

Abstract
3,4-Methylenedioxymethamphetamine (MDMA)-induced neurotoxicity and the protective role of monoamine oxidase-B (MAO-B) inhibition were evaluated at the mitochondrial level in various regions of the adolescent rat brain. Four groups of adolescent male Wistar rats were used: (1) saline control, (2) exposed to MDMA (4 × 10 mg/kg, i.p.; two hourly), (3) treated with selegiline (2 mg/kg, i.p.) 30 min before the same dosing of MDMA, and (4) treated with selegiline (2 mg/kg, i.p.). Body temperatures were monitored throughout the whole experiment. Animals were killed 2 weeks later, and mitochondria were isolated from several brain regions. Our results showed that “binge” MDMA administration causes, along with sustained hyperthermia, long-term alterations in brain mitochondria as evidenced by increased levels of lipid peroxides and protein carbonyls. Additionally, analysis of mitochondrial DNA (mtDNA) revealed that NDI nicotinamide adenine dinucleotide phosphate dehydrogenase subunit I and NDII (nicotinamide adenine dinucleotide phosphate dehydrogenase subunit II) subunits of mitochondrial complex I and cytochrome c oxidase subunit I of complex IV suffered deletions in MDMA-exposed animals. Inhibition of MAO-B by selegiline did not reduce hyperthermia but reversed MDMA-induced effects in the oxidative stress markers, mtDNA, and related protein expression. These results indicate that monoamine oxidation by MAO-B with subsequent mitochondrial damage may be an important contributing factor for MDMA-induced neurotoxicity.
Keywords: 3,4-methylenedioxymethamphetamine; neurotoxicity; adolescent rat model; brain mitochondria; monoamine oxidase B; oxidative stress
Introduction
Studies in rodents and nonhuman primates indicate that exposure to 3,4-methylenedioxymethamphetamine (MDMA) may elicit neurotoxicity to serotonin (5-HT)-containing axon terminals (Stone et al., 1986; Battaglia et al., 1987; Commins et al., 1987; Hatzidimitriou et al., 1999). MDMA binds to the 5-HT reuptake transporter and, inside axon terminals, causes an acute and powerful release of neurotransmitters (mainly 5-HT, but also noradrenaline and dopamine) from storage vesicles (Green et al., 1995; Sulzer et al., 2005). MDMA also contributes to a steady concentration of these neurotransmitters in the synaptic cleft by partially inhibiting their oxidation through monoamine oxidase (MAO) while inside nerve endings and by blocking their reuptake into nerve terminals (Leonardi and Azmitia, 1994; Green et al., 2003). During the phase of abrupt increase of the extravesicular levels of monoamine neurotransmitters inside nerve endings, a large amount is metabolized by MAO. Two isoforms of MAO exist: MAO-A and MAO-B. In the brain, MAO-A is expressed predominantly in catecholaminergic neurons, whereas MAO-B is expressed in serotoninergic neurons, astrocytes, and glia (Shih et al., 1999). 5-HT has been shown to be metabolized in vitro by MAO-A (Km, 178 ± 2 μm) and MAO-B (Km, 1170 ± 432 μm) (Shih et al., 1999). Although metabolism by MAO-B is only residual in the presence of MAO-A, it is fully effective in the absence of the latter, as it happens inside serotonergic nerves (Fowler and Tipton, 1982; Shih et al., 1999). Oxidative deamination of monoamine neurotransmitters by MAO produces hydrogen peroxide (H2O2), which subsequently may be converted into the highly reactive hydroxyl radical (HO), and consequent oxidative stress-related damage may occur. Corroborating this hypothesis, previous studies have shown that MDMA-induced chronic 5-HT loss requires the activity of MAO-B and involves oxidative stress (Schmidt, 1987; Sprague and Nichols, 1995; Sprague et al., 1998). More recently, it was demonstrated that MDMA-induced 5-HT depletion observed in wild-type mice did not occur in MAO-B-deficient mice (Fornai et al., 2001) and that an antisense oligonucleotide targeted at MAO-B attenuates rat striatal serotonergic neurotoxicity induced by MDMA (Falk et al., 2002). Moreover, it was also shown that dopamine uptake into serotonergic nerve endings, followed by MAO-B metabolism and subsequent oxidative stress also contributes to the neurotoxic effects of MDMA (Hrometz et al., 2004; Jones et al., 2004). Thus, it may be assumed that MAO-B-mediated metabolism of monoamine neurotransmitters following their vesicular release by MDMA is an important driving factor for MDMA-induced neurotoxicity. However, the mechanisms mediating the deleterious effects of MAO-B derived H2O2 at nerve endings are hitherto not known.
Considering that MAO enzymes are located at the outer membrane of mitochondria, our hypothesis is that H2O2 diffuses through mitochondrial membranes leading to oxidative damage on mitochondrial macromolecules, namely lipids, proteins, and DNA. Thus, the aim of the study was to expose an adolescent rat model to a neurotoxic “binge” administration of MDMA and to evaluate the neurotoxicity at the brain mitochondrial level. An additional important aim of this work was to verify whether inhibition of MAO-B by selegiline could prevent the toxic effects of MDMA.
Materials and Methods
Animal model.
Adolescent male Wistar rats were used. Animals were kept under controlled environmental conditions (temperature, 20 ± 2°C; relative humidity, 45–55%; 12 h light/dark cycle) and housed with food and water supplied ad libitum. All procedures were approved by the Portuguese Agency for Animal Welfare (General Board of Veterinary Medicine in compliance with the Institutional Guidelines and the European Convention).
On postnatal day 40 (P40), animals were divided into four experimental groups (MDMA group, selegiline plus MDMA group, selegiline control group, and an isovolumetric saline control group) and caged in pairs. Two days later, a subcutaneous probe (Implantable Programmable Temperature Transponder, IPTT-200; BMD, Einsteinweg, The Netherlands) (Kort et al., 1998) was inserted in the lumbar region to allow the measurement of body temperature throughout the experiment. On P45, the animals assigned to the MDMA group received a freshly prepared solution of 10 mg MDMA/kg body weight, intraperitoneally, every 2 h in a total of four injections. The selegiline plus MDMA group was submitted to the same MDMA dosing scheme, the first administration of MDMA preceded 30 min by the administration of seleginine (2 mg/kg, i.p.). Control animals received equal doses of saline vehicle (0.9% w/v) in the same protocol of administration, and the selegiline group was administrated a single dose of selegiline (2 mg/kg, i.p.). Body temperature was measured just before the first MDMA or isovolumetric saline injection, and then, every 15 min, for a period of 9 h. Body temperature was recorded daily throughout the course of the experiment, always between 10:00 and 12:00 A.M.
MDMA (HCl salt) was extracted and purified from high-purity MDMA tablets that were provided by the Portuguese Criminal Police Department. The obtained salt was pure and fully characterized by NMR and mass spectrometry methodologies. Selegiline was supplied by Sigma (Steinheim, Germany).
Mitochondria isolation for determination of lipid peroxides and protein carbonyls.
Two weeks after exposure, animals were killed by decapitation, and brains were removed rapidly on ice. Mitochondria were isolated from whole brain by the method of Rosenthal et al. (1987), with slight modifications. Briefly, the whole encephalon was washed, minced, and homogenized at 4°C in isolation medium (250 mm sucrose, 5 mm HEPES, 1 mm EGTA, 1 mg/ml BSA, pH 7.4) containing 5 mg of bacterial protease (P-5380; Sigma). Single brain homogenates were brought to 30 ml and centrifuged at 1260 × g (SORVAL RC 5B Plus; Kendro Laboratory Products, Newtown, CT) for 5 min. The supernatant was centrifuged at 20,000 × g for 10 min. The pellet including the fluffy synaptosomal layer was resuspended in 10 ml of isolation medium containing 0.02% digitonin (to release mitochondria from the synaptosomal fraction) and centrifuged at 20,000 × g for 10 min. The brown mitochondrial pellet (without the synaptosomal layer) was resuspended again in 10 ml of washing medium (225 mm sucrose, 5 mm HEPES, pH 7.4) and recentrifuged at 20,000 × g for 10 min. The main portion of the mitochondrial pellet was resuspended in 300 μl of washing medium. Mitochondrial proteins were determined by the biuret method, calibrated with BSA (Gornall et al., 1949).
Quantification of lipid peroxidation.
Lipid peroxidation was determined by measuring malondialdehyde (MDA) equivalents, using the thiobarbituric acid (TBA) assay, according to a modified procedure (Rohn et al., 1993). Mitochondrial protein (3 mg) was incubated for 30 min at 25°C in 3 ml of medium (175 mm KCL, 10 mm Tris, pH 7.4, and 3 μm rotenone). Samples of 0.3 ml were then incubated with 2.7 ml of TBA reactive substances reagent (9% TBA, 0.6 N HCl, and 0.0056% butylated hydroxyl toluene). The mixture was warmed to 80–90°C for 15 min and cooled by immersion in ice during 10 min before centrifuge at 1500 × g for 5 min. Lipid peroxidation was estimated by spectrophotometric determination, at 535 nm, of the MDA equivalents produced. The amount of MDA equivalents formed was calculated using a molar extinction coefficient of 1.56 × 105 mol−1cm−1 and expressed as nmol MDA equivalents/mg protein (Buege and Aust, 1978).
Quantification of protein carbonyls.
Protein carbonyls were quantified through the spectrophotometric method for carbonyl assay, using 2,4-dinitrophenylhydrazine (DNPH) (adapted from Reznick and Packer, 1994). Two samples of 1 ml of each 1 mg/ml mitochondrial extract were placed in glass tubes. To one tube, 4 ml of 10 mm DNPH in 2.5 m HCl solution was added, and to the other tube of the same sample, only 4 ml of 2.5 m HCl (blank tube) was added. Tubes were left for 1 h at room temperature in the dark and vortexed every 15 min. At this point, 5 ml of 20% trichloroacetic acid (TCA) (w/v) solution was added to both DNPH and HCl samples to a final concentration of 10% (w/v) TCA. The tubes were left in ice for 10 min and then centrifuged for 5 min. The resultant supernatant was discarded. Next, another wash was performed with 4 ml of 10% TCA, and the protein pellets were broken mechanically. The protein pellets were washed three times with ethanol-ethyl acetate (1:1) (v/v). The final pellet was dissolved in 6 m guanidine hydrochloride solution and left for 10 min at 37°C in agitation in a water bath. All samples were centrifuged to remove any insoluble material remaining in suspension. The concentration of DNPH was determined at 360 nm, and the molar absorption coefficient of 22 × 103 mol−1cm−1 was used to quantify the levels of protein carbonyls. Protein concentration in the samples was calculated by determining the absorbance at 280 nm. Protein carbonyl content was expressed as nmol protein carbonyl formed/mg mitochondrial protein (Reznick and Packer, 1994).
DNA isolation for PCR.
Two weeks after exposure, animals were killed by decapitation, brains were rapidly removed, and dissection of the different brain regions (prefrontal cortex, striatum, hippocampus, amygdala, ventral mesencephalon [comprising substantia nigra and ventral tegmental area (VTA/SN)] and raphe nuclei) was performed on ice. DNA from the different brain regions was extracted with GenomicPrep Cells and Tissue DNA Isolation kit (GE Healthcare, Buckinghamshire, UK) according to the instructions of the manufacturer. No pools of animals were necessary. Extracted DNA (5 μl) was applied on a 1% agarose gel to quantify the amount of DNA used on the subsequent PCR protocols.
Photographs were taken under UV transillumination (Typhoon 8600; Molecular Dynamics, Buckinghamshire, UK), and the semiquantitative analysis of extracted DNA was made with the software Image Quant 5.1.
PCR.
Previously isolated brain areas were analyzed for a deletion between direct repeats corresponding to base pairs 1095–4095 of rat mitochondrial DNA (mtDNA). Deletion primers were designed based on the sequence of the rat mtDNA (GenBank accession number NC-001665) to detect a deletion corresponding to nicotinamide adenine dinucleotide phosphate dehydrogenase subunit I (NDI) and nicotinamide adenine dinucleotide phosphate dehydrogenase subunit II (NDII) subunits of mitochondrial complex I (NADH dehydrogenase) and COXI subunit of mitochondrial complex IV (cytochrome c oxidase) (Suliman et al., 2003).
The following mtDNA deletion primers were used: 5′-AGTCGTAACAAGGTAAGCAT-3′ (base pairs 982–1001) mtf1 primer and 5′-ATTTCTACTCTTTTAGCAT-3′ (base pairs 5632–5651) mtr2 primer (Suliman et al., 2003). The reaction mixture consisted of primers in a concentration of 400 μm (1 μl of stock 20 pmol; MWG-Biotech, Ebersberg, Germany), 40 ng of template DNA, 1.5 mm MgCl2 (50 mm stock; Bio-Rad, Hercules, CA), 1 μl of 10 mm PCR nucleotide mix (Eppendorf, Hamburg, Germany), 0.25 μl of Taq Polymerase (5 U/μl) (Bio-Rad), and 5 μl of enzyme buffer 10× (Bio-Rad). The final volume of the PCR was 50 μl, and the program used was 94°C for 2 min, 50°C for 30 s, 72°C for 2 min (35 cycles), and 72°C for 7 min (1 cycle) (MyCyclerthermocycler; Bio-Rad).
Negative controls were included containing all the above mentioned PCR components except template DNA. Ten microliter aliquots of the PCR products were electrophoresed through a 1–1.5% agarose gel in Tris-acetate containing ethidium bromide at 45 V/cm. Photographs were taken under UV transillumination (Typhoon; Molecular Dynamics), and the semiquantitative analysis of amplified DNA was made with the software Image Quant 5.1.
Western blot analysis of isolated whole-brain mitochondria.
Two weeks after exposure, animals were killed by decapitation, and brains were removed rapidly on ice as described above. Isolated whole-brain mitochondria were resuspended in extraction buffer (20 mm Tris-HCl, pH 7.6, 250 mm sucrose, 40 mm KCl, 2 mm EGTA). The homogenate was centrifuged at 600 × g for 10 min at 4°C, and the supernatant was taken for mitochondrial Western blot analysis, 15 μg of protein was loaded per lane and separated on 10% SDS-polyacrylamide gels. The gels were transferred to a polyvinylidene fluoride membrane for protein blotting (0.2 μm; Bio-Rad) membranes by electroblotting 1 h at 150 mA. The filters were blocked in 5% nonfat dry milk and 0.1% Tween 20 overnight at 4°C. Blots were then incubated with mouse monoclonal antibody against COXI (2 μg/ml) (Invitrogen, Eugene, OR) or NDII (0.5 μg/ml) (Invitrogen) diluted in 0.1% TBS Tween 20 (20 mm Tris, 137 mm NaCl, pH 7.6) for 1 h at room temperature. Membranes were washed three times for 10 min in the same buffer and incubated for 1 h with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Imun-Star, 1:20,000; Bio-Rad). Immunoreactive proteins were revealed using enhanced chemiluminescence method (Immun-Star HRP Chemiluminescent kit; Bio-Rad). Blots were analyzed with Quantity One Software version 4.5 (Bio-Rad).
Statistical analyses.
Data concerning evolution of body temperature were analyzed using a two-way ANOVA (treatment vs time). Significant differences were further tested using the post hoc Tukey's HSD for unequal n.
Data concerning MDA equivalents, protein carbonyls, mtDNA deletions, and Western blot quantifications were analyzed using a one-way ANOVA (treatment). Significant main effects and interactions were further explored using the post hoc Scheffé's test. The statistical level of significance was considered at p < 0.05.
Results
Effects of MDMA administration on body temperature
Hyperthermia is one of the most dangerous clinical symptoms of MDMA intoxication. Accordingly, in the present study, MDMA administration resulted in thermoregulation alterations. Analysis of data obtained for body temperature profile throughout the day of exposure shows that rats treated with MDMA presented significantly higher average body temperatures 30 min after the first injection and until the end of the measuring period, when compared with saline controls (p < 0.001 for most values measured) (Fig. 1). Animals exposed to MDMA plus selegiline also presented a higher average body temperature when compared with the control group (p < 0.001) or the group exposed only to selegiline (p < 0.001) (Fig. 1). However, no significant differences were observed between body temperatures of MDMA and MDMA plus selegiline exposed rats. Selegiline administered animals did not show significant differences in body temperature when compared with the control group (Fig. 1). Although after the third injection, the effects of increased core temperature were less evident in MDMA group, rats treated with MDMA preceded by selegiline continued to exhibit, to a certain extent, temperatures similar to the ones observed after the first and the second injections (Fig. 1).
Figure 1.

MDMA induced hyperthermia in adolescent male Wistar rats. The image represents body temperature evolution (measured by scanning a subcutaneously inserted probe every 15 min for 9 h) throughout the period of exposure to the following: MDMA (4 × 10 mg/kg), selegiline (2 mg/kg) plus MDMA (4 × 10 mg/kg), control saline (isovolumetric saline), and selegiline (2 mg/kg). Selegiline was administered 30 min before exposure to MDMA. Results are reported as mean ± SEM for n = 10 in control and MDMA exposed rats and n = 6 in the other groups. Evolution curves for MDMA and MDMA plus selegiline exposed rats started to display significantly higher values than the curves of control and selegiline exposed rats 30 min after the first dose (p < 0.001; two-way ANOVA followed by a post hoc Tukey's HSD for unequal n). Arrows indicate injection timings.
MDMA-treated animals demonstrated a chronic alteration in thermoregulation throughout the 13 d after the day of exposure, showing small but significantly increased values (p < 0.05) that were prevented by selegiline (data not shown).
Effects of MDMA administration on lipid peroxidation
The extension of lipid peroxidation was measured in the mitochondrial fraction of whole brain homogenates of adolescent rats 2 weeks after exposure to MDMA, MDMA plus selegiline, selegiline, and isovolumetric saline. MDMA-treated rats had significantly higher levels of malondialdehyde equivalents (p < 0.001), when compared with saline controls, selegiline, or MDMA plus selegiline. The levels of lipid peroxidation on animals injected with MDMA plus selegiline were not significantly different from the control groups (selegiline and saline) (Fig. 2).
Figure 2.

MDMA induced lipid peroxidation in whole-brain mitochondria and the protective effect of selegiline. Lipid peroxidation was determined by measuring MDA equivalents, using the TBA assay. Animals were killed 14 d after exposure to: MDMA (4 × 10 mg/kg), selegiline (Sel.) (2 mg/kg) plus MDMA (4 × 10 mg/kg), saline (isovolumetric saline), or selegiline (2 mg/kg). Selegiline was administered 30 min before exposure to MDMA. Columns represent mean + SEM, expressed in nanomoles MDA equivalents per milligram of protein for each experimental group (n = 10 for control and MDMA; n = 6 for MDMA plus selegiline and selegiline). Animals exposed to MDMA presented significantly higher values of MDA equivalents than all other groups (*p < 0.001, one-way ANOVA followed by a post hoc Scheffé's test).
Effects of MDMA administration on the formation of protein carbonyls
Two weeks after exposure, the levels of protein carbonyls were evaluated in the mitochondrial fraction of whole brain homogenates of adolescent male rats injected with MDMA, MDMA plus selegiline, selegiline, and isovolumetric saline. The administration of a neurotoxic dose of MDMA produced a significant increase on protein carbonyls of whole-brain mitochondria (p < 0.001) comparatively to all of the other groups. Animals treated with selegiline before MDMA administration presented carbonyl levels that were similar to those measured in control groups (Fig. 3).
Figure 3.

MDMA induced carbonyl formation in whole-brain mitochondria and the protective effect of selegiline. Protein carbonyls were quantified by reaction with DNPH using a spectrophotometric assay for carbonyls. Animals were killed 14 d after exposure to: MDMA (4 × 10 mg/kg), selegiline (Sel.) (2 mg/kg) plus MDMA (4 × 10 mg/kg), saline (isovolumetric saline), or selegiline (2 mg/kg). Selegiline was administered 30 min before exposure to MDMA. Columns represent mean + SEM, expressed in nanomoles of protein carbonyls per milligram of total protein for each experimental group (n = 10 for control and MDMA; n = 6 for MDMA plus selegiline and selegiline). Animals exposed to MDMA presented significantly higher levels of carbonyl formation than all other groups (*p < 0.001, one-way ANOVA followed by a post hoc Scheffé's test).
Effects of MDMA administration on mtDNA
PCR
DNA from different brain areas (frontal cortex, striatum, amygdala, VTA/SN, hippocampus, and raphe nuclei) were submitted to an amplification procedure with TaqDNA polymerase to verify the presence of a deletion corresponding to the genes NDI and NDII of the mitochondrial complex I and COXI of complex IV. With the set of primers used, a band corresponding to NDI, NDII, and COXI deletion was expected. This deletion was substantially more evident on animals injected with MDMA (p < 0.001 for all tested areas except the prefrontal cortex) comparatively to all the other groups. In animals treated with selegiline, there was a significant attenuation on the level of deletion (p < 0.001) in most brain areas, although these levels were still higher than those observed for the control and the selegiline groups (Fig. 4).
Figure 4.
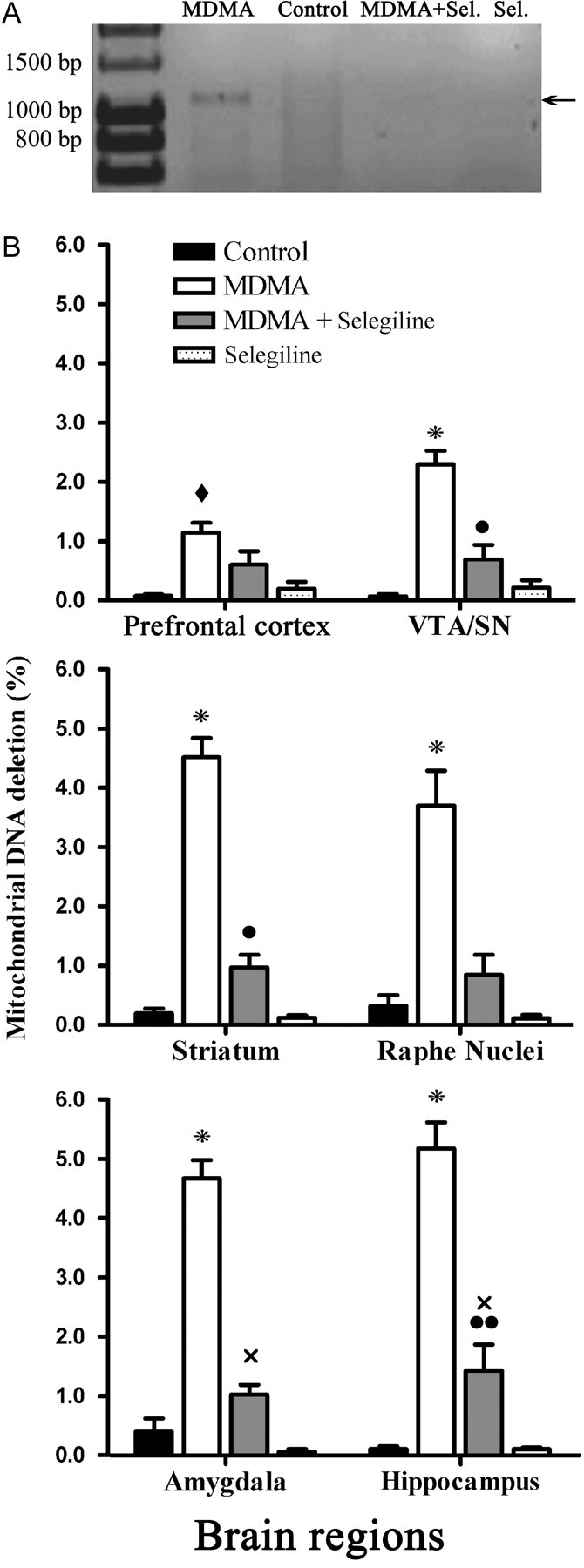
Protective effects of selegiline over the deletion of the mitochondrial gene sequences for NDI, NDII, and COXI in male Wistar rats exposed to a neurotoxic dose of MDMA. A, A representative densitometry analysis is shown for the raphe nuclei; lanes, from left to right, correspond to standard Hyper ladder I, MDMA, control, MDMA plus selegiline (Sel.), and selegiline. The PCR was performed with the mtf1 and mtr2 primers, amplifying a fragment correspondent to the deletion of the sequences for NDI, NDII, and COXI. Animals were killed 14 d after exposure to MDMA (4 × 10 mg/kg), selegiline (2 mg/kg) plus MDMA (4 × 10 mg/kg), saline (isovolumetric saline), or selegiline (2 mg/kg). Selegiline was administered 30 min before exposure to MDMA. B, Graphic representation of the levels of deletion of the mitochondrial gene sequences for NDI, NDII, and COXI for the four experimental groups, in different brain regions: prefrontal cortex, VTA/SN, striatum, raphe nuclei, amygdala, and hippocampus. Columns represent mean + SEM, expressed as percentage of DNA deletion, for each experimental group (n = 6). Significant differences between treatments are represented as: *p < 0.001, used for MDMA, significantly higher than all other groups; ♦p < 0.01, used for MDMA, significantly higher than control and selegiline groups; ●p < 0.05 and ●●p < 0.01, used for MDMA plus selegiline, significantly higher than control; xp < 0.01, used for MDMA plus selegiline, significantly higher than selegiline alone (one-way ANOVA followed by a post hoc Scheffé's test).
Figure 4A is representative of the PCR for the mentioned deletion (the image shown corresponds to a raphe nuclei sample). In the prefrontal cortex, animals treated with MDMA presented lower levels of the deletion than the other tested areas, although the difference between MDMA exposed animals and the other groups was still highly significant (p < 0.01) (Fig. 4B). In the VTA/SN, striatum, raphe nuclei, amygdala, and hippocampus, the group exposed to MDMA was significantly more affected than the other, presenting increased levels of deletion (p < 0.001) (Fig. 4B). A significant protection was obtained in animals previously dosed with selegiline, where levels of deletion, although higher than in control and selegiline groups, were significantly lower than for the MDMA group in all tested regions except the prefrontal cortex (Fig. 4B).
Western blot analysis for the expression of mitochondrial subunit NDII and COXI
Expression of NDII was considerably diminished (p < 0.001) on MDMA group compared with control groups (isovolumetric saline and selegiline) and with MDMA plus selegiline groups. Animals treated with MDMA plus selegiline presented values of expression that were not significantly different from controls (Fig. 5).
Figure 5.

MDMA induced decreased expression of the subunit NDII in whole-brain mitochondria and the protective effect of selegiline. NDII expression was determined by Western blot analysis performed with isolated whole-brain mitochondria of male Wistar rats, using a mouse monoclonal antibody anti-NDII subunit. Animals were killed 14 d after exposure to: MDMA (4 × 10 mg/kg), selegiline (Sel.) (2 mg/kg) plus MDMA (4 × 10 mg/kg), saline (isovolumetric saline), or selegiline (2 mg/kg). Selegiline was administered 30 min before exposure to MDMA. Columns represent densitometric analysis of Western blots (mean + SEM; n = 6) expressed in outer diameter/area (mm2). Densitometric analysis of α-tubulin served as loading control. Expression of NDII in the MDMA group was significantly decreased when compared with the other experimental groups (*p < 0.001, one-way ANOVA followed by a post hoc Scheffé's test).
COXI was also less expressed on MDMA administered animals relatively to controls (isovolumetric saline and selegiline) (Fig. 6). Selegiline protected, to some extent, the deficient expression of these two subunits caused by exposure to MDMA.
Figure 6.

MDMA induced decreased expression of the subunit COXI in whole-brain mitochondria and the protective effect of selegiline. COXI expression was determined by Western blot analysis performed with isolated whole-brain mitochondria of male Wistar rats, using a mouse monoclonal antibody anti-COXI subunit. Animals were killed 14 d after exposure to: MDMA (4 × 10 mg/kg), selegiline (Sel.) (2 mg/kg) plus MDMA (4 × 10 mg/kg), saline (isovolumetric saline), or selegiline (2 mg/kg). Selegiline was administered 30 min before exposure to MDMA. Columns represent densitometric analysis of Western blots (mean + SEM; n = 6) expressed in outer diameter/area (mm2). Densitometric analysis of α-tubulin served as loading control. Expression of COXI in the MDMA group was more significantly decreased (*p < 0.001) when compared with control and selegiline groups than when compared with the MDMA plus selegiline group (♦p < 0.01, one-way ANOVA followed by a post hoc Scheffé's test).
Discussion
The neurotoxic effects that may result from MDMA consumption have been a matter of continuous research in the last decades. Several factors may contribute to MDMA-induced neurotoxicity, namely MDMA metabolism, sustained receptor stimulation, hyperthermia, enzymatic and nonenzymatic oxidation of neurotransmitters, inhibition of neurotransmitters synthesis, inflammation, and oxidative stress. The results obtained in the present study provide a new insight on the deleterious effects of MDMA to the CNS. To our knowledge, this is the first study to report a MAO-B-dependent, MDMA-induced damage on mitochondrial macromolecules, especially to mtDNA and subsequent impairment in the correspondent protein expression.
The exposure of the adolescence rat model to a neurotoxic “binge” administration of MDMA induced an oxidative stress status in brain mitochondria, as reflected by a robust increase in mitochondrial lipid peroxidation and protein carbonylation, 14 d after administration. Additionally, more studies in the prefrontal cortex, striatum, amygdala, ventral mesencephalon, hippocampus, cerebellum, and raphe nuclei revealed that mtDNA from these regions codifying the NDI and NDII subunits of the mitochondrial complex I (NADH dehydrogenase) and COXI of complex IV (cytochrome c oxidase) were partially deleted in MDMA-exposed animals, leading to the subsequent decrease of protein expression for these enzymatic systems. The contribution of MAO-B to MDMA-induced mitochondrial damage in the CNS was also clearly evidenced, because the inhibition of this enzyme by selegiline protected brain mitochondria against lipid peroxidation and protein carbonylation, as well as partially prevented mtDNA deletion and consequent decrease of protein expression. Selegiline was administered in a dose that is known to inhibit MAO-B. Although some authors argue that the often observed protective role of selegiline may also be attributable to its hydroxyl radical scavenging activity (Muralikrishnan et al., 2003; Sharma et al., 2003), others had not found such effect (Ferger et al., 1998).
MAO is localized in the outer membrane of mitochondria. Thus, the formation of H2O2 resulting from the metabolism of MDMA-released monoamine neurotransmitters by MAO will mostly affect mitochondria itself. When H2O2 removal pathways are lacking or inactivated, H2O2 accumulates and is converted into the highly reactive HO, inducing intense oxidative stress in mitochondria (Mathai and Sitaramam, 1994; Agostinelli et al., 2004). Rat mtDNA, like human mtDNA, is vulnerable to reactive oxygen species (ROS), because it lacks the protection of histones and DNA-binding proteins (Shigenaga et al., 1994). Recently, a new oxidant-sensitive deletion in hepatic mtDNA of 3.81 kb was identified in the region between 1095 and 4905 bp involving two direct repeats in rat mtDNA (Suliman et al., 2002; Suliman et al., 2003). In the present study, the same type of mtDNA depletion was observed, for the first time, in MDMA-treated rats. The deleted portion of the mitochondrial genome includes codons for NADH dehydrogenase (NDI, NDII), cytochrome c oxidase (COX1), and tRNA 16S and rRNA for six amino acids (Suliman et al., 2002, 2003). Mitochondria carrying this deletion in sufficient copy number would have both impaired entry of reducing equivalents into the respiratory chain at complex I and functional alterations in molecular O2 reduction at complex IV, which is also indicated in our study through the decreased expression of NDII and COXI. As the percentage of mtDNA deletions increases, mitochondrial energy output declines, ROS production increases further, and mitochondrial decline and mtDNA damage accumulate within the cells, leading to activation of apoptotic and necrotic cell death.
In addition to the immediate metabolism of vesicular released 5-HT, reuptake of dopamine into the 5-HT terminal may also contribute to MDMA-induced neurotoxicity. As discussed by Hrometz et al. (2004), in vivo, MDMA administration leads to a rapid release of 5-HT and inhibition of tryptophan hydroxylase, which results in the depletion of neuronal 5-HT. The initially released 5-HT activates 5-HT2A postsynaptic receptors that lead to the upregulation of dopamine biosynthesis and release. Under these circumstances, dopamine may be transported into serotonergic nerve terminals by 5-HT transporter (SERT), which means that further MAO-B metabolism of dopamine may also contribute to the oxidative stress-related neurotoxic effect of MDMA (Sprague and Nichols, 1995). Although this hypothesis was not tested in the present study, it was demonstrated previously that a correlation exists between the extent of dopamine release and the extent of 5-HT depletion induced by MDMA, these effects being prevented by treatment with dopamine uptake inhibitors or inhibition of dopamine synthesis (Gudelsky and Yamamoto, 2003).
The differential effects in various brain regions have been a characteristic feature of MDMA neurotoxicity. MDMA is known to induce 5-HT depletion in selected brain regions in various animal species (Schmidt, 1987; Finnegan et al., 1988; O'Hearn et al., 1988; Ricaurte et al., 1988). In a previous study, O'Hearn et al. (1988) performed immunocytochemical analysis of regional brain sections 2 weeks after high-dose MDMA administration to rats and observed that the terminal portions of 5-HT axons were selectively vulnerable to the neurotoxic effects of MDMA, whereas preterminal axons, fibers of passage, and raphe cell bodies were not damaged. Regional differences in neurotoxicity were manifested by a greater loss of 5-HT axons in several forebrain areas, particularly neocortex, striatum, and thalamus, whereas more fibers were spared in hippocampus, septum, and amygdala. Battaglia et al. (1991) also demonstrated that, in rat, brain regions containing 5-HT pathways or perikarya were little affected by MDMA, the predominant effects being mediated on axons and terminals. Later, Aguirre et al. (1995) corroborated the previous data by showing the lack of 5-HT depletion in the dorsal raphe region of the brain stem, which includes part of the serotonergic cell bodies. The regional data obtained in our study comprises the striatum and the hippocampus as prone to MDMA-induced mtDNA deletion, but this time a deleterious effect with similar potency was observed for raphe nuclei and the hippocampus, whereas the frontal cortex and the VTA/SN were less affected. Our data imply that the classical view of MDMA neurotoxicity may not be applied to mitochondrial effects because other areas were also strongly affected. Recently, it was demonstrated that, in the dorsal raphe nucleus, serotonin-positive neuronal cell bodies were positive for MAO-B, which was present in all mitochondria. In contrast, within the neuronal cell bodies and dendrites that were positive for MAO-B, whereas most mitochondria contained this enzyme, a substantial quantity of mitochondria lacked it (Arai et al., 2002). Thus, the localization of MAO-B in the mitochondrial outer membrane of the neuronal cell bodies may help to understand the mitochondrial damage occurring at these sites.
Another potentially deleterious effect mediated by MDMA, hyperthermia, was observed in the present study but, under the present experimental conditions, did not substantially contribute to the changes in the measured toxicological biomarkers. The exposure of the adolescence rat model to a neurotoxic binge administration of MDMA provoked acute and long-term hyperthermia. Acute hyperthermia corresponds to a characteristic MDMA effect on laboratory animals maintained at ambient temperatures of approximately ≥20°C and is aggravated if ambient temperature increases (Malberg and Seiden, 1998; Carvalho et al., 2002; Green et al., 2005). Note that hyperthermia has been shown to potentiate the neurotoxic effects of MDMA both in vivo (Malberg and Seiden, 1998) and in vitro (Capela et al., 2006a,b). However, considering that the strong neuroprotective effect of MAO-B inhibition was independent of body temperature, the obtained data also contribute to a better clarification of MDMA effects, showing that the mechanism of MDMA neurotoxicity extends beyond hyperthermia.
In this study, we used a rat adolescence model. The impact of MDMA on adolescent abusers may be particularly harmful because of the vulnerability of their cerebral and hormonal systems, which are still undergoing crucial maturational changes (Montoya et al., 2002). For example, the density of the 5-HT transporter (SERT) rises during adolescence (Moll et al., 2000). A few experimental studies using adolescent rat models have shown that MDMA induces changes in several behavioral and physiological markers (Fone et al., 2002; Koenig et al., 2005; Piper et al., 2005) and revealed differences in the response when adolescence models are compared with adult models, namely in terms of the thermoregulation (Piper et al., 2005).
In conclusion, the results obtained in the present study demonstrate, for the first time, that the exposure of an adolescence rat model to a neurotoxic binge administration of MDMA induces not only an oxidative stress status in brain mitochondria but also deletion of mtDNA and subsequent impairment in the genes required for protein synthesis, which are essential complexes of the mitochondrial respiratory system required for energy production. The tissue-specific manifestations induced by MDMA may result from varying energetic roles and needs of the different tissues. The contribution of MAO-B to MDMA-induced mitochondrial damage in CNS was clearly evidenced, given that the inhibition of this enzyme by selegiline completely protected brain mitochondria against lipid peroxidation and protein carbonylation and significantly prevented mtDNA deletion and correspondent protein expression.
Footnotes
This work was supported by Fundação Calouste Gulbenkian (Principal Investigator, Teresa Summavielle) and “Programa de Financiamento Plurianual do Instituto de Biologia Molecular e Celular.” E.A., T.S., and C.J.A. were supported by Fundação para a Ciência e Tecnologia Grants SFRH/BD/12176/2003, SFRH/BPD/20997/2004, and SFRH/BD/17195/2004.
References
- Agostinelli E, Arancia G, Vedova LD, Belli F, Marra M, Salvi M, Toninello A. The biological functions of polyamine oxidation products by amine oxidases: perspectives of clinical applications. Amino Acids. 2004;27:347–358. doi: 10.1007/s00726-004-0114-4. [DOI] [PubMed] [Google Scholar]
- Aguirre N, Galbete JL, Lasheras B, Del Rio J. Methylenedioxymethamphetamine induces opposite changes in central pre- and postsynaptic 5-HT1A receptors in rats. Eur J Pharmacol. 1995;281:101–105. doi: 10.1016/0014-2999(95)00303-3. [DOI] [PubMed] [Google Scholar]
- Arai R, Karasawa N, Kurokawa K, Kanai H, Horiike K, Ito A. Differential subcellular location of mitochondria in rat serotonergic neurons depends on the presence and the absence of monoamine oxidase type B. Neuroscience. 2002;114:825–835. doi: 10.1016/s0306-4522(02)00351-2. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Yeh SY, O'Hearn E, Molliver ME, Kuhar MJ, De Souza EB. 3,4-Methylenedioxymethamphetamine and 3,4-methylenedioxyamphetamine destroy serotonin terminals in rat brain: quantification of neurodegeneration by measurement of [3H]paroxetine-labeled serotonin uptake sites. J Pharmacol Exp Ther. 1987;242:911–916. [PubMed] [Google Scholar]
- Battaglia G, Sharkey J, Kuhar MJ, de Souza EB. Neuroanatomic specificity and time course of alterations in rat brain serotonergic pathways induced by MDMA (3,4-methylenedioxymethamphetamine): assessment using quantitative autoradiography. Synapse. 1991;8:249–260. doi: 10.1002/syn.890080403. [DOI] [PubMed] [Google Scholar]
- Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- Capela JP, Ruscher K, Lautenschlager M, Freyer D, Dirnagl U, Gaio AR, Bastos ML, Meisel A, Carvalho F. Ecstasy-induced cell death in cortical neuronal cultures is serotonin 2A-receptor-dependent and potentiated under hyperthermia. Neuroscience. 2006a;139:1069–1081. doi: 10.1016/j.neuroscience.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Capela JP, Meisel A, Abreu AR, Branco PS, Ferreira LM, Lobo AM, Remiao F, Bastos ML, Carvalho F. Neurotoxicity of ecstasy metabolites in rat cortical neurons, and influence of hyperthermia. J Pharmacol Exp Ther. 2006b;316:53–61. doi: 10.1124/jpet.105.092577. [DOI] [PubMed] [Google Scholar]
- Carvalho M, Carvalho F, Pereira ML, Pires-das-Neves R, Bastos ML. Effect of 3,4-methylenedioxymethamphetamine (“ecstacy”) on body temperature and liver antioxidant status in mice. Influence of ambient temperature. Arch Toxicol. 2002;76:166–172. doi: 10.1007/s00204-002-0324-z. [DOI] [PubMed] [Google Scholar]
- Commins DL, Vosmer G, Virus RM, Woolverton WL, Schuster CR, Seiden LS. Biochemical and histological evidence that methylenedioxymethylamphetamine (MDMA) is toxic to neurons in the rat brain. J Pharmacol Exp Ther. 1987;241:338–345. [PubMed] [Google Scholar]
- Falk EM, Cook VJ, Nichols DE, Sprague JE. An antisense oligonucleotide targeted at MAO-B attenuates rat striatal serotonergic neurotoxicity induced by MDMA. Pharmacol Biochem Behav. 2002;72:617–622. doi: 10.1016/s0091-3057(02)00728-1. [DOI] [PubMed] [Google Scholar]
- Ferger B, van Amsterdam C, Seyfried C, Kuschinsky K. Effects of alpha-phenyl-tert-butylnitrone and selegiline on hydroxyl free radicals in rat striatum produced by local application of glutamate. J Neurochem. 1998;70:276–280. doi: 10.1046/j.1471-4159.1998.70010276.x. [DOI] [PubMed] [Google Scholar]
- Finnegan KT, Ricaurte GA, Ritchie LD, Irwin I, Peroutka SJ, Langston JW. Orally administered MDMA causes a long-term depletion of serotonin in rat brain. Brain Res. 1988;447:141–144. doi: 10.1016/0006-8993(88)90974-2. [DOI] [PubMed] [Google Scholar]
- Fone KC, Beckett SR, Topham IA, Swettenham J, Ball M, Maddocks L. Long-term changes in social interaction and reward following repeated MDMA administration to adolescent rats without accompanying serotonergic neurotoxicity. Psychopharmacology (Berl) 2002;159:437–444. doi: 10.1007/s00213-001-0931-z. [DOI] [PubMed] [Google Scholar]
- Fornai F, Giorgi FS, Gesi M, Chen K, Alessri MG, Shih JC. Biochemical effects of the monoamine neurotoxins DSP-4 and MDMA in specific brain regions of MAO-B-deficient mice. Synapse. 2001;39:213–221. doi: 10.1002/1098-2396(20010301)39:3<213::AID-SYN1002>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Tipton KF. Deamination of 5-hydroxytryptamine by both forms of monoamine oxidase in the rat brain. J Neurochem. 1982;38:733–736. doi: 10.1111/j.1471-4159.1982.tb08692.x. [DOI] [PubMed] [Google Scholar]
- Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- Green AR, Cross AJ, Goodwin GM. Rev of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or “ecstasy”) Psychopharmacology (Berl) 1995;119:247–260. doi: 10.1007/BF02246288. [DOI] [PubMed] [Google Scholar]
- Green AR, Mechan AO, Elliott JM, O'Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) Pharmacol Rev. 2003;55:463–508. doi: 10.1124/pr.55.3.3. [DOI] [PubMed] [Google Scholar]
- Green AR, O'Shea E, Saadat KS, Elliott JM, Colado MI. Studies on the effect of MDMA (“ecstasy”) on the body temperature of rats housed at different ambient room temperatures. Br J Pharmacol. 2005;146:306–312. doi: 10.1038/sj.bjp.0706318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudelsky GA, Yamamoto BK. Neuropharmacology and neurotoxicity of 3,4-methylenedioxymethamphetamine. Methods Mol Med. 2003;79:55–73. doi: 10.1385/1-59259-358-5:55. [DOI] [PubMed] [Google Scholar]
- Hatzidimitriou G, McCann UD, Ricaurte GA. Altered serotonin innervation patterns in the forebrain of monkeys treated with (+/-)3,4-methylenedioxymethamphetamine seven years previously: factors influencing abnormal recovery. J Neurosci. 1999;19:5096–5107. doi: 10.1523/JNEUROSCI.19-12-05096.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrometz SL, Brown AW, Nichols DE, Sprague JE. 3,4-Methylenedioxymethamphetamine (MDMA, ecstasy)-mediated production of hydrogen peroxide in an in vitro model: the role of dopamine, the serotonin-reuptake transporter, and monoamine oxidase-B. Neurosci Lett. 2004;367:56–59. doi: 10.1016/j.neulet.2004.05.075. [DOI] [PubMed] [Google Scholar]
- Jones DC, Lau SS, Monks TJ. Thioether metabolites of 3,4-methylenedioxyamphetamine and 3,4-methylenedioxymethamphetamine inhibit human serotonin transporter (hSERT) function and simultaneously stimulate dopamine uptake into hSERT-expressing SK-N-MC cells. J Pharmacol Exp Ther. 2004;311:298–306. doi: 10.1124/jpet.104.069260. [DOI] [PubMed] [Google Scholar]
- Koenig J, Lazarus C, Jeltsch H, Ben Hamida S, Riegert C, Kelche C, Jones BC, Cassel JC. MDMA (ecstasy) effects in pubescent rats: males are more sensitive than females. Pharmacol Biochem Behav. 2005;81:635–644. doi: 10.1016/j.pbb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Kort WJ, Hekking-Weijma JM, TenKate MT, Sorm V, VanStrik R. A microchip implant system as a method to determine body temperature of terminally ill rats and mice. Lab Anim. 1998;32:260–269. doi: 10.1258/002367798780559329. [DOI] [PubMed] [Google Scholar]
- Leonardi ET, Azmitia EC. MDMA (ecstasy) inhibition of MAO type A and type B: comparisons with fenfluramine and fluoxetine (Prozac) Neuropsychopharmacology. 1994;10:231–238. doi: 10.1038/npp.1994.26. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Seiden LS. Small changes in ambient temperature cause large changes in 3,4-methylenedioxymethamphetamine (MDMA)-induced serotonin neurotoxicity and core body temperature in the rat. J Neurosci. 1998;18:5086–5094. doi: 10.1523/JNEUROSCI.18-13-05086.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai JC, Sitaramam V. Stretch sensitivity of transmembrane mobility of hydrogen peroxide through voids in the bilayer. Role of cardiolipin. J Biol Chem. 1994;269:17784–17793. [PubMed] [Google Scholar]
- Moll GH, Mehnert C, Wicker M, Bock N, Rothenberger A, Ruther E, Huether G. Age-associated changes in the densities of presynaptic monoamine transporters in different regions of the rat brain from early juvenile life to late adulthood. Dev Brain Res. 2000;119:251–257. doi: 10.1016/s0165-3806(99)00182-0. [DOI] [PubMed] [Google Scholar]
- Montoya AG, Sorrentino R, Lukas SE, Price BH. Long-term neuropsychiatric consequences of “ecstasy” (MDMA): a review. Harv Rev Psychiatry. 2002;10:212–220. [PubMed] [Google Scholar]
- Muralikrishnan D, Samantaray S, Mohanakumar KP. D-deprenyl protects nigrostriatal neurons against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurotoxicity. Synapse. 2003;50:7–13. doi: 10.1002/syn.10239. [DOI] [PubMed] [Google Scholar]
- O'Hearn E, Battaglia G, De Souza EB, Kuhar MJ, Molliver ME. Methylenedioxyamphetamine (MDA) and methylenedioxymethamphetamine (MDMA) cause selective ablation of serotonergic axon terminals in forebrain: immunocytochemical evidence for neurotoxicity. J Neurosci. 1988;8:2788–2803. doi: 10.1523/JNEUROSCI.08-08-02788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper BJ, Fraiman JB, Meyer JS. Repeated MDMA (“ecstasy”) exposure in adolescent male rats alters temperature regulation, spontaneous motor activity, attention, and serotonin transporter binding. Dev Psychobiol. 2005;47:145–157. doi: 10.1002/dev.20085. [DOI] [PubMed] [Google Scholar]
- Reznick AZ, Packer L. Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. 1994;233:357–363. doi: 10.1016/s0076-6879(94)33041-7. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Forno LS, Wilson MA, DeLanney LE, Irwin I, Molliver ME, Langston JW. (+/-)3,4-Methylenedioxymethamphetamine selectively damages central serotonergic neurons in nonhuman primates. JAMA. 1988;260:51–55. [PubMed] [Google Scholar]
- Rohn TT, Hinds TR, Vincenzi FF. Ion transport ATPases as targets for free radical damage. Protection by an aminosteroid of the Ca2+ pump ATPase and Na+/K+ pump ATPase of human red blood cell membranes. Biochem Pharmacol. 1993;46:525–534. doi: 10.1016/0006-2952(93)90530-a. [DOI] [PubMed] [Google Scholar]
- Rosenthal RE, Hamud F, Fiskum G, Varghese PJ, Sharpe S. Cerebral ischemia and reperfusion: prevention of brain mitochondrial injury by lidoflazine. J Cereb Blood Flow Metab. 1987;7:752–758. doi: 10.1038/jcbfm.1987.130. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ. Neurotoxicity of the psychedelic amphetamine, methylenedioxymethamphetamine. J Pharmacol Exp Ther. 1987;240:1–7. [PubMed] [Google Scholar]
- Sharma SK, Carlson EC, Ebadi M. Neuroprotective actions of Selegiline in inhibiting 1-methyl, 4-phenyl, pyridinium ion (MPP+)-induced apoptosis in SK-N-SH neurons. J Neurocytol. 2003;32:329–343. doi: 10.1023/B:NEUR.0000011327.23739.1b. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC, Grimsby J, Chen K. Molecular biology of monoamine oxidase A and B: their role in the degradation of serotonin. In: Gothert HB, editor. Serotoninergic neurons and 5-HT receptors in the SNC. Berlin: Springer; 1999. pp. 655–670. [Google Scholar]
- Sprague JE, Nichols DE. The monoamine oxidase-B inhibitor L-deprenyl protects against 3,4-methylenedioxymethamphetamine-induced lipid peroxidation and long-term serotonergic deficits. J Phar-macol Exp Ther. 1995;273:667–673. [PubMed] [Google Scholar]
- Sprague JE, Everman SL, Nichols DE. An integrated hypothesis for the serotonergic axonal loss induced by 3,4-methylenedioxymethamphetamine. Neurotoxicology. 1998;19:427–441. [PubMed] [Google Scholar]
- Stone DM, Stahl DC, Hanson GR, Gibb JW. The effects of 3,4-methylenedioxymethamphetamine (MDMA) and 3,4-methylenedioxyamphetamine (MDA) on monoaminergic systems in the rat brain. Eur J Pharmacol. 1986;128:41–48. doi: 10.1016/0014-2999(86)90555-8. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Velsor LW, Day BJ, Ghio AJ, Piantadosi CA. Rapid mtDNA deletion by oxidants in rat liver mitochondria after hemin exposure. Free Radic Biol Med. 2002;32:246–256. doi: 10.1016/s0891-5849(01)00797-3. [DOI] [PubMed] [Google Scholar]
- Suliman HB, Carraway MS, Piantadosi CA. Postlipopolysaccharide oxidative damage of mitochondrial DNA. Am J Respir Crit Care Med. 2003;167:570–579. doi: 10.1164/rccm.200206-518OC. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]