

Abstract
Vascular endothelial growth factor (VEGF) plays a neuroprotective role in mice harboring mutations of copper–zinc superoxide dismutase 1 (SOD1) in familial amyotrophic lateral sclerosis (ALS). Conversely, the loss of VEGF expression through genetic depletion can give rise to a phenotype resembling ALS independent of SOD1 mutations. Here, we observe a profound downregulation of VEGF mRNA expression in spinal cords of G93A SOD1 mice that occurred early in the course of the disease. Using an in vitro culture model of glial cells expressing mutant SOD1, we demonstrate destabilization and downregulation of VEGF RNA with concomitant loss of protein expression that correlates with level of transgene expression. Using a luciferase reporter assay, we show that this molecular effect is mediated through a portion of the VEGF 3′-untranslated region (UTR) that harbors a class II adenylate/uridylate-rich element. Other mutant forms of SOD1 produced a similar negative effect on luciferase RNA and protein expression. Mobility shift assay with a VEGF 3′-UTR probe reveals an aberrantly migrating complex that contains mutant SOD1. We further show that the RNA stabilizing protein, HuR (human antigen R), is translocated from nucleus to cytoplasm in mutant SOD1 cells in vitro and mouse motor neurons in vivo. In summary, our data suggest that mutant SOD1 gains a novel function, possibly by altering the ribonucleoprotein complex with the VEGF 3′-UTR. We postulate that the resultant dysregulation of VEGF posttranscriptional processing critically reduces the level of this neuroprotective growth factor and accelerates the neurodegenerative process in ALS.
Keywords: AU-rich element, 3′-untranslated region, RNA-binding proteins, posttranscriptional gene regulation, HuR translocation, motor neurons
Introduction
Vascular endothelial growth factor (VEGF), known mainly for its angiogenic properties, was serendipitously linked to amyotrophic lateral sclerosis (ALS) when a motor neuron disease phenotype developed in mice genetically deficient for the hypoxia-response elements of the VEGF promoter (Oosthuyse et al., 2001). Since this observation, mounting evidence indicates a direct role for VEGF as a neuroprotective factor, independent of its effect on angiogenesis, that attenuates the phenotype of ALS mice expressing the copper–zinc superoxide dismutase 1 (SOD1) mutation (Lambrechts et al., 2003; Azzouz et al., 2004; Storkebaum and Carmeliet, 2004; Storkebaum et al., 2004, 2005; Wang et al., 2007). The regulation of VEGF expression is complex and occurs at multiple levels, extending from transcription to translation (Brockington et al., 2004; Mukhopadhyay and Datta, 2004). RNA stabilization represents an important posttranscriptional control point for the upregulation of VEGF and other growth factor expression, and may serve as a survival pathway in response to cellular stress (Ross, 1995; Brennan and Steitz, 2001; Guhaniyogi and Brewer, 2001). Hypoxia, inflammatory cytokines, and oxidative stress, for example, all of which have been linked to motor neuron death in ALS (Cleveland and Rothstein, 2001; Ischiropoulos and Beckman, 2003; Shaw, 2005) are strong activators of RNA stabilization. A dysfunction in this pathway may therefore impair the intrinsic defense of a cell against stress. Here, we show that expression of mutant SOD1 protein destabilizes VEGF mRNA and markedly downregulates protein production. This dysregulation is mediated through the adenylate/uridylate-rich elements (AREs) in the VEGF 3′-untranslated region (UTR), where an aberrant ribonucleoprotein (RNP) complex is formed in the presence of mutant SOD1. These findings point to a possible new “toxic” gain of function of mutant SOD1 that may promote motor neuron death.
Materials and Methods
Constructs.
All oligonucleotides were purchased from Sigma Genosys (The Woodlands, TX). The following SOD1-specific oligonucleotides were used: upstream, 5′-TCAAGCTTGCGGCCGCATGGACTACAA-GGACGACGATGACAAGGCGACGAAGGCCGTGTGC-3′, and downstream, 5′-TAGACCTTGATATCTTATTGGGCGATCCCAATT-3′. PCR primers were used to amplify mutant and wild-type (WT) SOD1 cDNAs from established clones. PCR products were digested with NotI and EcoRV and then ligated into pTRE2 (Clontech, Mountain View, CA) or pcDNA3.1 (Invitrogen, Carlsbad, CA). For FLAG-human antigen R (HuR), the cDNA from a pTRE2 construct previously made (Nabors et al., 2003) was excised with NotI and EcoRV and ligated into pcDNA3.1. A FLAG-KH-type splicing regulatory protein (KSRP) construct in pcDNA was generously provided by Dr. Ching-Yi Chen (University of Alabama at Birmingham, Birmingham, AL). The following PCR primers were used for green fluorescent fusion (GFP) protein cloning: HuR upstream, 5′-AACAGTTACTCGAGTTCTAATGGTTATGAAGACCA-3′, and downstream, 5′-AGGATTGGTGAATTCTTATTTGTGGGACTT-GTTG-3′, and KSRP upstream, 5′-AACAGTTACTCGAGTGGCGGCGGCCCGGGCGGGGGGT-3′, and downstream, 5′-AGGATTGGTGAATTCTCATTGAGCCTGCTGCTGTCCCT-3′ (Sigma Genosys). PCR products were digested with XhoI and EcoR1, and then ligated into pEGFP (Clontech). The cloning of luciferase-3′-UTR constructs has been described previously (Nabors et al., 2003; Suswam et al., 2005a). All plasmids were verified by sequencing.
Cell culture, transfections, and luciferase assays.
The propagation, maintenance, and transfection of U251 Tet-On cells have been described previously (Nabors et al., 2003). After transfection with pTRE2 or pTRE2-SOD1 plasmids, U251 Tet-On clones were selected with hygromycin (Mediatech, Herndon, VA) and expanded for analysis. For transgene expression, cells were treated with doxycycline (Dox) (Sigma, St. Louis, MO) for 48 h. Control cells were treated with vehicle (ethanol) alone. For cytokine stimulations, cells were treated with tumor necrosis factor α (TNF-α) (R&D Systems, Minneapolis, MN) at 10 ng/ml for 24 h before kinetic analysis. For transient transfections, pcDNA-SOD1 and PGL2–3′-UTR clones were cotransfected into U251 MG cells using Trans IT (Mirus, Madison, WI) and according to manufacturer's protocol. A β-galactosidase reporter was used to control for transfection efficiency. Luciferase and β-galactosidase activities were quantified as described previously (Nabors et al., 2003). For RNA analysis, luciferase and β-galactosidase RNA levels were measured by quantitative reverse transcription-PCR (qRT-PCR). The luciferase RNA value was then divided by the respective β-galactosidase RNA value, and then expressed as a percentage of empty pcDNA vector (control).
Protein preparation, Western blotting, antibodies, immunofluorescence, and ELISA.
Cytosolic and nuclear extracts from cultured cells and mouse tissues were prepared and quantitated as described previously (Suswam et al., 2005a). For mouse, occipital cortex, cervical or lumbar spinal cord tissues were used. Whole-cell extracts were prepared using the MPER kit (Pierce Endogen, Rockford, IL) and quantitated using a BCA assay (Pierce Endogen). For Western blot, 30 μg (cultured cell) or 50 μg (tissue) extract was mixed with SDS sample buffer, boiled for 10 min, and separated on a 12% SDS-polyacrylamide gel. The proteins were transferred to a nitrocellulose membrane (Pierce Endogen). Western blots were probed and developed as previously detailed (Suswam et al., 2005a). The following antibodies were used: Cu/Zn-SOD1 (StressGen, Victoria, British Columbia, Canada), FLAG (Sigma), HuR 3A2 (Santa Cruz Biotechnology, Santa Cruz, CA), KSRP (generously provided by Dr. Ching-Yi Chen), lamin A/C (Santa Cruz Biotechnology), α-tubulin (Santa Cruz Biotechnology). For fluorescence microscopy, G93A and WT SOD1 U251 Tet-On clones were transiently transfected with 2.0 μg of GFP-HuR or GFP-KSRP plasmid and grown on 50 mm glass-bottom dishes (MatTek, Ashland, MA) with or without Dox (1.0 μg/ml) treatment for 48 h. For nuclear staining, a 1:40 dilution of 1 m HEPES and 10 μg/ml Hoechst 33258 (Sigma) was added. Live cells were visualized with an Olympus (Tokyo, Japan) 1 × 70 inverted microscope for GFP-HuR or GFP-KSRP (green) and Hoechst (blue). Monochrome images from each color channel were acquired separately and then colorized and merged. The Hoechst-stained nuclei were pseudocolored red for better contrast against the corresponding images from the green channel. The image was processed with Photoshop (Adobe, San Jose, CA). VEGF ELISA was performed on culture media using a kit (R&D Systems). The values were normalized to total protein concentration of the cell pellet.
Immunohistochemistry.
Spinal cords from G93A and WT SOD1 transgenic (Tg) mice at 40, 90, and 120 d of age were fixed by immersion in 4% paraformaldehyde and flash frozen in liquid nitrogen. Ten micrometer sections were prepared with a cryostat and washed in PBS for 10 min. Sections were blocked with AffiniPure Fab Fragments (Jackson ImmunoResearch Laboratories, West Grove, PA) at a dilution of 1:30 in PBS-Tween for 2 h at room temperature. Sections were washed in PBS and fixed in 4% paraformaldehyde for 10 min. Sections were incubated for 1 h in PBS with 5% goat serum and 1% BSA. At a 1:100 dilution, the primary antibody for HuR (3A2) and human SOD1 (provided by Joseph S. Beckman, Oregon State University, Corvallis, OR) were added and incubated overnight at 4°C. The sections were washed in PBS, and then incubated at room temperature for 1 h with Alexa Fluor 568 goat anti-mouse or Alexa Fluor 488 goat anti-rabbit (Invitrogen) at a 1:400 dilution. Sections were washed with PBS followed by distilled H2O, and then mounted with ProLong Gold antifade reagent with DAPI (4′,6′-diamidino-2-phenylindole) (Invitrogen).
RNA isolation, RT-PCR, RNA kinetics, and quantitative real-time PCR.
RNA was isolated using RNAeasy columns (Qiagen, Valencia, CA) and quantitated by the RiboGreen fluorometric assay (Invitrogen). RT-PCR of mouse tissue RNA was performed with 5 μm random hexamers and 15 U of AMV reverse transcriptase (Invitrogen) in RT buffer (50 mm Tris acetate, pH 8.4, 55 mm potassium acetate, 8 mm magnesium acetate, stabilizer, 4 μg/ml BSA, 1 mm dNTP). PCR primers for mouse VEGF were as follows: upstream, 5′-AACCATGAACTTTCTGCT-3′, and downstream, 5′-CATCTTCCAGGAGTACCC-3′. The internal housekeeping control, S9, was used in the same PCR: upstream, 5′-AGC-TGGGTTTGTCGCAAA-3′, and downstream, 5′-ATCTTGGCCAG-GGTAAA-3′. For kinetic analysis, we used actinomycin D (ActD) and followed our previously published protocol (Nabors et al., 2003). Degradation curves and estimated half-lives were generated with Graph Pad (Graph Pad Software, San Diego, CA) using a model of exponential decay (Nabors et al., 2003). For RNA analysis of the Dox dose–response, cells were treated with ActD for 2 h, and then RNA was harvested and assayed for VEGF expression. Values were normalized to S9 for each sample, and then expressed as a percentage of the RNA in cells before ActD treatment. Methods for qRT-PCR methods as well as primer/probe sequences for luciferase and VEGF have been described previously (Nabors et al., 2003; Suswam et al., 2005a). Standard real-time PCR amplification curves were generated for each target mRNA and S9 control using the threshold cycle (Ct) method. These curves were linear (r2 > 0.98). Primers for β-galactosidase were as follows: upstream, 5′-ATCAGGATATGTG-GCGGATGA-3′, and downstream, 5′-TGATTTGTGTAGTCGGTTT-ATGCA-3′. TaqMan probe was 5′-CGGCATTTTCCGTGACGTCTCG-TT-3′ (Applied Biosystems, Foster City, CA). All qRT-PCR analyses were done on an ABI 7900 PCR instrument (Applied Biosystems) in triplicate.
RNA probe, electrophoretic mobility shift assay, and UV cross-linking.
The VEGF 3′-UTR probe was labeled as detailed previously (King, 2000; Suswam et al., 2005b). Gel-shift and UV cross-linking assays were done based on methods described previously (Dixon et al., 2001; Suswam et al., 2005b). For gel shift assays, 30 μg of whole-cell extract was used. For UV cross-linking, 50 μg of cytoplasmic extract was used. For supershift analysis, antibody (1 μl) was added to the reaction and incubated for an additional 30 min at room temperature. Loading buffer was added, and the samples were electrophoresed in a non-denaturing 6% polyacrylamide gel (Suswam et al., 2005b). The gels were dried and visualized by a PhosphorImager (Storm 820; Amersham Biosciences, Piscataway, NJ).
Statistics.
Mann–Whitney or Student's t test was used for two-group comparisons and Kruskal–Wallis test was used for multiple-group comparisons.
Results
VEGF RNA is downregulated in spinal cords of ALS mice
We compared patterns of VEGF mRNA expression in spinal cord and brain in G93A SOD1 Tg, WT SOD1 Tg, and age-matched control mice using RT-PCR. Starting at an age before the onset of disease (60 d), there was a decline in VEGF band intensity from spinal cord mRNA of G93A Tg mice compared with WT Tg and age-matched controls (Fig. 1A). This relative decline progressed such that no VEGF band was detected in the middle and end stages of the disease. Band intensities of VEGF from G93A Tg brain appeared to increase in these later stages, whereas those from WT Tg and age-matched control samples remained relatively constant. For accurate quantitative assessment of VEGF RNA levels, we performed quantitative real-time PCR on brain and spinal cord tissues (Fig. 1B). In the G93A SOD1 mice, there was a >60-fold drop in VEGF RNA levels in the spinal cord compared with brain at end stage, and a 50-fold decline compared with the WT Tg control. The decline of VEGF RNA in G93A Tg spinal cord, relative to brain, was significant at early, middle, and end stages. A decrease in VEGF expression was appreciable at 60 d of age, which is before clinical manifestations of weakness, but this was not statistically significant. Interestingly, as the disease progressed, there was a gradual, but nonsignificant, increase in brain VEGF RNA expression over the preclinical stage. No appreciable changes in VEGF expression were noted in the WT SOD1 Tg or age-matched controls. Thus, the negative effect of G93A mutant SOD1 on VEGF RNA expression appeared limited to spinal cord tissues.
Figure 1.

VEGF RNA is downregulated in G93A SOD1 transgenic mice. A, Ethidium-stained agarose gel showing results of RT-PCR for VEGF and S9 (housekeeping control) mRNA targets in brain (Br) or spinal cord (SC) tissues. B, Mean VEGF RNA levels (±SE), as determined by qRT-PCR, in mutant or control mice, analyzed in triplicate. Ages for matched controls were as follows: day 60 (Pre), day 83 (early), 106 (middle), and 124 (end). Data were derived from four to six independent experiments, each analyzed in duplicate or triplicate. *p < 0.004 and **p < 0.0001 comparing brain to spinal cord levels of VEGF in the G93A Tg mice.
VEGF mRNA is destabilized in SOD1 mutant cells
Because posttranscriptional gene regulation substantially influences VEGF mRNA expression, we hypothesized that there may be a defect in VEGF RNA stabilization contributing to the decline in VEGF RNA levels in SOD1 mutant mice. To investigate that hypothesis, we stably transfected FLAG-tagged G93A mutant and wild-type SOD1 transgenes into a tetracycline (tet)-inducible glioma cell line (Nabors et al., 2003). This cell lineage typically expresses moderate levels of VEGF and has active RNA stabilization pathways (Tsai et al., 1995; Ryuto et al., 1996; Liu et al., 2002; Nabors et al., 2003). With Dox treatment, we observed marked induction of transgene expression in two independent mutant and wild-type clones using an anti-FLAG antibody (Fig. 2A). In the absence of Dox, there was no detectable band. The endogenous and ectopic forms were identified with a SOD1 antibody. We compared VEGF RNA expression in WT SOD1 no. 1 and G93A SOD1 no. 1 using qRT-PCR after 48 h of Dox treatment (Fig. 2B). A clone expressing empty pTRE vector and the parent U251 cell line were used as additional controls. With Dox treatment, there was a striking and significant decline in VEGF RNA levels in the G93A clone compared with the wild-type clone (p < 0.01) and with the other controls (p < 0.001). With no Dox stimulation, the mean VEGF RNA level in the mutant clone was lower than WT or the controls, but this did not reach statistical significance. We next compared VEGF RNA half-lives of these clones (Fig. 3). In the absence of Dox, the half-lives were comparable (1.8 h). With the addition of Dox, however, there was a clear separation in the half-lives, with the mutant clone showing a 2.3-fold decrement compared with the wild-type clone and a 2-fold decrement compared with Dox (−) cells. In the wild-type clone, the half-life increased marginally to 2.1 h with Dox stimulation. Similar RNA kinetics were observed in G93A SOD1 no. 2 (data not shown). To ensure that the Dox had no effect on VEGF RNA stabilization, we tested the parent U251MG cell line and found a half-life similar to the Dox-negative controls above, even up to 2.0 μg/ml Dox (data not shown). Because neuroinflammation is an important component to motor neuron degeneration, and RNA stabilization can be induced by cytokine stimulation (Chen and Shyu, 1995; Guhaniyogi and Brewer, 2001; Nabors et al., 2003; Dean et al., 2004), we next tested the effects of the proinflammatory cytokine, TNF-α, on VEGF half-life. After a 24 h stimulation period, we saw modest increments in half-life of both the wild-type and mutant clones in the absence of Dox treatment. This incremental pattern is similar to that observed previously in this cell line (Nabors et al., 2003). With Dox stimulation, however, there was a twofold decline in the half-life, similar to the change in unstimulated cells. We analyzed the kinetics of IL-8 and TNF-α, two other mRNAs that contain AREs in the 3′-UTR, and found no effect on half-life in unstimulated cells (data not shown).
Figure 2.
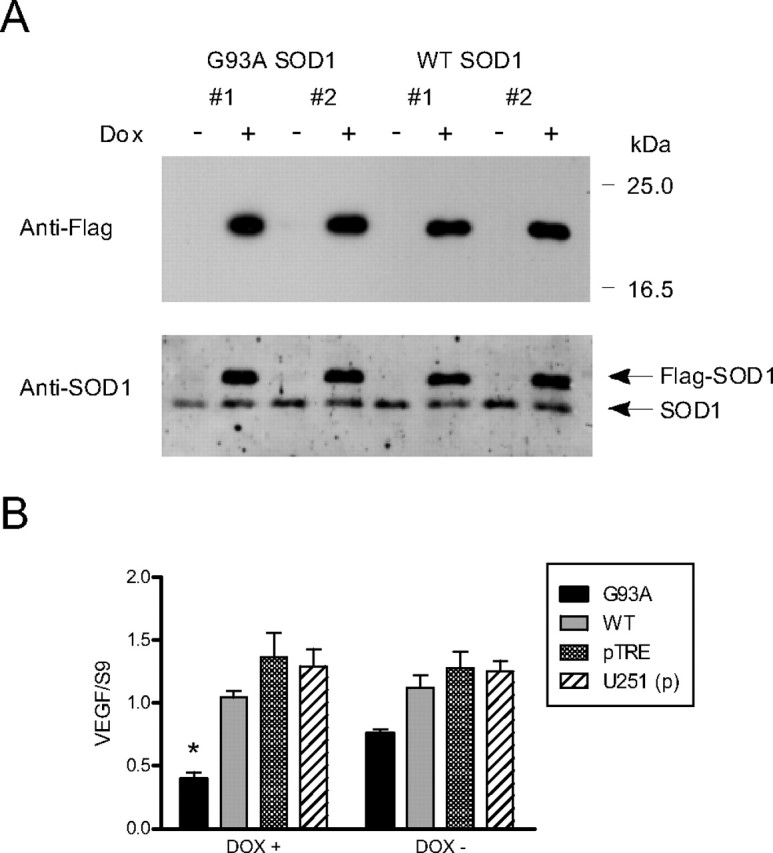
VEGF is downregulated in cells overexpressing G93A SOD1 protein. A, Western blot of U251 tet-on clones expressing wild-type or G93A mutant SOD1 FLAG fusion proteins. Ectopic proteins are detected when the cells are stimulated with Dox (1.0 μg/ml). B, VEGF RNA levels in clone 1 for WT and G93A SOD1 as determined by qRT-PCR. pTRE is a clone expressing empty vector and U251 (p) is the parent U251 tet-on cell line. Results are the mean (±SE) of five to nine independent Dox stimulations for each clone, analyzed in triplicate. *p < 0.001 compared with pTre and U251 (p) controls and p < 0.01 compared with the WT clone.
Figure 3.

VEGF mRNA kinetic analysis of G93A mutant and wild-type SOD1 clones. Percentage of RNA remaining represents the amount of VEGF mRNA relative to the amount just before the addition of actinomycin D (0 h). One-phase exponential decay curves were generated as described previously (Nabors et al., 2003; Suswam et al., 2005a), and RNA half-lives were extrapolated from the curves (shown in legend). The bottom panels represent decay curves after TNF-α stimulation. The data are the mean (±SE) of four to six independent experiments.
The tet-inducible system permits dosing the level of transgene expression by varying the amount of Dox added to the culture media. With a range of 0.01–1.0 μg/ml Dox, there was a dose-dependent increase in wild-type and mutant FLAG-SOD1 protein expression by Western blot (Fig. 4A). With those dose increments, VEGF RNA levels were quantitated 2 h after the administration of ActD and compared with baseline levels (Fig. 4B). In contrast to the wild-type clone, there was a marked and progressive decrease in VEGF mRNA in the G93A SOD1 mutant clone. At a dose of 1.0 μg/ml Dox, VEGF RNA levels were at 33% of baseline compared with 82% for the wild-type clone. The relative decline was significant at doses of 0.25–1.00 μg/ml (p < 0.005). VEGF RNA levels in the parent cell line were similar to the wild-type SOD1 clone. To determine whether the VEGF protein levels were concomitantly decreased, we performed an ELISA analysis on the culture media from cells treated with varying doses of Dox (Fig. 4C). Similar to the mRNA, there was a progressive decline in VEGF protein with increasing doses of Dox in mutant, but not wild-type, cells. Because the basal level of VEGF in G93A SOD1 mutant cells was approximately one-half that of WT SOD1, we compared VEGF expression as percentage of baseline (Dox 0.00). At the highest dose of Dox (1.0 μg/ml), there was a threefold decline compared with baseline, whereas the wild-type SOD1 clone or the parent cell line, at a similar dose, showed no appreciable decline. The differences in expression between WT and G93A SOD1 clones were significant at the 0.25–1.00 μg/ml dose range (p < 0.01 for the 0.25 μg/ml dose and p < 0.006 for the higher doses). In summary, these data indicate that expression of G93A SOD1 had a marked negative effect on VEGF RNA stability, RNA expression, and protein expression.
Figure 4.

Reduction of VEGF RNA and protein expression is dependent on level of G93A SOD1 expression. A, Western blot of clones treated with escalating doses of Dox, probed with an anti-FLAG antibody. B, Percentage of VEGF RNA after 2 h of ActD treatment compared with baseline VEGF RNA levels of cells treated with no Dox (0.00). **p < 0.005. C, VEGF protein expression in culture media of SOD1 clones as determined by ELISA. Values were normalized to total cellular protein and expressed as a percentage of Dox (0.00). The values for VEGF in Dox (0.00) cells are shown above the bar in picograms per milligram total protein. *p < 0.01 and **p < 0.006 comparing WT versus G93A SOD1 clones. Data for RNA and protein expression represent the mean (±SE) of six independent experiments.
G93A SOD1 exerts the negative regulatory effect on VEGF expression via the ARE in the VEGF 3′-UTR
We next used a luciferase reporter assay to determine the impact of the 3′-UTR on the observed RNA destabilization and downregulation of VEGF (Nabors et al., 2003). Mutant or wild-type SOD1 plasmids were cotransfected into U251MG cells along with a luciferase reporter plasmid containing the ARE segment of the VEGF 3′-UTR or a control fragment (Fig. 5A) (King, 2000; Nabors et al., 2001). A β-galactosidase reporter was used to control for transfection efficiency. For wild-type SOD1, there was no difference in luciferase activity with the VEGF 3′-UTR compared with control 3′-UTR or vector alone (Fig. 5B). For G93A SOD1, however, there was a significant decline (p < 0.003) in luciferase activity with the VEGF 3′-UTR compared with control 3′-UTR. We then determined whether this negative effect occurred with other mutant forms of SOD1 associated with motor neuron degeneration. We performed cotransfection with G37R and L38V mutant SOD1 plasmids and also found a significant and equivalent decline in luciferase expression (p < 0.003) compared with the control 3′-UTR. We next evaluated RNA levels of luciferase under the same conditions and found a significant (p < 0.02) decrease in expression of the reporter bearing the VEGF 3′-UTR when cotransfected with each of the three mutant SOD1 plasmids (Fig. 5C). Once again, no effect was observed with wild-type SOD1 or the control 3′-UTR fragment. In summary, the ARE led to significantly decreased luciferase activity and RNA expression levels in the presence of mutant SOD1, and thus recapitulated the effect observed with endogenous VEGF.
Figure 5.

VEGF 3′-UTR mediates the negative regulatory effect of G93A SOD1. A, Schematic diagram of the luciferase reporter plasmid used. The VEGF ARE and control 3′-UTR were cloned into the 3′-UTR of a luciferase expression plasmid (Nabors et al., 2003). B, Luciferase activity measured in U251 MG cells cotransfected with wild-type or mutant SOD1 plasmids as indicated. The activity is expressed as a percentage of empty pcDNA vector. Results are the mean ± SE of five to seven independent transfections. **p < 0.003 (VEGF vs control 3′-UTR). C, Same as B except Luciferase RNA was measured by qRT-PCR after transfection (see Materials and Methods). **p < 0.02 (VEGF vs control 3′-UTR).
We next analyzed the VEGF 3′-UTR RNP complex by electrophoretic mobility shift assay (EMSA) to determine whether there were any differences between G93A and wild-type SOD1 tet-on clones (Fig. 6A). Cellular extracts were prepared from these cells and the parent U251MG tet-on cell line and incubated with a radiolabeled probe derived from the VEGF ARE (shown in Fig. 5A). Recombinant T-cell internal antigen 1-related protein (TIAR) was used as a positive control for binding (Suswam et al., 2005b). With the parent line (P, lane 2) we observed two shifts that were similar to that found in the wild-type SOD1 clone (lane 7). The shifts were not altered by addition of an anti-FLAG antibody either in the parent cell line (lane 3) or the wild-type SOD1 clone (lane 8). TIAR produced a shift consistent with its known binding affinity for VEGF 3′-UTR (lane 4) (Suswam et al., 2005b). We next evaluated mutant SOD1 and observed a loss of the upper shift, and the appearance of a novel shift with greater mobility (lane 5). This complex was partially supershifted by an anti-FLAG antibody (lane 6). A nearly complete supershift of the altered complex was observed with an anti-SOD1 antibody (lanes 11 and 12). No supershift was observed with wild-type SOD1 extract (lanes 9 and 10). Using extract from mutant SOD1 cells, we next performed UV cross-linking with the same probe and observed a spectrum of bands similar to what we have reported previously (Fig. 6B, lane 1) (Nabors et al., 2003; Suswam et al., 2005b). Immunoprecipitation of cross-linked extract with anti-FLAG or two different anti-SOD1 antibodies failed to show that mutant or endogenous SOD1 bound directly to RNA (lanes 2–4). However, HuR (lane 5), TIAR (lane 6), and KSRP (lane 7) all bound to the probe and were readily immunoprecipitated. As a positive control, the FLAG antibody immunoprecipitated FLAG-tristetraprolin, an ARE-binding protein that was cross-linked to the probe in extract derived from a clone expressing that fusion protein (lane 8). A similar pattern was observed for wild-type SOD1 extract (data not shown). Because UV cross-linking preferentially identifies RNA-binding proteins (RBPs) binding to uridylates, however, we cannot determine whether the presence of mutant SOD1 in the RNP complex (seen by EMSA) was attributable to RNA–protein or protein–protein interactions.
Figure 6.

G93A SOD1 expression produces an altered RNP complex with VEGF 3′-UTR. A, Electrophoretic mobility shift assay using a probe containing the VEGF ARE. Sources of extract are as follows: P, parent cell line; Tr, recombinant TIAR; Wt, wild-type SOD1 clone; Mt, mutant G93A SOD1 clone. Antibodies added are shown above the gel. The light arrows indicate the two major shifts observed with Wt and P cell extracts; the black arrow indicates the altered shift observed in Mt extract. B, UV cross-linking and immunoprecipitation of the VEGF 3′-UTR riboprobe using cell extract from the G93A SOD1clone. Lane 1, Total extract; 2, anti-FLAG antibody; 3, anti-SOD1 antibody (StressGen); 4, anti-SOD1 antibody (Santa Cruz Biotechnology); 5, anti-HuR; 6, anti-TIAR; 7, anti-KSRP; 8, anti-FLAG antibody using cross-linked extract from a clone expressing FLAG-TTP.
HuR is translocated to the cytoplasm in SOD1 mutant cells and motor neurons
With VEGF RNA being destabilized in mutant SOD1 cells, we postulated that changes in location or function of the RNA stabilizing protein HuR may be implicated in the biochemical phenotype. We transiently transfected GFP-HuR plasmid into the SOD1 clones and analyzed the cells by fluorescence microscopy (Fig. 7). With Dox stimulation, there was a striking translocation of HuR into the cytoplasm in mutant SOD1 cells that did not occur in wild-type SOD1 cells. In the absence of Dox, however, HuR maintained its usual nuclear location in mutant cells (Brennan and Steitz, 2001). To determine whether this was a specific effect on HuR, we tested another ARE-binding protein, KSRP, that also resides in the nucleus and can translocate (Min et al., 1997; Gu et al., 2002; Briata et al., 2003; Hall et al., 2004). We observed no shift in GFP-KSRP in mutant cells treated with Dox, indicating that the effect of mutant SOD1 on HuR localization was specific. To confirm this finding, we transiently transfected the SOD1 clones with FLAG-HuR or FLAG-KSRP and prepared nuclear and cytoplasmic extracts (Fig. 8). Because of the size differences among SOD1, HuR, and KSRP, we were able to distinguish the simultaneous expression of FLAG fusion proteins by Western blot. Similar to the results of fluorescence cytochemistry, there was a shift of HuR to the cytoplasm in cells expressing mutant SOD1 compared with wild-type control (left top panel). There was no shift, however, with KSRP (right top panel). The FLAG-SOD1 proteins were similarly expressed in all four transfections. Equal loading and the quality of nuclear and cytoplasmic extracts was confirmed with α-tubulin and lamin antibodies (bottom panels).
Figure 7.
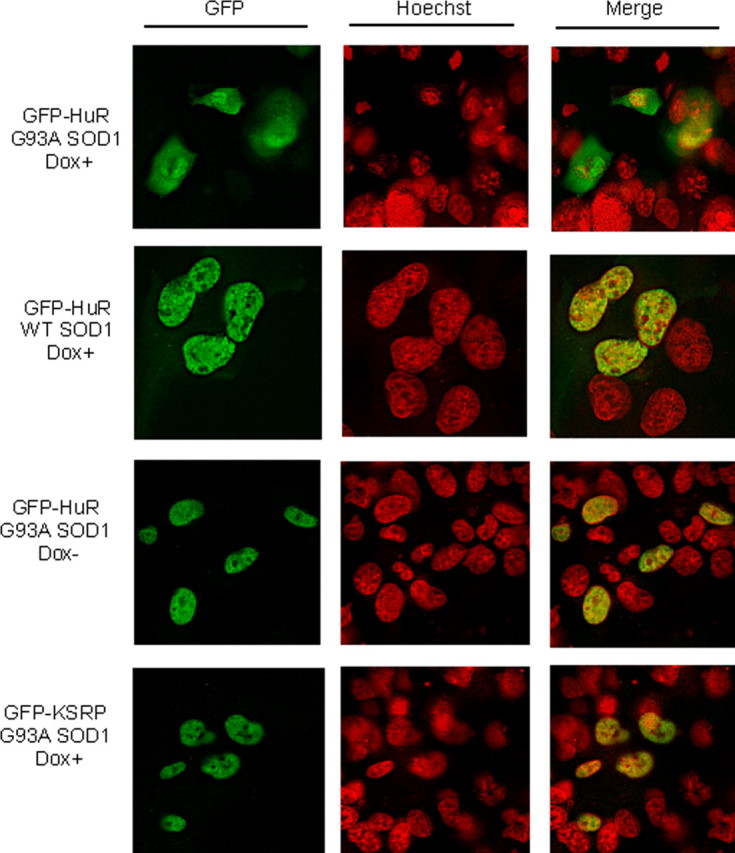
HuR translocates to the cytoplasm when G93A SOD1 is expressed. Fluorescence microscopy of wild-type and G93A SOD1 clones transiently transfected with GFP-HuR or GFP-KSRP plasmids. Cells were stimulated with doxycycline (Dox+) at a dose of 1.0 μg/ml or vehicle (Dox−) for 24 h.
Figure 8.

Western blot showing cytoplasmic translocation of HuR in mutant SOD1 cells. G93A mutant (Mt) or wild-type (Wt) SOD1 cells were transiently transfected with FLAG-HuR or FLAG-KSRP (depicted above the blots), and then treated with Dox. The blots were probed with an anti-FLAG antibody (top three panels) and control antibodies (bottom two panels).
We next determined the relevance of our in vitro observations by studying the distribution of HuR in spinal cord tissue from transgenic ALS mice with G93A mutant SOD1 and age-matched transgenic mice harboring human wild-type SOD1 as a control. In the SOD1 mutant mice, HuR was localized in the cytoplasm (in addition to the nucleus) of motor neurons from preclinical to late stages of the disease (Fig. 9A). In contrast, HuR was mainly detected in the nuclei of motor neurons from control mice. Staining with an anti-human SOD1 antibody confirmed cytoplasmic expression of the SOD1 transgene in both groups. These results were supported by Western blot analysis of nuclear and cytoplasmic extracts from spinal cords of similarly aged mice (Fig. 9B). HuR bands were prominent in the cytosol fraction of mutant, but not wild-type, extracts. Nuclear HuR is seen in all samples. Thus, the translocation of HuR observed in the SOD clones also occurred in motor neurons of SOD1 transgenic mouse.
Figure 9.

HuR translocates to the cytoplasm in motor neurons of G93A transgenic mice in vivo. A, Immunohistochemistry of spinal cords from human G93A or wild-type SOD1 Tg mice. Antibodies are depicted to the left. The hSOD1 antibody is specific for the human form. Ages of the mice are shown above the panels. B, Western blots of spinal cord extracts from G93A or wild-type SOD1 Tg mice. C, Cytoplasm; N, nuclear. Antibodies used are shown to the left of the blots.
Discussion
Our findings indicate that mutant SOD1 can disrupt posttranscriptional regulation of VEGF, leading to decreased production of this neuroprotective growth factor. This disruption supports the hypothesis that mutant SOD1 exerts its deleterious effect on motor neurons through a toxic gain of function (Pasinelli et al., 2004). Although the malignant glioma cell line used in this study may not reflect normal VEGF biology, our model is supported by correlations in the SOD1 mutant mouse, including the downregulation of VEGF mRNA expression and cytoplasmic translocation of HuR.
Posttranscriptional gene regulation is part of the defense of a cell against intrinsic or extrinsic stressors such as hypoxia, inflammation, or oxidative stress. It can work in concert with transcription, as with VEGF and hypoxia, to upregulate gene expression (Brennan and Steitz, 2001; Mukhopadhyay and Datta, 2004). Because some of these stressors are present within a motor neuron or its milieu in ALS (Cluskey and Ramsden, 2001; Lambrechts et al., 2003; Shaw, 2005), we were surprised to see a marked downregulation of VEGF RNA in spinal cords of mutant SOD1 mice. This decline was apparent before the onset of weakness (60 d of age), indicating an early event. The decline in VEGF RNA levels was also restricted to spinal cord, with brain mRNA levels exceeding control mice. Variations in VEGF RNA expression have previously been reported in mice exposed to hypoxia, suggesting that stress response capacities differ among cell populations (Marti and Risau, 1998). In motor neurons and astrocytes of normal human and mouse spinal cords, there is typically strong expression of VEGF and its receptors (Oosthuyse et al., 2001; Brockington et al., 2006). The decrease in VEGF RNA may therefore have resulted either from loss of motor neurons (or other cell types) or an overall decrease in production. The modest change in VEGF RNA levels from early to late clinical stage suggested an impairment of VEGF production. A study of spinal cord tissues from sporadic ALS patients indicated lower levels of VEGF in anterior horn cells compared with normal controls (Brockington et al., 2006). In another study, VEGF production in cultured glial cells derived from spinal cords of G93A SOD1 mice was lower than WT SOD1 cells, but the difference did not reach significance (Van Den Bosch et al., 2004). The cells were derived from embryonic tissues, however, which may minimize the difference if there is an age-dependent decline [as our data suggested (Fig. 1)].
The reduction in VEGF RNA prompted us to investigate posttranscriptional regulation of this gene. Not only did we find a reduction in VEGF RNA and RNA half-life in mutant SOD1 cells, but the effect was not reversed with TNF-α. In this cell line, we previously observed an increase in VEGF RNA stabilization with TNF-α stimulation (Nabors et al., 2003). This cytokine and others are abundant in spinal cord tissues of mutant SOD1 mice and correlate with production of oxidative stress and motor neuron degeneration (Elliott, 2001; Hensley et al., 2002, 2003; Mhatre et al., 2004). Although a recent report indicated no differences in motor neuron survival when TNF-α was genetically deleted (Gowing et al., 2006), our findings suggest a more generalized impairment in stress response at the level of RNA stabilization. Interestingly, we did not observe a change in IL-8 or TNF-α mRNA half-lives, suggesting that the negative effect was specific for VEGF. Although these mRNAs contain AREs similar to VEGF, differential stabilization does occur in response to certain stimuli or cellular conditions (Schuler and Cole, 1988; Lindsten et al., 1989; Bohjanen et al., 1991; Chen et al., 2002, 2006; Nabors et al., 2003; Tebo et al., 2003; Suswam et al., 2005a). Thus, the twofold to threefold decrement in RNA half-life and the nearly threefold drop in RNA production could explain the changes in VEGF RNA expression we observed in spinal cord tissues of mutant mice. Moreover, the impact on VEGF RNA expression was exquisitely gene dose dependent, which is consistent with the observation that multiple copies of the mutant gene are necessary to produce the disease.
Our luciferase data suggested that the negative impact on VEGF expression was mediated through cis-elements in the 3′-UTR. Moreover, the effect was observed with other disease-producing mutant forms of SOD1 (Rosen et al., 1993). The VEGF 3′-UTR locus contains a class II ARE critical for regulating RNA stability (Chen and Shyu, 1995; Levy et al., 1996, 1998; Nabors et al., 2003). The ARE governs both stability and instability through RNP complex formation with cellular RBPs (Chen and Shyu, 1995; Ross, 1995; Brennan and Steitz, 2001; Guhaniyogi and Brewer, 2001). The ARE has been implicated in translational control, either positively or negatively, independent of RNA stability (Jain et al., 1997; Mazan-Mamczarz et al., 2003; Grosset et al., 2004; Barreau et al., 2006). The altered mobility shift and the presence of mutant SOD1 in the RNP complex indicate a change in either RBP–VEGF RNA interactions, protein–protein interactions, or both. Strong and colleagues have shown that mutant SOD1 can destabilize low-molecular-weight NFL (neurofilament) mRNA by binding directly to the 3′-UTR (Ge et al., 2005). We did not observe direct binding of mutant SOD1 to the VEGF 3′-UTR by UV cross-linking. Because this assay identifies uridylate binding, mutant SOD1 may have bound to a non-U-rich region within the 3′-UTR probe.
Although the presence of mutant SOD1 altered the RNP complex, the mechanism by which VEGF RNA becomes destabilized and downregulated is not clear. There are a number of RBPs with similar binding affinities to AREs in the 3′-UTR, but with opposing action. HuR targets AREs and stabilizes transcripts, whereas KSRP, tristetraprolin, and ARE binding factor 1 (AUF1) bind to the same loci but function as destabilizers. RBPs with opposing effects on RNA stability also have overlapping expression patterns (e.g., HuR, KSRP, and AUF1), including the glioma cell line used in this study (Fig. 6B) (Lafon et al., 1998; Gouble and Morello, 2000; Chen et al., 2002; Lal et al., 2004; Lu and Schneider, 2004; Suswam et al., 2005a). Thus, the determinants of stabilization and translation may relate to subcellular localization of the RBP in relationship to the target RNA (Gallouzi et al., 2000; Wilkie et al., 2003; Lal et al., 2004; Barreau et al., 2006). Cytoplasmic translocation of HuR, which can be triggered by cell stresses such as heat shock and UV radiation, is strongly linked to its positive effect on stability and translation (Keene, 1999, 2001; Brennan and Steitz, 2001; Sheflin et al., 2001; Cherradi et al., 2006; Kawai et al., 2006; Wang et al., 2006). In our study, the translocation of HuR was associated with VEGF RNA destabilization and decreased protein expression. One explanation is that HuR translocation may be a normal response to stresses induced by mutant SOD1 that then becomes dissociated from RNA stabilization and translation. One hypothesis for cellular toxicity is the capacity for mutant SOD1 to sequester essential cellular factors, thereby reducing or eliminating their function (Boillee et al., 2006). Mutant SOD1, for example, coprecipitates with heat shock proteins, αB-crystallin, and BclII (Shinder et al., 2001; Okado-Matsumoto and Fridovich, 2002; Pasinelli et al., 2004). HuR may thus be sequestered by mutant SOD1. A second explanation for the cytoplasmic accumulation of HuR is an abnormality of nuclear localization. HuR shuttles between the nucleus and cytoplasm, and mutant SOD1 may interfere with that process. This dysfunction would have to be selective because KSRP properly localized to the nucleus (Figs. 7, 8).
What is the relevance of our findings to ALS? Motor neuron degeneration in transgenic mice (and possibly humans) is quite sensitive to VEGF levels. Both depletion and repletion of VEGF can modify the disease (Lambrechts et al., 2003, 2004; Storkebaum et al., 2005). Mice expressing the SOD1G93A mutation with a VEGFHRE−/− background, for example, showed more severe weakness and died earlier (Lambrechts et al., 2003). Conversely, when mice with a SOD1G93A background were genetically engineered to overexpress VEGF in neurons, there was delayed motor neuron degeneration and prolonged survival (Wang et al., 2007). Exogenous VEGF also delayed onset of disease and improved survival in mutant mice (Azzouz et al., 2004; Storkebaum et al., 2005). Some studies have indicated low VEGF levels in serum and CSF of sporadic ALS patients, possibly because of polymorphisms in the 5′-UTR or promoter elements (Lambrechts et al., 2003; Devos et al., 2004). Other studies have shown either no difference, an increase, or variable levels of VEGF protein depending on disease subtype (Nygren et al., 2002; Ilzecka, 2004). Our finding that posttranscriptional gene regulation of VEGF was negatively affected at baseline suggests that mutant SOD1 may increase the vulnerability of certain cells to even minor stressors. Lower baseline VEGF levels in VEGFHRE−/− mice, for example, sensitized them to ischemic insults that, in wild-type mice, would not lead to permanent neurological dysfunction (Lambrechts et al., 2003). Thus, we postulate that the impairment of posttranscriptional processing of VEGF RNA by mutant SOD1 blocks an essential neuroprotective pathway and accelerates motor neuron degeneration.
Footnotes
This work was supported by the Department of Neurology, University of Alabama at Birmingham, and Birmingham Veterans Affairs Medical Center (L.L.); by National Institutes of Health Grants NS36761 and NS42834 (A.G.E.) and CA97247 (E.S.); and by a Merit Review from the Department of Veterans Affairs (P.H.K.). We thank Albert Tousson for his assistance with fluorescence microscopy, Xiuhua Yang for technical assistance, and the tissue culture and sequencing core facilities of the University of Alabama at Birmingham Comprehensive Cancer Center for their support of this research.
References
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2006;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohjanen PR, Petryniak B, June CH, Thompson CB, Lindsten T. An inducible cytoplasmic factor (AU-B) binds selectively to AUUUA multimers in the 3′ untranslated region of lymphokine mRNA. Mol Cell Biol. 1991;11:3288–3295. doi: 10.1128/mcb.11.6.3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland D. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R. The Wnt/β-catenin–Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell. 2003;12:1201–1211. doi: 10.1016/s1097-2765(03)00407-6. [DOI] [PubMed] [Google Scholar]
- Brockington A, Lewis C, Wharton S, Shaw PJ. Vascular endothelial growth factor and the nervous system. Neuropath Appl Neurol. 2004;30:427–446. doi: 10.1111/j.1365-2990.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- Brockington A, Wharton SB, Fernando M, Gelsthorpe CH, Baxter L, Ince PG, Lewis CE, Shaw PJ. Expression of vascular endothelial growth factor and its receptors in the central nervous system in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:26–36. doi: 10.1097/01.jnen.0000196134.51217.74. [DOI] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen C-YA, Xu N, Shyu A-B. Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol Cell Biol. 2002;22:7268–7278. doi: 10.1128/MCB.22.20.7268-7278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-L, Huang Y-L, Lin N-Y, Chen H-C, Chiu W-C, Chang C-J. Differential regulation of ARE-mediated TNFα and IL-1β mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochem Biophys Res Commun. 2006;346:160–168. doi: 10.1016/j.bbrc.2006.05.093. [DOI] [PubMed] [Google Scholar]
- Cherradi N, Lejczak C, Desroches-Castan A, Feige J-J. Antagonistic functions of tetradecanoyl phorbol acetate-inducible-sequence 11b and HuR in the hormonal regulation of vascular endothelial growth factor messenger ribonucleic acid stability by adrenocorticotropin. Mol Endocrinol. 2006;20:916–930. doi: 10.1210/me.2005-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in amyotrophic lateral sclerosis. Mol Pathol. 2001;54:386–392. [PMC free article] [PubMed] [Google Scholar]
- Dean JL, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 2004;16:1113–1121. doi: 10.1016/j.cellsig.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Devos D, Moreau C, Lassalle P, Perez T, De Seze J, Brunaud-Danel V, Destee A, Tonnel AB, Just N. Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 2004;62:2127–2129. doi: 10.1212/01.wnl.0000129913.44351.a3. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JL. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Mol Brain Res. 2001;95:172–178. doi: 10.1016/s0169-328x(01)00242-x. [DOI] [PubMed] [Google Scholar]
- Gallouzi IE, Brennan CM, Stenberg MG, Swanson MS, Eversole A, Maizels N, Steitz JA. HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci USA. 2000;97:3073–3078. doi: 10.1073/pnas.97.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W-W, Wen W, Strong W, Leystra-Lantz C, Strong MJ. Mutant copper-zinc superoxide dismutase binds to and destabilizes human low molecular weight neurofilament mRNA. J Biol Chem. 2005;280:118–124. doi: 10.1074/jbc.M405065200. [DOI] [PubMed] [Google Scholar]
- Gouble A, Morello D. Synchronous and regulated expression of two AU-binding proteins, AUF1 and HuR, throughout murine development. Oncogene. 2000;19:5377–5384. doi: 10.1038/sj.onc.1203910. [DOI] [PubMed] [Google Scholar]
- Gowing G, Dequen F, Soucy G, Julien J-P. Absence of tumor necrosis factor-α does not affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci. 2006;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosset C, Boniface R, Duchez P, Solanilla A, Cosson B, Ripoche J. In vivo studies of translational repression mediated by the granulocyte-macrophage colony-stimulating factor AU-rich element. J Biol Chem. 2004;279:13354–13362. doi: 10.1074/jbc.M308003200. [DOI] [PubMed] [Google Scholar]
- Gu W, Pan F, Zhang H, Bassell GJ, Singer RH. A predominantly nuclear protein affecting cytoplasmic localization of β-actin mRNA in fibroblasts and neurons. J Cell Biol. 2002;156:41–52. doi: 10.1083/jcb.200105133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene. 2001;265:11–23. doi: 10.1016/s0378-1119(01)00350-x. [DOI] [PubMed] [Google Scholar]
- Hall MP, Huang S, Black DL. Differentiation-induced colocalization of the KH-type splicing regulatory protein with polypyrimidine tract binding protein and the c-src pre-mRNA. Mol Biol Cell. 2004;15:774–786. doi: 10.1091/mbc.E03-09-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Floyd RA, Gordon B, Mou S, Pye QN, Stewart C, West M, Williamson K. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem. 2002;82:365–374. doi: 10.1046/j.1471-4159.2002.00968.x. [DOI] [PubMed] [Google Scholar]
- Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, Hamdheydari L, Mhatre M, Mou S, Pye QN. Message and protein-level elevation of tumor necrosis factorα (TNFα) and TNFα-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol Dis. 2003;14:74–80. doi: 10.1016/s0969-9961(03)00087-1. [DOI] [PubMed] [Google Scholar]
- Ilzecka J. Cerebrospinal fluid vascular endothelial growth factor in patients with amyotrophic lateral sclerosis. Clin Neurol Neurosurg. 2004;106:289–293. doi: 10.1016/j.clineuro.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RG, Andrews LG, McGowan KM, Pekala PH, Keene JD. Ectopic expression of Hel-N1, an RNA binding protein, increases glucose transporter (GLUT1) expression in 3T3–L1 adipocytes. Mol Cell Biol. 1997;17:954–962. doi: 10.1128/mcb.17.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Lal A, Yang X, Galban S, Mazan-Mamczarz K, Gorospe M. Translational control of cytochrome c by RNA-binding proteins TIA-1 and HuR. Mol Cell Biol. 2006;26:3295–3307. doi: 10.1128/MCB.26.8.3295-3307.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD. Why is Hu where? Shuttling of early-response-gene messenger RNA subsets. Proc Natl Acad Sci USA. 1999;96:5–7. doi: 10.1073/pnas.96.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc Natl Acad Sci USA. 2001;98:7018–7024. doi: 10.1073/pnas.111145598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King PH. RNA-binding analyses of HuC and HuD with the VEGF and c-myc 3′-untranslated regions using a novel ELISA-based assay. Nucleic Acids Res. 2000;28:e20. doi: 10.1093/nar/28.7.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon I, Carballes F, Brewer G, Poiret M, Morello D. Developmental expression of AUF1 and HuR, two c-myc mRNA binding proteins. Oncogene. 1998;16:3413–3421. doi: 10.1038/sj.onc.1201895. [DOI] [PubMed] [Google Scholar]
- Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Carmeliet P. VEGF: necessary to prevent motoneuron degeneration, sufficient to treat ALS? Trends Mol Med. 2004;10:275–282. doi: 10.1016/j.molmed.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Levy AP, Kevy NS, Goldberg MA. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271:2746–2753. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- Levy NS, Chung S, Furneaux H, Levy AP. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998;273:6417–6423. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Liu LX, Lu H, Luo Y, Date T, Belanger AJ, Vincent KA, Akita GY, Goldberg M, Cheng SH, Gregory RJ, Jiang C. Stabilization of vascular endothelial growth factor mRNA by hypoxia-inducible factor 1. Biochem Biophys Res Commun. 2002;291:908–914. doi: 10.1006/bbrc.2002.6551. [DOI] [PubMed] [Google Scholar]
- Lu J-Y, Schneider RJ. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. J Biol Chem. 2004;279:12974–12979. doi: 10.1074/jbc.M310433200. [DOI] [PubMed] [Google Scholar]
- Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci USA. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Galban S, de Silanes IL, Martindale JL, Atasoy U, Keene JD, Gorospe M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA. 2003;100:8354–8359. doi: 10.1073/pnas.1432104100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhatre M, Floyd RA, Hensley K. Oxidative stress and neuroinflammation in Alzheimer's disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. J Alzheimers Dis. 2004;6:147–157. doi: 10.3233/jad-2004-6206. [DOI] [PubMed] [Google Scholar]
- Min H, Turck CW, Nikolic JM, Black DL. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 1997;11:1023–1036. doi: 10.1101/gad.11.8.1023. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin Cancer Biol. 2004;14:123–130. doi: 10.1016/j.semcancer.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Nabors LB, Gillespie GY, Harkins L, King PH. HuR, an RNA stability factor, is expressed in malignant brain tumors and binds to adenine and uridine-rich elements within the 3′-untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res. 2001;61:2154–2161. [PubMed] [Google Scholar]
- Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ, King PH. Tumor necrosis factor-α induces angiogenic factor up-regulation in malignant glioma cells: a role for RNA stabilization and HuR. Cancer Res. 2003;63:4181–4187. [PubMed] [Google Scholar]
- Nygren I, Larsson A, Johansson A, Askmark H. VEGF is increased in serum but not in spinal cord from patients with amyotrophic lateral sclerosis. NeuroReport. 2002;13:2199–2201. doi: 10.1097/00001756-200212030-00007. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci USA. 2002;99:9010–9014. doi: 10.1073/pnas.132260399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown JRH. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hantati A. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ross J. mRNA stability in mammalian cells. Microbiol Rev. 1995;59:423–450. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryuto M, Ono M, Izumi H, Yoshida S, Weich HA, Kohno K, Kuwano M. Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP-1. J Biol Chem. 1996;271:28220–28228. doi: 10.1074/jbc.271.45.28220. [DOI] [PubMed] [Google Scholar]
- Schuler GD, Cole MD. GM-CSF and oncogene mRNA stabilities are independently regulated in trans in a mouse monocytic tumor. Cell. 1988;55:1115–1122. doi: 10.1016/0092-8674(88)90256-5. [DOI] [PubMed] [Google Scholar]
- Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry. 2005;76:1046–1057. doi: 10.1136/jnnp.2004.048652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheflin LG, Zhang W, Spaulding SW. Androgen regulates the level and subcellular distribution of the AU-rich ribonucleic acid-binding protein HuR both in vitro and in vivo. Endocrinology. 2001;142:2361–2368. doi: 10.1210/endo.142.6.8164. [DOI] [PubMed] [Google Scholar]
- Shinder GA, Lacourse M-C, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Carmeliet P. VEGF: a critical player in neurodegeneration. J Clin Invest. 2004;113:14–18. doi: 10.1172/JCI200420682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. BioEssays. 2004;26:943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Suswam EA, Nabors LB, Huang Y, Yang X, King PH. IL-1β induces stabilization of IL-8 mRNA in malignant breast cancer cells via the 3′ untranslated region: involvement of divergent RNA-binding factors HuR, KSRP and TIAR. Int J Cancer. 2005a;113:911–919. doi: 10.1002/ijc.20675. [DOI] [PubMed] [Google Scholar]
- Suswam EA, Li YY, Mahtani H, King PH. Novel DNA-binding properties of the RNA-binding protein TIAR. Nucleic Acids Res. 2005b;33:4507–4518. doi: 10.1093/nar/gki763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebo J, Der S, Frevel M, Khabar KSA, Williams BRG, Hamilton TA. Heterogeneity in control of mRNA stability by AU-rich elements. J Biol Chem. 2003;278:12085–12093. doi: 10.1074/jbc.M212992200. [DOI] [PubMed] [Google Scholar]
- Tsai J, Goldman CK, Gillespie GY. Vascular endothelial growth factor in human glioma cell lines: induced by EGF, PDGF-BB, and bFGF. J Neurosurg. 1995;82:864–873. doi: 10.3171/jns.1995.82.5.0864. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Storkebaum E, Vleminckx V, Moons L, Vanopdenbosch L, Scheveneels W, Carmeliet P, Robberecht W. Effects of vascular endothelial growth factor (VEGF) on motor neuron degeneration. Neurobiol Dis. 2004;17:21–28. doi: 10.1016/j.nbd.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Wang JG, Collinge M, Ramgolam V, Ayalon O, Fan XC, Pardi R, Bender JR. LFA-1-dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J Immunol. 2006;176:2105–2113. doi: 10.4049/jimmunol.176.4.2105. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ou Mao X, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007;27:304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie GS, Dickson KS, Gray NK. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem Sci. 2003;28:182–188. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]