

Abstract
Metabotropic glutamate receptors (mGluRs) modulate neural excitability and network tone in many brain regions. Expression of mGluRs is particularly high in the accessory olfactory bulb (AOB), a CNS structure critical for detecting chemicals that identify kin and conspecifics. Because of its relative simplicity and its direct projection to the hypothalamus, the AOB provides a model system for studying how mGluRs affect the flow of encoded sensory information to downstream areas. We investigated the role of group I mGluRs in synaptic processing in AOB slices and found that under control conditions, recurrent inhibition of principal neurons (mitral cells) was completely eliminated by the mGluR1 antagonist LY367385 [(S)-(+)-α-amino-4-carboxy-2 methylbenzeneacetic acid]. In addition, the group I mGluR agonist DHPG [(S)-3,5-dihydroxyphenylglycine; 20 μm] induced a dramatic increase in the rate of spontaneous IPSCs. This increase was dependent on voltage-gated calcium channels but persisted even after blockade of ionotropic glutamatergic transmission and sodium channels. Together, these results indicate that mGluR1 plays a critical role in controlling information flow through the AOB and suggest that mGluR1 may be an important locus for experience-dependent changes in synaptic function.
Keywords: AOB, vomeronasal, GABA, granule cell, pheromone, olfaction
Introduction
The mammalian accessory olfactory bulb (AOB) is a CNS structure involved in identifying chemicals that signal the social and reproductive status of conspecifics (Stowers et al., 2002; Dulac and Torello, 2003). Individual AOB principal neurons (mitral cells) receive excitatory inputs from vomeronasal axons in a small number of glomeruli (Meisami and Bhatnagar, 1998). In contrast to the broadly tuned mitral cells of the main olfactory bulb (MOB), AOB mitral cells are exquisitely selective for specific, socially relevant ligands or mixtures (Luo et al., 2003). Such selectivity likely indicates a sparse representation of pheromonal information; that is, any given mitral cell is activated only rarely, and the responses of a small number of mitral cells are likely to be behaviorally relevant. Consistent with this, AOB mitral cells are immediately presynaptic to brain areas that cause the changes in behavioral, hormonal, and metabolic states associated with pheromones, including the hypothalamus and amygdala (Kevetter and Winans, 1981).
The throughput of information from the AOB to these downstream structures is controlled, in large part, by recurrent inhibition (RI) of mitral cells by granule cells, a population of axonless, spiny, GABAergic interneurons that inhibit mitral cells via reciprocal dendrodendritic synapses. (Isaacson and Strowbridge, 1998; Schoppa et al., 1998; Taniguchi and Kaba, 2001; Urban, 2002). This relatively simple relay-like relationship between the AOB and downstream structures motivates the hypothesis that RI serves a sensory “gating” function (Brennan et al., 1990; Kaba et al., 1994; Brennan and Keverne, 1997; Best et al., 2005). Behavioral and physiological experiments support this notion, as changes in the strength of RI are thought to underlie specific forms of olfactory learning (Hayashi et al., 1993; Kaba et al., 1994; Brennan and Keverne, 1997). Moreover, these changes depend critically on activation of metabotropic glutamate receptors (mGluRs), which are richly expressed in the AOB (Ohishi et al., 1993, 1998; Sahara et al., 2001).
In the present study, we examined the role of mGluRs in mediating RI of AOB mitral cells. Given the high expression levels of mGluRs in the MOB and AOB, and the fact that activation of these receptors is proposed to be important in the induction of experience-dependent changes in information processing in the AOB, we reasoned that they might play an important role in regulating RI. Surprisingly, and in contrast to what we and others (Dong et al., 2007) observe in the MOB, we found that RI of AOB mitral cells requires activation of the group I mGluR, mGluR1. This requirement for mGluR1 was dramatically reduced when extracellular magnesium concentration was lowered, suggesting that mGluR1 activation enhances the coupling of NMDA receptors (NMDARs) to inhibition. Inhibition was also induced by direct application of the group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG), even after blockade of ionotropic glutamatergic synaptic transmission and sodium spikes. These results suggest that mGluR activation is critical for regulating granule cell excitability and that the mechanisms of RI show interesting differences in the MOB and AOB. Finally, these data point to group I mGluRs as a critical locus for controlling information flow through the AOB.
Materials and Methods
Slice preparation.
The methods are as described previously by Urban and Castro (2005). Briefly, sagittal olfactory bulb slices (300–350 μm thick) were prepared from young mice [postnatal day 14 (P14) to P28]. Mice were anesthetized (0.1% ketamine/0.1% xylazine; ∼3 mg/kg, i.p.) and decapitated. Olfactory bulbs were removed and sectioned on a vibratome while submerged in ice-cold oxygenated Ringer's solution containing the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 25 glucose, 2 CaCl2. In some experiments, 0.5 ascorbate, 1 pyruvate, and 2 mm myo-inositol were added to the slicing medium. All animal care was in accordance with the guidelines of the Institutional Animal Care and Use Committee of Carnegie Mellon University.
Electrophysiology.
Whole-cell voltage recordings were obtained from the somata of identified AOB mitral cells (Stuart and Spruston, 1995). Slices were superfused with the oxygenated Ringer's solution described above and warmed to 34–36°C. Whole-cell recordings were established using pipettes (resistances of 2–8 MΩ) filled with a solution containing the following (in mm): 120 postassium gluconate, 2 KCl, 10 HEPES, 10 sodium phosphocreatine, 4 MgATP, and 0.3 Na3GTP, adjusted to pH 7.3 with KOH. Voltage- and current-clamp recordings were performed using a MultiClamp 700B amplifier (Molecular Devices, Union City, CA). Data were filtered (4 kHz low pass) and digitized at 10 kHz using an ITC-18 (Instrutech, Mineola, NY) controlled by custom software written in IgorPro (Wavemetrics, Lake Oswego, OR). In all experiments in which membrane current was recorded, mitral cells were held at −40 mV to facilitate the recording of GABAA-mediated IPSCs.
Data Analysis.
Miniature IPSCs were detected using custom-written functions in IgorPro (Wavemetrics), which implemented the algorithm described by Kudoh and Taguchi (2002). Event rates for control conditions were calculated in a 1 min window before the addition of drug. For drug conditions, a window 3 min after drug addition was used to calculate event rate. For analysis of the onset kinetics of the enhanced rate of IPSCs, sigmoids were fit to plots of IPSC rate (calculated in 500 ms bins) versus time. These sigmoids were normalized by their maxima and aligned to the first point at which the derivative of a given fitted sigmoid was nonzero. From these aligned curves, a mean sigmoid was generated according to the following equation: y = 1/(exp(thalf− t)/rate), where the parameters thalf and rate were averages of these same parameters from the individual sigmoids. For more standard measures of time-to-peak of these same IPSC rate data (the values reported in the text), an alternative procedure was used. The SD of IPSC event rate (also calculated in 500 ms bins) was calculated for a 1 min period before DHPG addition, and onset was defined as the first time point when IPSC event rate exceeded the mean of the baseline period by 2 SDs. The mean event rate and SD were then calculated for a period of 2 min after the onset (defined as above), and peak time was defined as the first point after onset when this mean event rate was achieved and maintained within 1 SD of this value for ≥10 s (20 samplings of the rate). Data are reported as mean ± SEM unless otherwise indicated. Statistical significance was assessed using paired or unpaired t tests as appropriate.
Drugs.
APV, CNQX, bicuculline, and cyclopiazonic acid (CPA) were all obtained from Sigma-Aldrich (St. Louis, MO) and used at concentrations of 50, 20, 10, and 30 μm, respectively. Gabazine, DHPG, and (S)-(+)-α-amino-4-carboxy-2 methylbenzeneacetic acid (LY367385; LY) were obtained from Tocris (Ellisville, MO), and used at final concentrations of 10, 20, and 100 μm, respectively. 3-((2-Methyl-1,3-thiazol-4-yl)ethynyl)pyridine (MTEP) (Calbiochem, La Jolla, CA) was used at 2 μm and prepared from a 25 mm stock in 100% DMSO. CPA and LY were prepared in stocks of 18 and 100 mm, respectively, both in 100% DMSO. The DHPG stock was prepared in dH20 at 100 mm. The voltage-gated channel blockers TTX, nickel, and cadmium were used at concentrations of 1, 100, and 30 μm, respectively.
Results
RI of AOB mitral cells requires mGluR1 activation
In mitral cells of the MOB and AOB, action potentials or large membrane depolarizations evoke long-lasting inhibition mediated by GABAA receptors (Isaacson and Strowbridge, 1998; Schoppa et al., 1998; Margrie et al., 2001; Taniguchi and Kaba, 2001). This form of inhibition, in which activity in an excitatory neuron results in self-inhibition dependent on dendrodendritic synapses with inhibitory interneurons, is typically termed RI. We adhere to this nomenclature in the remainder of the text to differentiate RI from inhibition that is evoked pharmacologically and not strictly by spiking in a presynaptic excitatory neuron. In an initial set of experiments, we characterized RI in AOB mitral cells. In voltage clamp, a 100 ms depolarization of mitral cells from −40 to +20 mV evoked a slow outward current (Fig. 1A) consisting of many discrete outward synaptic events superimposed on the slower current (Fig. 1A). This current was eliminated by addition of the GABAA-specific antagonist gabazine (SR95531; 10 μm), indicating that these voltage steps evoke RI (Fig. 1A). In similar experiments, trains of seven action potentials at 40 Hz were evoked in mitral cells by current injection, and RI was observed as a slow, gabazine-sensitive hyperpolarization (Fig. 1B). RI in this case was quantified as the voltage integral after the final spike in the series of action potentials (from 70 ms after the final injected current step to 1130 ms after this step; this latter number corresponded to the end of the sweep). We typically saw asynchronous IPSP-like events lasting most or all of this period (Fig. 1B, both control cases). These results demonstrate that both action potentials and membrane depolarization evoke long-lasting RI in AOB mitral cells, consistent with previous results (Taniguchi and Kaba, 2001).
Figure 1.
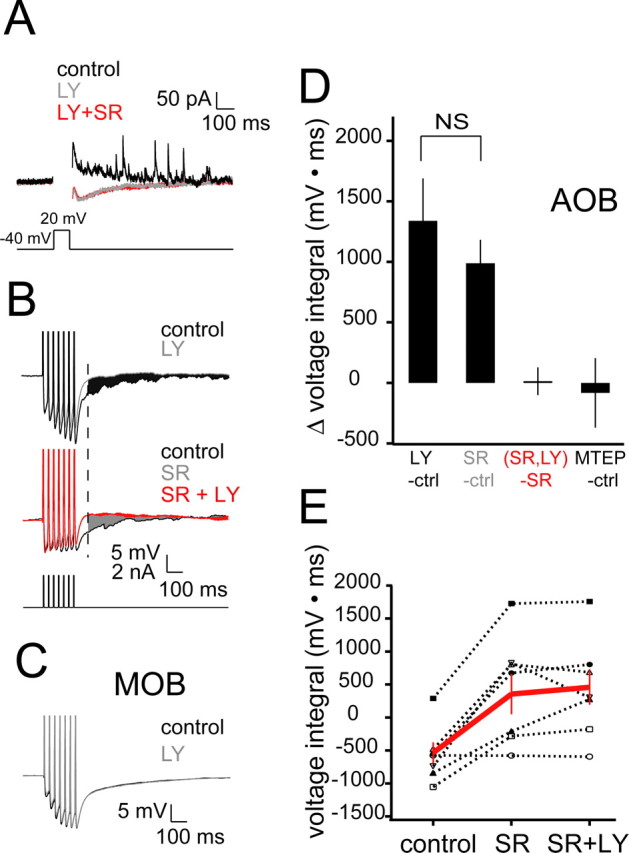
The mGluR1 antagonist LY eliminates RI in AOB mitral cells. A, Single sweeps showing currents evoked by depolarization of a mitral cell from −40 to 20 mV. Action currents during the depolarization are blanked for better visibility. Black, control sweep showing slow outward component and discrete outward events; gray, sweep showing blockade of the slow and discrete outward currents with addition of LY (100 μm); red, sweep showing lack of effect of SR95531 (gabazine; 10 μm) added after LY. B, Single sweeps from current-clamp experiments in which RI was elicited by current pulses (3 ms × 40 Hz). Top, Traces showing the effect of LY on RI. Note the discrete events evident in the control case that are eliminated by LY addition. The black shaded area corresponds to the difference in voltage integrals used to measure changes in RI between the control and LY cases. Middle, Occlusion-type experiments in which RI was first completely blocked by SR and LY subsequently added. The gray shaded area corresponds to the difference between voltage integrals for the control and SR cases. The red shaded area corresponds to the difference between voltage integrals for the SR and SR plus LY cases. The dashed line indicates the time from which the integral was calculated. C, Traces (each an average of 10 sweeps) demonstrating the lack of effect of LY on RI in the MOB (n = 5). Stimulation protocol and integral measurement are identical to those shown in B. Traces below are averages of 10 sweeps under control (black) and LY cases. D, Summary data for experiments similar to those shown in B. The colors of the group labels correspond to the colors of the shaded regions shown in B (n = 8 for LY control; n = 7 for [(SR + LY) − SR alone] group), except for the MTEP control group (n = 7), which has no corresponding trace in B. E, Summary of individual experiments showing the occlusion effect of SR on LY. Red lines and markers indicate mean ± SEM for all experiments. Control, −536.36 ± 160.52 mV · ms; SR, 355.90 ± 306.67 mV · ms; SR plus LY, 459.14 ± 264.49 mV · ms; n = 7. ctrl, Control; SR, SR95531.
We next tested whether RI was modulated by activation of mGluRs in AOB slices. Adding the mGluR1 antagonist LY (100 μm) to the bathing medium resulted in a decrease in RI that was comparable with the decrease seen with the addition of gabazine [Δ voltage integral, 1338.48 ± 351.74 mV · ms (LY; n = 7); 988.63 ± 193.79 mV · ms (gabazine; n = 8; p = 0.40)], suggesting that a large fraction of RI required activation of mGluR1 (Fig. 1B,C). In contrast, the selective mGluR5 antagonist MTEP (2 μm) had no effect on RI (Fig. 1D).
It is possible, although unlikely, that blockade of mGluR1 upregulates a depolarization-evoked inward current that masks RI. To test whether the mGluR1-dependent component of RI is the same as the GABAA-dependent component, we first blocked inhibition with gabazine and then added LY (Fig. 1B, an example is shown at the bottom). Under these conditions, addition of gabazine completely occluded the subsequent effect of LY (Fig. 1A–C). Together, these data demonstrate that mGluR1 activation is necessary for RI in AOB mitral cells in control conditions. In contrast, in experiments in MOB mitral cells, LY had no effect on RI (control voltage integral, −1377.19 ± 452.51 mV · ms; LY voltage integral, −1360.51 ± 481.80 mV · ms; p = 0.92; n = 4) (Fig. 1C) (see also Heinbockel et al., 2006), showing that the specific involvement of mGluR1 in RI is unique to AOB mitral cells.
Activation of mGluRs evokes spontaneous IPSCs in mitral cells
Given the strong dependence of RI on mGluR1, we next sought to determine whether direct activation of group I mGluRs is sufficient to evoke IPSCs in mitral cells. Mitral cells were voltage clamped at −40 mV during addition of the broad-spectrum group I mGluR agonist DHPG (20 μm). DHPG resulted in a more than 10-fold increase in the rate of spontaneous outward events, with the event rate increasing from an average of 0.41 ± 0.20 to 5.86 ± 1.26 Hz (n = 6; p < 0.01; t test) (Fig. 2A,B). These events were completely blocked by 10 μm bicuculline (Fig. 3B,C), indicating that they are mediated by GABAA receptors. In addition, the kinetics and amplitudes of these events were nearly identical for the baseline and DHPG cases (see cumulative amplitude distribution in Fig. 2C, corresponding traces in the inset), suggesting that DHPG is causing release of GABA at reciprocal synapses onto mitral cells. Amplitude, rise-time, and decay-time histograms of observed events were unimodal (data not shown), consistent with a single presynaptic source. The effect of DHPG persisted in the presence of the glutamate receptor blockers APV (50 μm) and CNQX (20 μm) (Fig. 3A), indicating that the effect was at least partially independent of glutamate release from mitral cells. However, the rate of DHPG-evoked IPSCs was lower when ionotropic glutamate receptors were blocked than in the control case (control plus DHPG, 5.89 ± 1.26; APV plus CNQX plus DHPG, 2.40 ± 0.72 Hz; n = 6; p < 0.03).
Figure 2.

DHPG enhances the rate of spontaneous IPSCs in mitral cells. A, Bath application of DHPG (20 μm) greatly increases the occurrence of spontaneous IPSCs in mitral cells. Addition of DHPG in general evoked a slow inward or outward current followed by a rapid transition to a high rate of IPSCs. Traces on the bottom of A show expanded versions of the control and DHPG conditions. B, Cumulative probability plot of interevent intervals for control and DHPG conditions (n = 6). Inset shows mean event rate for the two conditions (control, 0.41 ± 0.2 Hz; DHPG, 5.86 ± 1.26 Hz; n = 6; p < 0.01). C, Cumulative probability plot of event amplitudes for control and DHPG conditions indicating no significant differences. Inset shows average IPSC waveforms for each of the conditions. D, Mean increase in IPSC rate across all mitral cells (n = 6). The mean trace was generated from normalized and aligned sigmoid fits to plots of IPSC rate versus time, as described in the text. Error bars indicate ±SEM. E, Examples of sigmoid fits to IPSC event rates for three representative mitral cells. F, Raw data of the cell indicated by the gray line in E (different cell than in A). Data segment is 20 s after the earliest observed increase in IPSC event rate (as defined in Materials and Methods). Ctrl, Control.
Figure 3.

The increase in IPSC rate with DHPG addition occurs via a mechanism presynaptic to mitral cells. A, Sample sweep in which APV (50 μm) and CNQX (20 μm) were bath applied for 10 min before addition of 20 μm DHPG (baseline period is truncated). B, Sample sweep showing complete absence of spontaneous and DHPG-evoked IPSCs in 10 μm bicuculline. As above, bicuculline addition preceded DHPG addition by 10 min (baseline is truncated). C, Summary data of effects of synaptic blockers on DHPG-induced IPSCs. BIC, Bicuculline.
It is worth noting that the ∼15 pA slow inward current observed in Figure 2A was not observed in all mitral cells. Changes in steady-state current evoked by DHPG were heterogeneous: in some cells DHPG evoked slow inward currents and in others slow outward currents. The mean change in mitral cell holding current was −9.4 ± 10.6 pA (n = 8), which is a smaller and less reliable effect than observed in MOB mitral cells (Heinbockel et al., 2004). In addition, we failed to find a strong relationship between the magnitude of DHPG-evoked current and the DHPG-evoked increase in IPSC rate within cells (R2 = 0.16 for event rate vs holding current). In contrast, the very rapid onset of enhanced IPSCs was seen in all mitral cells tested (Fig. 2D–F). The mean time-to-peak event rate from onset across all cells was 24.3 ± 6.3 s (n = 6) (see Materials and Methods for definitions of onset and peak).
The effect of the mGluR-evoked increase in the rate of IPSCs is primarily mediated by mGluR1
DHPG is a broad spectrum group I mGluR agonist (Ito et al., 1992), and therefore activates both receptors in this class, mGluR1 and mGluR5 (Conn and Pin, 1997). To determine which of these mGluRs contributes to the increase in mitral cell IPSCs, we performed experiments in which DHPG was added in the presence of LY or MTEP, specific blockers of mGluR1 and mGluR5, respectively. When DHPG (20 μm) was added to the bathing medium in the presence of 100 μm LY, the rate of mitral cell IPSCs did not increase (LY alone, 0.35 ± 0.23 Hz; LY plus DHPG, 0.19 ± 0.08 Hz; n = 5; p = 0.53) (Fig. 4A,D). In contrast, the rate of IPSCs was enhanced when DHPG was added in the presence of 2 μm MTEP (MTEP alone, 0.27 ± 0.10 Hz; MTEP plus DHPG, 2.88 ± 0.46 Hz; n = 4; p = 0.0009) (Fig. 4B,D). These results indicate that DHPG-evoked IPSCs are because of the effect of DHPG on mGluR1.
Figure 4.

The effect of the mGluR-evoked increase in the rate of IPSCs is mediated primarily by mGluR1. A, Bath application of DHPG (20 μm) in the presence of mGluR1 antagonist LY (100 μm) eliminated the increase of spontaneous IPSCs in mitral cells. B, Bath application of DHPG in the presence of mGluR5 antagonist MTEP (2 μm) still showed a significant increase in the occurrence of IPSCs in mitral cells. In both cases, slices were bathed in the individual antagonist for 15 min before addition of the antagonist plus DHPG. C, Expanded version of traces showing individual events in two conditions: IPSCs in the presence of the individual antagonist alone (left) and with addition of DHPG along with the antagonist (right). D, Cumulative probability plot of interevent intervals for control DHPG conditions (black line), DHPG in the presence of MTEP (dashed line), and DHPG in the presence of LY (gray line). Inset shows mean event rates for all conditions [LY baseline, 0.35 ± 0.23 Hz; LY plus DHPG, 0.19 ± 0.08 Hz (n = 5); MTEP baseline, 0.27 ± 0.10 Hz; MTEP plus DHPG, 2.88 ± 0.46 Hz (n = 4)].
DHPG-evoked IPSCs require calcium influx but not sodium spikes
As noted above and similar to what others have seen in the MOB (Heinbockel et al., 2004), we sometimes observed a slow DHPG-evoked inward current in mitral cells that accompanied the increase in IPSCs (Fig. 2A). Hence, a potential mechanism for the increased barrage of spontaneous IPSCs is direct depolarization of mitral cells, with concomitant elevated mitral cell firing rates. Thus, we next tested whether action potentials are required for the increase in IPSC rate by mGluR agonists by including 1 μm TTX in the bath solution before addition of DHPG. Under these conditions, the rate of mitral cell IPSCs was still increased (baseline, 0.04 ± 0.02 Hz; DHPG, 1.99 ± 0.76 Hz; p < 0.05; n = 4) (Fig. 5), indicating that sodium spikes are not required for mGluR activation to evoke IPSCs. Because the IPSC rate was lower in TTX than control conditions, sodium channels and spontaneous spiking by granule cells are likely to play a role in setting the rate of IPSCs. In contrast, when the calcium channel blockers cadmium (30 μm) and nickel (100 μm) were included in the bath, DHPG did not cause a significant increase in IPSC rate (baseline, 0.34 ± 0.09Hz; DHPG, 0.33 ± 0.17 Hz; p > 0.05; n = 5) (Fig. 5).
Figure 5.
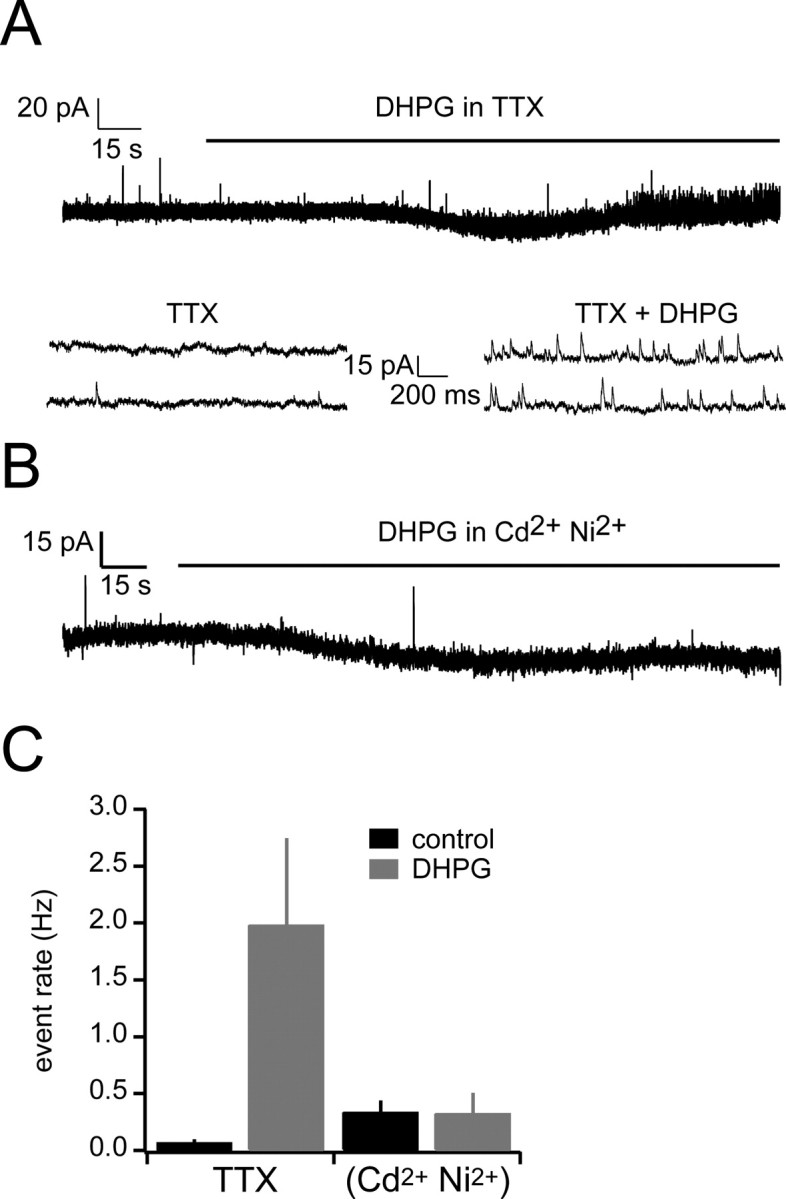
The increase in spontaneous IPSCs is dependent on VGCCs but not sodium channels. A, Sample sweep in which DHPG was bath applied after addition of TTX. Bottom shows expanded sections from the baseline period and TTX addition period. B, Sample sweep in which DHPG was bath applied after addition of cadmium and nickel. C, Summary data for the effects of voltage-gated channel blockers on DHPG-induced IPSCs.
Release from internal stores is not required for DHPG-evoked inhibition
In a number of cell types, activation of mGluRs is coupled to release of calcium from internal stores either via second messengers or calcium influx through voltage-gated calcium channels (VGCCs) (Chavis et al., 1996; Finch and Augustine, 1998; Takechi et al., 1998; del Rio et al., 1999). The experiments above indicate that calcium influx through VGCCs is necessary to evoke recurrent IPSCs but does not address the question of whether GABA release also depends on release of calcium from internal stores. To test this possibility, we bath applied the sarcoplasmic reticulum calcium pump inhibitor CPA (30 μm) to deplete internal calcium stores before adding DHPG. Enhanced rates of IPSCs were still observed under these conditions, with a mean DHPG-evoked rate of 8.23 ± 2.27 Hz (n = 5) (Fig. 6), comparable with that observed under control conditions (5.86 ±.1.26 Hz; n = 6). This indicates that DHPG likely acts by causing a direct, calcium-dependent depolarization in granule cells, possibly activation of a calcium conductance, or closure of a potassium channel (Schoppa and Westbrook, 1997) and that GABA release is then triggered by voltage-gated calcium current in granule cells.
Figure 6.

DHPG-evoked IPSCs do not depend on internal calcium stores. A, Application of DHPG in the presence of CPA (100 μm) still induced a large increase in the occurrence of IPSCs in mitral cells. The slice was bathed in CPA for 20 min before the application of CPA plus DHPG. B, Cumulative probability plot of interevent intervals for CPG plus DHPG (black line) and control CPA (gray line). Inset shows event rates for both conditions (CPA only, 0.37 ± 0.17 Hz; CPA plus DHPG, 8.23 ± 2.27 Hz; n = 5).
Mechanisms of mGluR1 action in granule cells
In the MOB and AOB, RI evoked by mitral cell spiking is mediated predominantly by granule cells, the major interneuron population (Price and Powell, 1970a,b; Isaacson and Strowbridge, 1998; Schoppa et al., 1998; Isaacson, 2001). Given that we saw complete elimination of RI in AOB mitral cells with blockade of mGluR1 (Fig. 1) and that DHPG-evoked IPSCs in mitral cells persisted when ionotropic glutamatergic transmission was blocked (Fig. 3), we reasoned that granule cells are likely to be the major cell type contributing to the large increase in miniature IPSCs observed in our experiments. In addition, the persistence of DHPG-evoked spontaneous IPSCs in the presence of TTX, but their elimination by blockers of VGCCs, is consistent with studies in granule cells demonstrating that local or global calcium spikes are potential triggers of transmitter release (Egger et al., 2003, 2005; Pinato and Midtgaard, 2003, 2005; Zelles et al., 2006). Thus, one possible mechanism for the action of DHPG is via direct depolarization of granule cells. In this scenario, glutamate released from mitral cells would bind to metabotropic receptors on granule cells and cause a local or global calcium elevation sufficient to evoke GABA release from granule cells. To test this possibility, we recorded from granule cell somata in the presence of blockers of fast glutamatergic transmission (APV, 50 μm; CNQX, 20 μm) and added DHPG (20 μm). In four granule cells tested, we observed a depolarization of 6.79 ± 0.79 mV by the third minute after DHPG addition, an effect that persisted for the duration of drug addition (Fig. 7A). Granule cells were also depolarized by 5.15 ± 0.42 mV (n = 5) by DHPG when MTEP (2 μm) was included in the bath solution (Fig. 7B), indicating that activation of mGluR1 is sufficient for the effect. In contrast, no significant depolarization was observed when mGluR1 was blocked by LY before DHPG addition (n = 5) (Fig. 7B). It is worth noting granule cell spines are electrotonically isolated from the granule cell soma (Rall and Shepherd, 1968; Woolf et al., 1991; Egger and Urban, 2006), and that depolarization observed at the soma may be an underestimate of voltage changes more local to spines and associated release machinery.
Figure 7.

DHPG depolarizes granule cells. A, Left, Group data showing granule cell depolarization with addition of DHPG (20 μm) starting at t = 0 (n = 4; mean depolarization, 6.79 ± 0.79; shaded areas indicate ±SEM). Right, An example experiment is shown. Negative-going deflections are hyperpolarizing pulses used to monitor input resistance. B, Same layout and experimental design as in A, only with 2 μm MTEP (n = 5) or 100 μm LY (n = 5) included in the bath to block mGluR5 or mGluR1, respectively. Right, Example experiment showing persistence of DHPG-evoked granule cell depolarization when mGluR5 is antagonized. C, Effectiveness of LY in blocking RI at normal (1.0 mm; n = 7) and low (0.2 mm; n = 6) magnesium concentrations. D, Single sample sweeps showing RI after seven evoked spikes at 40 Hz in 0.2 mm Mg2+. Gray, Control; red, after LY alone; black, LY plus gabazine.
Studies in both the MOB and AOB have demonstrated that NMDARs, likely those present on granule cell spines, are important for RI of mitral cells (Chen et al., 2000). In control conditions, blockade of NMDARs results in near complete elimination of action potential-evoked RI; some of these findings also suggest that calcium influx through NMDARs can be directly coupled to transmitter release (Chen et al., 2000; Halabisky et al., 2000). Given that NMDAR activation is require for RI in control conditions, our results (Fig. 1) suggest that activation of mGluR1 may play a modulatory but still critical role in evoking transmitter release from granule cells.
One possibility we considered was that because of its depolarizing effect on granule cells, mGluR1 may facilitate voltage-dependent calcium influx that is directly coupled to GABA release. Specifically, mGluR1 may depolarize granule cells by an amount sufficient to relieve magnesium blockade of NMDARs and facilitate reciprocal communication via the “classical” ionotropic pathway. One prediction of this model is that under conditions permissive for NMDAR activation, mGluR1 antagonists should be relatively less effective at eliminating spike-evoked RI. To test this, we evoked action potentials (seven spikes at 40 Hz) in mitral cells under low magnesium (0.2 mm) conditions and measured the magnitude of RI before and after addition of LY. In agreement with our proposed mechanism, LY did not cause a significant change in RI in 0.2 mm magnesium (Fig. 7C,D) (76 ± 24% of control post-LY) compared with the change observed in 1.0 mm magnesium (Figs. 1, 7C) (28 ± 17% of control post-LY; p = 0.012; n = 6).
Discussion
We report two main findings in this study. First, under physiological conditions, mGluR1 is required for RI of AOB mitral cells. Second, direct activation of group I mGluRs evokes robust GABAergic inhibition of mitral cells via a presynaptic mechanism that depends on VGCCs.
Metabotropic glutamate receptors are expressed throughout the CNS and couple glutamate binding to a host of changes in the intrinsic and synaptic properties of neurons. These changes include spatially localized increases in intracellular calcium via activation of ryanodine receptors (Chavis et al., 1996; Finch and Augustine, 1998; Takechi et al., 1998; del et al., 1999), up or downregulation of potassium and calcium conductances (Charpak et al., 1990; Schoppa and Westbrook, 1997; Chavis et al., 1998; Fagni et al., 2000; Heinbockel et al., 2006), and reduction in the strength of electrical synapses (Landisman and Connors, 2005). These cellular responses often serve to regulate network “tone” and modulate the propagation and extent of neural activity. In the MOB, activation of mGluR1 is known to increase mitral cell excitability, and contributes a slow, phasic component to the responses of mitral cells to olfactory nerve stimulation (Heinbockel et al., 2004; De Saint and Westbrook, 2005; Ennis et al., 2006).
Much of the interest in metabotropic glutamate receptors in the olfactory bulb stems from their dense expression in this structure, and from reports of their role in regulation of AOB-dependent behaviors. Mitral cells express at least four mGluRs including mGluR1, 2, 7, and 8 (Masu et al., 1991; Martin et al., 1992; Shigemoto et al., 1992). mGluR2s have been localized to granule cells and their activation is reported to underlie suppression of granule cell inhibition of mitral cell inhibition that is linked to forms of olfactory memory (Kaba et al., 1994). Despite the considerable attention that has been given to the role of specific activation of mGluR2 in this form of olfactory memory (Hayashi et al., 1993; Kaba et al., 1994), synaptic release of glutamate from mitral cells is likely to activate both class 1 and class 2 mGluRs, and thus, understanding the role that both of these receptor types play in regulation of olfactory bulb neurons is critical. In this regard, our observation that activation of mGluR1 results in effects on granule cell activity and inhibition at reciprocal synapses that oppose the effects reported for mGluR2 activation raises interesting questions about how these receptors influence mitral cell activity.
mGluR1 immunoreactivity in the AOB is concentrated largely in the mitral-tufted cell layer, with less robust staining observed in the granule cell layer (Sahara et al., 2001). Given that we observed granule cell depolarization in response to DHPG (Fig. 7) and that RI was eliminated by an antagonist of mGluR1 but not mGluR5 (Fig. 1), our results are consistent with an expression pattern in which mGluR1 is highly localized to granule cell spines contacting mitral cell dendrites.
Our observations that mGluR activation is required for RI is particularly surprising, because experiments in both MOB and AOB have demonstrated that reciprocal inhibition of mitral cells is eliminated by blockade of AMPA receptors and NMDARs. Our interpretation of this finding is that activation of mGluR1, by glutamate released from mitral/tufted cells, is necessary to create conditions favorable for dendrodendritic inhibition via the “classical” reciprocal ionotropic pathway, a result supported by the low-magnesium experiments of Figure 7. This may result from direct, mGluR-dependent depolarization of granule cells, similar to what we observed in Figure 7. With typical magnesium concentrations (our bath solution contained 1 mm Mg2+), NMDARs in granule cells will be blocked by Mg2+, especially at the hyperpolarized resting potentials (approximately −80 mV) typical of these cells. The depolarization of granule cells by mGluR activation may thus be required to relieve this Mg2+ block, which is in turn required to elicit GABA release from granule cells.
It is interesting that the direct effects of DHPG (depolarization of granule cells, inconsistent changes in mitral cell holding current) occur on the timescale of minutes, whereas the synaptic effects of DHPG (increase in mitral cell IPSC rate) occur with considerable delay but rapid onset (∼20 s). Although we did not test directly how these two effects are inter-related, we offer two speculations on how the timescales can be reconciled. First, assuming that granule cell voltage gates RI by the NMDAR-dependent mechanism described in the Results (Fig. 7C), DHPG-evoked IPSCs will not begin until relatively depolarized potentials are reached. However, given the negative slope conductance of the NMDAR occurring at approximately −65 mV, when these potentials are reached, a large fraction of NMDARs may become available very rapidly, resulting in rapid onset of the increase in spontaneous IPSCs. Another possibility is that the rapid onset of IPSCs may reflect a population-level phenomenon, such as the onset of synchrony among granule cells.
We also observed that direct application of DHPG evoked IPSCs in mitral cells, even when excitatory synaptic transmission was blocked (Fig. 3). This observation indicates that activation of mGluR1 by synaptically released glutamate may result directly in GABA release without requiring activation of AMPA receptors and NMDARs. However, bath application of an mGluR agonist is likely to cause stronger mGluR1 activity than glutamate supplied by physiological release from mitral cells. Nevertheless, the results from these agonist experiments are important for understanding the cellular consequences of activating mGluR1. In particular, the results of the experiments shown in Figures 5 and 6 indicate that activation of group I mGluRs evokes release in a manner that is dependent on VGCCs but independent of sodium spikes and internal calcium stores. These observations suggest that mGluRs are likely to elicit the kind of large depolarization required to open VGCCs coupled to release. However, because we observe only ∼7 mV depolarization in granule cell somatic recordings, it is possible that granule cell dendrites are substantially more depolarized by mGluR activation than are granule cell somata. Alternatively, mGluRs may facilitate activation of calcium channels by voltage-independent mechanisms. Several studies have shown that granule cells exhibit low threshold calcium spikes that can be localized to granule cell spines or can propagate along the granule cell apical dendrite (Egger et al., 2003, 2005). In summary, we propose that mGluR1 activation in granule cells may act in two capacities: first, they may play a facilitating role in RI by partially relieving magnesium blockage of NMDARs, and second, under conditions of high network activity, they may be capable of evoking release in a manner that is independent of activation of ionotropic glutamate receptors.
A number of studies have examined the extent to which metabotropic glutamate receptors control information throughput in sensory systems, a function sometimes referred to as “gating.” In the thalamus, for example, mGluRs are coupled to presynaptic release from GABA-containing interneurons, and strongly modulate feedforward information flow to higher cortical areas (Cox and Sherman, 2000; Govindaiah and Cox, 2004). Similarly, in the main olfactory system, a presynaptic group II mGluR has been shown to be critical for habituation of the odor-induced heart-rate orienting response (Best et al., 2005). The AOB provides perhaps the most tractable example of a feedforward system in which information flow is controlled by mGluRs. Classically, work has addressed how activation of mGluR2 in granule cell spines results in diminished RI of mitral cells. This reduction in inhibition is believed to be a neural substrate of the Bruce effect, the failure of embryo implantation after exposure of a female rodent to pheromones of nonstud males (Hayashi et al., 1993; Kaba et al., 1994; Brennan and Keverne, 1997). Regardless of the precise mechanisms underlying this behavior, the Bruce effect highlights the importance of controlling the propagation of activity in the AOB, because presumably small subsets of active mitral cells can communicate signals that result in terminated pregnancy. The requirement for mGluR1 in RI observed in the present study suggests that this receptor plays a powerful role in controlling how pheromone detection is coupled to behavioral and endocrine responses.
Footnotes
This work was supported by the National Institute on Deafness and Other Communication Disorders Grants R01 DC005798 and R21 DC006631 (N.N.U.), National Research Service Award predoctoral fellowship F31 DC08230 (J.B.C.), and Human Frontiers Science Program Grant RGY79/2005. We thank Greg LaRocca for excellent technical assistance.
References
- Best AR, Thompson JV, Fletcher ML, Wilson DA. Cortical metabotropic glutamate receptors contribute to habituation of a simple odor-evoked behavior. J Neurosci. 2005;25:2513–2517. doi: 10.1523/JNEUROSCI.5298-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PA, Keverne EB. Neural mechanisms of mammalian olfactory learning. Prog Neurobiol. 1997;51:457–481. doi: 10.1016/s0301-0082(96)00069-x. [DOI] [PubMed] [Google Scholar]
- Brennan P, Kaba H, Keverne EB. Olfactory recognition: a simple memory system. Science. 1990;250:1223–1226. doi: 10.1126/science.2147078. [DOI] [PubMed] [Google Scholar]
- Charpak S, Gahwiler BH, Do KQ, Knopfel T. Potassium conductances in hippocampal neurons blocked by excitatory amino-acid transmitters. Nature. 1990;347:765–767. doi: 10.1038/347765a0. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Chavis P, Ango F, Michel JM, Bockaert J, Fagni L. Modulation of big K+ channel activity by ryanodine receptors and L-type Ca2+ channels in neurons. Eur J Neurosci. 1998;10:2322–2327. doi: 10.1046/j.1460-9568.1998.00243.x. [DOI] [PubMed] [Google Scholar]
- Chen WR, Xiong W, Shepherd GM. Analysis of relations between NMDA receptors and GABA release at olfactory bulb reciprocal synapses. Neuron. 2000;25:625–633. doi: 10.1016/s0896-6273(00)81065-x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Cox CL, Sherman SM. Control of dendritic outputs of inhibitory interneurons in the lateral geniculate nucleus. Neuron. 2000;27:597–610. doi: 10.1016/s0896-6273(00)00069-6. [DOI] [PubMed] [Google Scholar]
- De Saint JD, Westbrook GL. Detecting activity in olfactory bulb glomeruli with astrocyte recording. J Neurosci. 2005;25:2917–2924. doi: 10.1523/JNEUROSCI.5042-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rio E, McLaughlin M, Downes CP, Nicholls DG. Differential coupling of G-protein-linked receptors to Ca2+ mobilization through inositol(1,4,5)trisphosphate or ryanodine receptors in cerebellar granule cells in primary culture. Eur J Neurosci. 1999;11:3015–3022. doi: 10.1046/j.1460-9568.1999.00714.x. [DOI] [PubMed] [Google Scholar]
- Dong H, Hayar A, Ennis M. Activation of group 1 metabotropic glutamate receptors on main olfactory bulb granule and periglomerular cells enhances synaptic inhibition of mitral cells. J Neurosci. 2007;27:5654–5663. doi: 10.1523/JNEUROSCI.5495-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulac C, Torello AT. Molecular detection of pheromone signals in mammals: from genes to behaviour. Nat Rev Neurosci. 2003;4:551–562. doi: 10.1038/nrn1140. [DOI] [PubMed] [Google Scholar]
- Egger V, Urban NN. Dynamic connectivity in the mitral cell-granule cell microcircuit. Semin Cell Dev Biol. 2006;17:424–432. doi: 10.1016/j.semcdb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Egger V, Svoboda K, Mainen ZF. Mechanisms of lateral inhibition in the olfactory bulb: efficiency and modulation of spike-evoked calcium influx into granule cells. J Neurosci. 2003;23:7551–7558. doi: 10.1523/JNEUROSCI.23-20-07551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Svoboda K, Mainen ZF. Dendrodendritic synaptic signals in olfactory bulb granule cells: local spine boost and global low-threshold spike. J Neurosci. 2005;25:3521–3530. doi: 10.1523/JNEUROSCI.4746-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis M, Zhu M, Heinbockel T, Hayar A. Olfactory nerve-evoked, metabotropic glutamate receptor-mediated synaptic responses in rat olfactory bulb mitral cells. J Neurophysiol. 2006;95:2233–2241. doi: 10.1152/jn.01150.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J. Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci. 2000;23:80–88. doi: 10.1016/s0166-2236(99)01492-7. [DOI] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Govindaiah , Cox CL. Synaptic activation of metabotropic glutamate receptors regulates dendritic outputs of thalamic interneurons. Neuron. 2004;41:611–623. doi: 10.1016/s0896-6273(04)00013-3. [DOI] [PubMed] [Google Scholar]
- Halabisky B, Friedman D, Radojicic M, Strowbridge BW. Calcium influx through NMDA receptors directly evokes GABA release in olfactory bulb granule cells. J Neurosci. 2000;20:5124–5134. doi: 10.1523/JNEUROSCI.20-13-05124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Momiyama A, Takahashi T, Ohishi H, Ogawa-Meguro R, Shigemoto R, Mizuno N, Nakanishi S. Role of a metabotropic glutamate receptor in synaptic modulation in the accessory olfactory bulb. Nature. 1993;366:687–690. doi: 10.1038/366687a0. [DOI] [PubMed] [Google Scholar]
- Heinbockel T, Heyward P, Conquet F, Ennis M. Regulation of main olfactory bulb mitral cell excitability by metabotropic glutamate receptor mGluR1. J Neurophysiol. 2004;92:3085–3096. doi: 10.1152/jn.00349.2004. [DOI] [PubMed] [Google Scholar]
- Heinbockel T, Laaris N, Ennis M. Metabotropic glutamate receptors in the main olfactory bulb drive granule cell-mediated inhibition. J Neurophysiol. 2006;97:858–870. doi: 10.1152/jn.00884.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS. Mechanisms governing dendritic gamma-aminobutyric acid (GABA) release in the rat olfactory bulb. Proc Natl Acad Sci USA. 2001;98:337–342. doi: 10.1073/pnas.021445798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron. 1998;20:749–761. doi: 10.1016/s0896-6273(00)81013-2. [DOI] [PubMed] [Google Scholar]
- Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S, Sugiyama H. 3,5-Dihydroxyphenyl-glycine: a potent agonist of metabotropic glutamate receptors. NeuroReport. 1992;3:1013–1016. [PubMed] [Google Scholar]
- Kaba H, Hayashi Y, Higuchi T, Nakanishi S. Induction of an olfactory memory by the activation of a metabotropic glutamate receptor. Science. 1994;265:262–264. doi: 10.1126/science.8023145. [DOI] [PubMed] [Google Scholar]
- Kevetter GA, Winans SS. Connections of the corticomedial amygdala in the golden hamster. I. Efferents of the “vomeronasal amygdala.”. J Comp Neurol. 1981;197:81–98. doi: 10.1002/cne.901970107. [DOI] [PubMed] [Google Scholar]
- Kudoh SN, Taguchi T. A simple exploratory algorithm for the accurate and fast detection of spontaneous synaptic events. Biosens Bioelectron. 2002;17:773–782. doi: 10.1016/s0956-5663(02)00053-2. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Connors BW. Long-term modulation of electrical synapses in the mammalian thalamus. Science. 2005;310:1809–1813. doi: 10.1126/science.1114655. [DOI] [PubMed] [Google Scholar]
- Luo M, Fee MS, Katz LC. Encoding pheromonal signals in the accessory olfactory bulb of behaving mice. Science. 2003;299:1196–1201. doi: 10.1126/science.1082133. [DOI] [PubMed] [Google Scholar]
- Margrie TW, Sakmann B, Urban NN. Action potential propagation in mitral cell lateral dendrites is decremental and controls recurrent and lateral inhibition in the mammalian olfactory bulb. Proc Natl Acad Sci USA. 2001;98:319–324. doi: 10.1073/pnas.011523098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Blackstone CD, Huganir RL, Price DL. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron. 1992;9:259–270. doi: 10.1016/0896-6273(92)90165-a. [DOI] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Meisami E, Bhatnagar KP. Structure and diversity in mammalian accessory olfactory bulb. Microsc Res Tech. 1998;43:476–499. doi: 10.1002/(SICI)1097-0029(19981215)43:6<476::AID-JEMT2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the messenger RNA for a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat. Neuroscience. 1993;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Neki A, Mizuno N. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res. 1998;30:65–82. doi: 10.1016/s0168-0102(97)00120-x. [DOI] [PubMed] [Google Scholar]
- Pinato G, Midtgaard J. Regulation of granule cell excitability by a low-threshold calcium spike in turtle olfactory bulb. J Neurophysiol. 2003;90:3341–3351. doi: 10.1152/jn.00560.2003. [DOI] [PubMed] [Google Scholar]
- Pinato G, Midtgaard J. Dendritic sodium spikelets and low-threshold calcium spikes in turtle olfactory bulb granule cells. J Neurophysiol. 2005;93:1285–1294. doi: 10.1152/jn.00807.2004. [DOI] [PubMed] [Google Scholar]
- Price JL, Powell TP. The morphology of the granule cells of the olfactory bulb. J Cell Sci. 1970a;7:91–123. doi: 10.1242/jcs.7.1.91. [DOI] [PubMed] [Google Scholar]
- Price JL, Powell TP. The synaptology of the granule cells of the olfactory bulb. J Cell Sci. 1970b;7:125–155. doi: 10.1242/jcs.7.1.125. [DOI] [PubMed] [Google Scholar]
- Rall W, Shepherd GM. Theoretical reconstruction of field potentials and dendrodendritic synaptic interactions in olfactory bulb. J Neurophys. 1968;31:884–915. doi: 10.1152/jn.1968.31.6.884. [DOI] [PubMed] [Google Scholar]
- Sahara Y, Kubota T, Ichikawa M. Cellular localization of metabotropic glutamate receptors mGluR1, 2/3, 5 and 7 in the main and accessory olfactory bulb of the rat. Neurosci Lett. 2001;312:59–62. doi: 10.1016/s0304-3940(01)02184-x. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Modulation of mEPSCs in olfactory bulb mitral cells by metabotropic glutamate receptors. J Neurophysiol. 1997;78:1468–1475. doi: 10.1152/jn.1997.78.3.1468. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, Westbrook GL. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci. 1998;18:6790–6802. doi: 10.1523/JNEUROSCI.18-17-06790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurol. 1992;322:121–135. doi: 10.1002/cne.903220110. [DOI] [PubMed] [Google Scholar]
- Stowers L, Holy TE, Meister M, Dulac C, Koentges G. Loss of sex discrimination and male-male aggression in mice deficient for TRP2. Science. 2002;295:1493–1500. doi: 10.1126/science.1069259. [DOI] [PubMed] [Google Scholar]
- Stuart G, Spruston N. Probing dendritic function with patch pipettes. Curr Opin Neurobiol. 1995;5:389–394. doi: 10.1016/0959-4388(95)80053-0. [DOI] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Kaba H. Properties of reciprocal synapses in the mouse accessory olfactory bulb. Neuroscience. 2001;108:365–370. doi: 10.1016/s0306-4522(01)00427-4. [DOI] [PubMed] [Google Scholar]
- Urban NN. Lateral inhibition in the olfactory bulb and in olfaction. Physiol Behav. 2002;77:607–612. doi: 10.1016/s0031-9384(02)00895-8. [DOI] [PubMed] [Google Scholar]
- Urban NN, Castro JB. Tuft calcium spikes in accessory olfactory bulb mitral cells. J Neurosci. 2005;25:5024–5028. doi: 10.1523/JNEUROSCI.0297-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf TB, Shepherd GM, Greer CA. Local information processing in dendritic trees: subsets of spines in granule cells of the mammalian olfactory bulb. J Neurosci. 1991;11:1837–1854. doi: 10.1523/JNEUROSCI.11-06-01837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelles T, Boyd JD, Hardy AB, Delaney KR. Branch-specific Ca2+ influx from Na+-dependent dendritic spikes in olfactory granule cells. J Neurosci. 2006;26:30–40. doi: 10.1523/JNEUROSCI.1419-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]