

Abstract
Peripheral nerve section promotes regenerative, elongated neuritic growth of adult sensory neurons. Although the role of chloride homeostasis, through the regulation of ionotropic GABA receptors, in the growth status of immature neurons in the CNS begins to emerge, nothing is known of its role in the regenerative growth of injured adult neurons. To analyze the intracellular Cl− variation after a sciatic nerve section in vivo, gramicidin perforated-patch recordings were used to study muscimol-induced currents in mice dorsal root ganglion neurons isolated from control and axotomized neurons. We show that the reversal potential of muscimol-induced current, EGABA-A, was shifted toward depolarized potentials in axotomized neurons. This was attributable to Cl− influx because removal of extracellular Cl− prevented this shift. Application of bumetanide, an inhibitor of NKCC1 cotransporter and EGABA-A recordings in sensory neurons from NKCC1−/− mice, identified NKCC1 as being responsible for the increase in intracellular Cl− in axotomized neurons. In addition, we demonstrate with a phospho-NKCC1 antibody that nerve injury induces an increase in the phosphorylated form of NKCC1 in dorsal root ganglia that could account for intracellular Cl− accumulation. Time-lapse recordings of the neuritic growth of axotomized neurons show a faster growth velocity compared with control. Bumetanide, the intrathecal injection of NKCC1 small interfering RNA, and the use of NKCC1−/− mice demonstrated that NKCC1 is involved in determining the velocity of elongated growth of axotomized neurons. Our results clearly show that NKCC1-induced increase in intracellular chloride concentration is a major event accompanying peripheral nerve regeneration.
Keywords: NKCC1, phosphorylation, regenerative growth, injury, chloride homeostasis, DRG
Introduction
Axotomy of adult peripheral neurons induces rapid axon regeneration. This has been demonstrated in vivo (Tanaka et al., 1992; Jacob and McQuarrie, 1993) and in vitro (Smith and Skene, 1997; Lankford et al., 1998; Andre et al., 2003) in which a conditioning lesion increases the ability of the associated primary afferent neuron to regenerate successfully. Molecular mechanisms responsible for this increased neuronal growth ability are accompanied by a shift in cellular organization, such as the appearance of growth cones at the proximal tip of the lesioned axons and the swelling of the neuronal cell body associated with a strong increase in cellular metabolism and protein synthesis (Makwana and Raivich, 2005). Transcriptomic analysis of gene expression after peripheral nerve injury has led to the identification of many injury-related and regeneration-associated genes, such as regulatory proteins including growth factor receptors and transcription factors (Araki et al., 2001; Xiao et al., 2002; Mechaly et al., 2006). The role and the mechanisms controlling the intracellular ionic strength accompanying the increase in cell size necessary to neurite growth are less known.
Among the mechanisms controlling the intracellular ionic strength, the regulation of cell volume is a complex process that involves the activity of membrane ion channels and cotransporters (Lang et al., 1998). The ubiquitously expressed protein NKCC1 (encoded by the slc12a2 gene), plays a “housekeeping” role in cell volume regulation. NKCC1 is an electroneutral transporter responsible for the entry of Na+, K+, and Cl− into the cell and is expressed in dorsal root ganglion (DRG) neurons (Plotkin et al., 1997). Its activity is believed to be primarily responsible for the high intracellular Cl− concentration, [Cl−]i, in peripheral nervous system (Sung et al., 2000; Alvarez-Leefmans et al., 2001). Most studies on the functional role of NKCC1 in sensory neurons are related to the control of cellular excitability through chloride channels such as the GABAA receptor (Sung et al., 2000; Laird et al., 2004). In agreement with its role in presynaptic inhibition, disruption of NKCC1 in mice leads to altered pain perception (Delpire et al., 1999). However, studies on non-neuronal cells have demonstrated that, in addition to maintaining cell volume during hypo-osmotic or hyperosmotic challenge, volume regulatory mechanisms are also used to initiate changes in cell volume, as required for cell growth and differentiation (O'Neill, 1999). Conceptually, besides its role in cell excitability, activation of NKCC1 in the absence of cell shrinkage could lead to cell enlargement. Moreover, the rapid activation of NKCC1 through phosphorylation by growth factors in non-neuronal cells lends credence to a role of NKCC1 in cell growth (Waldegger et al., 1998; Jiang et al., 2001).
The contribution of NKCC1 to the process of neurite growth has to our knowledge never been addressed. To examine this, we analyzed the role and regulation of NKCC1 in the regenerative growth of sensory neurons in vitro conditioned with a peripheral nerve injury in vivo.
Materials and Methods
Animals and surgery
Adult Swiss mice (CERJ, Le Genest St. Isle, France) and C57BL/6 mice (6–8 weeks of age) were housed in cages with a 12 h light/dark cycle and fed food and water ad libitum. The care and use of mice conformed to institutional policies and guidelines. Mice were deeply anesthetized by isoflurane inhalation. The left sciatic nerve was exposed at the midthigh level and sectioned, and a 3–5 mm fragment of nerve was removed. Mice were kept alive for 4–10 d and then were killed by CO2 inhalation, followed by cervical dislocation, and their lumbar DRG L4–L5 were removed.
Gene knockdown experiments
Small interfering RNAs.
Pooled nontargeting control small interfering (siRNA) or specific siRNA against NKCC1 (slc12a2) used in this study were the on-target plus SMART pools from Dharmacon [Perbio Science, Brebières, France; Dharmacon catalog #L-044448-01 targeting NKCC1 (GenBank accession number NM_009194)].
Preparation of the RNA–polymer complex.
The method used for intrathecal delivery was adapted from one reported previously (Tan et al., 2005). For polyethylenimine (PEI) complexation, 5 μg of specific or nonspecific siRNAs were complexed with 1.8 μl of 200 μm linear low-molecular-weight PEI ExGen 500 (Euromedex, Souffelweyersheim, France). RNA–polymer complexes were allowed 10 min to form at room temperature. To allow visualization of transfected cells, 3 mm dextran–tetramethylrhodamine (Invitrogen, Cergy Pontoise, France) were added to the 5% glucose solution containing the RNA–polymer complex.
In vivo delivery of siRNA.
Six to 8 μl of the final solution were injected at the S1 level of adult mice once a day for 5 d. Animals were allowed 2 d recovery and then were killed, and lumbar L4–L5 DRG was collected and processed for either immunochemistry or dissociation for time-lapse video microscopy and electrophysiological recordings. In preliminary experiments, intrathecal transfection efficiency was evaluated with a green fluorescent protein (GFP) siRNA tagged with rhodamine (Qiagen, Courtaboeuf, France) injected in an actin–GFP mice. Fluorescence analysis of rhodamine-tagged neurons on DRG slices demonstrated that 50–80% of neurons were transfected with this method and that transfected neurons did not express GFP, evaluated as a lack of costaining between green GFP and red siRNA (data not shown).
Genotyping.
This was performed using PCR on toe DNA. For NKCC1−/− mice, primers for the mutant gene were forward 5′-TGCAACTGGTATTCTAGCTGGAGC-3′ and reverse 5′-TACAACACACACTCCAACCTCCG-3′ and for control gene were forward 5′-TATCTCAGGTGATCTTGC-3′ and reverse 5′-ACACTGCAATTCCTATGTAAACC-3′.
Cell dissociation.
Neuron cultures were established from lumbar (L4–L5) DRG. Ganglia were successively treated by two incubations with collagenase A (1 mg/ml; Roche Diagnostics, Meylan, France) for 45 min each at 37°C and then with 0.25% trypsin–EDTA (Sigma, St. Quentin Fallavier, France) for 30 min. They were then mechanically dissociated, and isolated cells were collected by centrifugation, followed by suspension in neurobasal culture medium supplemented with 2% B27 (Invitrogen), 2 mm glutamine, and penicillin/streptomycin (20 U/ml, 0.2 mg/ml; Sigma). Dissociated neurons were plated on d-polyornithine (0.5 mg/ml)–laminin (5 μg/ml)-coated glass coverslips and kept at 35°C in an incubator with a humidified 95% air/5% CO2 atmosphere. Two hours after plating, the culture medium was carefully removed and replaced to eliminate dead cells and tissue debris. For survival experiments with bumetanide incubation, cells were counted 2 and 24 h after plating, using phase contrast under a Zeiss (Le Pecq, France) photonic microscope.
Electrophysiological recordings.
The ionotropic GABAA current was recorded at 20–22°C using the gramicidin-perforated patch-clamp technique in DRG neurons after 1 d in vitro using an Axopatch 200B amplifier (Dipsi Industrie, Chatillon, France). The bath solution contained 140 mm tetraethylammonium (TEA)-Cl, 2 mm CaCl2, 1.5 mm MgCl2, 10 mm glucose, and 10 mm HEPES, at pH 7.4 with CsOH, and the pipette solution contained 30 mm CsCl and 110 mm Cs-methanesulfonate, or 140 mm CsCl, 1.5 mm Mg-ATP, 0.5 mm Na-GTP, 0.1 mm EGTA, and 10 mm HEPES, at pH 7.35. To determine [Cl−]i, gramicidin A (Fluka, St. Quentin Fallavier, France) (50 μg/ml) was added to the pipette solution. The progress of gramicidin perforation was evaluated by on-line monitoring with pClamp version 8.2 software (Dipsi Industrie) the capacitive current transient produced by a 10 ms depolarizing voltage step (10 mV) from a −80 mV holding potential. With 50 μg/ml gramicidin, the access resistance dropped to 30–40 MΩ within 15 min of seal formation (Kyrozis and Reichling, 1995). Puff application of muscimol (Sigma) activated a chloride current attributable to GABAA receptor activation. The [Cl−]i was calculated using the following Nernst equation: Erev = RT/ZF × (log ([Cl−]i/[Cl−]e), where RT/ZF was 58 mV at room temperature, and [Cl−]e was 147 mm. The contribution of the HCO3− anion to IGABA-A was minimized by using HEPES-buffered solutions.
Video microscopy.
For time-lapse video microscopy, neuronal cultures were observed with an inverted Zeiss Axiovert 200M equipped with a CCD camera (Micromax; Roper Scientific, Evry, France) and a motorized platine driven with MetaMorph 7.0 software (Molecular Devices, Downingtown, PA). Temperature and atmosphere were controlled (37°C, 5% CO2). Neurons were seeded in four-well chambers at a density of 1000 neurons per well. At 2 h after plating, neurons were placed in the recording chamber and left to grow in the culture medium. Phase-contrast images of several neurons per well were collected with an LD A-Plan 20×/0.3 objective every 30 min for 24 h and analyzed off-line with the MetaMorph software. Neurite growth of each neuron was calculated as the length of a neurite reached each hour for 24 h and expressed as a velocity in micrometers per hour.
Total RNA isolation, reverse transcriptase-PCR, and real time reverse transcriptase-PCR.
RNA of lumbar L4–L5 DRG was isolated and purified using the TriReagent solution (Sigma). cDNA synthesis was performed by reverse transcription with 100 U of Superscript II reverse transcriptase (Invitrogen) and 5 μm hexamer random primers (Roche Diagnostics), 0.5 mm of each dNTP, 10 mm of DTT, and 20 U of RNase-OUT. PCR amplification was performed using 0.2 μm of each primer, 10× PCR buffer, 0.2 mm of dNTP, 1.5 mm of MgCl2, and 0.5 U of Platinum TaqDNA polymerase (Invitrogen). Negative control consisted of DNase-treated and non-reverse-transcribed total RNA. The reverse transcriptase (RT)-PCR product was identified by direct sequence analysis (Genome Express, Meylan, France). One set of specific primers for NKCC1 amplification was used: NKCC1 sense, 5′ CATGGTGTCAGGATTTGCAC; and antisense, 5′ CGTTCAATTCAGCAATCAGG. The expected length of resulting amplification product was 236 bp.
Real-time PCR was performed using SYBR Green I dye detection on the LightCycler system (Roche Diagnostics) as described previously (Mechaly et al., 2006). Briefly, PCRs were performed in glass capillaries in a 10 μl volume containing 3 μl of RT product (final dilution of 1:30), 0.5 μm of forward and reverse primers, and 5 μl of QuantiTect SYBR Green PCR Master Mix (Qiagen). Negative control using water instead of template and/or non-reverse-transcribed RNA was performed simultaneously. After an initial activation step of 15 min at 95°C, 45 cycles consisting of 94°C for 15 s, 55°C for 20 s, and 72°C for 35 s were performed. The identity of RT-PCR amplified products was confirmed at least once by sequencing (Genome Express) and by routinely finishing PCR runs with a melting curve analysis (70–95°C at 0.2°C increments), allowing the melting behavior of amplicons to be verified. Primers used were the same as those used for RT-PCR. As described previously (Mechaly et al., 2006), the relative amounts of specifically amplified cDNAs were calculated using the ΔCT method on three independent experimental replicates and normalized by two stable control genes, Polr2j and Ddx48, the expression of which does not change after axotomy.
Electrophoresis and Western blotting.
Total proteins from lumbar L4–L5 DRG were prepared by addition of 100 μl of PBS with protease inhibitors (60 mm sodium fluoride, 30 mm sodium pyrophosphate, 0.2 mm sodium orthovanadate, and 0.5 μm calyculin A) and homogenized at 4°C using a Dounce homogenizer in 5 vol of radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 2 mm EDTA, and 0.5% desoxycholic acid) containing protease and phosphatase inhibitors. Cellular debris and nuclei were removed by centrifugation at 14,000 rpm for 20 min at 4°C. Supernatants were collected, and total protein amounts were measured using the BCA assay method (Pierce). A total of 20 μg of total protein of the lysate from control and axotomized DRG were diluted into 5× Laemmli's SDS sample buffer and boiled for 2 h at 40°C. Loads of 20 μg of protein per well were then separated by 10% SDS-PAGE (120 V for 1 h 30 min). Proteins were then transferred (13 V for 1 h 30 min) to Immobilon-P membranes (GE Healthcare, Little Chalfont, UK) and blocked with 5% dry milk for 2 h at room temperature.
NKCC1 was detected with a T4 monoclonal antibody generated against a fusion protein fragment encompassing the C-terminus (S760–S1212) amino acid of the human colon NKCC1 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). The phosphorylated form of NKCC1 was detected using rabbit R5 polyclonal antibody (kindly provided by Dr. B. Forbush, Yale University, New Haven, CT) raised against a diphosphopeptide containing Thr212 and Thr217 of human NKCC1. Both primary antibodies T4 and R5 were used at the concentration of 1:1000 overnight at 4°C. The following day, membrane were extensively washed with TBST buffer [0.1% (v/v) Tween 20, 25 mm Tris, and 150 mm NaCl, pH 7.5] and incubated for 1 h with a secondary antibody, either goat anti-mouse peroxidase conjugated or goat anti-rabbit peroxidase conjugated (1:5000; GE Healthcare). After washing with TBST, the immune complex was detected by BM-Chemiluminescence blotting substrate (POD) (Roche Diagnostics) on chemiluminescent film. Blots were reblotted with anti-actin antibody (Santa Cruz Biotechnology via Tebu, Le Perray en Yvelines, France) as a loading control.
Immunohistochemistry.
Frozen transverse sections (14 μm thick) were prepared from adult lumbar L5 DRG fixed with 4% paraformaldehyde in PBS and cryopreserved in 25% sucrose in PBS, before embedding in O.C.T. compound (Sakura Finetek, Zoeterwoude, The Netherlands). The sections were treated with 1% SDS and 8% 2-mercaptoethanol for 5 min and then washed in PBS and incubated with 15% goat serum in PBS for 30 min at room temperature. Sections were then incubated overnight at 4°C with the R5 primary antibody against phospho-NKCC1 (1:500 in PBS) diluted in 0.3% goat serum. After washing the primary antibody with PBS, sections were incubated for 1 h at room temperature with a goat anti-rabbit secondary antibody conjugated with cyanine 3 (1:3000). Omission of the primary antibody or its replacement with rabbit non-immune serum were used for control experiments. The slides were then washed in PBS before mounting with aqueous medium. Images were collected using a PL-Apochromat 20×/0.8 objective on a upright Zeiss microscope equipped with a CCD camera AxioCam MRm (Zeiss). Image acquisition and analysis were done using Axio Vision (Zeiss).
Statistical analysis.
All data are expressed as the mean ± SEM. Unpaired Student's t test or one-way or two-way ANOVA when appropriate were used for comparison between groups for all experiments, except for quantitative RT (qRT)-PCR experiments (Mann–Whitney U test). p < 0.05 was considered statistically significant.
Results
Peripheral nerve injury induces an increase in [Cl−]i in sensory neurons
Four to 10 d after an in vivo sciatic nerve section, dissociated axotomized neurons were selected according to their regenerative, elongated growth as was described previously after 1 d in vitro (Andre et al., 2003). This morphological criterion ensured that these neurons had been conditioned by a lesion (Smith and Skene, 1997). The [Cl−]i was determined by activating the ligand-gated Cl− channel GABAA receptor with muscimol. To measure the reversal potential of the muscimol-induced current, EGABA-A, the external recording solution was designed to inhibit the Na+, Ca2+, and K+ currents, which had the advantage of determining EGABA-A with a ramp protocol needing only one application of muscimol. Under whole-cell patch clamp with 30 or 140 mm intracellular Cl−, EGABA-A averaged −33.0 ± 2.2 mV, n = 7 and −0.4 ± 4.6 mV, n = 10, respectively, in control neurons (Fig. 1Aa,Ab). These values were close to the expected values for the reversal potential of chloride ions in a 147 mm external Cl− (−40.0 mV for 30 mm internal Cl−; −1.2 mV for 140 mm internal Cl−) and demonstrated the accuracy of our method of measurement. To estimate the actual [Cl−]i in sensory neurons, we used the gramicidin-perforated patch-clamp method. Under these conditions, EGABA-A averaged −39.9 ± 1.3 mV, n = 16 in control neurons. In axotomized neurons, EGABA-A was −19.7 ± 1.2 mV, n = 16 (Fig. 1B). Thus, EGABA-A was significantly more depolarized in axotomized neurons compared with control (Fig. 1C) (p < 0.001).
Figure 1.

Reversal potential of GABAA receptor-mediated chloride current in control and axotomized mouse sensory neurons. A, Conventional whole-cell patch-clamp recordings with a patch electrode containing 30 mm Cl− (a) and 140 mm Cl− (b). Ramp protocols from −80 to +40 mV were applied every 5 s in a Na-free, K-free, 147 mm TEA-Cl extracellular solution (c). To reduce Ca2+ currents amplitude, 100 μm NiCl2 and CdCl2 were added. Under these experimental conditions, the voltage ramp elicited a small-amplitude outwardly rectifying conductance. Application of 100 μm muscimol to activate the GABAA current induced an increase in slope conductance. Reversal potential of GABAA current, EGABA-A, was determined at the intersection point between control and muscimol conductance. Consistent with a muscimol-induced chloride current, EGABA-A was −32 mV in 30 mm (a) and +4 mV in 140 mm intracellular Cl− (b) in these neurons. B, Gramicidin-perforated patch recordings were used to determine [Cl−]i. EGABA-A was − 40 mV in a control neuron (a) and −20 mV in an axotomized neuron (b). C, The mean EGABA-A in control was significantly less depolarized than in axotomized neurons (***p < 0.001, Student's t test). The Nernst equation was used to calculate [Cl−]i, averaging 30.7 ± 1.6 mm, n = 16 and 68.4 ± 3.2 mm, n = 16 in control and axotomized neurons, respectively.
Efflux of HCO3− through the anion pore can have a significant contribution to the measurement of the reversal potential of IGABA-A (Kaila and Voipio, 1987). Although our recording conditions were designed to minimize such contribution, we performed a series of experiments in which a low external chloride solution was used to evaluate the contribution of Cl− ions to IGABA-A. To deplete intracellular Cl−, axotomized neurons were incubated overnight in a solution containing 5 mm Cl− ions instead of 147 mm. Afterward, EGABA-A measurements were performed in the standard recording solution. Under these experimental conditions, EGABA-A averaged −33.0 ± 0.7 mV, n = 6, suggesting that Cl− ions are the main contributors of IGABA-A recorded in axotomized neurons (data not shown).
We calculated [Cl−]i in each neuron based on the Nernst equation using EGABA-A and the [Cl−]e set to 147 mm in the extracellular solution (see Materials and Methods). [Cl−]i values were 30.7 ± 1.6 mm in control neurons (n = 16) and 68.4 ± 3.2 mm in axotomized neurons (n = 16). The increase in [Cl−]i was observed in axotomized neurons regardless of their somatic size. Moreover, pretreatment with a low external Cl− solution induced a decrease in [Cl−]i to 40.0 ± 2.3 mm, n = 6, in axotomized neurons.
These data reveal that a conditioning sciatic nerve injury promotes an increase in [Cl−]i of axotomized sensory neurons in vitro.
NKCC1 is responsible for [Cl−]I increase in axotomized neurons
To determine whether NKCC1 is responsible for the increase in [Cl−]i in sensory neurons after a peripheral nerve section, we first used a pharmacological inhibitor of NKCC1, the diuretic bumetanide. Pretreatment of sensory neurons with 10 μm bumetanide, 1 h before cell recordings, induced a shift in the reversal potential of IGABA-A toward a slightly more negative value in control neurons (−46.7 ± 1.5 mV; n = 10; p < 0.001 compared with the control value). In axotomized neurons, bumetanide reduced [Cl−]i increase. Under these conditions, EGABA-A was −36.4 ± 1.4 mV, n = 10, significantly hyperpolarized relative to nontreated axotomized neurons (p < 0.001) (Fig. 2A). Under bumetamide treatment, the increase in [Cl−]i amounted 11 mm (p < 0.001) instead of 38 mm.
Figure 2.
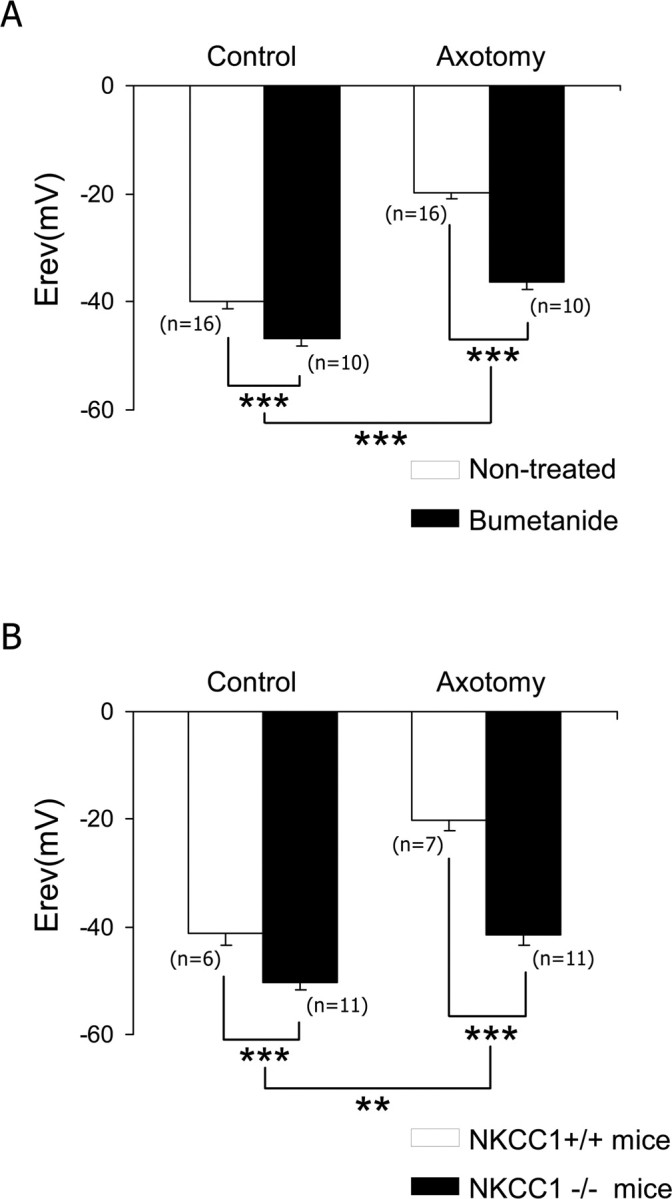
NKCC1 is responsible for [Cl−]i increase in axotomized neurons. A, Neuronal cultures pretreated for 1 h with 10 μm bumetanide showed a shift of EGABA-A toward hyperpolarized values in both control and axotomized neurons, and the main effect of bumetanide was associated with axotomy. EGABA-A remains significantly more depolarized in bumetanide-treated axotomized neurons than in bumetanide-treated control neurons (p < 0.001; data not shown). B, Analysis of the NKCC1−/− mouse demonstrated a shift in EGABA-A toward hyperpolarized values in control and axotomized neurons, and the main effect of NKCC1 deletion was associated with axotomy. EGABA-A remains significantly more depolarized in NKCC1−/− axotomized neurons than in NKCC1−/− control neurons (p < 0.001; data not shown). **p < 0.01, ***p < 0.001, two-way ANOVA.
To ensure that the increase in [Cl−]i after nerve injury is related to the NKCC1 cotransporter, we next performed gramicidin-perforated patch recordings in sensory neurons from NKCC1−/− mice. As reported previously (Sung et al., 2000), the EGABA-A was slightly more hyperpolarized in NKCC1−/− mice compared with wild type. The reversal potential averaged −41.3 ± 1.9 mV in wild-type C57BL/6 mice (n = 6) and −50.3 ± 1.5 mV, n = 11, in NKCC1−/− mice, which corresponds to an [Cl−]i of 29.2 ± 2.6 and 20.4 ± 1.3 mm, respectively (Fig. 2B) (p < 0.001). In axotomized neurons from NKCC1−/− mice, the increase in [Cl−]i was nearly abolished. The reversal potential averaged −20.1 ± 1.8 mV, n = 7, in wild-type mice and −41.4 ± 1.9 mV in NKCC1−/− mice, n = 11 (p < 0.001), which corresponded to [Cl−]i of 67.1 ± 4.9 and 29.3 ± 2.1 mm, respectively (Fig. 2B). Thus, in NKCC1−/− mice neurons, the basal [Cl−]i was significantly decreased by 9 mm compared with their control littermate, and the increase in [Cl−]i after injury amounted 9 mm (p < 0.001) instead of 38 mm and was not significantly different from bumetanide-treated axotomized neurons (p < 0.05, one-way ANOVA).
Because bumetanide is reported to inhibit other transporters and also GABAA receptors (Sung et al., 2000) and because adult knock-out mice may develop functional compensatory effects, we confirmed the involvement of NKCC1 in the control of [Cl−]i in axotomized neurons by using RNA interference for gene silencing. We performed a series of experiments in which NKCC1 was inhibited by intrathecal injections of NKCC1 siRNA for 5 d (see Materials and Methods). On day 3 of injection, the left sciatic nerve was sectioned, and, 4 d later, L4–L5 DRG were removed and cell cultures were prepared. To examine the specificity and efficiency of NKCC1 siRNA injection on gene expression, we performed real-time RT-PCR and functional analysis of NKCC1 by electrophysiological measurements of [Cl−]i. Evaluation of the percentage of transfected neurons was done by counting rhodamine-labeled neurons on DRG sections. Figure 3A shows the percentage of labeled neurons in a L5 DRG that varied between 40 and 50%. To ensure the effectiveness of NKCC1 siRNA, gene expression of NKCC1 was compared in noninjected mice, in mice with intrathecal injection of control siRNA, and mice injected with the NKCC1 siRNA (Fig. 3Ba,Bb). Although PEI-delivered siRNA-negative control did not significantly modify NKCC1 mRNA compared with control (Fig. 3Ba), PEI-delivered siRNA NKCC1 resulted in 40 ± 3% (n = 4) decrease of NKCC1 mRNA in lumbar DRG compared with thoracic DRG (Fig. 3Bb), demonstrating the efficiency of the NKCC1 siRNA pool close to the site injection (S1 level).
Figure 3.

NKCC1 is responsible for [Cl−]i increase in axotomized neurons. Gene inhibition of NKCC1 was achieved after NKCC1 siRNA/dextran–rhodamine intrathecal injection. Transfection was estimated by visualization of fluorescent neurons in DRG or in culture. A, Neuron counting in a slice of L5 DRG shows a near 40% of bright fluorescent neurons. Scale bar, 100 μm. B, qRT-PCR analysis of NKCC1 transcripts in lumbar and thoracic DRG. a, Control siRNA-injected mice shows similar level of NKCC1 transcripts than noninjected mice (n = 4). b, Intrathecal injection of NKCC1 siRNA demonstrates the decrease in NKCC1 transcripts in lumbar compared with thoracic DRG (n = 4; ***p < 0.001, Mann–Whitney U test). C, Phase-contrast photograph of axotomized neurons at 1 d in vitro from L4–L5 DRG after intrathecal siRNA/dextran injection (a). Half of these neurons express rhodamine fluorescence (red) (b). Scale bar, 20 μm. Recordings of [Cl−]i with the gramicidin-perforated patch-clamp method demonstrating that most of the fluorescent axotomized neurons showed EGABA-A to be significantly more hyperpolarized in NKCC1 siRNA than in control siRNA-treated animals (c). ***p < 0.001, one-way ANOVA.
Efficiency of the cotransfection of dextran and siRNA and validation of NKCC1 as being responsible for Cl− accumulation in axotomized neurons were analyzed at the functional protein level by electrophysiological recordings of [Cl−]i. Primary cultures were composed of rhodamine-labeled and nonlabeled neurons (Fig. 3Ca,Cb). Experiments performed on control siRNA-treated mice showed that rhodamine-labeled axotomized neurons had an EGABA-A not significantly different from nontreated axotomized neurons (−20.1 ± 2.5 mV, n = 7 compared with −19.7 ± 1.2 mV, n = 16, respectively). Recordings from NKCC1 siRNA-treated mice demonstrated a significant decrease in EGABA-A of axotomized neurons compared with control siRNA-injected animals (−40.0 ± 1.4 mV, n = 8 compared with −20.1 ± 2.5 mV, n = 7, respectively; p < 0.001) (Fig. 3Cc).
Altogether, these data clearly demonstrate that NKCC1 is responsible for the increase in [Cl−]i induced by nerve injury.
NKCC1 phosphorylation is essential for [Cl−]i increase
To elucidate the molecular mechanisms leading to the increase in [Cl−]i after peripheral nerve injury, we first analyzed the changes in transcriptional level of NKCC1 in whole DRG. NKCC1 transcript was detected in control and in axotomized DRG (Fig. 4A). Real-time RT-PCR analysis showed no significant change at the transcriptional level of NKCC1 cotransporter after axotomy (Fig. 4B). From these data, it appeared that [Cl−]i increase could be related to an increase in the activity of the cotransporter NKCC1. Actually, the NKCC1 cotransporter is phosphorylated under a variety of conditions, and phosphorylation of threonine residues leads to an increase in its activity (Flemmer et al., 2002). To test the possibility of NKCC1 phosphorylation being the molecular basis for [Cl−]i increase, we first analyzed the total protein expression of NKCC1 with the T4 antibody and then phosphorylated expression of NKCC1 with the phospho-specific antibody R5. We show that axotomy does not change the total amount of NKCC1 in whole DRG but promotes phosphorylation of NKCC1 (Fig. 4C).
Figure 4.

Nerve injury induces NKCC1 phosphorylation. A, RT-PCR was performed on control and axotomized DRG. The NKCC1 primer amplified a product of the expected size (236 bp) in control (Ct) and axotomized (Ax) DRG whose molecular identity was confirmed by sequencing. B, qRT-PCR analysis of NKCC1 transcript levels in lumbar L4–L5 DRG from control and axotomized mice shows that peripheral nerve injury does not quantitatively modify NKCC1 mRNA (n = 3). C, Western blot of protein expression with T4 (NKCC1) and R5 (phospho-NKCC1) antibodies shows an increase in protein phosphorylation in axotomized DRG. SDS-PAGE was conducted using 20 μg of protein extract from uninjured DRG (control) and 5 d after sciatic nerve section (axotomized DRG). The apparent molecular size markers are indicated on the right. The same extracts were probed for β-actin.
To further analyze the cellular distribution of phospho-NKCC1 in the DRG, immunohistochemistry was performed on DRG slices (a phospho-NKCC1 signal could not be resolved in primary cultures). To analyze specific staining of R5, we used DRG from NKCC1−/− mice. Compared with the null staining in NKCC1−/− mice, a weak phospho-NKCC1 staining was observed in control DRG (Fig. 5A,B). After nerve injury, a strong R5 staining was observed in the cytoplasm and at the neuron membrane (Fig. 5C).
Figure 5.

Phospho-NKCC1 immunohistochemistry in adult DRG. Phospho-NKCC1 protein expression was studied by immunohistochemistry using the rabbit polyclonal antibody R5 directed against a diphosphopeptide containing Thr212 and Thr217. A, R5 staining in DRG from NKCC1−/− mice is used as negative control for specific staining. B, Control DRG neurons show a weak staining of phospho-NKCC1 (red) in their cytoplasm and at the cell membrane. C, At 5 d after peripheral nerve injury, intense R5 staining is expressed in sensory neurons. The R5 signal (red) is located in the cytoplasm and at the cell membrane. Scale bar, 50 μm.
Altogether, these data demonstrate that nerve injury induces an increase in phosphorylation of NKCC1 in sensory neurons. At the functional level, many studies have established that phosphorylation of NKCC1 at threonine sites increases its activity, measured as an increase in [Cl−]i (Lytle, 1997; Flemmer et al., 2002). To test this hypothesis in sensory neurons, we checked whether the cellular phosphorylation–dephosphorylation state could change [Cl−]i in sensory neurons. On one hand, control neurons were incubated for 15 min with 20 nm calyculin A, a potent protein phosphatase inhibitor 1 that promotes NKCC1 phosphorylation (Lytle, 1997). This treatment induced a depolarization in EGABA-A to −21.6 ± 5.4 mV, n = 10, which corresponded to an increase in [Cl−]i amounting to 63.6 ± 12.9 mm, a value close to those recorded in conditioned regenerating neurons. In contrast, incubation of axotomized sensory neurons with 20 nm staurosporine, a kinase inhibitor with broad selectivity, reduced the depolarization of EGABA-A to −29.8 ± 2.8 mV, n = 7 (p < 0.001 compared with nontreated axotomized neurons), which corresponded to 45.2 ± 4.7 mm intracellular Cl−.
NKCC1 increases neurite growth velocity of regenerating neurons
As shown previously, the regenerative growth of axotomized neurons in vitro was characterized by an elongated, sparsely branched (<1.5 branches per 100 μm) neurite growth. Contrary to axotomized neurons, control growth was characterized by >1.5 branches per 100 μm or had neurites shorter than one cell diameter (Andre et al., 2003). The percentage of axotomized neurons accounted for ∼60% of neuronal population in primary culture from L4–L5 DRG, and the morphological characteristic of elongated growth was used for off-line analysis of axotomized neuron neurite growth. The role of NKCC1 on the regenerative process was evaluated with time-lapse video microscopy, allowing the measurement on-line of the initiation and the velocity of the elongated regenerative neuritic growth in vitro (Fig. 6Aa,Ab) (supplemental movie, available at www.jneurosci.org as supplemental material). As shown in Figure 6A, neurite initiation measured as a neurite length longer than one cell diameter was evaluated 7 h after plating. The percentage of neurons displaying neurite initiation was significantly higher in axotomized neurons [54.0 ± 3.8%, n = 8 wells (276 total neurons) compared with 8.6 ± 3.0%, n = 8 wells (245 total neurons) in control (p < 0.001] (Fig. 6B). Moreover, neurite growth velocity was significantly increased in elongated axotomized neurons (48.5 ± 6.7 μm/h, n = 45 compared with 24.8 ± 4.3 μm/h, n = 43 in control; p < 0.001) (Fig. 6C).
Figure 6.

Analysis of the regenerative neurite growth. Time-lapse video microscopy was used to analyze the neuritic growth velocity of control and axotomized sensory neurons. A, Phase-contrast photograph showing neurons from a sciatic nerve injury conditioned culture 3 h (a) and 24 h (b) after plating. Scale bar 20 μm. B, Neurite initiation was evaluated 7 h after plating and estimated as a neurite length longer than one cell diameter. The total number of neurons counted per well ranged in between 20 and 30; eight wells were analyzed per conditions. The percentage of neurons having initiated neurite growth was significantly greater after an axotomy. C, Growth velocity, estimated as the length of a neurite measured each hour for 24 h, is significantly faster in axotomized neurons compared with control neurons. ***p < 0.001, Student's t test.
In the presence of bumetanide, growth velocity was not significantly different in control neurons (23.0 ± 3.0 μm/h; data not shown) but showed a significantly lower increase in axotomized neurons (32.0 ± 4.0 μm/h, n = 40) (Fig. 7A). Moreover, the percentage of axotomized neurons with neurite initiation, at 7 h, was decreased under bumetanide treatment [36.8 ± 4.4%, n = 8 wells (240 total neurons); p < 0.001; data not shown] but remained higher than in control neurons (p < 0.01). This effect of bumetanide on neurite initiation is probably unrelated to NKCC1 inhibition (see below). Despite inducing a neurite growth velocity in axotomized neurons similar to control neurons and decreasing the percentage of neurons showing neurite initiation, bumetanide did not change the elongated mode of growth in axotomized neurons. Cell counting indicated that bumetanide did not induce cell death until 72 h in either control or axotomized neurons (three cultures; data not shown).
Figure 7.

NKCC1 is involved in the regenerative neurite growth. A, Bumetanide at 10 μm was added 2 h after plating, and the culture was processed for time-lapse microscopy. Bumetanide treatment significantly decreased growth velocity of regenerating neurons. B, Axotomized neuron growth velocity was significantly decreased when recorded in a low external Cl− solution (in mm: 140 Na-methanesulfonate, 2 CaCl2, 1.5 MgCl2, 5 KCl, 10 glucose, and 10 HEPES). C, Regenerative neuritic growth velocity from NKCC1−/− is significantly faster compared with NKCC1+/+ mice. D, Silencing NKCC1 expression with an intrathecal injection of NKCC1 siRNA once a day for 5 d significantly decreased growth velocity of rhodamine-labeled regenerating neurons. No modification of growth velocity was seen with control siRNA compared with nontreated axotomized neurons. **p < 0.01, ***p < 0.001, one way ANOVA.
To see whether intracellular Cl− ions were involved in neurite initiation and growth after axotomy, intracellular Cl− was depleted by incubating axotomized neurons in a solution containing 12 mm Cl− ions instead of 150 mm. Under these conditions, growth velocity was significantly decreased compared with normal Cl− conditions (34.0 ± 7.3 μm/h, n = 31 compared with 59.2 ± 10.1 μm/h, n = 29; p < 0.001) (Fig. 7B). Contrary to bumetanide, the percentage of neurons with neurite initiation remained unchanged compared with normal external Cl− concentration [63.7 ± 4.9%, n = 8 wells (230 total neurons) in low external Cl− concentration]. These data suggest that regulation of intracellular chloride concentration is an important factor involved in the regenerative growth velocity.
Analysis of neurite growth in C57BL/6 mice shows that the elongated mode of growth induced by peripheral nerve injury remains in the C57BL/6 mice (NKCC1+/+) and in the genetically modified mice (NKCC1−/−). Growth velocity of axotomized neurons from NKCC1+/+ mice was 49.6 ± 7.6 μm/h, n = 15. In NKCC1−/− neurons, growth velocity was significantly decreased compared with NKCC1+/+ mice (30.5 ± 8.7 μm/h; n = 40; p < 0.001) (Fig. 7C). Contrary to bumetanide, neurite initiation in axotomized neurons from NKCC1−/− mice was not significantly different from that of wild-type mice [48.7 ± 5.3%, n = 6 wells (180 total neurons 180)].
When using NKCC1 silencing with RNA interference, time-lapse recordings were performed on both rhodamine-labeled and nonfluorescent neurons. First, the effects of the intrathecal injection protocol on the growth velocity of axotomized neurons were evaluated with control siRNA. The neurite growth velocity of 56.3 ± 13.3 μm/h, n = 10, in labeled axotomized neurons was not significantly different from nontreated axotomized neurons. In vivo pretreatment with NKCC1 siRNA induced a significant decrease in growth velocity of fluorescent axotomized neurons to 28.5 ± 12.5 μm/h, n = 36 (Fig. 7D), but did not modify growth velocity in nonfluorescent axotomized neurons 51.9 ± 4.0 μm/h, n = 16 (data not shown). Neurite initiation of axotomized neurons was not significantly changed by NKCC1 siRNA transfection [40.5 ± 10.7%; n = 6 wells (200 total neurons)]. As with bumetanide and in NKCC1−/− mice, elongated mode of growth was unchanged with this NKCC1 knockdown strategy. Altogether, these data demonstrate that axotomized neurons treated with bumetanide, a low external Cl− solution, NKCC1 siRNA, or from NKCC1−/− mice display a similar growth velocity of ∼30 μm/h, i.e., a value close to control growth (p > 0.05, one-way ANOVA).
Discussion
The present study demonstrates that peripheral nerve injury leads to a substantial accumulation of Cl− in sensory neurons, which undergo a twofold to threefold fold increase in their [Cl−]i. Injury promotes phosphorylation of NKCC1, leading to increased activity without a change in transcript expression. Moreover, with the use of pharmacological inhibition, specific gene silencing with RNA interference, and NKCC1−/− mice, we show for the first time that this increase in intracellular chloride is involved in determining the velocity of regenerative neurite growth observed in vitro after a peripheral nerve injury in vivo.
It has been well established that sensory neurons display an earlier and enhanced pattern of neurite extension after a preconditioning injury in vivo compared with intact cells (Smith and Skene, 1997; Lankford et al., 1998; Andre et al., 2003). Elongated growth of neurons conditioned by in vivo sciatic nerve section, characterized by long, sparsely branched neurites, is a hallmark of the process of regeneration. In addition to these morphological analyses, we report that neurite growth initiation and velocity are faster in axotomized neurons than in control. There is an increasing amount of data aimed at identifying the factors responsible for efficient nerve regeneration. Among these factors, members of the gp130 family of cytokines as well the activating transcription factor 3 have been shown to be capable of recapitulating elongated growth and promoting neurite outgrowth (Cafferty et al., 2001; Seijffers et al., 2006). Our study adds a new dimension to this growing list of potential candidates by showing that chloride homeostasis and NKCC1 are also key elements involved in regeneration. Indeed, chloride homeostasis controls the velocity, but not the initiation or the mode, of elongated growth, pointing to the complexity of regenerative growth.
Cl− accumulation is the result of a decrease in outwardly directed Na+-independent K–Cl cotransporters of the KCC family and/or the increase of inwardly directed Na–K–Cl cotransporters. An increase in [Cl−]i attributed to downregulation of KCC2, a neuronal-specific isoform of the KCC family, has been reported in different types of adult central neurons after injury (Nabekura et al., 2002; Coull et al., 2003; Payne et al., 2003). In addition to NKCC1, members of the KCC (1–4) family are also expressed in sensory neurons. To date, Western blot analysis and immunostaining experiments have unequivocally identified KCC4 as being expressed in adult DRG (Karadsheh et al., 2004). We also detected KCC4 expression in DRG at the transcriptional level (our unpublished data), and a decrease in the activity of this cotransporter could contribute to the increase in [Cl−]i that we report after an injury. Our experiments with both pharmacological inhibition of NKCC1 with low concentration of bumetanide as well the use of knockdown strategies using NKCC1−/− mice or in vivo injection of NKCC1 siRNA strongly suggest that the main factor leading to [Cl−]i increase in axotomized sensory neurons is an increased activity of NKCC1. Besides the important lack of [Cl−]i increase observed in bumetanide-treated neurons and in NKCC1−/− mice, there remains a minor but significant increase in [Cl−]i under both conditions after axotomy. This effect could result from a downregulation of KCC induced by axotomy and contributing to the overall increase in [Cl−]i.
Contrary to the transcriptional regulation of KCC2 in central neurons, injury does not promote a change in NKCC1 transcript levels or in protein expression in peripheral neurons. Actually, our study demonstrates that there is an increase in the phosphorylated form of NKCC1 in sensory neurons after a nerve injury, as measured with Western blot and immunofluorescence with a specific phospho-NKCC1 antibody (Flemmer et al., 2002). In a recent work, an increase in phospho-NKCC1 was induced at central afferent terminal endings by a noxious visceral stimulus, confirming the posttranscriptional regulation of NKCC1 in sensory neurons (Galan and Cervero, 2005). From studies in non-neuronal cells, an influx of ions through NKCC1 is regulated by phosphorylation, causing a stimulation of NKCC1 activity (Darman and Forbush, 2002; Flatman, 2002). Although we could not directly demonstrate in vitro that such phosphorylation of NKCC1 is responsible for intracellular Cl− accumulation, we have shown that changes in phosphorylation, through either phosphatase inhibition or kinase inhibition, do result in [Cl−]i variations in vitro, consistent with activation or inactivation of NKCC1 activity. Our results are in agreement with previous reports showing that, in addition to transcriptional modifications, peripheral nerve injury induces early and long-term modifications in the phosphorylation state of various proteins. Among the most analyzed, the transcription factor STAT3 (signal transducer and activator of transcription 3), activated by the cytokine pathway, is phosphorylated. Interestingly, the phosphorylated STAT3 is associated with regenerative growth not only in vivo (Qiu et al., 2005) but also in vitro (Liu and Snider, 2001), confirming the long-term maintenance of some forms of phosphorylation. Heat shock protein 27 phosphorylation is also required for injured sensory neuron survival (Benn et al., 2002). Although the signaling pathways involved in NKCC1 phosphorylation after nerve injury remain to be determined, we have shown for the first time that a potential role of an increased activity of NKCC1 through phosphorylation is related to the regenerative growth of sensory neurons.
The increase in [Cl−]i reported in the present study could induce various cellular processes, ultimately leading to an accelerated neuritic growth. Activation of NKCC1 leads to cellular uptake of NaCl and KCl and thus to cell swelling. The extension of the plasma membrane and the addition of new elements to the actin filaments accompanying neuritic growth are thought to be facilitated by protrusion of the cell membrane, which may be caused by local osmotic swelling. The membrane tension hypothesis proposes that high tensions favor the recruitment of membrane to the surface, whereas low tensions favor retrieval (Dai et al., 1998). Increase in [Cl−]i is often correlated to an increase in osmotic tension (Chavas et al., 2004). According to the osmotic hypothesis, the tension associated with a volume increase could be a mechanism by which a cell regulates its growth.
Besides the membrane tension hypothesis that favors interactions between physical forces and membrane building, cell swelling is often associated with an increase in intracellular calcium concentration in many cell types, including sensory neurons (Okada et al., 2001; Viana et al., 2001; Alessandri-Haber et al., 2003). Calcium is a well known second messenger, and increase in its intracellular concentration regulates numerous kinases involved in growth (Henley and Poo, 2004). The best example for such positive crosstalk between chloride and calcium homeostasis in neurons is the excitatory action of the neurotransmitter GABA that occurs during the development of the CNS. The depolarizing effects of GABA, via the GABAA receptor, are attributed to an intracellular accumulation of Cl− in immature neurons attributable to a low level of expression of KCC2 (Ben-Ari, 2002; Stein and Nicoll, 2003). GABAA receptor-induced depolarization produces an increased concentration of intracellular calcium that could be the molecular basis for the trophic actions of GABA on neuronal development (Represa and Ben-Ari, 2005). Although it has been shown that growth of immature sensory neurons is activity dependent (Fields, 1998; Gomez and Spitzer, 1999; Gomez and Zheng, 2006), no data are available concerning the role of relationships between [Cl−]i and intracellular calcium transients on the regenerative growth of adult sensory neurons. Although it is tempting to speculate that molecular mechanisms involved in embryonic growth could be similar in adult regenerative growth, such experimental evidence is still lacking (Snider et al., 2002).
In conclusion, although much evidence indicates that changes in intracellular chloride profoundly affect cell excitability and contribute to painful behaviors after injury, our study emphasizes the importance of chloride homeostasis in the process of regenerative growth of sensory neurons. Future studies will be aimed at identifying whether NKCC1 affects regenerative growth via changes in osmotic tension or via secondary intracellular calcium effects.
Footnotes
This work was supported by the Association Française contre les Myopathies and the Ministère de la Recherche (S.P.) and National Institutes of Health Grant NS36758 (E.D.). We are very grateful to Dr. B. Forbush for the gift of anti-phospho-NKCC1 antibody R5; to Dr. H. Boukhaddaoui and the regional imaging platform, RIO, for image acquisition and analysis; to J. L. Pasquier for artwork; and to Dr. P. Carroll for critical comments on this paper.
References
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron. 2003;39:497–511. doi: 10.1016/s0896-6273(03)00462-8. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Leon-Olea M, Mendoza-Sotelo J, Alvarez FJ, Anton B, Garduno R. Immunolocalization of the Na+-K+-2Cl− cotransporter in peripheral nervous tissue of vertebrates. Neuroscience. 2001;104:569–582. doi: 10.1016/s0306-4522(01)00091-4. [DOI] [PubMed] [Google Scholar]
- Andre S, Boukhaddaoui H, Campo B, Al-Jumaily M, Mayeux V, Greuet D, Valmier J, Scamps F. Axotomy-induced expression of calcium-activated chloride current in subpopulations of mouse dorsal root ganglion neurons. J Neurophysiol. 2003;90:3764–3773. doi: 10.1152/jn.00449.2003. [DOI] [PubMed] [Google Scholar]
- Araki T, Nagarajan R, Milbrandt J. Identification of genes induced in peripheral nerve after injury. Expression profiling and novel gene discovery. J Biol Chem. 2001;276:34131–34141. doi: 10.1074/jbc.M104271200. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron. 2002;36:45–56. doi: 10.1016/s0896-6273(02)00941-8. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Gavazzi I, Powell J, McMahon SB, Heath JK, Munson J, Cohen J, Thompson SW. Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci. 2001;21:7161–7170. doi: 10.1523/JNEUROSCI.21-18-07161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavas J, Forero ME, Collin T, Llano I, Marty A. Osmotic tension as a possible link between GABA(A) receptor activation and intracellular calcium elevation. Neuron. 2004;44:701–713. doi: 10.1016/j.neuron.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Dai J, Sheetz MP, Wan X, Morris CE. Membrane tension in swelling and shrinking molluscan neurons. J Neurosci. 1998;18:6681–6692. doi: 10.1523/JNEUROSCI.18-17-06681.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Forbush B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J Biol Chem. 2002;277:37542–37550. doi: 10.1074/jbc.M206293200. [DOI] [PubMed] [Google Scholar]
- Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999;22:192–195. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- Fields RD. Effects of ion channel activity on development of dorsal root ganglion neurons. J Neurobiol. 1998;37:158–170. doi: 10.1002/(sici)1097-4695(199810)37:1<158::aid-neu12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Flatman PW. Regulation of Na-K-2Cl cotransport by phosphorylation and protein-protein interactions. Biochim Biophys Acta. 2002;1566:140–151. doi: 10.1016/s0005-2736(02)00586-2. [DOI] [PubMed] [Google Scholar]
- Flemmer AW, Gimenez I, Dowd BF, Darman RB, Forbush B. Activation of the Na-K-Cl otransporter NKCC1 detected with a phospho-specific antibody. J Biol Chem. 2002;277:37551–37558. doi: 10.1074/jbc.M206294200. [DOI] [PubMed] [Google Scholar]
- Galan A, Cervero F. Painful stimuli induce in vivo phosphorylation and membrane mobilization of mouse spinal cord NKCC1 co-transporter. Neuroscience. 2005;133:245–252. doi: 10.1016/j.neuroscience.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Spitzer NC. In vivo regulation of axon extension and pathfinding by growth-cone calcium transients. Nature. 1999;397:350–355. doi: 10.1038/16927. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- Henley J, Poo MM. Guiding neuronal growth cones using Ca2+ signals. Trends Cell Biol. 2004;14:320–330. doi: 10.1016/j.tcb.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob JM, McQuarrie IG. Acceleration of axonal outgrowth in rat sciatic nerve at one week after axotomy. J Neurobiol. 1993;24:356–367. doi: 10.1002/neu.480240308. [DOI] [PubMed] [Google Scholar]
- Jiang G, Klein JD, O'Neill WC. Growth factors stimulate the Na-K-2Cl cotransporter NKCC1 through a novel Cl−-dependent mechanism. Am J Physiol Cell Physiol. 2001;281:C1948–C1953. doi: 10.1152/ajpcell.2001.281.6.C1948. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- Karadsheh MF, Byun N, Mount DB, Delpire E. Localization of the KCC4 potassium-chloride cotransporter in the nervous system. Neuroscience. 2004;123:381–391. doi: 10.1016/j.neuroscience.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- Laird JM, Garcia-Nicas E, Delpire EJ, Cervero F. Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci Lett. 2004;361:200–203. doi: 10.1016/j.neulet.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Lang F, Busch GL, Volkl H. The diversity of volume regulatory mechanisms. Cell Physiol Biochem. 1998;8:1–45. doi: 10.1159/000016269. [DOI] [PubMed] [Google Scholar]
- Lankford KL, Waxman SG, Kocsis JD. Mechanisms of enhancement of neurite regeneration in vitro following a conditioning sciatic nerve lesion. J Comp Neurol. 1998;391:11–29. doi: 10.1002/(sici)1096-9861(19980202)391:1<11::aid-cne2>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RY, Snider WD. Different signaling pathways mediate regenerative versus developmental sensory axon growth. J Neurosci. 2001;21(RC164):1–5. doi: 10.1523/JNEUROSCI.21-17-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle C. Activation of the avian erythrocyte Na-K-Cl cotransport protein by cell shrinkage, cAMP, fluoride, and calyculin-A involves phosphorylation at common sites. J Biol Chem. 1997;272:15069–15077. doi: 10.1074/jbc.272.24.15069. [DOI] [PubMed] [Google Scholar]
- Makwana M, Raivich G. Molecular mechanisms in successful peripheral regeneration. FEBS J. 2005;272:2628–2638. doi: 10.1111/j.1742-4658.2005.04699.x. [DOI] [PubMed] [Google Scholar]
- Mechaly I, Bourane S, Piquemal D, Al-Jumaily M, Venteo S, Puech S, Scamps F, Valmier J, Carroll P. Gene profiling during development and after a peripheral nerve traumatism reveals genes specifically induced by injury in dorsal root ganglia. Mol Cell Neurosci. 2006;32:217–229. doi: 10.1016/j.mcn.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) J Physiol (Lond) 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill WC. Physiological significance of volume-regulatory transporters. Am J Physiol. 1999;276:C995–C1011. doi: 10.1152/ajpcell.1999.276.5.C995. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E. Expression of the Na+-K+-2Cl− cotransporter BSC2 in the nervous system. Am J Physiol. 1997;272:C173–C183. doi: 10.1152/ajpcell.1997.272.1.C173. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cafferty WB, McMahon SB, Thompson SW. Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J Neurosci. 2005;25:1645–1653. doi: 10.1523/JNEUROSCI.3269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28:278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Allchorne AJ, Woolf CJ. The transcription factor ATF-3 promotes neurite outgrowth. Mol Cell Neurosci. 2006;32:143–154. doi: 10.1016/j.mcn.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, Zhou FQ, Zhong J, Markus A. Signaling the pathway to regeneration. Neuron. 2002;35:13–16. doi: 10.1016/s0896-6273(02)00762-6. [DOI] [PubMed] [Google Scholar]
- Stein V, Nicoll RA. GABA generates excitement. Neuron. 2003;37:375–378. doi: 10.1016/s0896-6273(03)00056-4. [DOI] [PubMed] [Google Scholar]
- Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA receptor-mediated currents in dorsal root ganglion neurons isolated from Na–K–2Cl cotransporter null mice. J Neurosci. 2000;20:7531–7538. doi: 10.1523/JNEUROSCI.20-20-07531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Lan KC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Zhang QL, Webster HD. Myelinated fiber regeneration after sciatic nerve crush: morphometric observations in young adult and aging mice and the effects of macrophage suppression and conditioning lesions. Exp Neurol. 1992;118:53–61. doi: 10.1016/0014-4886(92)90022-i. [DOI] [PubMed] [Google Scholar]
- Viana F, de la Pena E, Pecson B, Schmidt RF, Belmonte C. Swelling-activated calcium signalling in cultured mouse primary sensory neurons. Eur J Neurosci. 2001;13:722–734. doi: 10.1046/j.0953-816x.2000.01441.x. [DOI] [PubMed] [Google Scholar]
- Waldegger S, Steuer S, Risler T, Heidland A, Capasso G, Massry S, Lang F. Mechanisms and clinical significance of cell volume regulation. Nephrol Dial Transplant. 1998;13:867–874. doi: 10.1093/ndt/13.4.867. [DOI] [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, Yang L, Huang WJ, Fu G, Xu SH, Cheng XP, Yan Q, Zhu ZD, Zhang X, Chen Z, Han ZG. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci USA. 2002;99:8360–8365. doi: 10.1073/pnas.122231899. [DOI] [PMC free article] [PubMed] [Google Scholar]