

Abstract
Accumulating evidence from clinical and preclinical studies shows that catechol-O-methyltransferase (COMT) plays a significant role in dopamine metabolism in the prefrontal cortex, but not in the striatum. However, to what extent dopamine overflow in the prefrontal cortex and striatum is controlled by enzymatic degradation versus reuptake is unknown. We used COMT deficient mice to investigate the role of COMT in these two brain regions with in vivo voltammetry. A real-time analysis of evoked dopamine overflow showed that removal of dopamine was twofold slower in the prefrontal cortex of mice lacking COMT than in wild-type mice, indicating that half of the dopamine decline in this brain region results from COMT-mediated enzymatic degradation. Lack of COMT did not influence dopamine overflow/decline in the dorsal striatum. COMT-deficient mice demonstrated a small (20–25%) but consistent increase in evoked dopamine release in the prefrontal cortex, but not in the dorsal striatum. Cocaine affected equally dopaminergic neurotransmission in the prefrontal cortex in both genotypes by prolonging 3–4 times dopamine elimination from extracellular space. Paradoxically, this happened without increase of the peak levels of evoked dopamine release. The present findings represent the first demonstration of the significant contribution of COMT in modulating the dynamics of dopamine overflow in the prefrontal cortex and underscore the therapeutic potential of manipulating COMT activity to alter dopaminergic neurotransmission in the prefrontal cortex.
Keywords: prefrontal cortex, caudate nucleus, COMT, cocaine, in vivo voltammetry, knock-out mice
Introduction
It is known that the density of dopamine transporter (DAT) is much lower in the prefrontal cortex than in the striatum (Scatton et al., 1985). In mice lacking dopamine transporter, dopamine processing is only slightly modified in the prefrontal cortex, but dopamine levels are significantly increased in the striatum (Mundorf et al., 2001). Voltammetric studies have also identified consistent differences between these structures, particularly a much higher rate of release and reuptake of dopamine in the caudate nucleus than in the prefrontal cortex (Garris et al., 1993; Garris and Wightman, 1994).
It appears that once released, dopamine persists for a long time in the extracellular space of the prefrontal cortex. Although the noradrenaline transporter (Mundorf et al., 2001; Carboni and Silvagni, 2004) and monoamine oxidase (MAO) (Westerink and Spaan, 1982) contribute to dopamine elimination, catechol-O-methyltransferase (COMT) evidently dominates in the regulation of dopamine levels in this cortical area. Indeed, formation of 3-methoxytyramine, a predominant dopamine metabolite, catalyzed by COMT, is higher in the prefrontal cortex than in the striatum, where homovanillic acid prevails (Karoum et al., 1994).
In majority of previous works, the role of COMT in regulation of extracellular dopamine has been investigated in striatum, using tolcapone, a brain penetrating COMT inhibitor (for review, see Männistö and Kaakkola, 1999). In one microdialysis study, the effects of tolcapone were analyzed in the prefrontal cortex. Acute tolcapone alone did not affect dopamine release, but it enhanced the effect of clozapine and K+-depolarization (Tunbridge et al., 2004). In the present work, Comt gene-disrupted mice (Gogos et al., 1998) were used to quantify the role of COMT in the dynamics of dopamine overflow in the prefrontal cortex and dorsal striatum.
COMT is genetically polymorphic having three levels of activity, low (COMTLL), intermediate (COMTLH), and high (COMTHH) (Weinshilboum and Raymond, 1977; Boudikova et al., 1990), which are determined primarily by a common Val108/158Met polymorphism (Lachman et al., 1998) that results from the thermolability of the Met variant. Although the frequencies of the Val and Met alleles vary fourfold among populations (DeMille et al., 2002), the COMT activities fluctuate much less (∼40% according to Chen et al. (2004). The impact of COMT polymorphism on cognition and in a progress of mental disorders has recently gained attention (Männistö and Kaakkola, 1999; Egan et al., 2001; Tunbridge et al., 2004; Gothelf et al., 2005; Mata et al., 2006; Tunbridge et al., 2006; Raux et al., 2007). Despite numerous reports, the role of COMT remains unclear because genetic association studies of clinical diagnosis have not been convincing (Munafo et al., 2005). Based on a large meta-analysis, a modest relationship between the Val108/158Met genotype and executive performance has been seen in healthy individuals, but not in schizophrenics (Barnett et al., 2007).
In this study, we describe for the first time the dynamics of evoked dopamine overflow in real-time, using in vivo voltammetry in the prefrontal cortex and in the caudate nucleus in COMT knock-out (KO) mice and their wild-type (WT) littermates. We also analyzed the effects of blocking dopamine transporter with cocaine in these mice.
Materials and Methods
Animals.
COMT KO mice were originally generated by Gogos et al. (1998). The mutated COMT allele was introduced into a mixed 129Sv/C57BL/6J genetic background and by four-generation backcrossing the mutation was introduced into a more homogenous C57BL/6J genetic background. Heterozygous (COMT+/−) male and female mice were bred in the National Laboratory Animal Center (Kuopio, Finland) to produce mice of all three genotypes. The mouse strain was regularly enriched by breeding C57BL/6J males or females with heterozygotes.
The genotype was determined by PCR. Genomic DNA was isolated from tail biopsies as described by Laird et al. (1991). For genotyping, a PCR method was developed using ACCATGGAGATTAACCCTGACTACG (sense), GTGTGTCTGGAAGGTAGCGGTC (antisense) primer set to detect wild-type alleles, and GTGTTCCGGCTGTCAGCGCA (sense), GTCCTGATAG-CGGTCCGCCA (antisense) primer set for the targeted alleles. Genomic PCR was completed using the Fail Safe PCR system (Epicenter Technologies, Madison, WI) using Fail Safe buffer B and the following thermal cycles: an initial denaturing at 98°C for 1 min and 35 cycles consisting of a denaturing temperature of 94°C for 30 s, an annealing temperature of 65°C for 1 min, and extension at 72°C for 3 min with a final extension of 70°C for 10 min. The amplified fragments were visualized by ethidium bromide staining under UV light after electrophoresis in a 2% agarose gel.
Preparation of animals.
Animals were kept under controlled conditions at the National Laboratory Animal Center (temperature +22 ± 1°C, lights on from 7:00 A.M. to 7:00 P.M.; humidity, 50–60%), with food and water available ad libitum. The experiments were conducted according to the European Community (Directive 86/609) guidelines, and approved by the Animal Ethics Committee at the University of Kuopio and local provincial government.
The 7- to 9-month-old male mice (28–36 g) were anesthetized with chloral hydrate (450 mg/kg, i.p.) and fixed in a stereotaxic frame. Rectal temperature was kept at 37°C with a heating lamp. The electrochemical probe (carbon fiber working electrode) was inserted through an opening in the skull first to the caudate nucleus [anterioposterior (AP), 0.5 mm; lateral (L), 2.2 mm and 6° from vertical; and ventral (V), 3.5 mm from bregma]. After measuring of dopamine overflow, the probe was removed and inserted in the prefrontal cortex (V, 1.8 mm) via another preliminary drilled opening in the skull with the following coordinates: AP, 2.47 mm; L, 0.5 mm. We slant the probe (6° from vertical) to eliminate bleeding from the sagittal sinus. The same angle of the probe was also applied for the dorsal striatum site to keep mechanical assembly of stereotaxic instrumentation (arm with probe) stable. A bipolar stimulating electrode was implanted in the medial forebrain bundle (AP, −2.1 mm; L, 1.1 mm; H, 5.0–5.2 mm) according to the mouse brain atlas (Paxinos and Franklin, 2000). The exact placement of the stimulating electrode in the dorsoventral coordinate was adjusted for maximal dopamine release in the caudate nucleus and the electrode was not moved during changing of the probe position. A small leak-free Ag/AgCl reference electrode in a saline bridge was placed on the skull. A stainless-steel screw was fixed onto the skull as the auxiliary electrode.
Electrochemical technique.
Stimulated release and reuptake of dopamine (termed here as dopamine overflow) was measured using constant potential amperometry (CPA) with a single Nafion-coated carbon fiber, 30 μm in diameter (World Precision Instruments, Sarasota, FL), insulated with epoxy glue in pulled capillary glass. A custom-built three-electrode potentiostat held the working electrode at 0.4 V against an Ag/AgCl reference electrode. The current at the working electrode was converted to voltage at a headstage converter. During registration of dopamine release in the prefrontal cortex, CPA was temporary switched to the fast cyclic voltammetry (FCV; from −0.4 to 0.9 V and back to −0.4 V, 50 V/s, at 0.2 s intervals). Five cyclic voltammograms were taken at equal intervals during the period τ2 on the descending part of the curve b (see Fig. 1), starting from the peak dopamine release, averaged and subtracted from the five cyclic voltammograms that were taken at the same intervals and averaged immediately before stimulation. Subtracted cyclic voltammograms (Fig. 1c), together with cocaine treatment (see below), were used for identification of released substances (Phillips and Wightman, 2003). The validity of the use of voltammetry for measurements of stimulated dopamine release in the prefrontal cortex was discussed previously (Garris et al., 1993). Working electrodes were calibrated before and after implantation except electrodes that were used for verification of the locations of the probes. Marking positions were made with anodic current (6 V, 15 s) applied via the working electrode.
Figure 1.

Dynamics of dopamine elimination from extracellular space in the caudate nucleus (a) and prefrontal cortex (b) in COMT knock-out and wild-type mice. a, b, Dopamine overflow after stimulation of the medial forebrain bundle. Stimulation (0.8 s, 50 Hz) was marked by a horizontal bar above the time scale. Dopamine elimination was characterized using parameter τ − time (in seconds) of the twofold decrease of the peak dopamine concentration (see Materials and Methods). Curves of dopamine decay in the caudate (a) and prefrontal cortex (b) were approximated by exponential decay function. c, Five cyclic voltammograms (CVs) were taken at equal intervals during period τ2 on the descending part of the curve b, starting from the peak dopamine release, averaged, and subtracted from the five CVs that were taken at the same intervals and averaged immediately before stimulation. Subtracted CVs demonstrated oxidation (downward, at ∼0.5 V) and reduction (upward, at about −0.2 V) peaks, which are specific for dopamine. d, Mean ± SEM (n = 7) of tau calculated for the caudate and prefrontal cortex in WT and COMT KO mice. *p < 0.05 (WT vs KO).
Electrical stimulation and experimental protocol.
A battery-operated constant current unit (A365; World Precision Instruments) run by a personal computer was used for the stimulation. Monophasic constant current pulses (1 ms in duration) of 180 μA were delivered to the stimulation electrode at different frequencies (10–60 Hz, 2 s in duration) and lengths (0.2–2 s, 50 Hz). These stimulations produced from barely measurable to near maximal dopamine release.
The intervals between stimulations varied in dependence on the intensity of stimulation from 1 min (after 10 Hz, 2 s stimulation) to 6 min (after 60 Hz, 2 s stimulation). This approach allowed to minimize influences of previous stimulation on the next one. Cocaine hydrochloride was dissolved in deionized water and injected intraperitoneally at a dose of 20 mg/kg. The effects of cocaine were analyzed 30 min after the treatment at different stimulation lengths and frequencies.
After lowering the probe to the caudate nucleus, finding an optimal location of the stimulating electrodes and stabilization of the baseline and responses, series of stimulations with different parameters as described above were applied on the medial forebrain bundle (MFB). After that, the probe was removed from the caudate and inserted in the prefrontal cortex. Similar series of stimulations (except stimulation at 10 Hz, which produced very low response in this structure) were applied on the MFB. Cocaine was injected after these measurements and consequences of the stimulations of the MFB were repeated again 30 min after treatment.
Data presentation and statistical analysis.
Dopamine release was expressed in molar concentrations. The effects of cocaine were calculated in molar concentrations as the peak dopamine response before and after treatment. The right slope of the dopamine overflow curve reflects the dynamics of dopamine elimination from the extracellular space (Wightman et al., 1988). This curve can be approximated by a standard exponential function f(t) = Ae−kt + C. Curve fitting was made with Clampfit version 8 (Molecular Devices, Sunnyvale, CA) using signals of similar amplitudes (e.g., after 20 Hz stimulation in the caudate and 60 Hz stimulation in the prefrontal cortex). We analyzed and depicted dopamine decay from the extracellular space using parameter tau (τ = 0.69 k −1), which is the time of the twofold decrease of the peak dopamine concentration after stimulation.
The effects of genotype on the peak dopamine release and dopamine elimination from the extracellular space were statistically analyzed with multivariate ANOVA (MANOVA; Prism 3; GraphPad Software, San Diego, CA). An anatomical structure (dorsal striatum and prefrontal cortex) or treatment (cocaine, saline) and the frequencies or lengths of stimulations were used as the within-subject factors and genotype as the between-subject factor. Bonferroni post hoc pairwise comparison was used to compare individual groups. Data are presented as mean ± SEM.
Results
Dopamine overflow in the caudate (Fig. 1a) was distinctly different from overflow in the prefrontal cortex (Fig. 1b). Specifically, stimulation of the MFB induced a sharp increase in the extracellular dopamine concentration in the caudate nucleus followed by a fast decay resulting from effective dopamine reuptake. In contrast, rise in dopamine levels after stimulation was considerably slower in the prefrontal cortex. Peak concentrations of dopamine in the prefrontal cortex were at least 10 times lower than in the caudate at high frequencies of stimulation. Dopamine removal was also significantly slower in this brain area, so that tau (τ) calculated for the WT mice was ∼10 times lower than in the caudate nucleus (1.92 ± 0.22 s vs 0.2 ± 0.03 s for the caudate nucleus and prefrontal cortex, respectively; F(1) = 61.78; p < 0.001) (Fig. 1a,b). The electrochemical approach used in this work (CPA) does not distinguish between dopamine, its metabolites, and ascorbic acid, thus raising questions regarding the specificity of the voltammetric signal in the prefrontal cortex. In addition to treatment of the electrode with anion repelling polymer (Nafion), which significantly improves selectivity (Gerhardt et al., 1984), we used FCV and obtained cyclic voltammograms typical for dopamine (Fig. 1c). Finally, response to cocaine (see below) was also taken as an additional verification of the specificity of the signal.
The role of COMT levels in dopamine overflow in the caudate and prefrontal cortex was studied by comparing data obtained in WT and COMT KO mice (Fig. 1). We found significant differences in the effects of genotype in these two brain regions. Specifically, COMT KO mice demonstrated a twofold slower decay of dopamine in the prefrontal cortex compared with WT littermates (anatomical structure by genotype interaction; F(1) = 7.56; p < 0.01), whereas no differences in dopamine decay were identified between COMT KO and WT mice in the caudate nucleus.
Genotypic differences were also observed in the amount of extracellular dopamine (peak dopamine release) after stimulation, likely reflecting the differences in neurotransmitter removal. COMT KO mice demonstrated a small (20–25%) but consistent increase in dopamine release in response to stimulation of increasing length in the prefrontal cortex in comparison with WT mice (Fig. 2b) (F(1) = 8.86; p < 0.005). No such differences between genotypes were observed in the caudate nucleus (Fig. 2a) (F(1) = 0.61; p < 0.5). Peak dopamine concentrations in response to stimulation at different frequencies (Fig. 2c,d) were also different between genotypes in the prefrontal cortex, so that COMT KO mice showed again slightly higher stimulated dopamine release (Fig. 2d), which was significant at low (20–30 Hz), but not at the high (50 and 60 Hz) frequencies of stimulations (F(1) = 12.08; p < 0.001). No differences in dopamine release were seen in the caudate nucleus (Fig. 2c).
Figure 2.
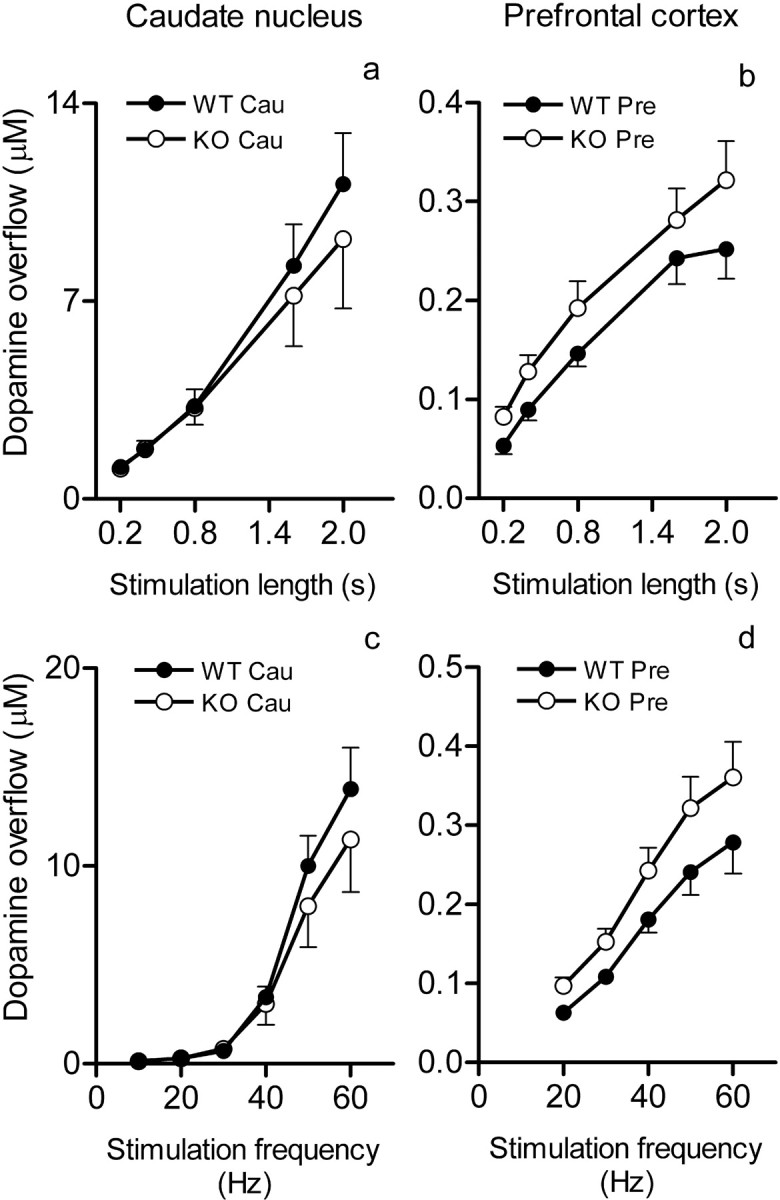
a–d, Dopamine overflow in the caudate nucleus (a, c) and prefrontal cortex (b, d) in dependence on the length and frequency of stimulation of the medial forebrain bundle and genotype. a, b, Peak dopamine overflow (mean ± SEM; 7 mice per group in all experiments) in response to increasing lengths (0.2–2 s) of 50 Hz stimulations. c, d, Peak dopamine overflow in response to increasing frequencies (10–60 Hz in the caudate and 20–60 Hz in the prefrontal cortex) of 2 s stimulations. Cau, Caudate nucleus; Pre, prefrontal cortex. Dopamine overflow in the prefrontal cortex was significantly higher in the COMT KO mice (for statistics, see Results).
Modified reuptake could account for changing dopamine decay in COMT KO mice. In experiments using cocaine to block DAT we found that, in the prefrontal cortex, cocaine prolongs transients of dopamine after neuronal bursts without increasing their amplitudes (Fig. 3). However, the effects of cocaine on the dopamine removal were similar between genotypes (Fig. 3) (genotype by treatment interaction; F(1) = 0.58; p < 0.45). This finding suggests that affinity of DAT is not different between the genotypes and, thus, differences in removal of dopamine in the prefrontal cortex resulted directly from the lack of COMT. Interestingly, despite the effects of cocaine on the reuptake of dopamine, we did not find any effects of the drug on the peak dopamine release in the prefrontal cortex in either genotypes (Fig. 4). This opposes with well established effects of cocaine in the caudate and nucleus accumbens, where it increases peak dopamine release (Stamford et al., 1988; Garris and Wightman, 1995; Wu et al., 2001; Greco and Garris, 2003; Walker et al., 2006).
Figure 3.

The effects of cocaine (20 mg/kg) on dynamics of dopamine elimination from extracellular space in the prefrontal cortex after short stimulation of the medial forebrain bundle in COMT KO and WT mice. The right panel shows typical example of the curves of dopamine release and removal after 2 s, 50 Hz stimulation before and after cocaine treatment. Recordings were filtrated with Clampfit (boxcar, 5 smoothing points) and approximated by an exponential decay function (see Materials and Methods). The width of the slope of dopamine decay at the half-height of the peak (τ) was calculated and presented as mean ± SEM (n = 7) in the left panel. Note that the large dose of cocaine significantly increased τ in both genotypes (*p < 0.05), but we did not find genotype-dependent differences in the effects of cocaine.
Figure 4.
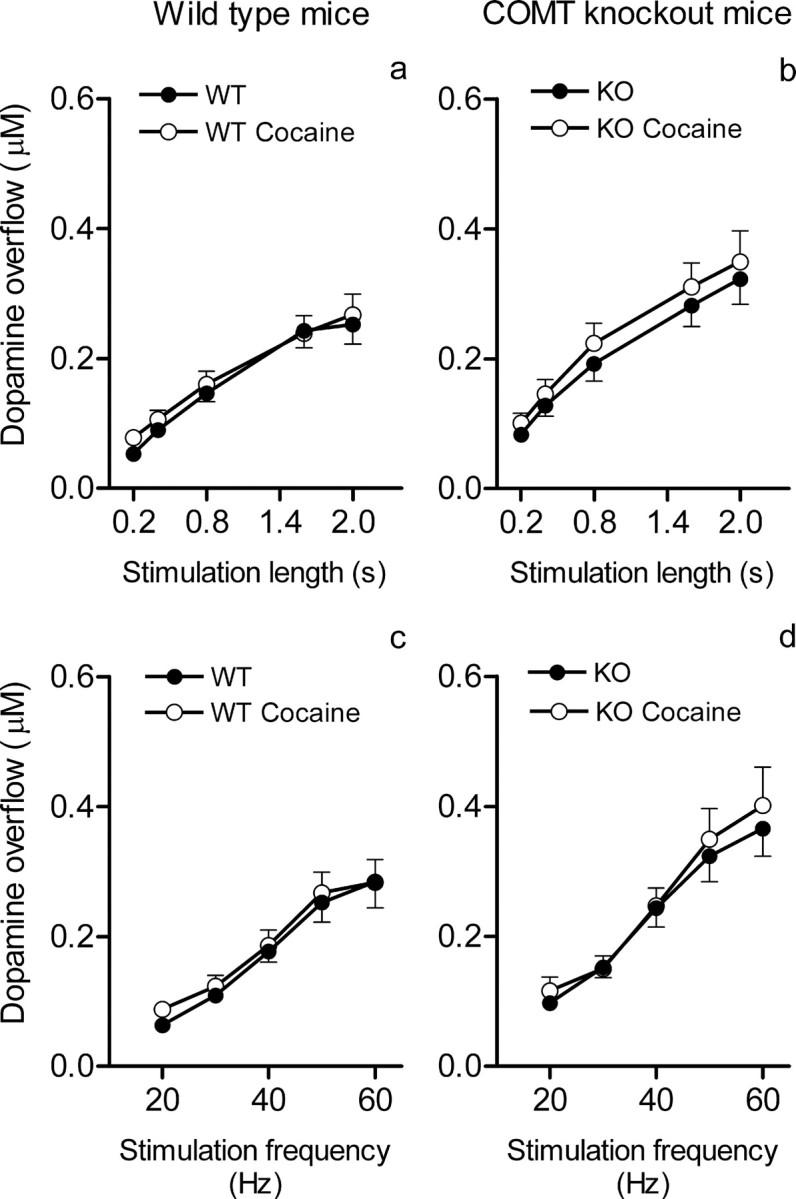
a–d, The effects of cocaine (20 mg/kg) on the peak dopamine overflow in the prefrontal cortex in dependence on the length (a, b) and frequency (c, d) of stimulation of the medial forebrain bundle and genotype. a, b, Peak dopamine overflow (mean ± SEM; 7 mice per group in all experiments) in response to increasing lengths (0.2–2 s) of 50 Hz stimulations. c, d, Peak dopamine overflow in response to increasing frequencies (20–60 Hz) of 2 s stimulations. The effects of cocaine on peak dopamine levels did not differ between genotypes (for statistics, see Results).
Discussion
Doubling of the time of elimination of dopamine from extracellular space in prefrontal cortex of COMT KO mice suggests that at least half of the extracellular dopamine is conjugated by COMT in the prefrontal cortex of WT mice. This represent the first quantitative estimation of the role of COMT in elimination of extracellular dopamine using in vivo voltammetry. Our results are consistent with qualitative predictions based on previous findings that reuptake in the prefrontal cortex is not as efficient as in the striatum (Garris et al., 1993; Garris and Wightman, 1994; Sesack et al., 1998; Mundorf et al., 2001). Indeed, our data clearly show that dopamine lingers in extracellular space in the prefrontal cortex 10 times longer than in the caudate nucleus.
Both MAO and COMT are intracellular enzymes; soluble COMT can be found in cytoplasm and nucleus whereas membrane-bound COMT is located in rough endoplasmic reticulum (Ulmanen et al., 1997). Cellular localization of COMT in the brain is still poorly characterized. Based on a few immunohistochemical studies (Kaplan et al., 1981; Karhunen et al., 1995) and studies assaying COMT activity (Silberstein et al., 1972; Garbarg et al., 1975; Hansson and Sellström, 1983; Spatz et al., 1986), COMT appears to be located in both glia and neurons. Previous COMT mRNA distribution studies suggest neuronal rather than glial localization in rats and humans (Matsumoto et al., 2003). Destruction of dopamine neurons by toxins, does not result in loss of striatal COMT activity, suggesting that nigrostriatal dopamine neurons do not contain significant amounts of COMT (Rivett et al., 1983; Kaakkola et al., 1987).
Assuming intracellular localization of COMT, dopamine should be first transported to the cells and only after that it can be metabolized by COMT and MAO-B (Trendelenburg, 1988). As a result, the duration of extracellular dopamine transients depends primarily on the efficacy of transportation of dopamine to glial cells or postsynaptic neurons and secondarily on the metabolizing ability of the enzymes. The capacity of glial uptake or postsynaptic neuronal uptake is 100 times higher (Vmax, 100 nmol/g/min) than capacity of presynaptic dopamine reuptake, and affinity is very low (Km, 250 μm vs ∼0.3 μm for DAT) (for review, see Trendelenburg, 1988; Männistö et al., 1992). We suggest that a lack of COMT produces intracellular accumulation of dopamine which results in slowing down further uptake-2 and prolongs the lifetime of extracellular dopamine transients. However, there is no direct experimental evidence that lack of COMT causes accumulation of dopamine in glial or postsynaptic cells yet.
Experiments in DAT-deficient mice show that reuptaken rather than newly synthesized dopamine contributes more significantly to the maintenance of vesicular stores of dopamine (Jones et al., 1998). Synthesis of dopamine is a relatively slow process (Besson et al., 1969). Therefore, in areas such as the striatum, dopamine reuptake represents the main mechanism for immediate replenishment of the readily releasable pool (RRP) of neurotransmitter during neuronal bursting.
In the prefrontal cortex, however, the low density of DAT (Scatton et al., 1985) and the apparent elimination of one-half of dopamine transient by uptake-2 and subsequent metabolism by COMT may seriously limit the RRP and ability of local dopaminergic terminals to respond to neuronal bursting which may have profound consequences on prefrontal dopaminergic transmission. The exact role of this apparently “uneconomical” approach to dopamine removal in prefrontal-based adaptive behaviors remains to be determined. However, it is clear that modulation of the COMT activity has a high impact on the function of the prefrontal cortex and thus a considerable therapeutic.
Parameters of dopamine overflow in the mouse prefrontal cortex obtained in the present study are very similar with those described previously in the rat prefrontal cortex where the release of dopamine per pulse of stimulation is approximately one-tenth of that in the caudate nucleus (Garris et al., 1993; Garris and Wightman, 1994).
We used cocaine to evaluate the contribution of reuptake in WT and COMT KO mice. We expected that cocaine would increase peak dopamine levels in both genotypes because extracellular dopamine levels after stimulation are a result of two opposing processes: release and reuptake (Wightman et al., 1988). Cocaine increases evoked dopamine levels in striatum (Greco and Garris, 2003). However, it did not affect the amplitude of the dopamine transient in either genotype in the prefrontal cortex. One explanation for this finding is the relatively small contribution of reuptake in prefrontal dopamine overflow, which may decrease the effect of cocaine on peak amplitudes to undetectable levels. Another possible explanation is a smaller RRP caused by a lower reuptake and subsequent decrease of the release per pulse of stimulation. We tested this hypothesis with the random walk model, which describes the dynamics of stimulated dopamine release and reuptake (Schmitz et al., 2001) and is incorporated in the Mini Analysis Program (Synaptosoft, Decatur, GA). Modeling showed that cocaine significantly decreased release per pulse of stimulation (from 0.013 ± 0.001 to 0.009 ± 0.001 μm) and increased Km (from 0.4 ± 0.05 to 3.54 ± 0.51 μm). However, because the model was created for analysis of dopamine dynamics after single-pulse stimulation (vs trains of pulses in our experiments), and the exact nature of uptake-2 in the prefrontal cortex is unknown, these results should be taken with caution.
Our data show that the effects of cocaine on the prefrontal cortex may be determined entirely by the increased time of presence of neurotransmitter in the extracellular space after transient release, an indication of the importance of volume transmission in the mechanism of action of cocaine in the prefrontal cortex, particularly in cocaine relapse (McFarland and Kalivas, 2001; Rebec and Sun, 2005).
In contrast to the effect of cocaine, lack of COMT resulted in an increase of the peak amplitudes of dopamine overflow (20–25%) in the prefrontal cortex. Inactivation of dopamine by COMT would decrease the amount of dopamine outside the neurons, and subsequently limit availability of dopamine for reuptake. This, in turn, would gradually decrease dopamine accumulation in the RRP and the amount of dopamine released per pulse. According to this hypothesis, lack of COMT in COMT KO mice conserves dopamine for reuptake by small amounts of DAT and permits accumulation of dopamine in the RRP. This may contribute to the moderately enhanced evoked dopamine release observed in the prefrontal cortex of the COMT KO mice.
The role of COMT in dopamine metabolism was studied in the rat striatum using COMT inhibitors. Tolcapone, a brain penetrating inhibitor, did not change basal dopamine levels, but increased dopamine releasing activity of l-dihydroxyphenylalanine, dopamine uptake blockers, and clorgyline (for review, see Männistö and Kaakkola, 1999). Striatal and prefrontal areas have not been compared directly, but in a single study tolcapone enhanced the effects of clozapine and K+-depolarization on dopamine release in the rat prefrontal cortex (Tunbridge et al., 2004). This increase appears to be consistent with the finding that a low COMT activity augmented the effect of olanzapine on working memory in schizophrenic patients (Bertolino et al., 2004). The importance of COMT in modulating cortical dopaminergic transmission is also highlighted in previous studies of proline dehydrogenase-deficient mice. These mice, are more sensitive than their wild-type littermates to the additive effect of amphetamine on dopamine release in the prefrontal cortex, but not in the striatum (Paterlini et al., 2005). As a result, transcriptional upregulation of COMT in prefrontal cortex is used as a homeostatic response, to control cortical dopaminergic hypersensitivity and attenuate a number of dopamine-related behavioral deficits. The impact of COMT variation on prefrontal cortical function and activation has been described in humans, in both normal individuals and in patients suffering from dopamine-related psychiatric disorders. A significant relationship between Val158/108Met genotype and the executive function has been seen in healthy individuals, but the importance of these findings in psychiatric illnesses remains unclear because genetic association studies have not been conclusive and interaction of multiple gene effects have seldom been taken into account (Barnett et al., 2007).
In summary, our real-time analysis of evoked dopamine overflow showed that, in the absence of COMT, removal of dopamine is twofold slower in the prefrontal cortex, but not in the caudate nucleus, indicating that 50% of the dopamine elimination results from COMT activity. In addition, we showed that COMT KO mice have a consistent 20–25% increase in evoked dopamine release in the prefrontal cortex. Finally, we showed that cocaine modulated dopaminergic neurotransmission in the prefrontal cortex similarly in both genotypes by prolonging, three to four times, dopamine elimination from extracellular space. Paradoxically, this occurred without an increase in the peak levels of evoked dopamine release. Our results show that COMT plays a key role in regulation of dopamine neurotransmission in the prefrontal cortex and provide a quantitative neurochemical foundation for differences in cognitive performance resulting from Comt gene polymorphism as well as for clinical findings, which may associate COMT activity with a number of psychiatric disorders.
Footnotes
This work was supported by the Sigrid Juselius Foundation, Helsinki University Research Funds, Academy of Finland Grants 210758/2004 and 117881/2006, and Tekes Grant 221853 (P.T.M.).
References
- Barnett JH, Jones PB, Robbins TW, Muller U. Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: a meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol Psychiatry. 2007;12:502–509. doi: 10.1038/sj.mp.4001973. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Caforio G, Blasi G, De Candia M, Latorre V, Petruzzella V, Altamura M, Nappi G, Papa S, Callicott JH, Mattay VS, Bellomo A, Scarabino T, Weinberger DR, Nardini M. Interaction of COMT Val108/158 Met genotype and olanzapine treatment on prefrontal cortical function in patients with schizophrenia. Am J Psychiatry. 2004;161:1798–1805. doi: 10.1176/ajp.161.10.1798. [DOI] [PubMed] [Google Scholar]
- Besson MJ, Cheramy A, Feltz P, Glowinski J. Release of newly synthesized dopamine from dopamine-containing terminals in the striatum of the rat. Proc Natl Acad Sci USA. 1969;62:741–748. doi: 10.1073/pnas.62.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudikova B, Szumlanski C, Maidak B, Weinshilboum R. Human liver catechol-O-methyltransferase pharmacogenetics. Clin Pharmacol Ther. 1990;48:381–389. doi: 10.1038/clpt.1990.166. [DOI] [PubMed] [Google Scholar]
- Carboni E, Silvagni A. Dopamine reuptake by norepinephrine neurons: exception or rule? Crit Rev Neurobiol. 2004;16:121–128. doi: 10.1615/critrevneurobiol.v16.i12.130. [DOI] [PubMed] [Google Scholar]
- Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, Kolachana BS, Hyde TM, Herman MM, Apud J, Egan MF, Kleinman JE, Weinberger DR. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMille MM, Kidd JR, Ruggeri V, Palmatier MA, Goldman D, Odunsi A, Okonofua F, Grigorenko E, Schulz LO, Bonne-Tamir B, Lu RB, Parnas J, Pakstis AJ, Kidd KK. Population variation in linkage disequilibrium across the COMT gene considering promoter region and coding region variation. Hum Genet. 2002;111:521–537. doi: 10.1007/s00439-002-0809-0. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbarg M, Baudry M, Benda P, Schwartz JC. Simultaneous presence of histamine-N-methyltransferase and catechol-O-methyltransferase in neuronal and glial cells in culture. Brain Res. 1975;83:538–541. doi: 10.1016/0006-8993(75)90850-1. [DOI] [PubMed] [Google Scholar]
- Garris PA, Wightman RM. Different kinetics govern dopaminergic transmission in the amygdala, prefrontal cortex, and striatum: an in vivo voltammetric study. J Neurosci. 1994;14:442–450. doi: 10.1523/JNEUROSCI.14-01-00442.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris PA, Wightman RM. Distinct pharmacological regulation of evoked dopamine efflux in the amygdala and striatum of the rat in vivo. Synapse. 1995;20:269–279. doi: 10.1002/syn.890200311. [DOI] [PubMed] [Google Scholar]
- Garris PA, Collins LB, Jones SR, Wightman RM. Evoked extracellular dopamine in vivo in the medial prefrontal cortex. J Neurochem. 1993;61:637–647. doi: 10.1111/j.1471-4159.1993.tb02168.x. [DOI] [PubMed] [Google Scholar]
- Gerhardt GA, Oke AF, Nagy G, Moghaddam B, Adams RN. Nafion-coated electrodes with high selectivity for CNS electrochemistry. Brain Res. 1984;290:390–395. doi: 10.1016/0006-8993(84)90963-6. [DOI] [PubMed] [Google Scholar]
- Gogos JA, Morgan M, Luine V, Santha M, Ogawa S, Pfaff D, Karayiorgou M. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc Natl Acad Sci USA. 1998;95:9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, Kwon H, Jin S, Jo B, Antonarakis SE, Morris MA, Reiss AL. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci. 2005;8:1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- Greco PG, Garris PA. In vivo interaction of cocaine with the dopamine transporter as measured by voltammetry. Eur J Pharmacol. 2003;479:117–125. doi: 10.1016/j.ejphar.2003.08.062. [DOI] [PubMed] [Google Scholar]
- Hansson E, Sellström A. MAO COMT, and GABA-T activities in primary astroglial cultures. J Neurochem. 1983;40:220–225. doi: 10.1111/j.1471-4159.1983.tb12674.x. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakkola S, Männistö PT, Nissinen E. Striatal membrane-bound and soluble catechol-O-methyl-transferase after selective neuronal lesions in the rat. J Neural Transm. 1987;69:221–228. doi: 10.1007/BF01244343. [DOI] [PubMed] [Google Scholar]
- Kaplan GP, Hartman BK, Creveling CR. Immunohistochemical localization of catechol-O-methyltransferase in circumventricular organs of the rat: potential variations in the blood–brain barrier to native catechols. Brain Res. 1981;229:323–335. doi: 10.1016/0006-8993(81)90997-5. [DOI] [PubMed] [Google Scholar]
- Karhunen T, Tilgmann C, Ulmanen I, Panula P. Catechol-O-methyltransferase (COMT) in rat brain: immunoelectron microscopic study with an antiserum against rat recombinant COMT protein. Neurosci Lett. 1995;187:57–60. doi: 10.1016/0304-3940(95)11337-v. [DOI] [PubMed] [Google Scholar]
- Karoum F, Chrapusta SJ, Egan MF. 3-Methoxytyramine is the major metabolite of released dopamine in the rat frontal cortex: reassessment of the effects of antipsychotics on the dynamics of dopamine release and metabolism in the frontal cortex, nucleus accumbens, and striatum by a simple two pool model. J Neurochem. 1994;63:972–979. doi: 10.1046/j.1471-4159.1994.63030972.x. [DOI] [PubMed] [Google Scholar]
- Lachman HM, Nolan KA, Mohr P, Saito T, Volavka J. Association between catechol O-methyltransferase genotype and violence in schizophrenia and schizoaffective disorder. Am J Psychiatry. 1998;155:835–837. doi: 10.1176/ajp.155.6.835. [DOI] [PubMed] [Google Scholar]
- Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Männistö PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51:593–628. [PubMed] [Google Scholar]
- Männistö PT, Ulmanen I, Lundström K, Taskinen J, Tenhunen J, Tilgmann C, Kaakkola S. Characteristics of catechol O-methyl-transferase (COMT) and properties of selective COMT inhibitors. Prog Drug Res. 1992;39:291–350. doi: 10.1007/978-3-0348-7144-0_9. [DOI] [PubMed] [Google Scholar]
- Mata I, Arranz MJ, Staddon S, Lopez-Ilundain JM, Tabares-Seisdedos R, Murray RM. The high-activity Val allele of the catechol-O-methyltransferase gene predicts greater cognitive deterioration in patients with psychosis. Psychiatr Genet. 2006;16:213–216. doi: 10.1097/01.ypg.0000218626.26622.a2. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Weickert CS, Akil M, Lipska BK, Hyde TM, Herman MM, Kleinman JE, Weinberger DR. Catechol O-methyltransferase mRNA expression in human and rat brain: evidence for a role in cortical neuronal function. Neuroscience. 2003;116:127–137. doi: 10.1016/s0306-4522(02)00556-0. [DOI] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21:8655–8663. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo MR, Bowes L, Clark TG, Flint J. Lack of association of the COMT (Val158/108 Met) gene and schizophrenia: a meta-analysis of case-control studies. Mol Psychiatry. 2005;10:765–770. doi: 10.1038/sj.mp.4001664. [DOI] [PubMed] [Google Scholar]
- Mundorf ML, Joseph JD, Austin CM, Caron MG, Wightman RM. Catecholamine release and uptake in the mouse prefrontal cortex. J Neurochem. 2001;79:130–142. doi: 10.1046/j.1471-4159.2001.00554.x. [DOI] [PubMed] [Google Scholar]
- Paterlini M, Zakharenko SS, Lai WS, Qin J, Zhang H, Mukai J, Westphal KG, Olivier B, Sulzer D, Pavlidis P, Siegelbaum SA, Karayiorgou M, Gogos JA. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2000. [Google Scholar]
- Phillips PE, Wightman RM. Critical guidelines for validation of the selectivity of in-vivo chemical microsensor. Trends Analyt Chem. 2003;22:509–514. [Google Scholar]
- Raux G, Bumsel E, Hecketsweiler B, van Amelsvoort T, Zinkstok J, Manouvrier-Hanu S, Fantini C, Breviere GM, Di Rosa G, Pustorino G, Vogels A, Swillen A, Legallic S, Bou J, Opolczynski G, Drouin-Garraud V, Lemarchand M, Philip N, Gerard-Desplanches A, Carlier M, et al. Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum Mol Genet. 2007;16:83–91. doi: 10.1093/hmg/ddl443. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Sun W. Neuronal substrates of relapse to cocaine-seeking behavior: role of prefrontal cortex. J Exp Anal Behav. 2005;84:653–666. doi: 10.1901/jeab.2005.105-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivett AJ, Francis A, Roth JA. Distinct cellular localization of membrane-bound and soluble forms of catechol-O-methyltransferase in brain. J Neurochem. 1983;40:215–219. doi: 10.1111/j.1471-4159.1983.tb12673.x. [DOI] [PubMed] [Google Scholar]
- Scatton B, Dubois A, Dubocovich ML, Zahniser NR, Fage D. Quantitative autoradiography of 3H-nomifensine binding sites in rat brain. Life Sci. 1985;36:815–822. doi: 10.1016/0024-3205(85)90204-8. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Hawrylak VA, Matus C, Guido MA, Levey AI. Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci. 1998;18:2697–2708. doi: 10.1523/JNEUROSCI.18-07-02697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein SD, Shein HM, Berv KR. Catechol-O-methyl transferase and monoamine oxidase activity in cultured rodent astrocytoma cells. Brain Res. 1972;41:245–248. doi: 10.1016/0006-8993(72)90638-5. [DOI] [PubMed] [Google Scholar]
- Spatz M, Kaneda N, Sumi C, Nagatsu I, Creveling CR, Nagatsu T. The presence of catechol-O-methyltransferase activity in separately cultured cerebromicrovascular endothelial and smooth muscle cells. Brain Res. 1986;381:363–367. doi: 10.1016/0006-8993(86)90090-9. [DOI] [PubMed] [Google Scholar]
- Stamford JA, Kruk ZL, Millar J. Stimulated limbic and striatal dopamine release measured by fast cyclic voltammetry: anatomical, electrochemical, and pharmacological characterisation. Brain Res. 1988;454:282–288. doi: 10.1016/0006-8993(88)90828-1. [DOI] [PubMed] [Google Scholar]
- Trendelenburg U. The extraneuronal uptake and metabolism of catecholamines. In: Trendelenburg U, Weiner N, editors. Handbook of experimental pharmacology. Berlin: Springer; 1988. pp. 279–319. [Google Scholar]
- Tunbridge EM, Bannerman DM, Sharp T, Harrison PJ. Catechol-O-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J Neurosci. 2004;24:5331–5335. doi: 10.1523/JNEUROSCI.1124-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-O-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2006;60:141–151. doi: 10.1016/j.biopsych.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Ulmanen I, Peränen J, Tenhunen J, Tilgmann C, Karhunen T, Panula P, Bernasconi L, Aubry JP, Lundström K. Expression and intracellular localization of catechol O-methyltransferase in transfected mammalian cells. Eur J Biochem. 1997;243:452–459. doi: 10.1111/j.1432-1033.1997.0452a.x. [DOI] [PubMed] [Google Scholar]
- Walker QD, Ray R, Kuhn CM. Sex differences in neurochemical effects of dopaminergic drugs in rat striatum. Neuropsychopharmacology. 2006;31:1193–1202. doi: 10.1038/sj.npp.1300915. [DOI] [PubMed] [Google Scholar]
- Weinshilboum RM, Raymond FA. Inheritance of low erythrocyte catechol-o-methyltransferase activity in man. Am J Hum Genet. 1977;29:125–135. [PMC free article] [PubMed] [Google Scholar]
- Westerink BH, Spaan SJ. Simultaneous determination of the formation rate of dopamine and its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) in various rat brain areas. Brain Res. 1982;252:239–245. doi: 10.1016/0006-8993(82)90391-2. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, May LJ. Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience. 1988;25:513–523. doi: 10.1016/0306-4522(88)90255-2. [DOI] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Kuhar MJ, Carroll FI, Garris PA. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J Neurosci. 2001;21:6338–6347. doi: 10.1523/JNEUROSCI.21-16-06338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]