

Abstract
Kainate-selective ionotropic glutamate receptors (GluRs) require external Na+ and Cl− as well as the neurotransmitter l-glutamate for activation. Although, external anions and cations apparently coactivate kainate receptors (KARs) in an identical manner, it has yet to be established how ions of opposite charge achieve this. An additional complication is that KARs are subject to other forms of cation modulation via extracellular acidification (i.e., protons) and divalent ions. Consequently, other cation species may compete with Na+ to regulate the time KARs remain in the open state. Here we designed experiments to unravel how external ions regulate GluR6 KARs. We show that GluR6 kinetics are unaffected by alterations in physiological pH but that divalent and alkali metal ions compete to determine the time course of KAR channel activity. Additionally, Na+ and Cl− ions coactivate GluR6 receptors by establishing a dipole, accounting for their common effect on KARs. Using charged amino acids as tethered ions, we further demonstrate that the docking order is fixed with cations binding first, followed by anions. Together, our findings identify the dipole as a novel gating feature that couples neurotransmitter binding to KAR activation.
Keywords: glutamate receptor, cation, anion, desensitization, gating, proton
Introduction
Numerous voltage- and ligand-gated ion channels have evolved allosteric binding sites to detect ions in the extracellular and intracellular milieu of the CNS. Often ion binding is a critical step in the gating process. For example, external protons activate a family of acid-sensing ion channels (Waldmann et al., 1997; Waldmann and Lazdunski, 1998), whereas cytosolic Ca2+ activates K+ channels in conjunction with membrane depolarization (Magleby, 2003). We have shown recently that kainate receptors (KARs) are unresponsive when all anions and cations are removed from the extracellular milieu (Wong et al., 2006), providing a mechanism for previous reports showing that KARs are sensitive to external ion substitutions (Bowie, 2002; Paternain et al., 2003). This observation cannot be explained by the failure of neurotransmitter binding; rather, external ions couple agonist binding to channel activation (Wong et al., 2006). The nature of the ion-binding site(s) remains to be fully elucidated; however, recent structural work argues for separate anion and cation binding sites (Plested and Mayer, 2007). However, previous ion-substitution experiments have shown that anions and cations regulate KARs in an apparently identical manner (Bowie, 2002; Paternain et al., 2003), suggesting that cations and anions may still regulate KARs through discrete, electrostatically coupled sites or via a dipole interaction (Bowie, 2002; Wong et al., 2006). Interestingly, closely related AMPA ionotropic glutamate receptors (iGluRs) gate normally when all external ions are removed (Wong et al., 2006), suggesting that amino acid residues critical to ion-dependent gating are unique to KARs.
The ion dependence of GluR6 KARs can be abolished by replacement of Met770 residue with the Lys found in the homologous position of AMPA receptors (AMPARs) (Paternain et al., 2003; Wong et al., 2006), although the mechanism by which it affects KAR function has not yet been resolved. Because Met770 is located outside the agonist-binding pocket (Mayer, 2005; Nanao et al., 2005), it has been speculated to participate in forming a novel ion-binding site on KARs (Paternain et al., 2003; Wong et al., 2006). In this case, the positively-charged Lys residue would render KARs insensitive to external ions through electrostatic repulsion much like a surrogate cation tethered within the ion-binding site(s). However, it remains possible that the Met770 residue is downstream of the ion-binding site(s) and is critical for conveying conformational changes in the agonist-binding domain (ABD) to the pore. KARs are also subject to regulation by other extracellular cations such as Ca2+ and Mg2+ (Ferrer-Montiel et al., 1996) as well as H+ (Mott et al., 2003). Although, H+ and divalent ions are thought to regulate KARs via mutually exclusive sites, the possibility of crosstalk between any of these ion-dependent mechanisms has not been examined.
Here, we designed experiments to resolve two outstanding issues. What is the mechanism by which external Na+ and Cl− coactivate KARs? And do other cations such as H+, Ca2+, or Mg2+ compete with extracellular Na+ as coactivators of KARs? Using GluR6M770K to disrupt ion effects, we show that external divalent ions, and not H+, substitute for Na+ in coactivating KARs. Furthermore, tethering a positive charge to the 770 site completely abolishes both cation and anion effects, demonstrating that Na+ and Cl− coactivate KARs via a dipole mechanism.
Materials and Methods
Cell culture and transfection.
tsA201 cells were transiently cotransfected with cDNA encoding wild-type (wt) or mutant GluR6(Q) KAR subunits and enhanced green fluorescent protein (eGFPS65T) as described previously (Bowie and Lange, 2002; Fay and Bowie, 2006). After transfection for 8–10 h using the calcium phosphate precipitation method, cells were washed with divalent-containing PBS and maintained in fresh medium. Electrophysiological recordings were performed 24–48 h later.
Electrophysiology.
All experiments were performed on outside-out patches excised from transfected cells, and agonist solutions were rapidly applied using a piezo-stack-driven perfusion system as described previously (Bowie and Lange, 2002). Solution exchange time was determined routinely at the end of each experiment by measuring the liquid junction current (10–90% rise time of 25–50 μs). All recordings were performed with an Axopatch 200B amplifier (Invitrogen, Palo Alto, CA) using thin-walled borosilicate glass pipettes (4–6 MΩ) coated with dental wax. Series resistances (7–12 MΩ) were compensated by 95% in all experiments. Current records were filtered at 10 kHz and digitized at 50–100 kHz, and, in all experiments, the reference electrode was connected to the bath via an agar bridge of 3 m KCl. Most recordings were performed at −20 mV to ensure adequate voltage-clamp control of peak currents. Data acquisition was performed using pClamp9 software (Invitrogen) and illustrated using Origin 7 (Microcal, Northampton, MA). All experiments were performed at room temperature.
Solutions.
Unless otherwise stated, external solutions contained the following: 150 mm NaCl, 5 mm HEPES, 0.1 mm CaCl2, 0.1 mm MgCl2, and 2% phenol red, with the osmotic pressure adjusted to 290 mOsm using sucrose. Agonist solutions were prepared by dissolving the Na+ salt of l-glutamate (l-Glu) into the external solution and adjusting the pH with 5N NaOH as necessary. For experiments illustrated in Figure 1, the control solution contained 2 mm Ca2+ and 1 mm Mg2+. pH was adjusted to 7.3 using 5N NaOH with the exception of experiments presented when different monovalent anions and cations were compared (see Figs. 2, 5–7). In this case, the pH was adjusted with the corresponding hydroxide solution (e.g., LiOH for LiCl). The internal solution contained 115 mm NaCl, 10 mm NaF, 5 mm HEPES, 5 mm Na4BAPTA, 0.5 mm CaCl2, 1 mm MgCl2, and 10 mm Na2ATP to chelate endogenous polyamines, pH was adjusted to 7.3 with 5N NaOH, and the osmotic pressure was adjusted to correspond with external solutions.
Figure 1.

Allosteric modulation of GluR6 kainate receptors. a, Membrane currents evoked by 10 mm Glu (Vm of −20 mV; patch 05714p1) in the presence of 150 mm NaCl, 2 mm Ca2+, and 1 mm Mg2+ (Control). Replacement of NaCl with CsCl, NaNO3, the removal of divalent ions, and external acidification (pH 5.5) are shown in the same patch recording. b, Summary plots of the experiment shown in a comparing the effect of ion modulation on the amplitude (black bars) and decay kinetics (white bars) of KARs. Data are expressed as the mean ± SEM; n = 5 patches for each condition. *p < 0.05, significant difference from control (Student's two-tailed, paired t test).
Figure 2.
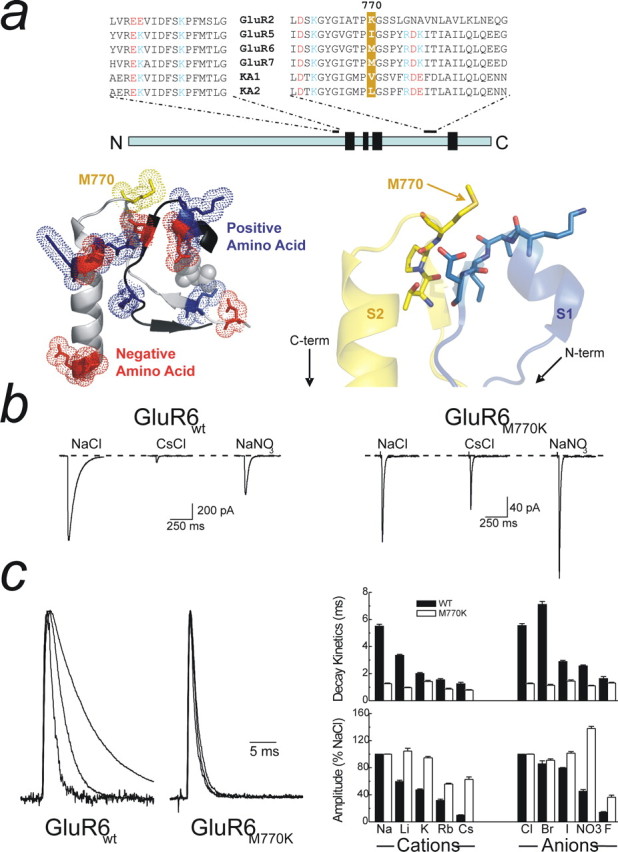
The M770K mutation delineates between channel amplitude and decay kinetics. a, Top, Sequence alignment for selected regions of the S1 and S2 subunits for AMPA and KARs, with the 770 position highlighted in yellow. Positive and negative residues are identified in blue and red, respectively. Note that the S1 and S2 domains are located in disparate parts of the primary sequence. Bottom left, Crystal structure (Protein Data Bank number 1S50) of the region surrounding M770 drawn using PyMol (DeLano Scientific, Palo Alto, CA) showing the interaction between the S1 and S2 domains. Note a nest of positive and negatively charged amino acids exists directly adjacent to M770 that may constitute an cation-binding site. Bottom right, Detailed view of M770 shows the close proximity of a number of exposed carbonyl oxygen atoms that may also represent putative Na+ binding sites. b, The effect of the M770K mutation on KAR modulation by external cations (Cs+) and anions (NO3−; patch 041008p3). c, Left, Normalized raw data traces (from b) showing that M770K abolishes the effect of ions on decay kinetics. The right shows the response profile of GluR6M770K for an extended series of anions and cations. There is no change in decay kinetics for all ions tested (top right), but response amplitude is still modulated (bottom right). Data are mean ± SEM for at least six patches in each ionic condition for both GluR6wt and GluR6M770K.
Figure 5.
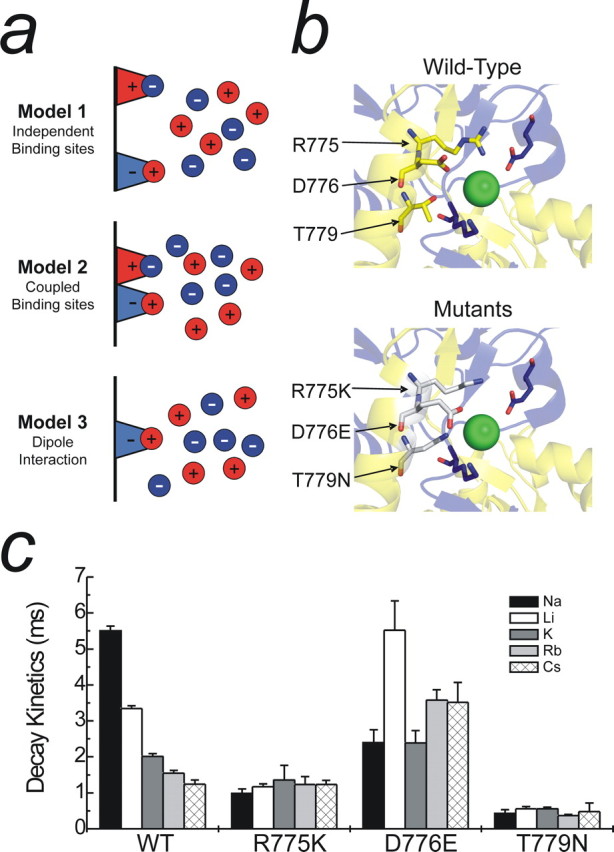
Possible mechanisms for monovalent ion interactions at KARs. a, Schematic showing three distinct models to explain the effect of monovalent anions and cations on GluR6 KARs. b, Top, Crystal structure showing critical amino acid residues that constitute the proposed anion binding site (Protein Data Bank number 2F34). Although only one dimer is shown, the ion is also conjugated by the corresponding amino acids from the adjacent subunit. b, Bottom, Proposed anion binding site containing point mutations (R775K, D776E, and T779N) that interfere with both anion and cation modulation of GluR KARs. Note that each mutation elicits an enlargement of the anion binding pocket through changes in the orientation of each residue. c, The effect of changing external cation species with the various anion binding site mutants. Both R775K and T779N abolish cation modulation, whereas cation modulation is markedly reduced in the D776E mutant. Data are mean ± SEM of at least three patches per mutant in each cation.
Figure 6.

A basic amino acid at the M770 position eliminates both anion and cation effects on KARs. a, Two basic (Lys and Arg; patch 041008p3 and patch 06520p3), a noncharged (Ala; patch 051202p3), and an acidic amino acid mutation (Glu; patch 06411p2) were made at Met770 to assess whether changes in charge or side chain length preferentially affected modulation by anions or cations. The raw data traces (GluR6wt; patch 04116p2) show the effect of external cation substitutions from Na+ to K+ and Cs+. Note that basic amino acid mutations lead to a substantial acceleration of decay kinetics in all ions tested. b, Pooled data for cation (top) and anion (bottom) substitutions with the various GluR6M770 point mutations. Addition of a positively charged amino acid (Lys or Arg) at this position abolishes both cation and anion modulation. However, external ions still modulate KARs with an aliphatic (Ala) or acidic (Glu) amino acid in place of Met770. Data are mean ± SEM of at least five patches for each mutation in each ionic condition.
Figure 7.
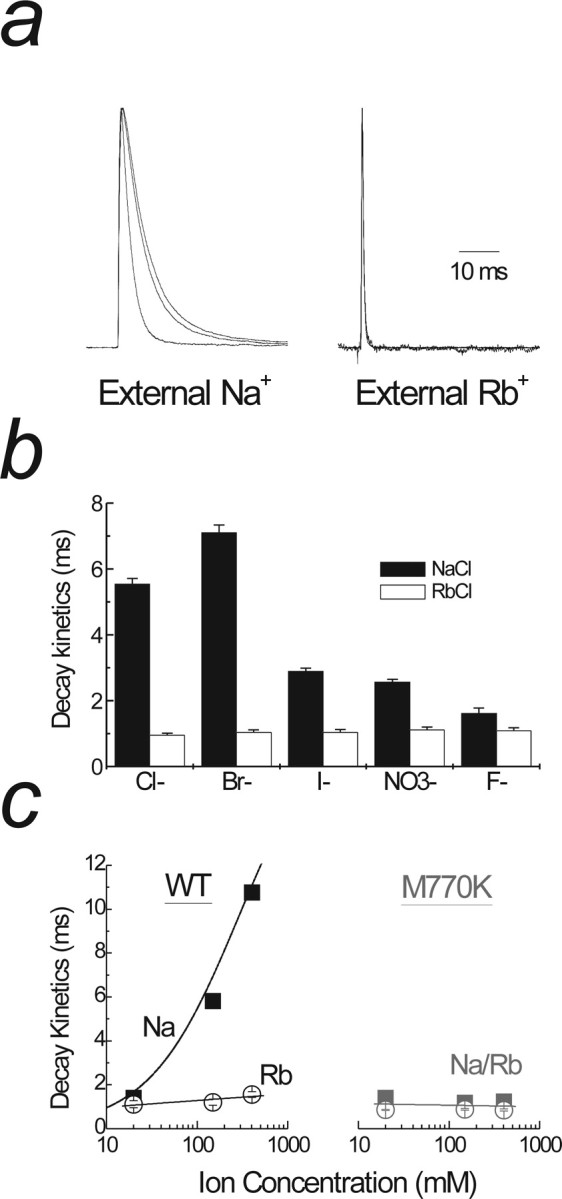
Basic amino acids at the M770 position act as surrogate cations. a, Left, Raw data traces showing anion substitutions in which Na+ is the major cation (patch 04130p2). There is an acceleration of decay kinetics when Br− is replaced with Cl− and NO3−. Right trace shows the same experiment as left but with Rb+ as the major cation (patch 06209p1). Note that anion substitutions do not change decay kinetics in the presence of Rb+. b, An extended series of anion substitutions with either Na+ (black bars) or Rb+ (white bars) as the dominant cation. There is no change in decay kinetics when anions are substituted in Rb+ in contrast to Na+. Data are mean ± SEM for at least six patches in each anion with Na+ or Rb+ as the major cation. c, The effect of changing the external Na+ (filled squares) and Rb+ (open circles) concentrations on the decay kinetics of wild-type GluR6 (left) and GluR6M770K (right) receptors. Note that the decay kinetics GluR6wt matches that of GluR6M770K only when Rb+ ions are used.
For experiments on pH changes (see Fig. 3), external HEPES was replaced with 5 mm 2-(cyclohexylamino)ethanesulphonic acid (for solutions between pH 8 and 9.5) or 5 mm MES (pH range of 4.5–6.2). The pH was adjusted as required with either 5N NaOH or 5N HCl. For agonist solutions, the Na+ salt of l-glutamate was dissolved in the corresponding pH solution. In experiments shown in Figure 8, solutions lacking external NaCl contained 0.1 mm CaCl2 and 0.1 mm MgCl2 to improve patch stability, 290 mm sucrose to maintain the osmotic pressure, and 5 mm Tris as a pH buffer. The pH was adjusted to 7.4 using 5N HCl. For agonist solutions, the free acid of l-glutamate was dissolved in NaCl-free solution, and the pH was adjusted using 2.5 m Tris (Wong et al., 2006).
Figure 3.

pH modulation of GluR6wt and GluR6M770K. a, Raw data showing the effect of lowering pH (i.e., increasing H+ concentration) on wild-type GluR6 receptors (patch 04615p3). b, The effect of lowering pH on GluR6M770K (patch 06525p1). External acidification leads to a decrease in amplitude but no change in the decay kinetics of both GluR6wt and GluR6M770K. c, Pooled data for an extended pH range for GluR6wt (black squares) and GluR6M770K (white circles). Dose–response curves were best fitted with a single binding site isotherm, with Imax constrained to 100%. The IC50 was pH 6 for GluR6wt, and pH 6.8 was for GluR6M770K. Hill slopes (nH) were 1 and 0.7, respectively. d, The effect of pH on channel decay kinetics. Data were fitted with a linear fit, and there was no significant change in decay kinetics with decreasing pH in both wild-type (black squares) and mutant (white circles) receptors. Data are mean ± SEM of at least six patches for each pH.
Figure 8.

A positively charged residue at the M770 position abolishes the absolute requirement for ions. a, Superimposed family of membrane currents evoked by 1 mm l-Glu between −100 and +110 mV in Na+-free solutions. KARs with Met770 substituted with either an aliphatic (M770A; patch 06724p3) or an acidic (M770D; patch 06727p2) amino acid still have an absolute requirement for external ions. In contrast, substitution of Met770 with a positively charged amino acid (Arg; patch 06713p2) results in an outward current in Na+-free solutions (middle). b, Mean current–voltage plots from at least three experiments in physiological Na+ (150 mm; open triangles) and Na+-free (0 mm; filled circles) solution, demonstrating that GluR6M770R is functional, whereas GluR6M770A and GluR6M770D are nonfunctional.
Site-directed mutagenesis.
Point mutations of the Met770 site was performed using the Stratagene (La Jolla, CA) Quickchange II XL site-directed mutagenesis kit using PfuUltra DNA polymerase and custom-made primers (Alpha DNA, Montreal, Quebec, Canada). Mutant cDNAs were amplified using XL10-Gold ultra-competent cells (Stratagene), purified with the QIAprep spin miniprep kit (Qiagen, Mississauga, Ontario, Canada), initially identified by restriction digest with BstBI/AfeI (for D528K mutation), AvaI (for M770R mutation), KpnI (M770D and M770A mutation), BstEII (for R775K mutation), NruI/EcoRV (for D776E mutation), and HphI (for T779N mutation; New England Biolabs, Beverly, MA) and confirmed by automated DNA sequencing (McGill University and Genome Quebec Innovation Center, Montreal, Quebec, Canada). To obtain large quantities of cDNA, mutant cDNA was amplified in bacterial culture (Top10 cells; Invitrogen) and purified using Qiafilter Maxiprep kits (Qiagen).
Data analysis.
The concentration–response curve to external pH (see Fig. 3c) was fit with the equation IpH = Imax/1 + ([H+]/IC50)N with Imax constrained to 100%. Imax represents the point at which H+ has no effect on the response amplitude, IpH represents the observed amplitude at any concentration of H+, and N is the slope. IC50 is the H+ concentration that blocks 50% of the observed response. Inhibition curves for increasing [Ca2+]o shown in Figure 4c were fit with a single binding site isotherm modified to account for incomplete block, which had the form ICa = (1 − Iss)/1 + ([Ca2+]/IC50)N + Iss, where Iss is the amplitude of the response that failed to be blocked at saturating Ca2+ concentrations.
Figure 4.

Ca2+ modulation of GluR6wt and GluR6M770K. a, Raw data showing the effect of increasing external Ca2+ on wild-type GluR6 KARs (patch 050920p1). b, The effect of Ca2+ on GluR6M770K (patch 06601p1). Insets to a and b show normalized raw data traces emphasizing an acceleration of decay kinetics with increasing [Ca2+]o in GluR6wt (inset, a) but not with GluR6M770K (inset, b). c, Dose–inhibition curve for Ca2+ for wild-type and M770K mutant KARs. Data were fit with a single binding site isotherm with a steady-state amplitude (Io, 22% of peak), with Imax constrained to 100%. The IC50 for Ca2+ was 1 mm for GluR6wt and 2 mm for GluR6M770K. The Hill slopes (nH) were 1.3 and 1.0, respectively. d, The acceleration in decay kinetics observed in GluR6wt with increasing Ca2+ concentration is abolished by M770K. Data are mean ± SEM of at least five patches at each calcium concentration.
When required, data were tested for statistical significance using a two-tailed, paired Student's t test. A p value of ≤0.05 was considered statistically significant.
Results
KARs are modulated by external pH and monovalent and divalent ions
The sensitivity of KARs to monovalent and divalent ions and pH was studied by varying the composition of the external solution and examining its effect on response amplitude and decay kinetics (Fig. 1a,b). Using an ultrafast agonist perfusion system, application of l-Glu (10 mm) rapidly activated GluR6 KARs in control conditions (150 mm NaCl, 2 mm Ca2+, and 1 mm Mg2+), producing a peak response that desensitized by 99 ± 0.3% in the continued presence of the agonist (Fig. 1a,b). The rate of onset of receptor desensitization was best fit with a double-exponential function with fast and slow time constants (and relative areas) of 3.96 ± 0.17 ms (97.4 ± 1.2%) and 45 ± 23 ms (2.6 ± 1.2%), respectively (Fig. 1a,b). As we reported previously (Bowie, 2002; Bowie and Lange, 2002; Wong et al., 2006), substitution of external Na+ or Cl− with an equimolar concentration of Cs+ or NO3− ions, respectively (Fig. 1a), resulted in a significant (p < 0.05, Student's two-tailed, paired t test) reduction in peak response amplitude (Cs+, 70 ± 3.2%; NO3−, 62 ± 2.2%) that was accompanied by a concomitant acceleration in decay kinetics (Cs+ τfast of 1.09 ± 0.15 ms; NO3− τfast of 2.14 ± 0.16 ms) (Fig. 1a,b). In contrast, changes to external divalent ions or pH only affected the peak response amplitude with little effect on decay kinetics (Fig. 1a,b). For example, removal of Ca2+ and Mg2+ from the external solution resulted in a 60 ± 10% increase in peak response amplitude, whereas lowering pH (i.e., increasing H+) from 7.4 to 5.5 decreased the peak response amplitude by 82 ± 1.2% (Fig. 1a,b). Decay kinetics of GluR6 receptors were similar before (see above) and after the removal of external divalents (τfast of 4.57 ± 0.14 ms) or after changes to pH 5.5 (τfast of 4.30 ± 0.15 ms) (Fig. 1a,b) as described previously (Mott et al., 2003).
Monovalent anions and cations coactivate GluR6 KARs with the neurotransmitter l-Glu by acting on a binding site(s) that is distinct from the ABD (Fig. 2a) (Paternain et al., 2003; Wong et al., 2006). In this context, two residues, Met770 and Asp528, were originally proposed to be critical for cation modulation of GluR6 KARs (Paternain et al., 2003). Although GluR6M770K receptors are indeed cation insensitive, modulation is essentially unaffected in GluR6D528K receptors (supplemental Fig. 1, available at www.jneurosci.org as supplemental material); consequently, we focused our analysis on the Met770 residue. We have shown previously that Met770, which also lies outside the ABD (Fig. 2a, bottom left), acts as a molecular switch to confer ion-independent gating behavior on KARs (Wong et al., 2006). Replacement of Met770 with the equivalent Lys residue found in AMPARs (Fig. 2a, top) eliminates the effect of external anions and cations on GluR6 KAR decay kinetics (Fig. 2b,c) (Wong et al., 2006). Importantly, external anions and cations continue to modulate the peak response of GluR6M770K receptors (Fig. 2b,c) contrary to the findings of a previous study (Paternain et al., 2003). This observation is not wholly unexpected given that external anions and cations also modulate peak responses of GluR1 AMPARs (Bowie, 2002). From a molecular standpoint, this finding suggests that the peak response amplitude is determined by amino acid residues common to both AMPARs and KARs, with only a modest contribution from the 770 site.
Whether the 770 residue represents the actual cation-binding site or is downstream of it has yet to be resolved. Interestingly, the electrostatic environment of the GluR6 crystal structure reveals that Met770 is juxtaposed between clusters of positively and negatively charged residues (Fig. 2a, bottom left), which may provide binding sites for anions and/or cations. Alternatively, close examination of Met770 reveals its close proximity to a number of exposed carbonyl oxygen atoms that, as discussed later, may also provide a binding site(s) for external cations (Fig. 2a, bottom right). We hypothesized previously that occupancy of the ion-binding site(s) affects the stability of neurotransmitter binding, which, in turn, affects decay kinetics (Wong et al., 2006). Thus, different ions stabilize the ABD to different degrees, which accounts for the distinct effects of Cs+ or NO3− compared with Na+ or Cl− on decay kinetics.
The molecular mechanism for the effects of external divalent ions (i.e., Ca2+ or Mg2+) or protons (i.e., pH) (Mott et al., 2003) on KARs is not yet known. Intuitively, it would be expected that all positively charged ions, such as external Ca2+ and Mg2+ or protons, regulate KARs by a similar mechanism as alkali metal ions such as Na+ or Cs+. However, the failure of external divalents and protons to affect KAR decay kinetics differs from our observations with monovalent cations (and anions) (Fig. 1b, right). These experiments, however, did not examine an extensive ion-concentration range. Therefore, it remains to be established whether external divalent ions or protons modulate GluR6wt receptors via the anion/cation binding site(s). As described below, we addressed this issue directly by comparing divalent ion and pH effects on the desensitization properties of GluR6M770K and GluR6wt KARs.
External divalent ions but not protons bind to the anion/cation site
To examine whether external divalent ions or protons regulate KARs by binding to the anion/cation site(s), we examined their effects on GluR6M770K receptors (see Figs. 3, 4). As mentioned above, the decay kinetics of GluR6M770K are unaffected by external anions and cations, unlike GluR6wt (Wong et al., 2006). Consequently, we reasoned that binding to the anion/cation site could be excluded if M770K mutation did not interfere with modulation by external divalents and protons.
Figure 3 summarizes experiments comparing the effect of pH on GluR6wt and GluR6M770K receptors. For wild-type receptors, lowering external pH (i.e., increasing protons) over an extended range elicited a concentration-dependent decrease in peak response amplitude (Fig. 3a,c), with little or no effect on channel decay kinetics (Fig. 3d). Changes in external pH similarly affected the peak response and decay kinetics of GluR6M770K receptors (Fig. 3b–d), demonstrating that external protons regulate GluR6 KARs via a mechanism independent of the anion/cation binding site consistent with mutational analysis of the putative H+ binding site (Mott et al., 2003). Concentration–inhibition relationships in each case were best fit with single binding site isotherms with IC50 values of 0.97 ± 0.08 μm (pH 6, n = 19; filled symbols) and 0.42 ± 0.09 μm (pH 6.6, n = 11; open symbols) for GluR6wt and GluR6M770K, respectively (Fig. 3c). The micromolar affinity of GluR6 KARs for external protons accounts for their lack of effect on the anion/cation site, which binds external alkali metal ions in the millimolar range (EC50 for Na+ of 106 ± 11 mm; n = 5) (Wong et al., 2006). Interestingly, there was a difference in Hill slope (nH) for proton block between GluR6M770K (nH = 0.8 ± 0.1) and GluR6wt (nH = 1.2 ± 0.1) receptors, which may be explained if discrete proton and anion/cation binding sites are allosterically coupled.
Like protons, external Ca2+ ions caused a dose-dependent reduction in the peak response amplitude of GluR6wt and GluR6M770K receptors (Fig. 4a–c). Interestingly, block of KARs by external Ca2+ was incomplete at saturating divalent ion concentrations (Fig. 4c), which most likely reflects the appreciable divalent ion permeation at high concentrations of external Ca2+ (Burnashev et al., 1995). The amplitude of the residual current response (Io) was identical between mutant (Io = 21 ± 5.9%) and wild-type (Io = 22 ± 2.1%) receptors, suggesting that the 770 residue of GluR6 is not critical to ion permeation. Fits of concentration–inhibition curves over an extended range of Ca2+ (100 μm to 30 mm) estimated the IC50 for Ca2+ block of GluR6M770K and GluR6wt receptors to be 2.31 ± 0.92 mm (nH = 1.0 ± 0.3) and 1.12 ± 0.10 mm (nH = 1.4 ± 0.1), respectively (n = 8 for each) (Fig. 4c). This finding demonstrates that KAR affinity for external divalent ions is intermediary in nature between the apparent binding affinities of protons and alkali metal ions. It is therefore possible that external Ca2+ ions bind to the monovalent anion/cation binding site(s). In support of this, decay kinetics of GluR6wt receptors were sensitive to external Ca2+ in a concentration-dependent manner (Fig. 4a, inset, d). For example, desensitization rates of GluR6wt accelerated from 5.03 ± 0.29 ms in 100 μm Ca2+ to 2.26 ± 0.22 ms in 30 mm Ca2+ (Fig. 4d). Importantly, the effect of external Ca2+ ions on decay kinetics was abolished in GluR6M770K receptors (Fig. 4d), demonstrating that divalent ions compete with external monovalent ions for the anion/cation binding site. In this case, the rate of onset of desensitization was 1.13 ± 0.1 ms in 100 μm Ca2+ and 0.93 ± 0.07 ms in 30 mm external Ca2+. Together, our data demonstrate that cation effects on KARs are distinguished by their apparent affinity.
Identifying electrostatic models of the anion/cation binding site(s)
The molecular nature by which anions and cations bind to KARs is not yet known, but three arrangements illustrated in Figure 5a may be considered. The first possibility is that both anions and cations operate through independent binding sites as suggested recently by Plested and Mayer (2007). This mechanism (model 1) would be supported experimentally if (1) GluR6 mutation analysis identified separate anion- and cation-binding sites and if (2) anions and cations were shown not to interact. The second possibility is that KARs possess separate binding sites that are electrostatically coupled. This arrangement (model 2) would be favored if (1) mutation experiments identified separate anion- and cation-binding sites and if (2) anion–cation interactions were established. The third and final possibility is that anions and cations affect receptor function via a dipole in which either an anion or cation sets up an electric field that attracts a counter ion. Although the KAR protein may also contribute to this electric field, for simplicity, we will assume that it is determined solely by the bound ion. A cation is shown bound in Figure 5a to illustrate this point, but, depending on the arrangement, an anion could also be shown. Model 3 would be favored if (1) mutation experiments eliminated both anion and cation effects and if (2) anions and cations were shown to interact.
To distinguish between independent (model 1) and coupled (i.e., models 2 and 3) ion-binding site models, we performed electrophysiological analysis of three GluR6 mutations (R775K, D776E, and T779N) that have been shown recently to affect anion binding to KARs (Plested and Mayer, 2007). Figure 5b (top) shows the molecular structure of the proposed anion binding pocket and how the location of each residue allows interaction with a bound chloride ion (green sphere). Mutation of any of these residues disrupts anion binding (Fig. 5b, bottom), which, importantly, has been proposed not to affect cation binding (Plested and Mayer, 2007). This latter observation led Plested and Mayer (2007) to conclude that KARs possess separate anion and cation binding sites (i.e., model 1). A significant caveat, however, was that Plested and Mayer (2007) limited their investigation to only Na+ and Cs+. Consequently, it remains to be established whether an extended range of cations would behave similarly. In view of this, we compared the effect of all five external cations (i.e., Na+, Li+, K+, Rb+, and Cs+) on GluR6wt and each mutant receptor (Fig. 5c). From these experiments, we observed that the decay kinetics of all mutant receptors to external cations was markedly distinct from GluR6wt (Fig. 5c). This observation demonstrates conclusively that disruption of the anion binding pocket also affects cation binding. Consequently, anions and cations do not affect GluR6 KARs through independent binding sites (i.e., model 1) as proposed by Plested and Mayer (2007).
Met770 is critical in determining ion effects on decay kinetics
To distinguish between models 2 and 3, we performed mutational analysis of the Met770 site by introducing amino acid residues with net positive (i.e., Arg or R, Lys or K) or negative (i.e., Asp or D) charge at physiological pH or with a residue that has no net charge (i.e., Ala or A) (Fig. 6). We reasoned that model 2 (coupled binding site model) would be favored if a single point mutation eliminated the effect of either anions or cations on GluR6 decay kinetics. Alternatively, model 3 (dipole model) would be favored if a single point mutation eliminated both anion and cation effects.
Figure 6 summarizes our findings comparing the desensitization properties of each mutant receptor in different external cations (Fig. 6b, top, Na+, K+, and Cs+) and anions (Fig. 6b, bottom, Cl−, I−, and F−). Introducing basic amino acids, Lys or Arg, at the 770 site imparted two important effects on the kinetic properties of GluR6 KARs. First, different external cations failed to regulate the decay kinetics of GluR6M770K or GluR6M770R receptors (Fig. 6a,b). For example, decay kinetics of GluR6M770R receptors in 150 mm external Na+ (τfast of 0.91 ± 0.10 ms) and Cs+ (τfast of 0.77 ± 0.07 ms) were almost identical, unlike the effects of Na+ (τfast of 5.50 ± 0.13 ms) and Cs+ (τfast of 1.24 ± 0.11 ms) on GluR6wt (Fig. 6a,b, top). Second, unlike GluR6wt receptors, changes in external anions also failed to affect channel kinetics of GluR6M770K or GluR6M770R receptors (Fig. 6a,b). These findings are consistent with the dipole arrangement outlined for model 3 (Fig. 5).
In contrast to the effect of basic amino acids, the introduction of a negatively charged Asp or uncharged Ala residue at the 770 site did not abolish the ability of external cations or anions to regulate GluR6 decay kinetics (Fig. 6a,b). Although the rank order of potency was unchanged for external cations, there was, however, attenuation in extent of modulation of each mutant receptor (Fig. 6b, top). For example, the rate of onset of desensitization accelerated approximately fourfold for GluR6wt channels when external Na+ was replaced by Cs+ (see above). In contrast, decay kinetics for GluR6M770D accelerated modestly when external Cs+ (τfast of 4.34 ± 0.33 ms) replaced Na+ (τfast of 5.81 ± 0.20 ms) (Fig. 6b, top). Similar results were observed with GluR6M770A receptors although to a lesser extent, suggesting that introduction of an Ala residue and particularly the negatively charged Asp at the 770 site may interfere with cation binding. Unlike cations, the effect of external anions on the kinetic properties of mutant receptors was similar to GluR6wt. For example, the rate of onset of GluR6wt desensitization in 150 mm Cl− (τfast of 5.50 ± 0.13 ms) and F− (τfast of 1.61 ± 0.16 ms) was almost identical to decay kinetics of GluR6M770D receptors in identical ionic conditions (Cl− τfast of 5.81 ± 0.20 ms; F− τfast of 1.38 ± 0.10 ms) (Fig. 6b, bottom).
Together, our observations favor the criteria we established previously for a dipole (i.e., model 3) rather than for coupled or independent binding sites (i.e., models 1 or 2) (Fig. 5). This conclusion is supported by several lines of evidence. First, mutation of amino acids that contribute to the anion binding pocket (i.e., R775, D76, or T779) also affect cation modulation of GluR6 KARs (Fig. 5c). Second, a single point mutation (i.e., GluR6M770K or GluR6M770R) is sufficient to render GluR6 KARs completely insensitive to both external anions and cations (Fig. 6b, top). Third, this effect is unique to positively charged residues (i.e., Arg or Lys) and not for negatively charged (i.e., Asp) or uncharged (i.e., Ala) residues. Furthermore, replacement of Met770 with an Asp or Ala residue still permits cation binding, accounting for their continued modulatory effect on GluR6M770D and GluR6M770A receptors. An alternative possibility is that Met770 may be downstream of the cation-binding site(s) and important for coupling it to the gating machinery. As described below, we were able to delineate between these possibilities by investigating anion effects on GluR6wt receptors.
Tethering a positive charge to the 770 site mimics Rb+ binding
In the dipole model, the lack of anion modulation can be explained if Arg or Lys residues at the 770 site interact with anions differently than freely diffusible Na+ ions. This is supported by the observation that GluR6M770K and GluR6M770R receptors desensitize more than fivefold faster (τfast of 1 ms) than GluR6wt (τfast of 5 ms) in 150 mm external NaCl (Fig. 6b, bottom). Alternatively, if the 770 residue is located downstream of the cation-binding site, anions fail to affect channel kinetics because Arg or Lys residues disrupt crosstalk between the ion-binding site and gating machinery.
To delineate between these mechanisms, we studied anion modulation of GluR6wt receptors and chose external Rb+ to match the decay kinetics observed with GluR6M770K and GluR6M770R receptors (Fig. 7). In external Rb+, GluR6wt receptors desensitize at an identical rate (τdecay of 1 ms) (Fig. 2c) to GluR6M770K and GluR6M770R receptors (Fig. 6b). We reasoned that, if Arg or Lys residues act at the ion-binding site, desensitization rates of GluR6wt in external Rb+ will also be anion insensitive. The data shown in Figure 7a compare desensitization rates of GluR6wt receptors in Na+ (left panel) or Rb+ (right panel) salts of 150 mm external Cl−, Br−, and NO3−. As we described previously (Bowie, 2002; Wong et al., 2006), external anions regulate the decay kinetics of GluR6wt receptors in 150 mm external Na+ (Fig. 7b). In contrast, however, the effect of a range of external anions on GluR6wt desensitization rates was abolished in 150 mm Rb+ (Fig. 7b); consistent with our hypothesis that Rb+ binding to wild-type GluR6 receptors mimics the effect of tethering a positive charge (i.e., Arg or Lys) to the 770 position. Importantly, this observation is not attributable to the fact that KAR desensitization has reached its maximal limit in Rb+ ions because GluR6wt receptors decay significantly faster in equimolar Cs+ (Cs+ τ of 0.65 ms; Rb+ τ of 1.22 ms) (Bowie, 2002).
To further demonstrate that Rb+ converts wild-type GluR6 to the phenotype of mutant receptors, we compared their desensitization kinetics in different concentrations of external cations (Fig. 7c). External Na+ has a concentration-dependent effect on the decay kinetics of wild-type GluR6 (Bowie, 2002; Bowie and Lange, 2002; Wong et al., 2006). Interestingly, this effect is abolished when the Met770 residue is replaced by a positively charged Arg (data not shown) or Lys (Fig. 7c). Consistent with our hypothesis, wild-type GluR6 desensitization kinetics were no longer affected by the external cation concentration when Na+ ions were replaced by Rb+ (Fig. 7c). This observation provides another line of evidence to support our central tenet that mutant GluR6 receptors have an identical phenotype to wild-type GluR6 bound by Rb+. This demonstrates unequivocally that Arg or Lys residues act like tethered Rb+ ions and therefore excludes the possibility that the 770 position is downstream of the cation-binding site.
Met770 residue is located within a cation-binding site
In a dipole arrangement, Lys or Arg residues may act as surrogate cations in the cation-binding pocket or tethered positive charges at its entrance, which would repel external cations. To delineate between each of these possibilities, we recorded responses from mutant GluR6 KARs in the absence of external ions (Fig. 8). We reasoned that, if the 770 residue was positioned at the entrance of the cation-binding site, GluR6M770R receptors would fail to gate in the absence of external ions. Alternatively, if the 770 site was located within a cation-binding pocket, GluR6M770R receptors would be responsive because the Arg residue would act as a surrogate cation. In agreement with this, l-Glu elicited outward membrane currents from GluR6M770R channels recorded in external ion-free solutions (Fig. 8, middle column), indicating that the 770 residue constitutes part of a binding pocket for external cations. As reported previously (Wong et al., 2006), GluR6M770K channels behave similarly, suggesting that it is the inclusion of a positive charge at the 770 site that is critical for conferring responsiveness. To strengthen this conclusion, we repeated experiments on GluR6M770A and GluR6M770D receptors in external ion-free conditions (Fig. 8). If the 770 site is part of a cation-binding pocket, tethering of an uncharged Ala or negatively charged Asp residue will not recover functionality. In agreement with this, data shown in Figure 8 reveal that GluR6M770A (Fig. 8, left) and GluR6M770D (Fig. 8, right) receptors fail to gate in the absence of external ions. That is, we did not observe outward membrane currents even at membrane potentials more positive than +100 mV. Together, these observations demonstrate that Met770 forms part of a cation-binding site on GluR6 KARs.
Discussion
Here we demonstrate that monovalent ions coactivate KARs via a dipole mechanism set up by cation binding (Fig. 5, Model 3). This conclusion is supported in several ways. First, a single point mutation is sufficient to abolish both anion and cation effects, ruling out the possibility of mutually exclusive ion-binding sites (Fig. 5a, Model 1). Second, the effect of Rb+ ions is mimicked by tethering a positive charge (i.e., Lys or Arg) to the 770 site, demonstrating that this residue is part of a cation-binding pocket (Fig. 7). Finally, the order of ion binding is fixed with cation binding first, followed by anions (Fig. 8). In summary, the dipole model provides a mechanism to explain how anions and cations exert similar effects on KARs (Bowie, 2002). Our observations also explain why AMPARs continue to gate when all external ions are removed (Wong et al., 2006). The presence of a Lys residue at the equivalent 770 position of AMPARs acts as a tethered cation and therefore underpins their lack of ion dependency.
Discriminating between ion effects on response amplitude and decay kinetics
The original observation identifying monovalent ions as regulators of GluR6 KARs noted that their effect on response amplitude and decay kinetics was apparently concomitant in nature (Bowie, 2002). In principle, peak response amplitude is governed by open-channel probability and/or unitary conductance(s), whereas macroscopic decay kinetics is a function of the stability of the open state(s). Therefore, the apparently causal relationship between ion effects on peak response and decay kinetics could have implied that all of these gating events were coupled through a common anion/cation binding site(s). This possibility, however, was dismissed because anions and cations also regulate peak responses elicited by GluR1 AMPARs (Bowie, 2002; Bowie and Lange, 2002), demonstrating that it is only their effect on decay kinetics that is unique to KARs. This prediction, however, was apparently overturned when Paternain et al. (2003) reported that Met770 of GluR6 KARs, which is absent in the equivalent position on AMPARs, was critical for conveying the effect of external cations on peak KAR responses. Because the authors did not examine decay kinetics, it was not known whether the 770 site was also critical for this property of the channel. However, the study by Paternain et al. (2003) was problematic because all agonist applications were performed in the whole-cell configuration, which is widely accepted to achieve solution exchange rates far slower than the gating properties of KARs (Bowie et al., 2003). Thus, their estimates of peak response amplitude were likely to be inaccurate. In support of this, when identical ion-substitution experiments were repeated in excised patches to achieve faster solution exchange, external cations and anions continued to modulate peak GluR6M770K responses (Wong et al., 2006), contrary to the findings of Paternain et al. (2003). Most notably, the effect of both cations and anions on GluR6 decay kinetics was abolished in GluR6M770K channels (Wong et al., 2006). Together with the present study, these findings suggest that the molecular determinants that govern peak response amplitude are common to both AMPARs and KARs, whereas the dipole that regulates decay kinetics is unique to KARs.
What is the nature of the anion/cation binding site?
Although the identification of monovalent ions as coactivators of iGluRs is a recent observation (Wong et al., 2006), their role in coactivating enzymes has been established for some time (Di Cera, 2006). Recent crystallographical data have directly demonstrated Na+ binding to enzymes such as β-galactosidase and thrombin (Di Cera, 2006) as well as in Na+-ATPases (Meier et al., 2005; Murata et al., 2005) and the bacterial Na+/Cl−-dependent neurotransmitter transporter (Yamashita et al., 2005). Examination of the crystal structures of Na+-dependent enzymes and transporters reveals Na+ binding directly to five or six backbone or side-chain carbonyl oxygen atoms (Pineda et al., 2004; Meier et al., 2005; Murata et al., 2005; Yamashita et al., 2005). The Na+ ion is stabilized in a binding pocket with ion-target distances of <2.5 Å conjugated with water molecules (Harding, 2002; Yamashita et al., 2005), which, in turn, can mediate long-distance interactions up to 15 Å (Pineda et al., 2004). Examination of the GluR6 crystal structure (1S50) (Mayer, 2005) reveals several backbone carbonyls adjacent to Met770, which may form a putative Na+ binding pocket(s) (Fig. 2a). However, the size of these pockets is >3 Å, suggesting that this region may bind cations of different size (Li+, 0.69 Å; Cs+, 1.69 Å) (Hille, 2001). It can also be hypothesized that the lack of charge screening in this region provides a mechanism through which the binding of a cation can attract an ion, in a manner analogous to that observed for Cu2+/Zn2+ superoxide dismutase (SOD) (Getzoff et al., 1983; Tainer et al., 1983). Here, a region of net positive charge is set up after Cu2+ binding, attracting an anionic superoxide moiety (Getzoff et al., 1983). Our data does not provide structural information on how the dipole is organized; however, recent identification of an anion-binding site (Plested and Mayer, 2007) suggests that anion–cation interactions are also stabilized by ion–protein interactions. This method of binding is observed in Cu2+/Zn2+–SOD, in which the superoxide anion is stabilized by both the Cu2+ and Arg141 (Getzoff et al., 1983). Clearly, additional experiments are required to resolve the molecular architecture of the cation binding site in GluR6 KARs.
It is surprising that a bound cation is not visible near Met770 in published crystal structures of GluR6 (Mayer, 2005; Nanao et al., 2005). This may be attributable to the low affinity by which cations bind to GluR6 KARs (106 mm for Na+) (Wong et al., 2006). In support of this, dehydrated cations that bind with high affinity to the selectivity filter of the KcsA ion channel are more easily resolved than lower-affinity, hydrated cations in the pore vestibule (Doyle et al., 1998). However, thrombin, which has approximately equal affinities for Na+ as GluR6 (110 mm in thrombin), has been crystallized with a bound Na+ ion (Pineda et al., 2004). In this case, the apparent discrepancy may be attributable to differences in crystallization conditions, with thrombin crystals grown in 200 mm Na+ (Pineda et al., 2004) and GluR6 grown in 20 mm Na+ (Mayer, 2005). Lower Na+ conditions may fail to saturate the cation-binding site on GluR6 KARs, accounting for the lack of a visible cation in published crystal structures.
Are KARs tonically blocked by pH or external divalents?
Large-scale fluctuations in the ionic composition of the extracellular milieu during physiological and pathological neuronal activity have been well documented (Nicholson et al., 1978; Chesler, 2003). Acidification of the interstitial space has been observed in response to pathological insult (Kraig et al., 1983; Somjen, 1984), whereas ischemia can produce a shift in interstitial pH to 6.5 or lower (Siemkowicz and Hansen, 1981). Native and recombinant NMDARs are modulated by physiological levels of protons (EC50 of pH 7.3) (Traynelis and Cull-Candy, 1991; Low et al., 2003; Banke et al., 2005), whereas AMPARs have a significantly lower sensitivity (EC50 of pH 6) (Traynelis and Cull-Candy, 1991; Lei et al., 2001). A recent study suggests that KARs are tonically blocked at physiological pH (Mott et al., 2003), whereas our data suggests GluR6 proton affinity is more comparable with AMPARs (IC50 of pH 6) (Fig. 3c). The reason for this discrepancy is unclear but may reflect differences in the agonist used in each study [domoate for Mott et al. (2003) vs glutamate for our study] or that Mott et al. (2003) measured the equilibrium response rather than the peak agonist response. Interestingly, KA1 and KA2 subunits shift GluR6 apparent affinity for protons (Mott et al., 2003), suggesting that pH regulation of native KARs may be more complex and determined by subunit composition.
In the CNS, decreases in bulk extracellular Ca2+ are observed (from 1.2 to 0.8 mm) after physiological and pathological neuronal activation (Nicholson et al., 1978). Ca2+ depletion is likely to be substantially higher in the synaptic cleft attributable to its small size and architecture (Vassilev et al., 1997), leading to profound inhibition of neurotransmitter release (Borst and Sakmann, 1999; Rusakov and Fine, 2003). Changes in extracellular Ca2+ modulate the degree of desensitization of NMDARs (Legendre et al., 1993; Vyklicky, 1993) and block AMPA receptors in a voltage-dependent manner (Bowie and Mayer, 1996). We demonstrate that KARs are tonically inhibited by physiologically relevant concentrations of external Ca2+ (IC50 of 1 mm) (Fig. 4c), suggesting that KAR-mediated synaptic currents may be finely tuned by extracellular Ca2+.
Conclusion
Here we identify a dipole mechanism as a critical design element that couples agonist binding to KAR activation. It is still unclear how external ions evolved as coactivators of KARs. Interestingly however, genomes of Caenorhabditis elegans and Caenorhabditis briggsae contain primitive iGluR-like sequences that possess a Lys residue at the homologous 770-position. It is therefore tempting to speculate that the gating mechanism of KARs evolved from an ancestral iGluR protein that behaved more like AMPARs. Tracing the evolutionary origin of iGluR gating may provide insight into the possible physiological role that external ions play in regulating KAR function in the vertebrate CNS.
Footnotes
This work was supported by operating grants from Canadian Institutes of Health Research (CIHR) (D.B.). A.Y.C.W was funded by the David T. W. Lin Fellowship, and D.M.M was supported by a predoctoral fellowship from CIHR. D.B. is the recipient of a Canada Research Chair award.
References
- Banke TG, Dravid SM, Traynelis SF. Protons trap NR1/NR2B NMDA receptors in a nonconducting state. J Neurosci. 2005;25:42–51. doi: 10.1523/JNEUROSCI.3154-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol (Lond) 1999;521:123–133. doi: 10.1111/j.1469-7793.1999.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D. External anions and cations distinguish between AMPA and kainate receptor gating mechanisms. J Physiol (Lond) 539. 2002;3:725–733. doi: 10.1113/jphysiol.2001.013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Lange GD. Functional stoichiometry of glutamate receptor desensitization. J Neurosci. 2002;22:3392–3403. doi: 10.1523/JNEUROSCI.22-09-03392.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Mayer ML. Dual blockade of glutamate receptors by external divalent cations and internal polyamines. Soc Neurosci Abstr. 1996;22:590. [Google Scholar]
- Bowie D, Garcia EP, Marshall J, Traynelis SF, Lange GD. Allosteric regulation and spatial distribution of kainate receptors bound to ancillary proteins. J Physiol (Lond) 2003;547:373–385. doi: 10.1113/jphysiol.2002.033076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N, Zhou Z, Neher E, Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol (Lond) 1995;485:403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Di Cera E. A structural perspective on enzymes activated by monovalent cations. J Biol Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fay AM, Bowie D. Concanavalin-A reports agonist-induced conformational changes in the intact GluR6 kainate receptor. J Physiol (Lond) 2006;572:201–213. doi: 10.1113/jphysiol.2005.103580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Montiel AV, Sun W, Montal M. A single tryptophan on M2 of glutamate receptor channels confers high permeability to divalent cations. Biophys J. 1996;71:749–758. doi: 10.1016/S0006-3495(96)79274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- Harding MM. Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr. 2002;58:872–874. doi: 10.1107/s0907444902003712. [DOI] [PubMed] [Google Scholar]
- Hille B. Sunderland, MA: Sinauer; 2001. Ionic channels of excitable membranes. [Google Scholar]
- Kraig RP, Ferreira-Filho CR, Nicholson C. Alkaline and acid transients in cerebellar microenvironment. J Neurophysiol. 1983;49:831–850. doi: 10.1152/jn.1983.49.3.831. [DOI] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C, Westbrook GL. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, Orser BA, Thatcher GR, Reynolds JN, MacDonald JF. Positive allosteric modulators of AMPA receptors reduce proton-induced receptor desensitization in rat hippocampal neurons. J Neurophysiol. 2001;85:2030–2038. doi: 10.1152/jn.2001.85.5.2030. [DOI] [PubMed] [Google Scholar]
- Low CM, Lyuboslavsky P, French A, Le P, Wyatte K, Thiel WH, Marchan EM, Igarashi K, Kashiwagi K, Gernert K, Williams K, Traynelis SF, Zheng F. Molecular determinants of proton-sensitive N-methyl-d-aspartate receptor gating. Mol Pharmacol. 2003;63:1212–1222. doi: 10.1124/mol.63.6.1212. [DOI] [PubMed] [Google Scholar]
- Magleby KL. Gating mechanism of BK (Slo1) channels: so near, yet so far. J Gen Physiol. 2003;121:81–96. doi: 10.1085/jgp.20028721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron. 2005;45:539–552. doi: 10.1016/j.neuron.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Meier T, Polzer P, Diederichs K, Welte W, Dimroth P. Structure of the rotor ring of F-type Na+-ATPase from Ilyobacter tartaricus. Science. 2005;308:659–662. doi: 10.1126/science.1111199. [DOI] [PubMed] [Google Scholar]
- Mott DD, Washburn MS, Zhang S, Dingledine RJ. Subunit-dependent modulation of kainate receptors by extracellular protons and polyamines. J Neurosci. 2003;23:1179–1188. doi: 10.1523/JNEUROSCI.23-04-01179.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Yamato I, Kakinuma Y, Leslie AG, Walker JE. Structure of the rotor of the V-type Na+-ATPase from Enterococcus hirae. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- Nanao MH, Green T, Stern-Bach Y, Heinemann SF, Choe S. Structure of the kainate receptor subunit GluR6 agonist-binding domain complexed with domoic acid. Proc Natl Acad Sci USA. 2005;102:1708–1713. doi: 10.1073/pnas.0409573102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, ten Bruggencate G, Stockle H, Steinberg R. Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J Neurophysiol. 1978;41:1026–1039. doi: 10.1152/jn.1978.41.4.1026. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Cohen A, Stern-Bach Y, Lerma J. A role for extracellular Na+ in the channel gating of native and recombinant kainate receptors. J Neurosci. 2003;23:8641–8648. doi: 10.1523/JNEUROSCI.23-25-08641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/s0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemkowicz E, Hansen AJ. Brain extracellular ion composition and EEG activity following 10 minutes ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–240. doi: 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Acidification of interstitial fluid in hippocampal formation caused by seizures and by spreading depression. Brain Res. 1984;311:186–188. doi: 10.1016/0006-8993(84)91416-1. [DOI] [PubMed] [Google Scholar]
- Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–287. doi: 10.1038/306284a0. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J Physiol (Lond) 1991;433:727–763. doi: 10.1113/jphysiol.1991.sp018453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys J. 1997;72:2103–2116. doi: 10.1016/S0006-3495(97)78853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L. Calcium-mediated modulation of N-methyl-d-aspartate (NMDA) responses in cultured rat hippocampal neurones. J Physiol (Lond) 1993;470:575–600. doi: 10.1113/jphysiol.1993.sp019876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H+-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Wong AY, Fay AM, Bowie D. External ions are coactivators of kainate receptors. J Neurosci. 2006;26:5750–5755. doi: 10.1523/JNEUROSCI.0301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]