

Abstract
The purpose of this study was to elucidate the cellular/biochemical pathway(s) by which interleukin-1β (IL-1β) contributes to the pathogenesis of hypoxic–ischemic brain damage. In vivo, IL-1 receptor type I (IL-1RI)-deficient mice showed smaller infarcts and less neurological deficits than wild-type animals after a 90 min reversible middle cerebral artery occlusion. In vitro, IL-1β mediated an enhancement of hypoxic neuronal injury in murine cortical cultures that was lacking in cultures derived from IL-1RI null mutant animals and was blocked by the IL-1 receptor antagonist or an IL-1RI blocking antibody. This IL-1β-mediated potentiation of hypoxic neuronal injury was associated with an increase in both cellular cystine uptake ([cystine]i) and extracellular glutamate levels ([glutamate]e) and was prevented by either ionotropic glutamate receptor antagonism or removal of l-cystine, suggesting a role for the cystine/glutamate antiporter (System xc−). Indeed, dual System xc−/metabotropic glutamate receptor subunit 1 (mGluR1) antagonism but not selective mGluR1 antagonism prevented neuronal injury. Additionally, cultures derived from mGluR1-deficient mice exhibited the same potentiation in injury after treatment with IL-1β as wild-type cultures, an effect prevented by System xc−/mGluR1 antagonism. Finally, assessment of System xc− function and kinetics in IL-1β-treated cultures revealed an increase in velocity of cystine transport (Vmax), in the absence of a change in affinity (Km). Neither the enhancement in [cystine]i, [glutamate]e, or neuronal injury were observed in chimeric cultures consisting of IL-1RI+/+ neurons plated on top of IL-1RI−/− astrocytes, highlighting the importance of astrocyte-mediated alterations in System xc− as a novel contributor to the development and progression of hypoxic neuronal injury.
Keywords: interleukin-1β, IL-1 receptor type I, hypoxia, excitotoxicity, astrocytes, System xc−
Introduction
Increased mRNA and protein expression for the proinflammatory cytokine interleukin-1β (IL-1β) has been demonstrated in rodent brains after experimental cerebral ischemia (Minami et al., 1992; Liu et al., 1993; Buttini et al., 1994; Yabuuchi et al., 1994; Sairanen et al., 1997). Moreover, numerous studies indicate that IL-1β plays an active role in the brain damage that accompanies ischemia in mice (Hara et al., 1997; Yang et al., 1997, 1998, 1999; Schielke et al., 1998; Liu et al., 1999; Ohtaki et al., 2003), in rats (Relton and Rothwell, 1992; Martin et al., 1994; Betz et al., 1995; Loddick and Rothwell, 1996; Mulcahy et al., 2003), and in humans (Emsley et al., 2005). Despite this, the cellular and biochemical pathway(s) by which this cytokine contributes to neuronal cell death after cerebral ischemia are still unknown.
The ability of IL-1β to influence cellular function depends on the expression of an appropriate receptor. Most evidence supports the contention that IL-1β exerts its action by binding to a specific plasma membrane receptor, designated IL-1 receptor type I (IL-1RI) (Sims et al., 1993). IL-1RI is expressed throughout the brain, with the highest levels found in cerebral cortex and hippocampus (Farrar et al., 1987; Takao et al., 1992; French et al., 1999). However, it has been suggested recently that an alternative signaling pathway (i.e., an unidentified functional receptor for IL-1) may exist (Desson and Ferguson, 2003; Diem et al., 2003; Andre et al., 2006). Additionally, cell-type-specific responses are likely induced in response to IL-1β. For instance, it has been demonstrated that different signaling pathways mediate IL-1β actions in neurons compared with astrocytes (Srinivasan et al., 2004). Thus, to develop successful clinical strategies based on the inhibition of IL-1β signaling for the treatment of cerebral ischemia, it will be important to identify the receptor, the cell type mediating the untoward effect, and the subsequently activated biochemical pathway(s) responsible.
Taking both in vivo and in vitro approaches, we investigated (1) whether the detrimental effects of IL-1β during cerebral ischemic injury are mediated via signaling through the canonical IL-1 receptor IL-1RI, (2) which cell type(s) participate in the response, and (3) whether and how glutamate receptor toxicity is involved. Herein, we demonstrate for the first time that IL-1RI signaling contributes to cerebral ischemic injury induced by middle cerebral artery occlusion (MCAO) in vivo. In vitro modeling confirms the role of IL-1RI, indicates the necessity of astrocyte activation, and demonstrates that an enhancement of glutamate excitotoxicity resulting from both increased cystine/glutamate antiporter (System xc−) function and loss of glutamate uptake underlies the neurotoxic effects of IL-1β.
Part of this work has been published previously in abstract form (Fogal et al., 2004, 2005b, 2006).
Materials and Methods
Animals.
This study was conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals and has been approved by the Institutional Animal Care and Use Committee of the University of Connecticut Health Center. CD1 mice were obtained from Charles River Laboratories (Wilmington, MA) and were used in all experiments unless indicated otherwise. IL-1RI null mutant animals (strain, B6.129S7-IL1r1tm1lmx/J) (Glaccum et al., 1997) were purchased from The Jackson Laboratory (Bar Harbor, ME). Because the mutant animals were bred to homozygosity, animals from the background strain (also purchased from The Jackson Laboratory) were bred in parallel, and their offspring were used as wild-type controls (C57BL/6). Mice deficient for all splice variants of metabotropic glutamate receptor subtype 1 (mGluR1) (Conquet et al., 1994) were provided by Dr. Francesco Ferraguti (Univesity of Innsbruck, Innsbruck, Austria) on behalf of GlaxoSmithKline (Harlin, Essex, UK) and rederived in the Gene Targeting and Transgenic Facility of the University of Connecticut Health Center. Parental strains heterozygous for the mutation were bred and cultures prepared from the homozygous offspring (mGluR1−/− and mGluR1+/+) via single embryo dissections.
Transient focal cerebral ischemia.
Transient focal cerebral ischemia was induced in male IL-1RI-deficient and wild-type mice weighing 22–25 g (10–12 weeks) by 90 min of reversible MCAO, followed by 70.5 h of reperfusion (total survival, 72 h) as described previously (McCullough et al., 2004). In short, under halothane anesthesia (5% for induction, 1% for maintenance), mice were spontaneously ventilated with oxygen-enriched (100%) air via a nose cone. Transient unilateral MCAO was performed by inserting a 6.0 nylon monofilament into the internal carotid artery 6 mm from the internal carotid/pterygopalatine artery bifurcation via an external carotid artery stump. The animal was then allowed to awaken from anesthesia, at which time intra-ischemic neurological deficits were confirmed as described below. At the end of a 90 min occlusion, mice were reanesthetized for removal of the suture. Rectal muscle temperatures were monitored and maintained at 37°C during surgery, ischemia, and reperfusion using a MONO-THERM system. At 70.5 h of reperfusion, animals were rapidly decapitated, and the brain was harvested for pathological examination.
Assessment of neurological deficits.
Neurological deficits were scored according to Longa et al. (1989) during ischemia (as criterion for inclusion), as well as 24 and 72 h after reperfusion as follows: 0, no deficit; 1, mild forelimb weakness on cage grasp; 2, torso turning to the ipsilateral side when held by tail with moderate forelimb weakness; 3, preferential circling to affected side; 4, forced circling to affected side; 5, unable to bear weight on affected side; 6, no spontaneous locomotor activity or barrel rolling. An animal showing no deficits (0) during the ischemic period was excluded from the study.
Quantification of infarct volume.
Animals were killed by decapitation, and brains were directly sectioned into five coronal 2 mm sections that were then stained with 1.5% 2,3,5-triphenyl-tetrazolium chloride (TTC) for 30 min. Infarct volumes were quantified using Sigma Scan Pro Image Analysis version 5.0.0 (SPSS, Chicago, IL). The area of infarct, identified by the lack of TTC staining, was measured on the rostral and caudal surfaces of each slice and numerically integrated across the thickness of the slice. Volumes from all slices were summed to calculate total infarct volume over the entire hemisphere. Infarct volumes were measured separately in the cerebral cortex, caudate–putamen, and hemisphere and were corrected for swelling by comparing the volume of interest in the infarcted hemisphere versus the non-infarcted hemisphere (Lin et al., 1993).
Laser Doppler flowmetry and physiological monitoring.
Monitoring of physiological variables along with laser Doppler flowmetry (LDF) was performed in a separate nonsurvival cohort of animals for all groups as described previously (McCullough et al., 2004). In short, the femoral artery was cannulated for measurement of arterial blood gases and mean arterial pressure (MAP). Arterial blood gases and glucose were measured at baseline and 30 min into MCAO. Intra-ischemic occlusion was monitored with an LDF probe (model MBF 3 D; Moor Instruments, Wilmington, DE), which was placed directly on the right parietal skull surface 2 mm posterior and 3 mm lateral to the bregma. After placement of the probe, the middle cerebral artery was occluded and the LDF was monitored both during ischemia and for 30 min after reperfusion. These animals were not allowed to wake during ischemia and were killed after 30 min of reperfusion. Therefore, all invasive blood pressure measurements were obtained in anesthetized animals.
Cell culture.
Mixed murine cortical cell cultures, containing neurons and astrocytes, were prepared from the respective mouse strains as described previously (Trackey et al., 2001; Fogal et al., 2005a). The cultures were prepared by a two-step plating method, whereby astrocytes were prepared first and grown to confluency and then cortical neurons were plated on top. To obtain cortical astrocytes, cerebral cortices from postnatal (1–3 d) animals were dissected under aseptic conditions, and cells were dissociated and plated into 15 mm multiwell dishes (Falcon Primaria; BD Biosciences, Lincoln Park, NJ) at a density of 1.0–1.2 hemispheres/10 ml per plate. Astrocyte plating medium consisted of glutamine-free Eagle's minimum essential medium (Earle's salts; Mediatech, Herndon, VA) supplemented with 2 mm glutamine, 20 mm glucose, and 4 mm bicarbonate [media stock (MS)], 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) or 10% bovine growth serum (BGS) (Hyclone), 10% iron-supplemented calf serum (CS) (Hyclone), 10 ng/ml epidermal growth factor (Invitrogen, Carlsbad, CA), 50 IU/ml penicillin, and 50 μg/ml streptomycin (Mediatech). Once confluent, and thus contact inhibited, astrocyte monolayers were treated once for 5–6 d with 8 μm cytosine β-d-arabinofuranoside (AraC) (Sigma, St. Louis, MO) to inhibit the growth of any rapidly dividing cells such as microglia. Cultures were then switched into maintenance medium (MS containing 10% CS and antibiotics). Cortical neurons were obtained similarly from the cerebral cortices of embryonic day 15 fetuses and plated at a density of 3.0–3.5 hemispheres/10 ml per plate on established astrocyte monolayers [14–28 d in vitro (DIV)] in MS supplemented with 5% CS and 5% FBS (or BGS) and antibiotics. At 6 DIV, mixed cultures were treated with 8 μm AraC for 2 d and then switched into maintenance medium. The medium was changed twice weekly, and cultures were used for experimentation between 13 and 15 DIV, at which time they are fully susceptible to NMDA receptor-mediated toxicity (Fogal et al., 2005c). One day before experimental manipulation, cells were shifted (250 μl of 400 μl exchanged) into MS. All cultures were maintained at 37°C in a humidified 6% CO2 containing incubator. These cultures contain an approximate 50:50 neuron to astrocyte ratio (Hewett et al., 2000).
IL-1β exposure.
Cells were exposed to IL-1β (recombinant mouse; R & D Systems, Minneapolis, MN) for 20–24 h before other experimental manipulations. Cultures were washed two times with MS (750 μl) to remove any residual serum, and then IL-1β solutions (1.5×, 250 μl) in an incubation buffer of MS plus 0.15% fatty acid free BSA or incubation buffer alone (250 μl) was added. The final well volume was 400 μl. Cells were then placed at 37°C in a humidified atmosphere containing 6% CO2. In experiments using recombinant mouse IL-1ra (rIL-1ra) (R & D Systems) or monoclonal anti-mouse IL-1RI antibody directed against the extracellular domain of mouse IL-1RI (MAB7711; R & D Systems), the respective solutions were prepared with IL-1β (1.5 ng/ml; 1.5×) and added as described above.
Hypoxia.
Cultures were placed into an anaerobic chamber (Forma Scientific, Marietta, OH) containing a gas mixture of 5% CO2, 10% H2, 85% N2 (<0.2% O2). Culture medium was replaced by thorough exchange with a deoxygenated balanced salt solution (BSS) containing 116 mm NaCl, 5.4 mm KCl, 0.8 mm MgSO4, 1 mm NaH2PO4, 26.2 mm NaHCO3, 1.8 mm CaCl2, 0.01 mm glycine, 2 mm l-glutamine, 1× MEM amino acids (Invitrogen, Carlsbad, CA), and 5 mm glucose (BSS5). Cell cultures were then placed in a 37°C incubator in the anaerobic chamber. Cells washed into a similar deoxygenated BSS but containing 20 mm glucose (BSS20) can resist neuronal injury even during long periods (>12 h) of oxygen deprivation (data not shown). Thus, parallel cultures within the same plate were placed into deoxygenated BSS20 to serve as sham controls. In experiments in which glutamate receptor antagonists were used, drugs were given at the initiation of the hypoxic insult (250 μl, 1.5×). Cultures were removed from the anaerobic chamber after 4–5 h and returned to a 37°C, 6% CO2-containing normoxic (21% O2) incubator. Cell death was assessed 20–24 h later.
Measurement of neuronal cell death in vitro.
Neuronal cell death was estimated in all experiments by examination of culture morphology under phase-contrast microscopy and quantitatively assessed by measurement of the lactate dehydrogenase (LDH) released into the culture supernatant by damaged or dead cells as described in detail previously (Koh and Choi, 1987; Uliasz and Hewett, 2000). The small amount of LDH (generally <15% of total) present in the supernatant of control sister cultures was subtracted from levels in experimental conditions to yield LDH release specific to experimental injury. Data were expressed as a percentage of total neuronal LDH activity (defined as 100%), determined in each experiment by assaying the supernatant of parallel cultures exposed to 250 μm NMDA for 20–24 h. Because cortical astrocytes do not contain NMDA receptors nor have been shown by us and others (Goldberg et al., 1987) to be injured by up to 12 h of oxygen deprivation, changes in LDH activity can be used as a specific marker of neuronal injury in this system. Additionally, we previously confirmed the LDH data with propidium iodide staining and the absence of astrocytic cell death via trypan blue staining (Fogal et al., 2005a), further ensuring the validity of LDH measurements in this paradigm.
Measurement of glutamate.
At various times after the onset of hypoxia, samples of the bathing media from the cell cultures were collected. After confirming absence of LDH leakage, which assessed for cell damage and hence nonspecific leakage of glutamate from dying cells, samples were stored at −20°C until analyzed. Two hundred microliters were assayed for glutamate via phenylisothiocyanate derivatization, HPLC (1100 series; Agilent, Palo Alto, CA) separation using a Hypersil-ODS reverse-phase column and ultraviolet detection at a wavelength of 254 nm, as described previously (Lobner and Choi, 1994; Hewett et al., 1996). Before the HPLC run, the samples were reconstituted in solvent consisting of 0.14 m sodium acetate, 0.05% tetraethylammonium, and 6% acetonitrile and brought to pH 6.4 with glacial acetic acid. The above solvent was used as the mobile phase with the column being washed between each sample run in 60% acetonitrile/40% water. Media glutamate levels were calculated by normalizing to glutamate standards, which were treated identically. Glutamate measurements are linear over the range 0.1–10 μm.
Measurement of glutamate transport.
Measurements of glutamate transport were performed as described and characterized previously (Drejer et al., 1982; Rosenberg et al., 1992; Hazell et al., 1997; Vidwans and Hewett, 2004). Sodium-dependent, high-affinity glutamate transporters (System XAG−) are not absolutely specific for l-glutamate but also transport d-aspartate with similar affinity (Danbolt, 2001). Hence, [3H]d-aspartate (PerkinElmer, Boston, MA) was used as the substrate for the high-affinity glutamate transporters because, unlike glutamate, it has the advantage of being non-metabolizable and is not a substrate for the synaptic vesicle uptake system (Drejer et al., 1982; Tabb and Ueda, 1991). We reported previously that [3H]d-aspartate uptake was sodium dependent, was in the linear range for up to 15 min of uptake, and was blocked by the glutamate transporter inhibitors pyrrolidine-2,4-dicarboxylate and dl-threo-β-benzyloxyaspartic acid (Vidwans and Hewett, 2004). To measure [3H]d-aspartate uptake under hypoxic conditions, cells were moved into the anaerobic chamber and washed into deoxygenated BSS5. At various times after the onset of hypoxia, 250 μl of BSS5 were exchanged with BSS5 containing [3H]d-aspartate (final concentration, 0.1 μCi/ml) and 50 μm unlabeled aspartate as a carrier. Uptake was terminated after 5 min by washing with ice-cold sodium-free choline stop buffer containing 116 mm choline chloride, 0.8 mm MgSO4, 1 mm KH2PO4, 10 mm HEPES, 5 mm KOH, 10 mm glucose, 0.9 mm CaCl2, and 5 mm non-radioactive d-aspartate. Culture wells were subsequently aspirated dry, and cells were lysed by addition of 400 μl of warm 0.2% SDS. The amount of accumulated radioactivity was estimated in 50% of the cell lysate via liquid scintillation counting (TriCarb 4000 scintillation counter; Packard Instruments, Downers Grove, IL). Readings of counts per minute were corrected back to the original volume of lysate and expressed as total counts per minute per well per 5 min. Equal cell density between wells was confirmed by measuring total protein content per well (as determined with BCA Assay; Pierce, Rockford, IL).
Measurement of System xc− activity.
System xc− activity was assessed by measuring [14C (U)]-l-cystine (14C-l-cystine) (PerkinElmer) uptake. Cells were moved into the anaerobic chamber and washed into deoxygenated BSS5 or treated equivalently but kept under normoxic conditions (control). Ninety minutes later, cells were washed with HBSS containing 120 mm NaCl, 5.4 mm KCl, 0.8 mm MgCl2, 1.8 mm CaCl2, 20 mm HEPES, 15 mm glucose, and 0.01 mm glycine, pH 7.4 (25°C). Then, 14C-l-cystine [3 μm 14C-l-cystine (S*) plus 27 μm unlabeled l-cystine (S)] was added in HBSS containing aspartate (1 mm) and acivicin (1 mm) to inhibit System XAG− and γ-glutamyl transpeptidase, respectively, thus isolating cystine uptake via System xc− (Knickelbein et al., 1997). Unless otherwise indicated, 14C-l-cystine uptake was terminated after 5 min by rapidly washing the cultures with ice-cold PBS. Uptake for this period is in the linear range as determined by a time course of 14C-l-cystine uptake (data not shown). Cultures were subsequently aspirated dry, and cells were lysed by addition of 400 μl of warm 0.2% SDS. The amount of accumulated radioactivity was estimated in 50% of cell lysate using a Packard Instruments Tricarb 4000 scintillation counter. To calculate the picomoles of cystine transported per minute, we first determined the specific activity, which is defined as the amount of radioactivity (in counts per minute) per picomoles of labeled cystine. Thus, radioactivity of known amounts of 14C-l-cystine (0, 2, 6, 10, and 20 pmol) was measured, the background (0) was subtracted from all readings, and the calculated individual counts per minute per picomoles values were averaged. Readings of counts per minute from experimental conditions were corrected back to the original volume of lysate, and the picomoles of cystine transported per minute was calculated using the following formula:
 |
where cpm is counts per minute, and S.A. is specific activity. Protein content for each well was determined using the BCA Assay (Pierce), and data were expressed as picomoles per minute per milligram protein.
To determine the kinetics of cystine uptake, 14C-l-cystine uptake was determined essentially as described above but measured over a range of cystine concentrations: 0.1 μm S*, 0.3 μm S*, 1 μm S*, 3 μm (1 μm S* plus 2 μm S), 10 μm (1 μm S* plus 9 μm S), and 30 μm (3 μm S* plus 27 μm S). Kinetic data of cystine/glutamate exchange were determined using the Hanes–Woolf plot in which [substrate]/velocity is plotted against [substrate]. The slope corresponds to 1/Vmax, whereas the x-intercept corresponds to Km. Kinetic data were further confirmed using the Woolf–Augustinsson–Hofstee plot, in which velocity is plotted versus velocity/[substrate]. The slope here corresponds to −Km, whereas the x-intercept corresponds to Vmax.
Statistical analysis.
All experiments were repeated at least three times, and statistical analyses were performed using Prism software (GraphPad Software, San Diego, CA) as described in each figure legend. When appropriate, percentage data were first transformed (arcsin square root) before analyses because, by nature, it is non-normally distributed (Steel and Torrie, 1980). Values of zero or less were set at 1 × 10−20 before transformation. In all experiments, significance was assessed at p < 0.05.
Results
IL-1RI-deficient mice are less susceptible to cerebral ischemic damage
Transient cerebral ischemia was induced by 90 min of reversible MCAO in IL-1RI-deficient mice (IL-1RI−/−) and their C57BL/6 wild-type controls (IL-1RI+/+). Compared with their wild-type counterparts, IL-1RI−/− animals had significantly smaller hemispheric (58.0 ± 5.4 vs 22.8 ± 5.5%), cerebral cortical (74.4 ± 6.5 vs 24.6 ± 8.9%), and striatal (94.2 ± 2.7 vs 64.4 ± 1.1%) infarct volumes 72 h after the onset of ischemia (Fig. 1A,B). IL-1RI−/− animals also had improved neurological scores over IL-1RI+/+ animals at 24 h after ischemia (p = 0.0315, Mann–Whitney test). Interestingly, scores remained stable in wild-type mice during the next 48 h, whereas IL-1RI null mutant animals demonstrated marked improvement (p < 0.0001) (Fig. 1C). Importantly, LDF measurements demonstrated comparable decreases in cortical perfusion throughout the ischemic period and similar increases in blood flow after reperfusion in both genotypes, suggesting that deletion of IL-1RI does not alter the severity of the ischemic insult (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Furthermore, there were no significant differences between IL-1RI null mutant and wild-type mice in MAP, pH, paCO2, paO2, or blood glucose levels before and during MCAO (supplemental Table 1, available at www.jneurosci.org as supplemental material). Hence, changes in these physiological parameters cannot account for the reduced susceptibility to cerebral ischemic damage in mice lacking IL-1RI.
Figure 1.
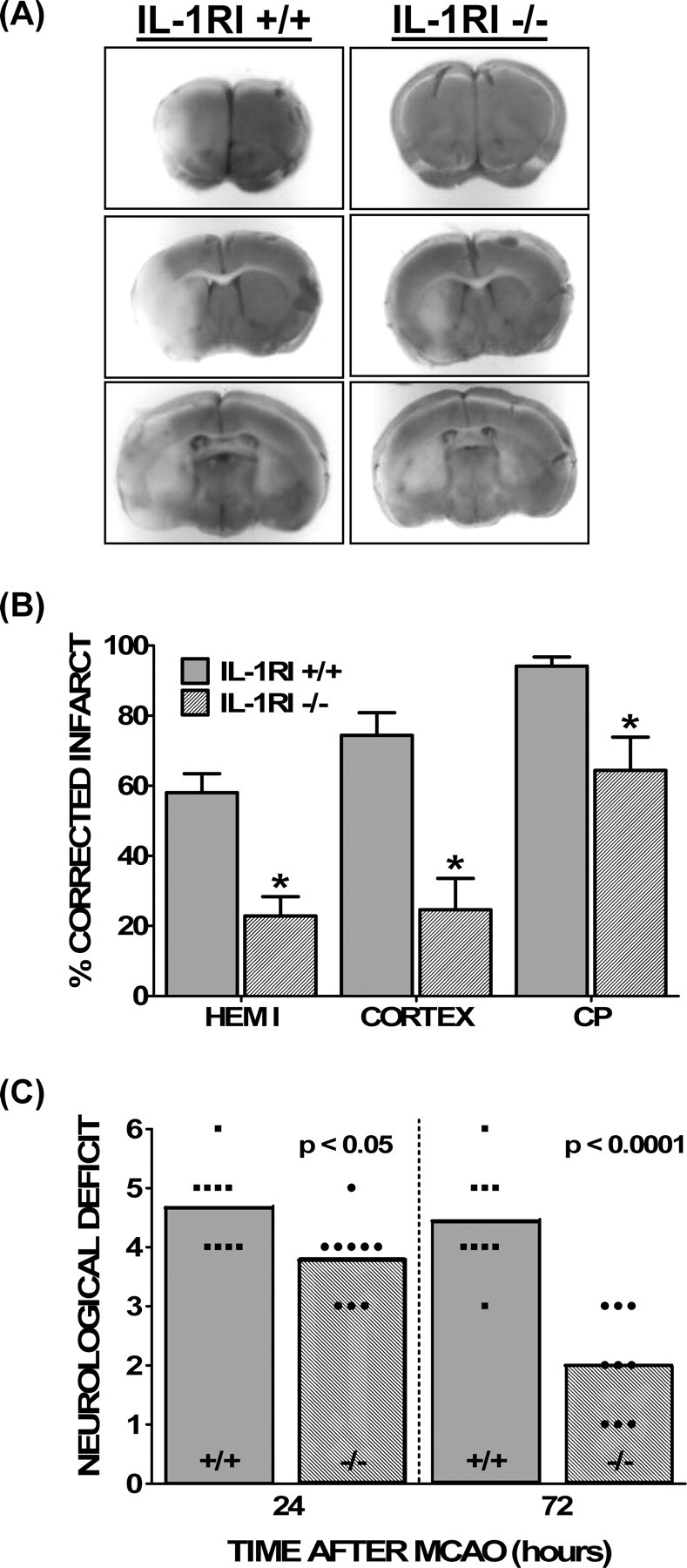
Comparison of ischemic brain damage between wild-type and IL-1RI null mutant animals. A, Representative TTC staining of coronal brain sections (2 mm) 70.5 h after a 90 min reversible MCAO for wild-type (IL-1RI+/+; left) and IL-1RI-deficient (IL-1RI−/−; right) male mice. The lack of TTC staining indicates an infarcted area. B, Infarct volumes in the hemisphere (hemi), cerebral cortex (cortex), and caudate–putamen (CP) of wild-type (IL-1RI+/+; gray bars) and IL-1RI knock-out (IL-1RI−/−; hatched bars) male mice were measured over five coronal sections. Infarct volumes are reported as the mean ± SEM percentage of non-ischemic hemisphere, corrected for edema. Actual infarct volumes for wild-type versus null mutant animals were as follows: hemisphere, 177.3 ± 18.0 versus 65 ± 16.4 mm3; cortex, 99.8 ± 8.2 versus 29.7 ± 11.3 mm3; caudate–putamen, 31.2 ± 2.0 versus 18.1 ± 2.9 mm3. An asterisk indicates a value significantly different from wild-type animals, as assessed via an unpaired t test (n = 9 animals per group). Significance was assessed at p < 0.05. C, Post-ischemic neurological deficits were scored as described in Materials and Methods at 24 and 72 h after MCAO in wild-type (IL-1RI+/+; squares; gray bars) and IL-1RI null mutant (IL-1RI−/−; circles; hatched bars) male mice. Neurological scores between groups were different at 24 and 72 h after MCAO, as determined by a Mann–Whitney U test (n = 9 animals per group, statistical significance was assessed at p < 0.05).
IL1-RI signaling is necessary for the IL-1β-mediated enhancement of hypoxia-induced neuronal cell death in vitro
To study the cellular and biochemical mechanisms by which IL-1β contributes to cerebral ischemic damage, it is advantageous to take an in vitro approach. Toward this end, we demonstrated previously that IL-1β alone (0.01–10 ng/ml) is not toxic to mixed cortical cell cultures but that the minimal neuronal death associated with a 4–5 h hypoxic insult was enhanced after IL-1β exposure in a concentration-dependent manner (20–24 h) (Fogal et al., 2005a). Additionally, under conditions of hypoxia, with or without IL-1β, we found no evidence of astrocyte injury as assessed by trypan blue exclusion via phase contrast microscopy (Fogal et al., 2005a). To determine whether this model reproduces the in vivo findings, we next assessed whether IL-1RI signaling was involved. Before the initiation of hypoxia, mixed cortical cell cultures were treated with IL-1β (1 ng/ml, 20–24 h) in the presence of increasing concentrations (10–1000 ng/ml) of rIL-1ra. rIL-1ra (10–1000 ng/ml), which antagonizes IL-1RI in mouse (Arend, 1993), prevented the potentiation of hypoxic neuronal injury in a concentration-dependent manner (Fig. 2A). Additionally, a blocking antibody directed against extracellular domain of IL-1RI (0.1–100 μg/ml) also suppressed the IL-1β-mediated enhancement of injury (Fig. 2B), whereas control IgG had no effect (data not shown). Together, these results indicate that our in vitro system recapitulates the deleterious effects of IL-1β found in vivo, hence representing a suitable model to study the cellular and biochemical mechanism(s) by which this cytokine contributes to hypoxic/ischemic neuronal injury.
Figure 2.

Role of IL-1RI signaling in the potentiation of hypoxic neuronal injury by IL-1β in vitro. A, B, Mixed cortical cell cultures were treated with 1 ng/ml IL-1β for 20–24 h in the presence or absence of rIL-1ra (10–1000 ng/ml; A) or anti-IL-1RI (0.1–100 μg/ml; B), washed, and then deprived of oxygen (5 h). The percentage of total neuronal cell death was determined 20–24 h later. An asterisk indicates values significantly greater than hypoxia alone, whereas # denotes a significant diminution of the IL-1β-mediated increase in injury (IL-1β) as determined by one-way ANOVA followed by a Student–Newman–Keuls t test. Significance was assessed at p < 0.05 (n = 3–9 cultures pooled from 2–3 different experiments).
IL-1β potentiates hypoxic neuronal cell death via glutamate receptor excitotoxicity
In addition to increasing hypoxia-induced neuronal cell death, treatment of the cultures with IL-1β increased the buildup of glutamate in the cell culture medium, which became statistically significant after 2 h of hypoxia (Fig. 3A). IL-1β had no such effect when cells were treated equivalently but kept under normoxic conditions (data not shown). Interestingly, overall glutamate transporter function was also significantly decreased after 2 h of hypoxia; however, there was no difference between IL-1β-treated and nontreated cultures (Fig. 3B), suggesting that a decrease in glutamate uptake that follows hypoxia cannot alone account for the IL-1β-specific effect. That this increase in extracellular glutamate levels ([glutamate]e) contributed to neuronal cell death was evident by the fact that ionotropic glutamate receptor antagonism prevented the deleterious effects of IL-1β (Fig. 4).
Figure 3.

Glutamate accumulation and glutamate transporter function under hypoxic conditions after IL-1β pretreatment. Mixed cortical cell cultures were treated with 1 ng/ml IL-1β for 20–24 h (black bars) or vehicle (hatched bars), washed, and then deprived of oxygen. A, After the period of time indicated, supernatant was collected to measure accumulation of glutamate in the bathing medium via HPLC. Data are expressed as the mean ± SEM glutamate accumulation in micromolar. An asterisk denotes a significant between group difference as assessed by two-way ANOVA followed by a Bonferroni's post hoc test (p < 0.001; n = 8 cultures pooled from 3 different experiments). B, After the times indicated, cells were washed, and [3H]d-aspartate (0.1 μCi) was added in the presence of 50 μm nonradioactive d-aspartate. Uptake was terminated after 5 min, and the amount of [3H]d-aspartate that accumulated into cells was measured via liquid scintillation counting. Data are expressed as mean ± SEM counts per minute per well per 5 min × 103. There was no statistically significant between-group difference in [3H]d-aspartate uptake as determined by two-way ANOVA. An asterisk indicates a significant decrease in [3H]d-aspartate uptake from the 30 min time point within each group as determined by two-way ANOVA followed by a Bonferroni's post hoc test for multiple comparisons. Significance was assessed at p < 0.05 (n = 8 cultures pooled from four different experiments).
Figure 4.

Effect of glutamate receptor antagonism on IL-1β-mediated potentiation of hypoxic neuronal cell death. Mixed cortical cell cultures were treated with 1 ng/ml IL-1β for 20–24 h, washed, and then deprived of oxygen (5 h) in presence or absence of the ionotropic glutamate receptor antagonists MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (10 μm) and CNQX (30 μm). The percentage of total neuronal cell death was determined 20–24 h later. The asterisk indicates values significantly greater than cells deprived of oxygen only (Hypoxia), whereas # denotes a significant decrease in IL-1β-mediated potentiation of hypoxic neuronal cell death as determined by one-way ANOVA followed by a Student–Newman–Keuls t test. Significance was assessed at p < 0.05 (n = 14 cultures pooled from four different experiments).
IL-1β enhances activity of the System xc−
Interestingly, we found that the potentiation of hypoxic neuronal injury by IL-1β occurred when the hypoxic insult was initiated in buffer supplemented with MEM amino acids but not in their absence (Fig. 5A). Furthermore, removal of l-cystine alone completely prevented the augmentation of hypoxic neuronal cell death by IL-1β (Fig. 5A), whereas addition of l-cystine to the oxygen deprivation medium enhanced both the IL-1β-mediated potentiation of hypoxic neuronal injury (Fig. 5B) and glutamate accumulation (Fig. 5C) in a concentration-dependent manner. Removal of l-methionine, necessary for protein synthesis initiation, or l-lysine, transported by alternative amino acid transporters (Christensen, 1990), had no effect, attesting to the specificity of the response for cystine (Fig. 5A). Notably, removal of l-cystine did not protect from hypoxic neuronal cell death initiated in the absence of IL-1β pretreatment (data not shown), suggesting that its necessity is specific to the IL-1β paradigm. The dependence of the IL-1β injury paradigm on both cystine and glutamate suggested a role for System xc−. Indeed, compared with control cultures, 14C-l-cystine uptake was significantly increased in IL-1β-treated cultures, and this increase was evident under both normoxic and hypoxic conditions (Fig. 6A,C) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Assessment of System xc− function and kinetics revealed an increase in velocity of cystine transport (Vmax) in the absence of a change in affinity (Km) (Fig. 6B,D) in IL-1β-treated cultures compared with control cultures. Kinetic data plotted using the Woolf–Augustinsson–Hofstee plot yielded similar results (data not shown).
Figure 5.
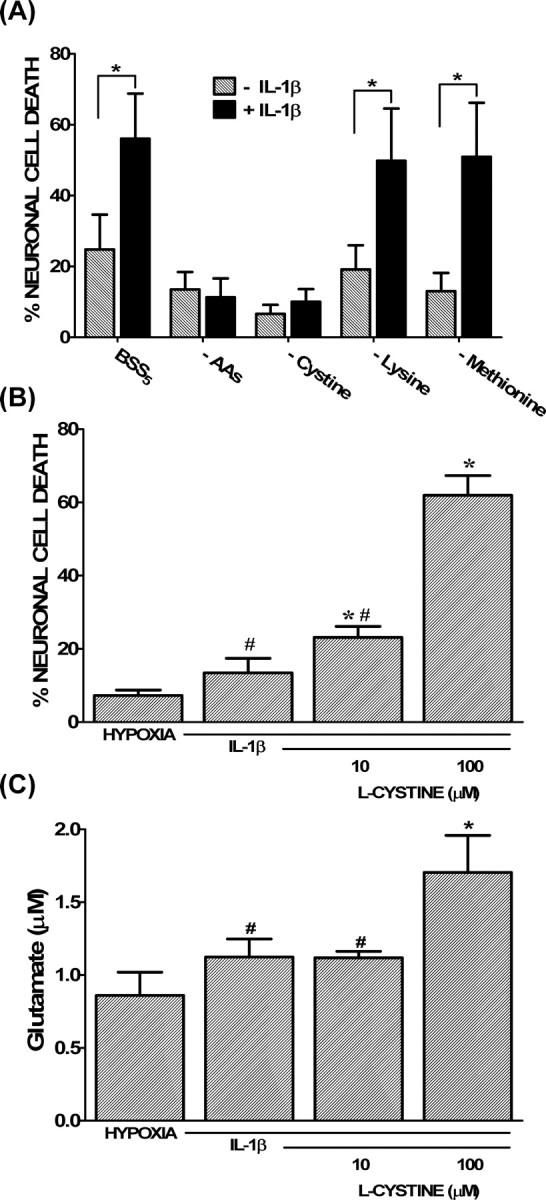
l-Cystine is necessary for the IL-1β-mediated potentiation of hypoxic neuronal cell death. Mixed cortical cell cultures were treated with 1 ng/ml IL-1β or vehicle for 20–24 h, after which cells were moved to the anaerobic chamber. A, Cells were deprived of oxygen (5 h) in a medium that contained MEM amino acids (BSS5), was devoid of MEM amino acids (−AAs), or one that lacked l-cystine (−Cystine), l-lysine (−Lysine), or l-methionine (−Methionine) only. The percentage of total neuronal cell loss was determined 20–24 h later. The asterisk indicates a significant between group difference as determined by two-way ANOVA followed by a Bonferroni's post hoc test (n = 8 cultures pooled from 3 different experiments). B, Cells treated with IL-1β were deprived of oxygen (5 h) in medium devoid or containing l-cystine (10 or 100 μm). The medium in which the control cells (HYPOXIA) were deprived of oxygen contained 100 μm l-cystine. The percentage of total neuronal cell death was determined 20–24 h later. An asterisk indicates a significant increase from hypoxia alone, whereas # denotes a significant difference from 100 μm l-cystine, the standard concentration of this amino acid in BSS5 medium, as determined via one-way ANOVA followed by a Student–Newman–Keuls test (n = 9 cultures pooled from 3 different experiments). C, Cells were treated as described in B, except supernatant was collected after 120 min of oxygen deprivation to measure accumulation of glutamate in the bathing medium via HPLC. Data are expressed as the mean ± SEM glutamate accumulation in micromolar (n = 4–5 cultures pooled from 2 different experiments). The asterisk indicates a value significantly greater than hypoxia alone, whereas # denotes a significant difference from 100 μm l-cystine, the standard concentration of this amino acid in BSS5 medium, as determined by one-way ANOVA followed by a Student–Newman–Keuls test. In all cases, significance was assessed at p < 0.05.
Figure 6.

Increase in cystine transporter activity follows IL-1β treatment. Mixed cortical cell cultures were treated with 1 ng/ml IL-1β or vehicle for 20–24 h. A–D, Cells were then moved to the anaerobic chamber, washed into deoxygenated BSS5, and deprived of oxygen for 0 min (A, C) or 90 min (B, D). Thereafter, cells were washed with HBSS, and 14C-l-cystine was added for 5 min. A, C, Data are expressed as 14C-l-cystine uptake in picomoles per minute per milligram protein. The asterisk indicates a significant between-group difference as determined by two-way ANOVA followed by a Bonferroni's post hoc test. Significance was assessed at p < 0.05 (n = 6 cultures pooled from 3 different experiments). C, D, Vmax and Km of 14C-l-cystine uptake in cultures were estimated using the Hanes–Woolf plot and are evident as 1/slope and −x-intercept, respectively. An asterisk indicates a significant increase from nontreated cultures (p < 0.05; n = 6 cultures pooled from 3 different experiments).
Inhibition of System xc− protects from the injury-enhancing effects of IL-1β
To assess the role of System xc− in IL-1β-mediated potentiation of hypoxic neuronal cell death, we took a pharmacological approach. The IL-1β-mediated increase in 14C-l-cystine transport as well as the enhanced neuronal cell death was inhibited by the dual System xc−/mGluR1 inhibitors (S)-4-carboxyphenylglycine (4-CPG) or (+)-2-methyl-4-carboxyphenylglycine (LY367385) but not the selective mGluR1 antagonist 6-amino-N-cyclohexyl-N,3-dimethylthiazolo[3,2-a]benzimi dazole-2-carboxamide hydrochloride (YM298198) (Fig. 7). Importantly, all antagonists protected against an enhancement of hypoxic neuronal cell death mediated by the mGluR1 agonist (RS)-3,5-dihydroxyphenylglycine (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), demonstrating their effectiveness. Finally, IL-1β augmented hypoxic neuronal cell death in cultures derived from mGluR1−/− animals to a similar level as in cultures derived from mGluR1+/+ animals (Fig. 8). Importantly, the ability of the System xc−/mGluR1 antagonist 4-CPG to protect persisted in mGluR1−/− cultures, whereas the selective mGluR1 antagonist YM298198 remained ineffective, further confirming the lack of contribution of mGluR1 activation to the injury process (Fig. 8).
Figure 7.
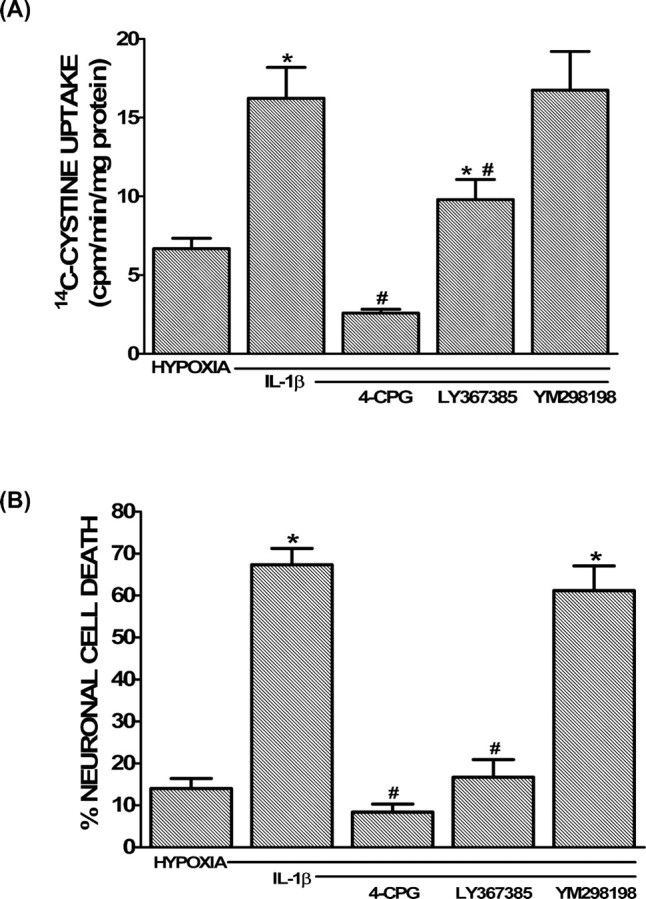
Prevention of the IL-1β-mediated increase in cystine uptake and neuronal injury by System xc−/mGluR1 antagonism. Mixed cortical cell cultures were treated with 1 ng/ml IL-1β for 20–24 h or vehicle (hypoxia control), washed, and then deprived of oxygen. A, After 60 min, cells were washed with HBSS, and 14C-l-cystine was added for 5 min in the absence or presence of the System xc−/mGluR1 antagonists 4-CPG (50 μm) or LY367385 (50 μm) or the selective mGluR1 antagonist YM298198 (10 μm). Intracellular 14C-l-cystine was measured via liquid scintillation counting as described in Materials and Methods. Data are expressed as 14C-l-cystine uptake in picomoles per minute per milligram protein (n = 6–12 cultures pooled from 2–4 different experiments). B, The percentage of total neuronal cell death in a parallel sister plate was determined 20–24 h later as described in Materials and Methods (n = 9 cultures pooled from 3 different experiments). An asterisk indicates a significant increase from hypoxia alone, whereas # denotes a significant diminution from the IL-1β-mediated enhancement of hypoxic neuronal cell death, as determined by one-way ANOVA followed by a Student–Newman–Keuls test. Significance was assessed at p < 0.05.
Figure 8.
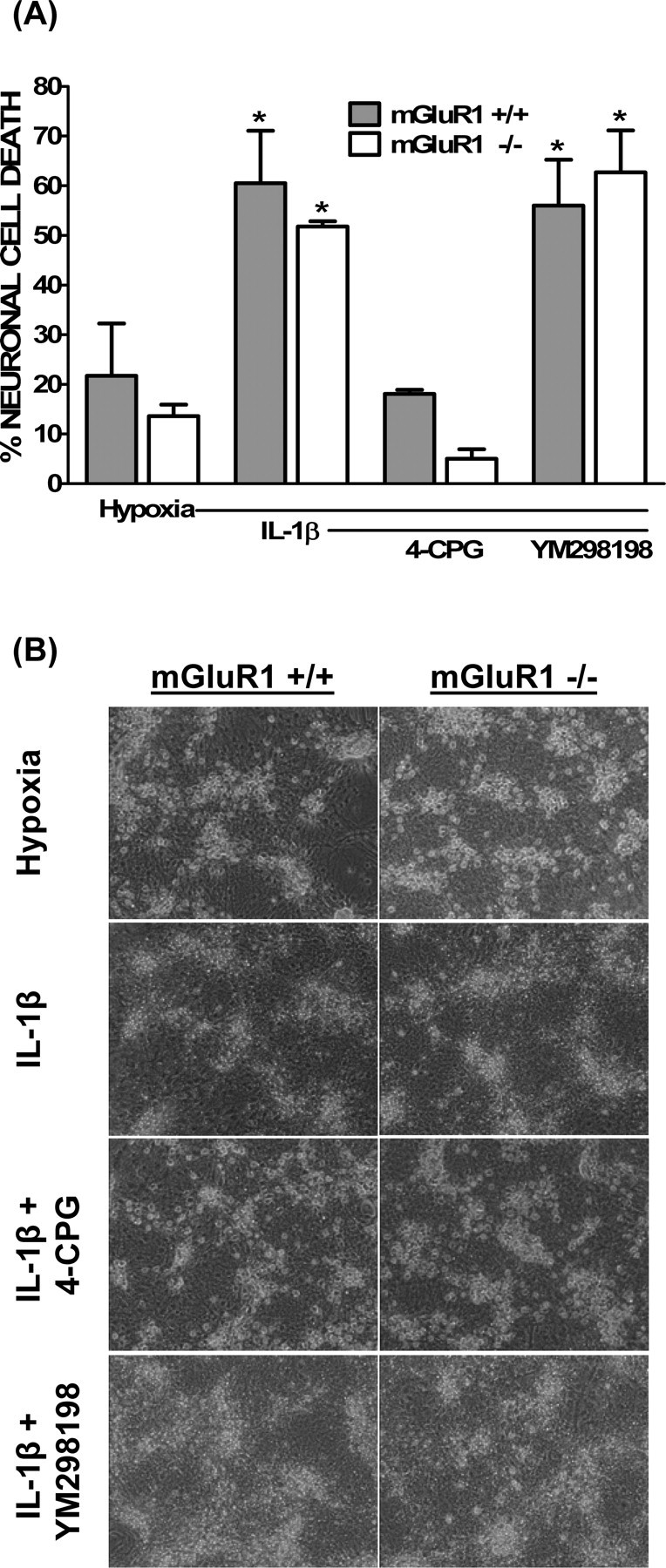
mGluR1 signaling is not required for the IL-1β-mediated potentiation of hypoxic neuronal cell death. Mixed cortical cell cultures were obtained from mGluR1−/− (white bars) or mGluR1+/+ (gray bars) animals. These cultures were treated with vehicle (hypoxia) or 1 ng/ml IL-1β for 20–24 h washed and then deprived of oxygen for 5 h in the absence or presence of 4-CPG (50 μm) or YM298198 (10 μm). A, The percentage of total neuronal cell death was determined 20–24 h later as described in Materials and Methods. An asterisk indicates a significant increase from control cultures of the respective genotype (hypoxia), whereas # denotes a significant between-group difference, as determined by two-way ANOVA followed by a Bonferroni's post hoc test. Significance was assessed at p < 0.05 (n = 4–6 cultures pooled from 2–3 different experiments). B, Representative phase contrast micrographs (20×) from cultures in A taken 20–24 h after a 5 h oxygen deprivation period.
Astrocyte IL1-RI signaling is necessary for the injury-enhancing effects of IL-1β
As mentioned, our cultures contain a near 50:50 mix of astrocytes and neurons, both of which have been shown to express IL-1RI (Ban et al., 1993; Wong and Licinio, 1994; Tomozawa et al., 1995; Hammond et al., 1999; Friedman, 2001). Thus, chimeric cultures containing a combination of neurons and astrocytes isolated from IL-1RI−/− and IL1-RI+/+ were next used to determine which cell type(s) contribute(s) to the IL-1β-mediated enhancement of hypoxic neuronal cell death. Importantly, just as in cultures isolated from CD1 mice, mixed cultures derived entirely from C57BL/6 wild-type mice (IL-1RI+/+) demonstrated the IL-1β-mediated enhancement in cellular cystine uptake ([cystine]i), [glutamate]e, and hypoxic neuronal injury (Fig. 9). As expected, IL-1β had no effect in cultures derived entirely from mice lacking the canonical signaling receptor IL-1RI (Fig. 9). Interestingly, in cultures in which IL-1RI−/− neurons were plated on top of IL-1RI+/+ astrocytes, IL-1β augmented [cystine]i, [glutamate]e, and neuronal injury to similar intensity as in cultures derived entirely from IL-1RI+/+ animals (Fig. 9). However, when IL1-RI+/+ neurons were plated on top of IL-1RI−/− astrocytes, neither the enhancement in [cystine]i, [glutamate]e, nor neuronal injury were observed (Fig. 9), indicating that astrocytic signaling is essential for the deleterious effects of IL-1β.
Figure 9.
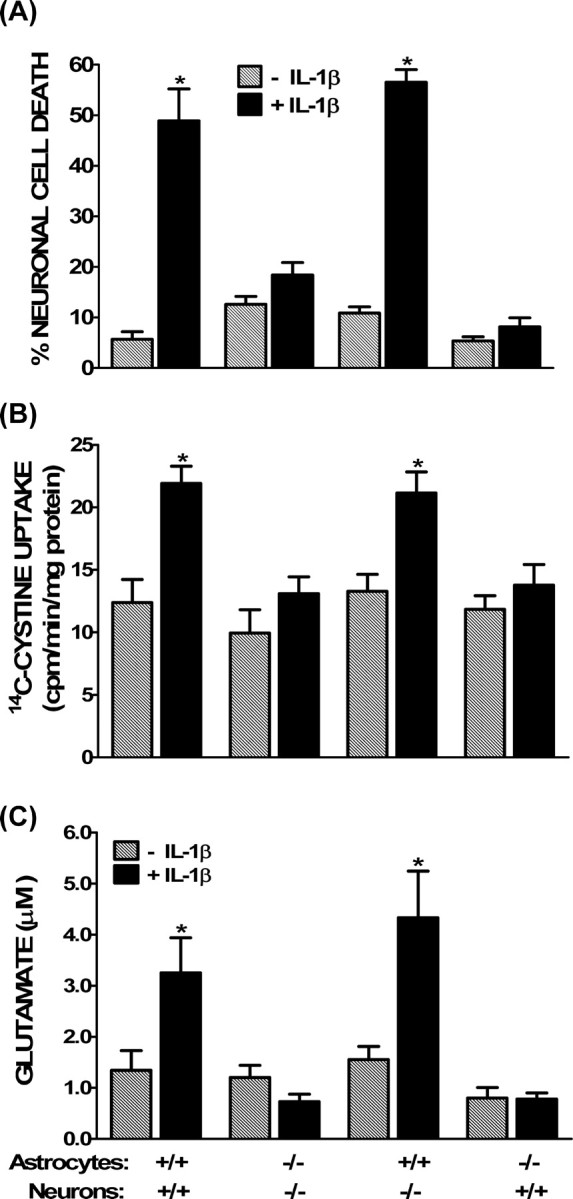
Astrocyte IL-1RI signaling is required for the mediation of IL-1β-induced hypoxic neuronal injury. Chimeric mixed cortical cell cultures were obtained by plating wild-type (IL-1RI+/+) or IL-1RI-deficient (IL-1RI−/−) neurons on either wild-type (IL-1RI+/+) or IL-1RI-null mutant (IL-1RI−/−) astrocytes These cultures were treated with 1 ng/ml IL-1β for 20–24 h (black bars) or vehicle (hatched bars), washed, and then deprived of oxygen. A, The percentage of total neuronal cell death was determined 20–24 h later as described in Materials and Methods (n = 18–24 cultures pooled from 4 different experiments). B, After 60 min, cells were washed with HBSS, and 14C-l-cystine was added for 5 min as described in Materials and Methods. Data are expressed as 14C-l-cystine uptake in picomoles per minute per milligram protein (n = 6–12 cultures pooled from 2–4 different experiments). C, After 120 min of hypoxia, supernatant was collected to measure accumulation of glutamate in the bathing medium via HPLC. Data are expressed as the mean ± SEM glutamate accumulation in micromolar (n = 6 cultures pooled from 3 different experiments). An asterisk indicates a value significantly greater than oxygen deprivation alone (−IL-1β), as determined by two-way ANOVA followed by a Bonferroni's t test for multiple comparisons. Significance was assessed at p < 0.05.
Discussion
Injury-induced increases in IL-1β (Minami et al., 1992; Liu et al., 1993; Buttini et al., 1994; Yabuuchi et al., 1994; Sairanen et al., 1997) are implicated in neuronal cell death that occurs subsequent to cerebral ischemia. (Relton and Rothwell, 1992; Martin et al., 1994; Betz et al., 1995; Garcia et al., 1995; Toulmond and Rothwell, 1995; Yamasaki et al., 1995; Loddick and Rothwell, 1996; Relton et al., 1996; Friedlander et al., 1997; Boutin et al., 2001). However, the mechanism(s) by which this occurs are poorly understood. After a transient MCAO, we demonstrate that IL-1RI null mutant mice are less susceptible to focal cerebral ischemic damage than wild-type control mice. This is significant for two reasons. First, it identifies the signaling receptor involved in the pathogenesis in vivo. Second, it affords us the opportunity to use cultures from these animals in a previously characterized in vitro model (Fogal et al., 2005a) to elucidate the cellular and biochemical pathways involved. In doing so, we show that IL-1β potentiates hypoxic neuronal injury in vitro in a manner that is dependent on astrocyte signaling and that ultimately results in the potentiation of glutamate excitotoxicity via increased activity of System xc−.
The role of IL-1RI signaling in the progression of injury after transient MCAO complements similar results found in a global ischemia (common carotid artery ligation plus hypoxia) model (Basu et al., 2005), perhaps suggesting a universal signaling pathway independent of the type of ischemic insult. However, a previous study using the standard monofilament model of MCAO used in this study reports negative results (Touzani et al., 2002). The reasons for the discrepancy between studies are likely multifaceted and could include the use of noncongenic animals in the former study (Wolfer et al., 2002) and a shorter duration of ischemia (30 vs 90 min used herein). The former time results in a very mild cerebral infarct that may have been insufficient to activate the IL-1 pathway. Finally, the time of infarct evaluation differed between the two studies (24 vs 72 h used herein) differed. The importance of assessing the role of inflammatory mediators at later time points is exemplified by the work of Iadecola et al. (1997), demonstrating a reduction in infarct size and an improvement of neurological deficits at 96 h, but not 24 h, after MCAO in nitric oxide synthase-2 null mutant animals. Likewise, in the present study, the severity of the neurological deficits in IL-1RI−/− animals improves with time, whereas scores remain stable in wild-type control mice. Similarly, in the global ischemia model, brain injury, assessed in individual animals via serial magnetic resonance imaging measurements, actually increases in wild-type mice when measured up to 2 months after the insult, whereas the lesion volume decreases in IL-1RI−/− over the same time frame (Basu et al., 2005).
Consistent with our in vivo results, we demonstrate that IL-1β pretreatment enhances hypoxic neuronal injury in a mixed cortical cell culture system in vitro (Fogal et al., 2005a) in an IL-1RI-dependent manner (this study). A pretreatment paradigm may at first appear incongruous to the in vivo situation, in which increased IL-1β expression follows the ischemic insult (Saito et al., 1996; Sairanen et al., 1997; Zhang et al., 1998). However, as mentioned, the tissue destruction that follows ischemia progresses over a delayed time frame (Garcia et al., 1993; Du et al., 1996; Baird et al., 1997; Iadecola et al., 1997; Schwamm et al., 1998) and IL-1β is produced both early and late after occlusion, the latter of which corresponds to the timing of the evolution of the infarct (Davies et al., 1999; Skifter et al., 2002).
We now demonstrate that the enhancement of hypoxic neuronal cell death by IL-1β in vitro correlates remarkably with an increase in both [cystine]i and [glutamate]e, the latter of which has been associated with ischemic brain injury in vivo (Benveniste et al., 1984). The fact that ionotropic glutamate receptor antagonism prevents the potentiation of hypoxic neuronal injury by IL-1β indicates that these changes are not merely correlative but represent the cause of injury. A link between glutamate receptor-mediated injury and IL-1β is further strengthened by in vivo evidence showing that intrastriatal injection of IL-1β enhances excitotoxic neuronal injury (Lawrence et al., 1998; Stroemer and Rothwell, 1998; Allan et al., 2000), whereas striatal and hippocampal neuronal injury induced by direct NMDA receptor activation is reduced after intrastriatal application of IL-1ra (Relton and Rothwell, 1992).
The codependence on cellular cystine indicates a link between IL-1β and System xc− function. Indeed, after IL-1β treatment, a significant increase in the velocity (Vmax) of System xc− was found. Whether this increased activity is associated with an upregulation of transporter expression (Sato et al., 1999, 2004; Gochenauer and Robinson, 2001; Sheldon et al., 2006; Goltz et al., 2007) or perhaps is a result of a posttranslational modification that alters transporter kinetics (Baker et al., 2002; Xi et al., 2003) remains to be determined. Although selective inhibitors for System xc− are not available at this time, several carboxyphenylglycine derivatives (i.e., 4-CPG and LY367385), mostly marketed as mGluR1 antagonists, were used to inhibit this transporter (Gochenauer and Robinson, 2001) and were found to be effective against the IL-1β-mediated potentiation of hypoxic neuronal cell death. A role for mGluR1 is unlikely, because the selective mGluR1 antagonists 7-(hydroxyimino)cyclopropa chromen-1a-carboxylate ethyl ester and YM298198 do not protect in this injury model, nor do mGluR1−/− cultures show decreased susceptibility to the IL-1β-mediated potentiation of hypoxic neuronal cell death when compared with wild-type cultures. Although a role for glutamate export via System xc− has been suggested to contribute to excitotoxic neuronal cell death associated with tumor growth (Ye and Sontheimer, 1999; Chung et al., 2005) and Alzheimer's disease (Barger and Basile, 2001; Qin et al., 2006), it has not been previously implicated in the progression of cerebral ischemic injury. However, systemic administration of LY367385 or an alternative carboxyphenylglycine derivative, MATIDA [(−)-3-methyl-5-carboxy-thien-2-yl-glycine], decreases the infarct volume after MCAO in rat (Moroni et al., 2002), a protective effect ascribed to inhibition of mGluR1. Yet curiously, compared with their wild-type controls, neuronal injury after MCAO is not reduced in mGluR1-deficient mice (Ferraguti et al., 1997). Thus, inhibition of System xc− might underlie the protection of so-called mGluR1 antagonist in the aforementioned studies.
Finally, using cultures consisting of a mixture of neurons and astrocytes from wild-type and IL-1RI−/− mice, we demonstrate that the IL-1β-mediated increase in [cystine]i, [glutamate]e, and hypoxic neuronal injury is dependent on astrocytic but not neuronal IL-1RI signaling. Cell-type-specific effects of IL-1β have been reported previously. IL-1β treatment of mixed hippocampal neuron–astrocyte cocultures results in nuclear translocation of the p65 nuclear factor-κB subunit in astrocytes but not neurons (Srinivasan et al., 2004). In contrast, hippocampal neurons respond to IL-1β with the activation of the cAMP response element binding protein (Srinivasan et al., 2004), which in some injury paradigms might be neuroprotective (Walton and Dragunow, 2000). Hence, IL-1RI signaling could result in harmful or beneficial effects with the prevailing effect dependent on the cell type involved. Indeed, the current study demonstrates that astrocyte activation mediates the potentiating effects of IL-1β in our injury paradigm via an enhancement of System xc− function. A role for microglia activation in our in vitro paradigm seems unlikely given that our cultures contain only a small fraction of contaminating microglial cells (<1%) (Hewett et al., 1993). Additionally, a recent study demonstrates that microglia, either in their resting state or after activation, do not respond directly to IL-1β, which is most likely attributable to a low ratio of signaling (i.e., IL-1RI) to decoy (i.e., IL-1RII) receptors (Pinteaux et al., 2002). Certainly a role for microglia in vivo is incontrovertible by virtue of their ability to secrete IL-1β after the ischemic insult (Hillhouse et al., 1998; Davies et al., 1999).
Why increase System xc− if only to cause injury? The well characterized role of System xc− is to provide cystine for the production of the major cellular antioxidant glutathione (Meister and Anderson, 1983). Intriguingly, it has been shown that astrocytes contain a greater concentration of glutathione and the enzymes involved in its synthesis than neurons (Makar et al., 1994; Wilson, 1997), indicating that they play an important role in scavenging of toxic reactive oxygen species that accumulate after cerebral ischemia (Cao et al., 1988; Traystman et al., 1991; Piantadosi and Zhang, 1996). Hence, it is intriguing to speculate that IL-1β is released as a protective mechanism to upregulate glutamate/cystine exchange to prevent oxidative stress. However, as our data demonstrate, this change becomes neurotoxic when found in combination with the decreased System XAG− function that occurs under hypoxic conditions, hence setting the stage for a toxic buildup of glutamate. In vitro data indicates that most functional System xc− and XAG− exists mainly in mature astrocytes (Kranich et al., 1998; Murphy et al., 1990; Chaudhry et al., 1995; Rothstein et al., 1996; Sonnewald et al., 1997; Schubert and Piasecki, 2001; Lewerenz et al., 2003). However, it should be noted that a recent immunohistochemical study reveals expression of xCT, the obligatory subunit of System xc−, in both astrocytes and neurons of the adult mouse cortex (Burdo et al., 2006); hence, a change in System xc− function in neurons cannot be completely excluded. Nevertheless, our data demonstrate that astrocytic IL-1R1 signaling mediates the increase System xc− function that proceeds IL-1β exposure, whereas the effect on System XAG− function occurs exclusively as a result of hypoxia. Overall, these results are especially exciting given the burgeoning respect astrocyte–neuron intercommunication in CNS physiology and pathology has been gaining. It is clear that the role of astrocytes in neuronal function and dysfunction has attained a new dimension in thought (Araque et al., 1999; Halassa et al., 2007). Although the active role that astrocytes play in ischemic neurodegeneration remains necessarily speculative, this study and others now suggest that the differential vulnerability of neuronal subpopulations to hypoxic/ischemic injury may in fact be mediated by differences in astrocyte function rather than differences in neuronal susceptibility (Ouyang et al., 2007).
In conclusion, we demonstrate for the first time that the detrimental effects of IL-1β during focal cerebral ischemic injury in vivo are mediated via signaling through IL-1RI. In vitro modeling confirms the role of IL-1RI, indicates the absolute necessity of astrocyte activation, and demonstrates that an enhancement of glutamate excitotoxicity, occurring via an increased accumulation of extracellular glutamate levels subsequent to an increase in System xc− velocity and a decrease in glutamate uptake, underlies the neurotoxic effects of IL-1β. Overall, these data highlight the potential importance of astrocytes to the pathology of cerebral ischemia and specifically identify alterations in System xc− as a novel contributor to the development and progression of injury. Hence, it is intriguing to consider the astrocyte a more viable upstream therapeutic target to modify excitotoxic/ischemic injury than inhibition of downstream neuronal effectors, the latter of which has so far proven ineffective.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke–National Institutes of Health Grants NS51445 (S.J.H.) and NS52061 (B.F.). We thank Dr. Leah A. Chase (Hope College, Holland, MI) for technical advice on measurements of System xc− activity. We are grateful to Dr. James Kew (GlaxoSmithKline, Harlin, Essex, UK) for his help in obtaining and Dr. Francesco Ferraguti (Department of Pharmacology, University of Innsbruck, Innsbruck, Austria) for providing the mGluR1 null mutants. Finally, we thank Dr. James A. Hewett for scientific advice and critique of this manuscript.
References
- Allan SM, Parker LC, Collins B, Davies R, Luheshi GN, Rothwell NJ. Cortical cell death induced by IL-1 is mediated via actions in the hypothalamus of the rat. Proc Natl Acad Sci USA. 2000;97:5580–5585. doi: 10.1073/pnas.090464197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre R, Moggs JG, Kimber I, Rothwell NJ, Pinteaux E. Gene regulation by IL-1beta independent of IL-1R1 in the mouse brain. Glia. 2006;53:477–483. doi: 10.1002/glia.20302. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Arend WP. Interleukin-1 receptor antagonist. Adv Immunol. 1993;54:167–227. doi: 10.1016/s0065-2776(08)60535-0. [DOI] [PubMed] [Google Scholar]
- Baird AE, Benfield A, Schlaug G, Siewert B, Lovblad KO, Edelman RR, Warach S. Enlargement of human cerebral ischemic lesion volumes measured by diffusion-weighted magnetic resonance imaging. Ann Neurol. 1997;41:581–589. doi: 10.1002/ana.410410506. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban EM, Sarlieve LL, Haour FG. Interleukin-1 binding sites on astrocytes. Neuroscience. 1993;52:725–733. doi: 10.1016/0306-4522(93)90421-b. [DOI] [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Basu A, Lazovic J, Krady JK, Mauger DT, Rothstein RP, Smith MB, Levison SW. Interleukin-1 and the interleukin-1 type 1 receptor are essential for the progressive neurodegeneration that ensues subsequent to a mild hypoxic/ischemic injury. J Cereb Blood Flow Metab. 2005;25:17–29. doi: 10.1038/sj.jcbfm.9600002. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Betz AL, Yang GY, Davidson BL. Attenuation of stroke size in rats using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist in brain. J Cereb Blood Flow Metab. 1995;15:547–551. doi: 10.1038/jcbfm.1995.68. [DOI] [PubMed] [Google Scholar]
- Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1α and IL-1β in ischemic brain damage. J Neurosci. 2001;21:5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc− in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54:549–557. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- Buttini M, Sauter A, Boddeke HW. Induction of interleukin-1 beta mRNA after focal cerebral ischaemia in the rat. Brain Res Mol Brain Res. 1994;23:126–134. doi: 10.1016/0169-328x(94)90218-6. [DOI] [PubMed] [Google Scholar]
- Cao W, Carney JM, Duchon A, Floyd RA, Chevion M. Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett. 1988;88:233–238. doi: 10.1016/0304-3940(88)90132-2. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Christensen HN. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev. 1990;70:43–77. doi: 10.1152/physrev.1990.70.1.43. [DOI] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, Collingridge G, Crépel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1999;19:87–98. doi: 10.1097/00004647-199901000-00010. [DOI] [PubMed] [Google Scholar]
- Desson SE, Ferguson AV. Interleukin 1beta modulates rat subfornical organ neurons as a result of activation of a non-selective cationic conductance. J Physiol (Lond) 2003;550:113–122. doi: 10.1113/jphysiol.2003.041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diem R, Hobom M, Grotsch P, Kramer B, Bahr M. Interleukin-1 beta protects neurons via the interleukin-1 (IL-1) receptor-mediated Akt pathway and by IL-1 receptor-independent decrease of transmembrane currents in vivo. Mol Cell Neurosci. 2003;22:487–500. doi: 10.1016/s1044-7431(02)00042-8. [DOI] [PubMed] [Google Scholar]
- Drejer J, Larsson OM, Schousboe A. Characterization of L-glutamate uptake into and release from astrocytes and neurons cultured from different brain regions. Exp Brain Res. 1982;47:259–269. doi: 10.1007/BF00239385. [DOI] [PubMed] [Google Scholar]
- Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Georgiou RF, Vail A, Hopkins SJ, Rothwell NJ, Tyrrell PJ. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar WL, Kilian PL, Ruff MR, Hill JM, Pert CB. Visualization and characterization of interleukin 1 receptors in brain. J Immunol. 1987;139:459–463. [PubMed] [Google Scholar]
- Ferraguti F, Pietra C, Valerio E, Corti C, Chiamulera C, Conquet F. Evidence against a permissive role of the metabotropic glutamate receptor 1 in acute excitotoxicity. Neuroscience. 1997;79:1–5. doi: 10.1016/s0306-4522(97)00074-2. [DOI] [PubMed] [Google Scholar]
- Fogal B, Hewett JA, Hewett SJ. Interleukin-1β potentiates hypoxic neuronal injury in vitro. Soc Neurosci Abstr. 2004;30:455–11. [Google Scholar]
- Fogal B, Hewett JA, Hewett SJ. Interleukin-1beta potentiates neuronal injury in a variety of injury models involving energy deprivation. J Neuroimmunol. 2005a;161:93–100. doi: 10.1016/j.jneuroim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Fogal B, Hewett JA, Hewett SJ. Astrocytic signaling is required for the potentiation of hypoxic neuronal injury by interleukin-1β. Soc Neurosci Abstr. 2005b;31:220–11. [Google Scholar]
- Fogal B, Trettel J, Uliasz TF, Levine ES, Hewett SJ. Changes in secondary glutamate release underlie the developmental regulation of excitotoxic neuronal cell death. Neuroscience. 2005c;132:929–942. doi: 10.1016/j.neuroscience.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Fogal B, Li J, Silakova JM, McCullough L, Hewett SJ. IL-1 receptor type I-deficient mice are less susceptible to cerebral ischemic and excitotoxic brain injury. Soc Neurosci Abstr. 2006;32:283–9. [Google Scholar]
- French RA, VanHoy RW, Chizzonite R, Zachary JF, Dantzer R, Parnet P, Bluthe RM, Kelley KW. Expression and localization of p80 and p68 interleukin-1 receptor proteins in the brain of adult mice. J Neuroimmunol. 1999;93:194–202. doi: 10.1016/s0165-5728(98)00224-0. [DOI] [PubMed] [Google Scholar]
- Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp Neurol. 2001;168:23–31. doi: 10.1006/exnr.2000.7595. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J, Chen S, Chopp M. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142:623–635. [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am J Pathol. 1995;147:1477–1486. [PMC free article] [PubMed] [Google Scholar]
- Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- Gochenauer GE, Robinson MB. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent l-[3H]glutamate transport and expression of both system xc(−) subunits. J Neurochem. 2001;78:276–286. doi: 10.1046/j.1471-4159.2001.00385.x. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Weiss JH, Pham PC, Choi DW. N-methyl-d-aspartate receptors mediate hypoxic neuronal injury in cortical culture. J Pharmacol Exp Ther. 1987;243:784–791. [PubMed] [Google Scholar]
- Goltz A, Costa N, Chase LA. Hydrogen peroxide regulates the trafficking of System xc− in a dopaminergic cell line. FASEB J. 2007;21:A244. [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hammond EA, Smart D, Toulmond S, Suman-Chauhan N, Hughes J, Hall MD. The interleukin-1 type I receptor is expressed in human hypothalamus. Brain. 1999;122:1697–1707. doi: 10.1093/brain/122.9.1697. [DOI] [PubMed] [Google Scholar]
- Hara H, Fink K, Endres M, Friedlander RM, Gagliardini V, Yuan J, Moskowitz MA. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997;17:370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Itzhak Y, Liu H, Norenberg MD. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) decreases glutamate uptake in cultured astrocytes. J Neurochem. 1997;68:2216–2219. doi: 10.1046/j.1471-4159.1997.68052216.x. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Corbett JA, McDaniel ML, Choi DW. Interferon-gamma and interleukin-1 beta induce nitric oxide formation from primary mouse astrocytes. Neurosci Lett. 1993;164:229–232. doi: 10.1016/0304-3940(93)90898-u. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Muir JK, Lobner D, Symons A, Choi DW. Potentiation of oxygen-glucose deprivation-induced neuronal death after induction of iNOS. Stroke. 1996;27:1586–1591. doi: 10.1161/01.str.27.9.1586. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-d-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- Hillhouse EW, Kida S, Iannotti F. Middle cerebral artery occlusion in the rat causes a biphasic production of immunoreactive interleukin-1beta in the cerebral cortex. Neurosci Lett. 1998;249:177–179. doi: 10.1016/s0304-3940(98)00392-9. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickelbein RG, Seres T, Lam G, Johnston RB, Jr, Warshaw JB. Characterization of multiple cysteine and cystine transporters in rat alveolar type II cells. Am J Physiol. 1997;273:L1147–L1155. doi: 10.1152/ajplung.1997.273.6.L1147. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Kranich O, Dringen R, Sandberg M, Hamprecht B. Utilization of cysteine and cysteine precursors for the synthesis of glutathione in astroglial cultures: preference for cystine. Glia. 1998;22:11–18. [PubMed] [Google Scholar]
- Lawrence CB, Allan SM, Rothwell NJ. Interleukin-1beta and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur J Neurosci. 1998;10:1188–1195. doi: 10.1046/j.1460-9568.1998.00136.x. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Letz J, Methner A. Activation of stimulatory heterotrimeric G proteins increases glutathione and protects neuronal cells against oxidative stress. J Neurochem. 2003;87:522–531. doi: 10.1046/j.1471-4159.2003.02019.x. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Liu T, McDonnell PC, Young PR, White RF, Siren AL, Hallenbeck JM, Barone FC, Feurestein GZ. Interleukin-1 beta mRNA expression in ischemic rat cortex. Stroke. 1993;24:1746–1750. doi: 10.1161/01.str.24.11.1746. discussion 1750–1741. [DOI] [PubMed] [Google Scholar]
- Liu XH, Kwon D, Schielke GP, Yang GY, Silverstein FS, Barks JD. Mice deficient in interleukin-1 converting enzyme are resistant to neonatal hypoxic-ischemic brain damage. J Cereb Blood Flow Metab. 1999;19:1099–1108. doi: 10.1097/00004647-199910000-00006. [DOI] [PubMed] [Google Scholar]
- Lobner D, Choi DW. Dipyridamole increases oxygen-glucose deprivation-induced injury in cortical cell culture. Stroke. 1994;25:2085–2089. doi: 10.1161/01.str.25.10.2085. discussion 2089–2090. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1996;16:932–940. doi: 10.1097/00004647-199609000-00017. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem. 1994;62:45–53. doi: 10.1046/j.1471-4159.1994.62010045.x. [DOI] [PubMed] [Google Scholar]
- Martin D, Chinookoswong N, Miller G. The interleukin-1 receptor antagonist (rhIL-1ra) protects against cerebral infarction in a rat model of hypoxia-ischemia. Exp Neurol. 1994;130:362–367. doi: 10.1006/exnr.1994.1215. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Minami M, Kuraishi Y, Yabuuchi K, Yamazaki A, Satoh M. Induction of interleukin-1 beta mRNA in rat brain after transient forebrain ischemia. J Neurochem. 1992;58:390–392. doi: 10.1111/j.1471-4159.1992.tb09324.x. [DOI] [PubMed] [Google Scholar]
- Moroni F, Attucci S, Cozzi A, Meli E, Picca R, Scheideler MA, Pellicciari R, Noe C, Sarichelou I, Pellegrini-Giampietro DE. The novel and systemically active metabotropic glutamate 1 (mGlu1) receptor antagonist 3-MATIDA reduces post-ischemic neuronal death. Neuropharmacology. 2002;42:741–751. doi: 10.1016/s0028-3908(02)00033-3. [DOI] [PubMed] [Google Scholar]
- Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA. Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol. 2003;140:471–476. doi: 10.1038/sj.bjp.0705462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Ohtaki H, Funahashi H, Dohi K, Oguro T, Horai R, Asano M, Iwakura Y, Yin L, Matsunaga M, Goto N, Shioda S. Suppression of oxidative neuronal damage after transient middle cerebral artery occlusion in mice lacking interleukin-1. Neurosci Res. 2003;45:313–324. doi: 10.1016/s0168-0102(02)00238-9. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. discussion 332. [DOI] [PubMed] [Google Scholar]
- Pinteaux E, Parker LC, Rothwell NJ, Luheshi GN. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem. 2002;83:754–763. doi: 10.1046/j.1471-4159.2002.01184.x. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc− and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-β peptide 1–40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- Relton JK, Martin D, Thompson RC, Russell DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp Neurol. 1996;138:206–213. doi: 10.1006/exnr.1996.0059. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Amin S, Leitner M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci. 1992;12:56–61. doi: 10.1523/JNEUROSCI.12-01-00056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sairanen TR, Lindsberg PJ, Brenner M, Siren AL. Global forebrain ischemia results in differential cellular expression of interleukin-1beta (IL-1beta) and its receptor at mRNA and protein level. J Cereb Blood Flow Metab. 1997;17:1107–1120. doi: 10.1097/00004647-199710000-00013. [DOI] [PubMed] [Google Scholar]
- Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206:149–152. doi: 10.1016/s0304-3940(96)12460-5. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun. 2004;325:109–116. doi: 10.1016/j.bbrc.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–7462. doi: 10.1523/JNEUROSCI.21-19-07455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamm LH, Koroshetz WJ, Sorensen AG, Wang B, Copen WA, Budzik R, Rordorf G, Buonanno FS, Schaefer PW, Gonzalez RG. Time course of lesion development in patients with acute stroke: serial diffusion- and hemodynamic-weighted magnetic resonance imaging. Stroke. 1998;29:2268–2276. doi: 10.1161/01.str.29.11.2268. [DOI] [PubMed] [Google Scholar]
- Sheldon N, VerHeulen M, DeYoung M, Chase L. Peroxide triggers the translocation of the transporter, System xc−, to the plasma membrane in cultured human glioma cells. FASEB J. 2006;20:A1366. [Google Scholar]
- Sims JE, Gayle MA, Slack JL, Alderson MR, Bird TA, Giri JG, Colotta F, Re F, Mantovani A, Shanebeck K, Grabstein KH, Dower SK. Interleukin 1 signaling occurs exclusively via the type I receptor. Proc Natl Acad Sci USA. 1993;90:6155–6159. doi: 10.1073/pnas.90.13.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skifter DA, Allegrini PR, Wiessner C, Mir AK. Similar time-course of interleukin-1 beta production and extracellular-signal-regulated kinase (ERK) activation in permanent focal brain ischemic injury. Metab Brain Dis. 2002;17:131–138. doi: 10.1023/a:1019917803470. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Schousboe A. Glutamate transport and metabolism in astrocytes. Glia. 1997;21:56–63. doi: 10.1002/(sici)1098-1136(199709)21:1<56::aid-glia6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Joseph DJ, Friedman W. Cell type-specific interleukin-1β signaling in the CNS. J Neurosci. 2004;24:6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel RGD, Torrie JH. Ed 2. New York: McGraw-Hill; 1980. Principles and procedures of statistics: a biometrical approach. [Google Scholar]
- Stroemer RP, Rothwell NJ. Exacerbation of ischemic brain damage by localized striatal injection of interleukin-1beta in the rat. J Cereb Blood Flow Metab. 1998;18:833–839. doi: 10.1097/00004647-199808000-00003. [DOI] [PubMed] [Google Scholar]
- Tabb JS, Ueda T. Phylogenetic studies on the synaptic vesicle glutamate transport system. J Neurosci. 1991;11:1822–1828. doi: 10.1523/JNEUROSCI.11-06-01822.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao T, Culp SG, Newton RC, De Souza EB. Type I interleukin-1 receptors in the mouse brain-endocrine-immune axis labelled with [125I]recombinant human interleukin-1 receptor antagonist. J Neuroimmunol. 1992;41:51–60. doi: 10.1016/0165-5728(92)90195-q. [DOI] [PubMed] [Google Scholar]
- Tomozawa Y, Inoue T, Satoh M. Expression of type I interleukin-1 receptor mRNA and its regulation in cultured astrocytes. Neurosci Lett. 1995;195:57–60. doi: 10.1016/0304-3940(95)11781-q. [DOI] [PubMed] [Google Scholar]
- Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- Touzani O, Boutin H, LeFeuvre R, Parker L, Miller A, Luheshi G, Rothwell N. Interleukin-1 influences ischemic brain damage in the mouse independently of the interleukin-1 type I receptor. J Neurosci. 2002;22:38–43. doi: 10.1523/JNEUROSCI.22-01-00038.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79:445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- Traystman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J Appl Physiol. 1991;71:1185–1195. doi: 10.1152/jappl.1991.71.4.1185. [DOI] [PubMed] [Google Scholar]
- Uliasz TF, Hewett SJ. A microtiter trypan blue absorbance assay for the quantitative determination of excitotoxic neuronal injury in cell culture. J Neurosci Methods. 2000;100:157–163. doi: 10.1016/s0165-0270(00)00248-x. [DOI] [PubMed] [Google Scholar]
- Vidwans AS, Hewett SJ. Enhanced release of synaptic glutamate underlies the potentiation of oxygen-glucose deprivation-induced neuronal injury after induction of NOS-2. Exp Neurol. 2004;190:91–101. doi: 10.1016/j.expneurol.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–340. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- Wong ML, Licinio J. Localization of interleukin 1 type I receptor mRNA in rat brain. Neuroimmunomodulation. 1994;1:110–115. doi: 10.1159/000097143. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Shen H, Baker DA, Kalivas PW. Inhibition of non-vesicular glutamate release by group III metabotropic glutamate receptors in the nucleus accumbens. J Neurochem. 2003;87:1204–1212. doi: 10.1046/j.1471-4159.2003.02093.x. [DOI] [PubMed] [Google Scholar]
- Yabuuchi K, Minami M, Katsumata S, Yamazaki A, Satoh M. An in situ hybridization study on interleukin-1 beta mRNA induced by transient forebrain ischemia in the rat brain. Brain Res Mol Brain Res. 1994;26:135–142. doi: 10.1016/0169-328x(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676–680. doi: 10.1161/01.str.26.4.676. discussion 681. [DOI] [PubMed] [Google Scholar]
- Yang GY, Zhao YJ, Davidson BL, Betz AL. Overexpression of interleukin-1 receptor antagonist in the mouse brain reduces ischemic brain injury. Brain Res. 1997;751:181–188. doi: 10.1016/s0006-8993(96)01277-2. [DOI] [PubMed] [Google Scholar]
- Yang GY, Liu XH, Kadoya C, Zhao YJ, Mao Y, Davidson BL, Betz AL. Attenuation of ischemic inflammatory response in mouse brain using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist. J Cereb Blood Flow Metab. 1998;18:840–847. doi: 10.1097/00004647-199808000-00004. [DOI] [PubMed] [Google Scholar]
- Yang GY, Mao Y, Zhou LF, Ye W, Liu XH, Gong C, Lorris Betz A. Attenuation of temporary focal cerebral ischemic injury in the mouse following transfection with interleukin-1 receptor antagonist. Brain Res Mol Brain Res. 1999;72:129–137. doi: 10.1016/s0169-328x(99)00205-3. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- Zhang Z, Chopp M, Goussev A, Powers C. Cerebral vessels express interleukin 1beta after focal cerebral ischemia. Brain Res. 1998;784:210–217. doi: 10.1016/s0006-8993(97)01317-6. [DOI] [PubMed] [Google Scholar]