz corner frequency. Data were digitized at 200 Hz using IGOR-National Instruments acquisition software (IgorPro for NIDAQ for Windows; Wavemetrics, Lake Oswego, OR). Rods were presented with 10 ms flashes of 500 nm light. The intensity of the light was controlled by using calibrated neutral density filters.
The form of the single-photon response was calculated by squaring the average response to 30 or more dim (linear) responses and scaling it to coincide with the time course of the ensemble light-dependent variance over the initial rising phase. Assuming the predominant source of variance over this time scale arises from Poisson fluctuations in the number of photoexcited rhodopsins, the inverse of this scaling factor corresponds to the average number of photoisomerizations per trial. The average linear response was then divided by the average number of photoisomerizations per trial to yield the average single-photon response. The effective collecting area was calculated by dividing the average number of photoisomerizations per trial by the flash strength (in photons · μm−2).
The time constant of recovery for dim flash responses was determined by least-squares fitting of a single exponential to the final falling phase of the response; for bright flash responses, a straight line was fitted to the relationship between the time in saturation and the natural log of the flash strength up to ln i = 9 (Krispel et al., 2006). The integration time was calculated as the time integral of the dim flash response divided by the peak amplitude.
Results
Expression and localization of GαtG2A mutant in transgenic rods
The G2A substitution was introduced into a transgene previously used for expression of the monoclonal antibody EE epitope-tagged transducin mutant (Fig. 1A) (Kerov et al., 2005; Moussaif et al., 2006). The EE tag facilitates determination of the expression levels and localization of the GαtG2A mutant in the presence of native Gαt in transgenic rods. The EE tag does not alter biochemical properties of transducin, and the flash responses from transgenic mouse rods expressing GαtEE are similar to those from the wild-type rods (Kerov et al., 2005; Moussaif et al., 2006). Several transgenic mouse lines expressing the EE-tagged GαtG2A mutant were established. The line with the highest expression level of GαtG2A was selected by immunoblot analysis with anti-EE antibody and used for breeding with Gαt knock-out mice (Calvert et al., 2000) to move the transgene into the hemizygous (Gαt+/−) and knock-out (Gαt−/−) backgrounds. Based on the immunoblot analysis of mouse retinal homogenates with anti-rod Gαt antibody, the total expression level of transducin-α (GαtG2A + native Gαt) in GαtG2A/Gαt+/− mice was similar to the Gαt level in wild-type (Gαt+/+) mice (Fig. 1B). The level of GαtG2A in the Gαt knock-out background was ∼30% of the wild-type level (Fig. 1B). The blot analysis with EE antibody indicated that expression of GαtG2A in the Gαt−/− background was ∼1.5-fold higher than in the Gαt+/− background (Fig. 1B,C), suggesting that GαtG2A/Gαt+/− mice express ∼20% GαtG2A and 80% native Gαt. Control immunoblot tests demonstrated that the levels of Gβ1 and several major phototransduction components such as rhodopsin, PDE6αβ, arrestin-1, and RGS9-1 were essentially unchanged in GαtG2A/Gαt−/− mice (Fig. 1D,E). Also, GαtG2A/Gαt+/− and GαtG2A/Gαt−/− mice showed normal gross morphology of the retina with no apparent signs of retinal degeneration (data not shown).
In dark adapted rods, native transducin localizes to the OS. We examined subcellular localization of GαtG2A in transgenic rods by immunofluorescence confocal microscopy of mouse retinal sections using anti-rod Gαt and anti-EE antibodies. In contrast to the native Gαt, GαtG2A displayed extensive mislocalization to the inner compartments of rod photoreceptor cells (Fig. 2A). Only a very weak EE signal was seen in the OS of GαtG2A/Gαt+/− and GαtG2A/Gαt−/− mice (Fig. 2A). ROS membranes were isolated from dark-adapted GαtG2A/Gαt−/− and control Gαt+/+ mice to determine the fraction of the mutant that correctly localized to the OS. The ROS immunoblot analysis revealed that the level of GαtG2A in the OS is ∼5–6% of the Gαt level in control mice (Fig. 2B,C). Thus, GαtG2A/Gαt−/− mice express GαtG2A at ∼30% of Gαt in wild-type mice, but only 15–20% of the mutant Gαt is able to reach the OS. This result is in agreement with the immunofluorescence staining of GαtG2A in the GαtG2A/Gαt−/− retinal sections (Fig. 2A).
Figure 2.

A, Localization of total Gαt and mutant GαtG2A in dark-adapted (DA) mouse retinas. GαtG2A/Gαt+/− and GαtG2A/Gαt−/− retina sections were stained with rabbit anti-rod Gαt antibody K-20 or a monoclonal anti-EE antibody and visualized with goat anti-rabbit AlexaFluor 555 and goat anti-mouse AlexaFluor 488 secondary antibodies using a Zeiss LSM 510 confocal microscope. Scale bar, 20 μm. IS, Inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer. B, Representative immunoblot (IB) analysis of relative levels of GαtG2A in GαtG2A/Gαt−/− ROS and native Gαt in WT ROS. C, Quantification of the relative protein levels from the immunoblot in B. The relative level of Gαt in WT ROS is ∼17-fold higher than the level of GαtG2A in transgenic ROSs (calculated as the ratio of the linear fit slopes). The average ratio of the slopes in three similar experiments is 18 ± 2 (mean ± SE).
Biochemical analysis of GαtG2A
The key signaling properties of GαtG2A such as its interaction with Gβ1γ1, R*-dependent binding of GTPγS, GTPase activity, and the ability to activate PDE6 were examined using preparations of ROS membranes isolated from GαtG2A/Gαt−/− and control wild-type (Gαt+/+) mice. To assay the ability of GαtG2A to bind Gβ1γ1, we measured the extent of pertussis toxin-induced ADP ribosylation of Gαt at Cys347 (van Dop et al., 1984; West et al., 1985; Watkins et al., 1985). Holotransducin, Gαtβ1γ1, is a much-preferred substrate in this reaction compared with Gαt alone (Watkins et al., 1985). The extent of ADP ribosylation of Gαt in wild-type ROS and GαtG2A in transgenic ROS was similar, and both were inhibited comparably by the addition of AlF4− (Fig. 3A). AlF4− ions activate GDP-bound Gαt subunits by mimicking the γ-phosphate of GTP and causing dissociation of Gβ1γ1. Thus, GαtG2A is capable of binding Gβ1γ1 and is present in ROS of GαtG2A/Gαt−/− mice as a heterotrimeric protein GαtG2Aβ1γ1. The binding of wild-type Gαtβ1γ1 to R* inhibited pertussis toxin-catalyzed ADP ribosylation of Gαt, apparently by masking the Cys residue (van Dop et al., 1984; Watkins et al., 1985; West et al., 1985). Likewise, ADP ribosylation of GαtG2Aβ1γ1 was inhibited by light, suggesting specific binding of the mutant transducin to R* (Fig. 3A).
Figure 3.

A, Pertussis toxin-catalyzed ADP ribosylation of Gαt and GαtG2A. Dark-isolated (D) or bleached (L) WT ROS (0.6 μm R) and GαtG2A/Gαt−/− ROS (6 μm R) were incubated with preactivated pertussis toxin (3 μg/ml) and 5 μm [32P]NAD for 1 h at 25°C in the presence or absence of 30 μm AlCl3 and 10 mm NaF. The reactions were stopped by the addition of SDS-PAGE sample buffer and analyzed by SDS-PAGE and autoradiography. B, Light-dependent binding of GTPγS to control WT and GαtG2A/Gαt−/− ROS. Mouse ROS (10 μl, 10 μm R) were mixed with 5 μl of 10 μm [35S]GTPγS under infrared illumination. After 10 s incubation, the amount of bound GTPγS was determined in a 6 μl aliquot by the nitrocellulose filter binding assay. The rest of the sample was bleached to determine the GTPγS binding in the light. Error bars indicate SE (n = 3). C, Transducin GTPase activities in ROS from control WT and GαtG2A/Gαt−/− mice. The single-turnover GTPase measurements were initiated by mixing photobleached ROS (12 μl) containing 10 μm rhodopsin with 3 μl of 0.1 μm [γ-32P]GTP. The time course of 32Pi formation was determined using the activated charcoal procedure after the reactions were stopped with perchloric acid. The GTPase rate constants from single-phase exponential fits for the control (■) and GαtG2A/Gαt−/− (▴) ROS were 0.15 ± 0.0 and 0.09 ± 0.00 s−1 (mean ± SE; n = 3). D, PDE6 activation in ROS membranes. cGMP hydrolysis was measured in suspensions of bleached ROS membranes (3 μm R) in the absence or presence of 10 μm GTPγS using a 30 s incubation with 4 mm [3H]cGMP. Trypsin (Tryp.)-treated ROS were prepared as described in Materials and Methods. Error bars indicate SE (n = 3).
The overall ability of GαtG2Aβ1γ1 to be activated by R* was determined by light-dependent binding of GTPγS to ROS membranes from GαtG2A/Gαt−/− mice. The level of light-dependent binding of [35S]GTPγS to GαtG2A/Gαt−/− ROS was ∼16-fold lower than (6% of) that of wild-type ROS (Fig. 3B). The decrease in [35S]GTPγS binding to GαtG2A ROS is very similar to the reduction in the expression level of the mutant Gαt in the ROS fraction (18-fold) (Fig. 2B), indicating that the entire pool of GαtG2Aβ1γ1 can be activated by R* in transgenic rods.
The GTPase activities of GαtG2A and native Gαt in GαtG2A/Gαt−/− and wild-type ROS membranes were measured under single turnover conditions ([GTP] < [Gt]). A low substrate concentration (20 nm [γ-32P]GTP) was used to ensure that the single turnover conditions held for the transgenic ROS with low levels of GαtG2A. The rates of 0.15 and 0.09 s−1 for control and GαtG2A/Gαt−/− ROS preparations, respectively, were determined from the single-exponential fits to the data (Fig. 3C). Hence, the G2A mutation caused an ∼1.7-fold decrease in the rate of GTP hydrolysis by Gαt.
The ability of GαtG2A to activate its effector, PDE6, was determined by measuring the rate of cGMP hydrolysis induced by the addition of GTPγS. The presence of GTPγS caused marked stimulation of cGMP hydrolysis in control ROS but had an approximately sevenfold smaller effect in GαtG2A/Gαt−/− ROS (Fig. 3D). As a control, ROS preparations were also treated with trypsin, which cleaves the inhibitory Pγ subunit and produces full activation of the PDE6 present in the assay. The addition of trypsin resulted in equivalent enzyme activity in control and transgenic ROS preparations (Fig. 3D), indicating that the lower GTPγS-induced PDE6 activation in GαtG2A/Gαt−/− ROS was not attributable to a reduction in PDE6 levels. Given the ∼18-fold reduction in GαtG2A expression in the transgenic rods, the sevenfold reduction in GTPγS-induced PDE6 activation indicates that the mutant Gαt not only is competent to activate its effector, but seems to do so even more efficiently than wild-type Gαt.
Physiology of GαtG2A rods
To assess the ability of nonacylated Gαt to mediate phototransduction in intact cells, suction electrodes were used to record the light-induced decrease in plasma membrane current mediated by the closure of cGMP-gated ion channels (for review, see Burns and Arshavsky, 2005). The amplitude and time course of the light response provides a quantitative measure of amplification and deactivation of the phototransduction cascade in the intact cell (for review, see Pugh and Lamb, 2000). GαtG2A/Gαt−/− rods responded to brief flashes of light in a manner similar to that of wild-type rods (Fig. 4A), indicating that GαtG2A indeed becomes activated by rhodopsin and in turn can bind and activate PDE6, generating sufficient cGMP hydrolysis to close cGMP-gated channels on the plasma membrane. However, GαtG2A/Gαt−/− rods were less sensitive than those of wild-type mice; the flash strength needed to generate a half-maximal response was sixfold brighter than that for wild-type rods (Fig. 4B, Table 1). This decrease in sensitivity was not associated with a change in the ability of GαtG2A/Gαt−/− rods to capture incident photons, because the effective collecting area of 0.17 ± 0.03 μm2 (n = 11) calculated by variance-to-mean analysis (see Materials and Methods) was similar to that of 0.23 ± 0.03 μm2 (n = 9) measured for wild-type rods (p = 0.20). Thus, the reduced sensitivity of GαtG2A/Gαt−/− rods was not caused by a decrease in photon capture, but rather reduced activation of GαtG2A by photoexcited rhodopsin and/or reduced activation of PDE6 by GαtG2A. Consistent with this idea, the average response generated by a single photoexcited rhodopsin was smaller for GαtG2A/Gαt−/− rods than wild-type rods (Fig. 5A, Table 1). The population average GαtG2A response deviated from that of the average wild-type response early on the rising phase of the response (Fig. 5B,C), reaching a twofold smaller peak slightly earlier. The early rising phase of the response time course, r(t), is well described by the activation theory of Pugh and Lamb (Lamb and Pugh, 1992; Pugh and Lamb, 1993):
 |
where rmax is the saturating amplitude, A is the amplification constant, Ö is the average number of photoexcited rhodopsins, and teff is an effective delay, which we assumed to be 2 ms. This equation was fitted to the population average single-photon responses (φ = 1) to yield calculated A values of 12.4 s−2 for wild-type and 4.10 s−2 for GαtG2A rods (Fig. 5C).
Figure 4.

GαtG2A rods are less sensitive than normal. A, Representative families of responses from a wild-type rod (top) and a transgenic GαtG2A/Gαt−/− rod. Flashes (10 ms, 500 nm) were delivered at t = 0. B, The flash strengths needed to generate the same relative response amplitudes were higher for the GαtG2A/Gαt−/− rod. Data are from the same cells as in A. Curves are saturating exponential fits.
Table 1.
Characteristics of wild-type and N-acylation-deficient Gα t rods
| Dark current (pA) | Ioa (photons/μm2) | Integration time (ms) | Dim flash τrec (ms) | Time to peak (ms) | Elementary amplitude (pA) | Bright flash τDb (ms) | |
|---|---|---|---|---|---|---|---|
| G2A−/− | 13.9 ± 0.7 (33) | 341.1 ± 24.2 (28) | 407 ± 30 (27) | 487 ± 32 (26) | 102 ± 6 (30) | 0.23 ± 0.03 (11) | 672 ± 98 (6) |
| G2A+/− | 10.4 ± 1.1 (11) | 65.6 ± 16.3 (7) | 259 ± 32 (9) | 245 ± 27 (10) | 116 ± 9 (10) | 0.58 ± 0.15 (5) | 184 ± 14 (6) |
| Wild type | 10.8 ± 0.9 (15) | 53.0 ± 3.9 (14) | 324 ± 30 (13) | 239 ± 19 (13) | 116 ± 7 (14) | 0.51 ± 0.07 (10) | 224+ 11 (10) |
Numbers in parentheses represent n.
aFlash strength that elicited a half-maximal response.
bDominant time constant for recovery of saturating flash responses.
Figure 5.
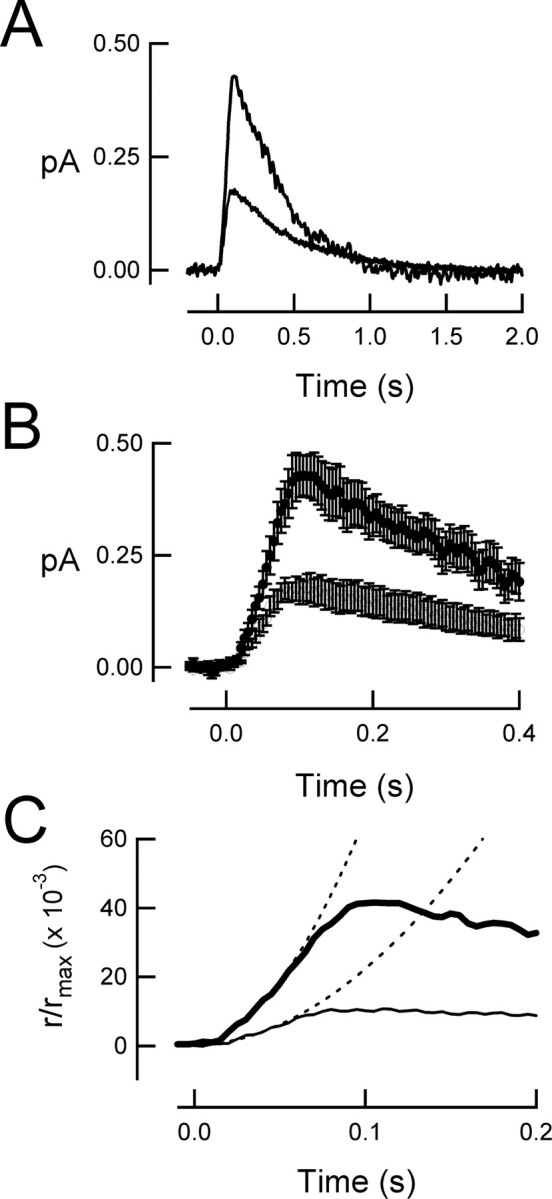
Population average single-photon responses reveal decrease in amplification in GαtG2A/Gαt−/− rods. A, Un-normalized average single-photon response from 11 GαtG2A/Gαt−/− rods (bottom trace) and 10 wild-type rods (top trace). The average dark current for the wild-type rods was 10.3 pA, and that for the GαtG2A rods was 16.6 pA. B, Same responses as in A, on an expanded time scale. Error bars indicate SEM. C, Same responses as in A, on a further expanded time scale. The dotted curves are the best parabolic fits of Equation 23 of Pugh and Lamb (1993) (see Results, Physiology of GαtG2A rods, Eq. 2), yielding amplification constants of 12.4 s−2 for the average wild-type response and 4.1 s−2 for the average GαtG2A/Gαt−/− response.
The light responses of GαtG2A/Gαt−/− rods also showed abnormal recovery kinetics. Whereas the dim flash responses of wild-type rods showed a final recovery time course approximated by a single exponential function with a 240 ms time constant, dim flash responses of GαtG2A/Gαt−/− rods recovered more slowly, with a time constant of ∼500 ms. Likewise, for bright flash responses, the dominant time constant of recovery (Nikonov et al., 1998) was approximately threefold longer for GαtG2A rods than for wild-type rods (Table 1). The slower recovery in the transgenic rods is consistent with the prolonged lifetime of activated GαtG2A resulting from impaired GTPase activity (Fig. 3C).
Discussion
This work represents the first investigation of the functional significance of N-terminal acylation in transducin function. The immunofluorescence analysis of retinal sections from transgenic mice expressing the N-acylation-deficient Gαt revealed a clear defect in GαtG2A localization. The major fraction of GαtG2A in the Gαt knock-out background was found in the inner compartments of transgenic rods instead of the OS. The marked mislocalization of GαtG2A did not result from the disruption of the Gαt–Gβ1γ1 interaction (Fig. 3A). Therefore, our results show for the first time the critical role of Gαt N-acylation in transducin localization. Native holo-transducin carries two lipid anchors, N-acyl on Gαt and C-terminal farnesyl on Gγ1, which appear to be necessary and sufficient for trafficking Gαtβ1γ1 to the OS in the dark-adapted retinas. Interestingly, Gβ1γ1 is relatively evenly distributed between the OS and the rod inner compartments in Gαt knock-out mice (Zhang et al., 2003). Hence, the lipophilic nature of the transducin molecules determined by the lipid modifications decreases in the same order as the ability to correctly localize to the OS: Gαtβ1γ1 > Gβ1γ1 > GαtG2Aβ1γ1. We surmise from this analysis that the lipid anchors on Gαtβ1γ1, particularly the N-terminal acylation of Gαt, represent key determinants for transducin compartmentalization to the OS.
The biochemical analysis of GαtG2A in transgenic ROS indicated that the lack of N-acylation of Gαt does not block the activation of mutant transducin by R*, nor the GTPγS-induced activation of PDE6 by GαtG2A, although the extent of each of these reactions was severely reduced. This reduction is likely attributable to the ∼18-fold lower than normal concentration of GαtG2A. In light of the greatly reduced levels of activatable GαtG2A, it is remarkable that the sensitivity of GαtG2A/Gαt−/− rods was not correspondingly reduced. Instead, this 18-fold reduction in heterotrimer levels was associated with an amplification constant that was reduced only by a factor of three (Fig. 5).
From a theoretical analysis of the phototransduction cascade, the amplification constant is proportional, among other things, to the rate of Gt activation per R* (νRG), the coupling efficiency (cGP; defined as the ratio of activated Gαt to activated PDE6 subunits), and βsub, the rate constant of cGMP hydrolysis by Gαt-activated PDE subunit (Pugh and Lamb, 1993). Of these parameters, only νRG is expected to be dependent on expression level, because it is thought to be limited by the rate of diffusional encounter between a single R* and transducin molecules on the disc membrane (Lamb and Pugh, 1992; Pugh and Lamb, 1993; Calvert et al., 2001). For the mammalian rod at 37°C, the diffusion limit on νRG can be described by a simplified equation:
where DR and DG are the lateral diffusion coefficients of R* and transducin on the disc membrane, and CG is the concentration (surface density) of transducin on the membrane (Lamb and Pugh, 1992; Pugh and Lamb, 1993). Thus, an ∼18-fold reduction of transducin concentration in the transgenic OS might be expected to cause an equivalent reduction in the amplification constant (and the rates of Gt and PDE6 activation per R*). Of course, if the G2A mutation also reduces cGP and/or βsub (which together define the ability of a given GαtG2A to activate a PDE6 subunit), then A would be expected to be even lower still.
How then do we explain the only threefold reduction in the amplification constant? One possibility is that the lack of N-acylation increases the lateral diffusion of Gt, thereby increasing its encounter rate with R* and the νRG rate. According to Equation 3 and taking the estimates DR ∼ 1.5 and DG ∼ 1.8 μm2 · s−1 at 37°C (Pugh and Lamb, 1993), an 18-fold reduction in CG accompanied by a threefold reduction in A implies a coincident tenfold increase in DG. On the other hand, GαtG2A may actually activate PDE6 more effectively than normal Gαt (i.e., couple to PDE6 with higher cGP and/or induce higher βsub). Support for this comes in part from the results of Figure 3D, which showed a sevenfold decrease in GTPγS-dependent PDE6 activity, compared with the 18-fold decrease in GαtG2A expression. Still, the PDE6 activation results in vitro do not necessarily indicate greater ability of GαtG2A to activate PDE6 in intact rods, because the assay was conducted in diluted suspensions of ROS membranes. Finally, it may be that the amplification constant does not scale linearly with transducin expression. Although previous studies have demonstrated a clear correlation between A and outer segment Gt levels (Sokolov et al., 2002), the linearity of this relationship has not been explicitly studied. Because the surface density of transducin is comparable with the Michaelis constant for rhodopsin-transducin binding at 34°C (Heck and Hofmann, 2001), the rate of activation by transducin might be less than linearly dependent on transducin expression.
Another clear effect of the G2A mutation is the slower than normal recovery of flash responses of transgenic rods. The time constants of recovery of dim and bright flash responses of GαtG2A/Gαt−/− rods are approximately two and three times slower than normal, respectively (Table 1), consistent with the reduced GTPase activity of GαtG2A in ROS membranes (Fig. 3C). In mouse rods, the time constant of dim flash response recovery and the dominant time constant for bright flash response recovery are both sensitive to the expression levels of the RGS9-1/Gβ5 complex (Krispel et al., 2006). The GAP activity of RGS9-1/Gβ5 is greatly accelerated by formation of the membrane-bound complex with the membrane anchor, R9AP (Lishko et al., 2002; Hu et al., 2003). We propose that the N-acylation of Gαt along with lipid modifications of PDE6 provides for tight tethering of the activated Gαt/PDE6 complex to the membrane, which enables a more efficient deactivation of Gαt by the GAP complex. Overall, this study demonstrates that in addition to the critical role in trafficking and subcellular localization, the N-acylation of transducin is essential for the normal recovery of rod photoresponses.
Footnotes
This work was supported by National Institutes of Health Grants R01 EY-12682 (N.O.A.) and EY14047 (M.E.B.). We thank Dr. Edward N. Pugh Jr for critical reading of this manuscript and valuable comments.
References
- Bourne HR. How receptors talk to trimeric G proteins. Curr Opin Cell Biol. 1997;9:134–142. doi: 10.1016/s0955-0674(97)80054-3. [DOI] [PubMed] [Google Scholar]
- Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48:387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Burns ME, Baylor DA. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu Rev Neurosci. 2001;24:779–805. doi: 10.1146/annurev.neuro.24.1.779. [DOI] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicolo M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN, Jr, Makino CL, Lem J. Phototransduction in transgenic mice after targeted deletion of the rod transducin α-subunit. Proc Natl Acad Sci USA. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, Makino CL. Membrane protein diffusion sets the speed of rod phototransduction. Nature. 2001;411:90–94. doi: 10.1038/35075083. [DOI] [PubMed] [Google Scholar]
- Catty P, Pfister C, Bruckert F, Deterre P. The cGMP phosphodiesterase-transducin complex of retinal rods. Membrane binding and subunits interactions. J Biol Chem. 1992;267:19489–19493. [PubMed] [Google Scholar]
- Chen CA, Manning DR. Regulation of G proteins by covalent modification. Oncogene. 2001;20:1643–1652. doi: 10.1038/sj.onc.1204185. [DOI] [PubMed] [Google Scholar]
- Clerc A, Bennett N. Activated cGMP phosphodiesterase of retinal rods. A complex with transducin α subunit. J Biol Chem. 1992;267:6620–6627. [PubMed] [Google Scholar]
- Escriba PV, Wedegaertner PB, Goni FM, Vogler O. Lipid-protein interactions in GPCR-associated signaling. Biochim Biophys Acta. 2006;1768:836–852. doi: 10.1016/j.bbamem.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Heck M, Hofmann KP. Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J Biol Chem. 2001;276:10000–10009. doi: 10.1074/jbc.M009475200. [DOI] [PubMed] [Google Scholar]
- Higgins JB, Casey PJ. The role of prenylation in G-protein assembly and function. Cell Signal. 1996;8:433–437. doi: 10.1016/s0898-6568(96)00071-x. [DOI] [PubMed] [Google Scholar]
- Hu G, Zhang Z, Wensel TG. Activation of RGS9-1GTPase acceleration by its membrane anchor, R9AP. J Biol Chem. 2003;278:14550–14554. doi: 10.1074/jbc.M212046200. [DOI] [PubMed] [Google Scholar]
- Kassai H, Aiba A, Nakao K, Nakamura K, Katsuki M, Xiong WH, Yau KW, Imai H, Shichida Y, Satomi Y, Takao T, Okano T, Fukada Y. Farnesylation of retinal transducin underlies its translocation during light adaptation. Neuron. 2005;47:529–539. doi: 10.1016/j.neuron.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerov V, Chen D, Moussaif M, Chen YJ, Chen CK, Artemyev NO. Transducin activation state controls its light-dependent translocation in rod photoreceptors. J Biol Chem. 2005;280:41069–41076. doi: 10.1074/jbc.M508849200. [DOI] [PubMed] [Google Scholar]
- Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen CK, Burns ME. RGS expression rate-limits recovery of rod photoresponses. Neuron. 2006;51:409–416. doi: 10.1016/j.neuron.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Lamb TD, Pugh EN., Jr A quantitative account of the activation steps involved in phototransduction in amphibian photoreceptors. J Physiol (Lond) 1992;449:719–758. doi: 10.1113/jphysiol.1992.sp019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lem J, Applebury ML, Falk JD, Flannery JG, Simon MI. Tissue-specific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron. 1991;6:201–210. doi: 10.1016/0896-6273(91)90356-5. [DOI] [PubMed] [Google Scholar]
- Lishko PV, Martemyanov KA, Hopp JA, Arshavsky VY. Specific binding of RGS9-Gβ5L to protein anchor in photoreceptor membranes greatly enhances its catalytic activity. J Biol Chem. 2002;277:24376–24381. doi: 10.1074/jbc.M203237200. [DOI] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Song H, Tsang SH, Chen CK, Sokolov M, Skiba NP, Arshavsky VY. Transducin translocation in rods is triggered by saturation of the GTPase-activating complex. J Neurosci. 2007;27:1151–1160. doi: 10.1523/JNEUROSCI.5010-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaif M, Rubin WW, Kerov V, Reh R, Chen D, Lem J, Chen C-K, Hurley JB, Burns ME, Artemyev NO. Phototransduction in a transgenic mouse model of Nougaret night blindness. J Neurosci. 2006;26:6863–6872. doi: 10.1523/JNEUROSCI.1322-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert TA, Hurley JB. Functional heterogeneity of transducin α subunits. FEBS Lett. 1998;422:343–345. doi: 10.1016/s0014-5793(98)00037-4. [DOI] [PubMed] [Google Scholar]
- Neubert TA, Johnson RS, Hurley JB, Walsh KA. The rod transducin α subunit amino terminus is heterogeneously fatty acylated. J Biol Chem. 1992;267:18274–18277. [PubMed] [Google Scholar]
- Nikonov S, Engheta N, Pugh EN., Jr Kinetics of recovery of the dark-adapted salamander rod photoresponse. J Gen Physiol. 1998;111:7–37. doi: 10.1085/jgp.111.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–1149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Phototransduction in vertebrate rods and cones: molecular mechanisms of amplification, recovery and light adaptation. In: Stavenga DG, de Grip WJ, Pugh EN, editors. Handbook of biological physics, Vol 3, Molecular mechanisms in visual transduction. Atlanta, GA: Elsevier; 2000. pp. 183–254. [Google Scholar]
- Raport CJ, Dere B, Hurley JB. Characterization of the mouse rod transducin α subunit gene. J Biol Chem. 1989;264:7122–7128. [PubMed] [Google Scholar]
- Sokolov M, Lyubarsky AL, Strissel KJ, Savchenko AB, Govardovskii VI, Pugh EN, Jr, Arshavsky VY. Massive light-driven translocation of transducin between the two major compartments of rod cells: a novel mechanism of light adaptation. Neuron. 2002;34:95–106. doi: 10.1016/s0896-6273(02)00636-0. [DOI] [PubMed] [Google Scholar]
- Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, Goff SP, Arshavsky VY. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- van Dop C, Yamanaka G, Steinberg F, Sekura RD, Manclark CR, Stryer L, Bourne HR. ADP-ribosylation of transducin by pertussis toxin blocks the light-stimulated hydrolysis of GTP and cGMP in retinal photoreceptors. J Biol Chem. 1984;259:23–26. [PubMed] [Google Scholar]
- Watkins PA, Burns DL, Kanaho Y, Liu TY, Hewlett EL, Moss J. ADP-ribosylation of transducin by pertussis toxin. J Biol Chem. 1985;260:13478–13482. [PubMed] [Google Scholar]
- Wedegaertner PB, Wilson PT, Bourne HR. Lipid modifications of trimeric G proteins. J Biol Chem. 1995;270:503–506. doi: 10.1074/jbc.270.2.503. [DOI] [PubMed] [Google Scholar]
- West RE, Jr, Moss J, Vaughan M, Liu T, Liu TY. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J Biol Chem. 1985;260:14428–14430. [PubMed] [Google Scholar]
- Zhang H, Huang W, Zhang H, Zhu X, Craft CM, Baehr W, Chen C-K. Light-dependent redistribution of visual arrestins and transducin subunits in mice with defective phototransduction. Mol Vis. 2003;9:231–237. [PubMed] [Google Scholar]