

Abstract
Inflammation and neurodegeneration coexist in many acute damage and chronic CNS disorders (e.g., stroke, Alzheimer's disease, Parkinson's disease). A well characterized animal model of brain damage involves administration of kainic acid, which causes limbic seizure activity and subsequent neuronal death, especially in the CA1 and CA3 pyramidal cells and interneurons in the hilus of the hippocampus. Our previous work demonstrated a potent anti-inflammatory and neuroprotective effect of two thiadiazolidinones compounds, NP00111 (2,4-dibenzyl-[1,2,4]thiadiazolidine-3,5-dione) and NP01138 (2-ethyl-4-phenyl-[1,2,4]thiadiazolidine-3,5-dione), in primary cultures of cortical neurons, astrocytes, and microglia. Here, we show that injection of NP031112, a more potent thiadiazolidinone derivative, into the rat hippocampus dramatically reduces kainic acid-induced inflammation, as measured by edema formation using T2-weighted magnetic resonance imaging and glial activation and has a neuroprotective effect in the damaged areas of the hippocampus. Last, NP031112-induced neuroprotection, both in vitro and in vivo, was substantially attenuated by cotreatment with GW9662 (2-chloro-5-nitrobenzanilide), a known antagonist of the nuclear receptor peroxisome proliferator-activated receptor γ, suggesting that the effects of NP031112 can be mediated through activation of this receptor. As such, these findings identify NP031112 as a potential therapeutic agent for the treatment of neurodegenerative disorders.
Keywords: excitotoxicity, neurodegenerative diseases, neuroinflammation, neuroprotection, peroxisome proliferator-activated receptor, thiadiazolidinones
Introduction
Excitotoxic brain damage is considered one of the major mechanisms by which neurons die in the adult CNS (Choi, 1988; Lipton and Nicotera, 1998). Studies using kainic acid (KA), an analog of glutamate, have provided major contributions to the understanding of neuronal cell death caused by excitotoxicity. Administration of KA in rats is known to induce a sequence of altered behavioral events characterized by epileptiform seizures (Ben-Ari et al., 1980; Sperk, 1994), which are followed by neurodegeneration in specific brain regions, such as the hippocampus, piriform cortex, thalamus, and amygdala. In the hippocampus, the CA3 pyramidal cells and interneurons in the hilus of the dentate gyrus are the most vulnerable, followed by CA1 pyramidal cells (Coyle, 1983; Sperk et al., 1985; Tauck and Nadler, 1985). An essential event in excitotoxicity is the direct and constant activation by KA of specific glutamate receptors, which results in neuronal cell death (Choi, 1988; Doble, 1999; Wang et al., 2005). In addition, glial cells, as mediators of the inflammatory response, also play an important role in the course of KA-induced hippocampal neurodegeneration. Activated astrocytes and microglial cells proliferate and increase the expression of genes implicated in the production of nitric oxide and cytokines. These agents, when released from activated glia, can contribute noticeably to the expansion of brain injury and the delayed loss of neurons (Barone and Feuerstein, 1999; del Zoppo et al., 2000).
Neuroprotection involves the use of agents to prevent disease or injury of the nervous system by inhibiting one or more events leading to neuronal death. Brain injury, as a consequence of vascular or traumatic accidents or neurodegenerative diseases, is a common event, and therefore, neuroprotection has emerged as an increasingly important segment of the biopharmaceutical market over the last years, representing a major source of untapped potential for development of new therapeutic products and strategies. Formerly, most therapy for neurological disorders was directed at symptoms; however, as the understanding of the molecular basis of neurological disease and injury increases, opportunities for new approaches appear. The peroxisome proliferator-activated receptor γ (PPARγ) has recently emerged as a therapeutic target for neuroprotection (Petrova et al., 1999; Bernardo et al., 2000; Landreth and Heneka, 2001; Diab et al., 2002; Townsend and Pratico, 2005). PPARγ ligands, including the antidiabetic thiazolidinediones (TZDs), nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, and the prostaglandin derivative 15-deoxy-Δ-12,14-prostaglandin J2, have been implicated in the anti-inflammatory process in diverse tissues, including the brain (Ricote et al., 1999; Kielian and Drew, 2003). In line with these results, we have recently shown that two thiadiazolidinone (TDZD) compounds, NP00111 (2,4-dibenzyl-[1,2,4]thiadiazolidine-3,5-dione) and NP01138 (2-ethyl-4-phenyl-[1,2,4]thiadiazolidine-3,5-dione), inhibited the activation of astrocytes and microglial cells and were neuroprotective in vitro through a mechanism apparently involving PPARγ activation (Luna-Medina et al., 2005), suggesting that TDZDs may act as anti-inflammatory and neuroprotective agents in the CNS.
In the present study, we demonstrate that NP031112, another TDZD compound, which is presently being developed for the treatment of Alzheimer's disease, is a potent anti-inflammatory and neuroprotective agent against KA-induced in vivo excitotoxicity. TDZD compounds have been shown to be non-ATP competitive inhibitors of glycogen synthase kinase 3β (GSK-3β) (Martinez et al., 2002a,b). It has also been shown that inactivation of GSK-3β protects against KA-induced neurotoxicity in vivo (Goodenough et al., 2004), and therefore this pathway could be involved in the NP031112 response to the excitotoxin in vivo. However, our results suggest that NP031112 may act as well through its ability to activate the PPARγ nuclear receptor.
Materials and Methods
Cell culture, transfection, and treatment.
Rat primary astrocytes, microglia, and neurons were harvested and cultured as described previously (Luna-Medina et al., 2005). The purity of the cultures was >95%, as determined by immunofluorescence analysis using anti-cd11b (OX-42) to detect microglial cells, glial fibrillary acidic protein (GFAP) to identify astrocytes, and anti-microtubule-associated protein 2 (MAP2) to identify neurons. NP031112 (2.5 μm) was added to the culture medium of astrocytes and microglia 1 h before exposure to glutamate (500 μm), cells were incubated for 24 h before tissue culture medium was collected, and the cells were evaluated for tumor necrosis factor-α (TNF-α) and cyclooxygenase type 2 (COX-2) expression. For transient transfection experiments, primary cultures of astrocytes were transfected with the reporter plasmid pPPRE-tk-luc, containing three PPARγ consensus binding sites upstream of a minimal promoter using Transfast (Promega, Madison, WI) according to the manufacturer's guidelines. Typically, cells received 0.2 μg of luciferase reporter plasmid and were harvested 24 h after treatment with different concentrations of NP031112 for determination of luciferase and β-galactosidase (to determine transfection efficiency) activities. Each transient transfection experiment was repeated at least three times in triplicate.
Antibodies.
Mouse monoclonal antibodies to MAP2 and GFAP were from Sigma (St. Louis, MO). Mouse OX-42 and anti-neuN monoclonal antibodies were purchased from Serotec (Duseldorf, Germany), Millipore (Bedford, MA), respectively. Polyclonal anti-TNF-α and anti-COX-2 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunocytochemistry.
At the end of the treatment period, the cultures, grown on glass coverslips in 24-well cell culture plates, were washed with PBS and fixed for 30 min with 4% paraformaldehyde at 25°C and permeabilized with 0.1% Triton X-100 for 30 min at 37°C. After 1 h incubation with the corresponding primary antibody, cells were washed with PBS and incubated with an Alexa-labeled secondary antibody (Invitrogen, San Diego, CA) for 45 min at 37°C. Images were acquired using a Radiance 2100 confocal microscope (Bio-Rad, Hercules, CA), with a 350 nm diode laser to excite DAPI (4,6,diamidino-2-phenylindole) and 647 nm laser line to excite Alexa 647. Confocal microscope settings were adjusted to produce the optimum signal-to-noise ratio. To compare fluorescence signals from different preparations, settings were fixed for all samples within the same analysis. Fluorescence analysis was performed using LaserPix software (Bio-Rad). A quantitative analysis of labeled cells was undertaken using the image analySIS software (Soft Imaging System, Münster, Germany) and normalized to total nuclei. Areas to be counted were traced at high power (400×), and at least four different counting fields were selected at random per culture.
Measurement of apoptosis.
To calculate the extent of apoptotic cell death, cortical neuronal cultures were treated or not with NP031112 and incubated with glutamate (100 μm), and phosphatydilserine exposure on the surface of apoptotic cells was detected by confocal microscopy after staining with Annexin V-FITC (Bender MedSystems, Vienna, Austria). Neuronal cell death was assessed by counting the percentage of Annexin-V-positive cells in four independent high-magnification (200×) fields per culture, as described above.
KA administration.
Adult male Wistar rats (8–12 weeks old) were used in this study. Adequate measures were taken to minimize pain or discomfort of animals. Experiments were performed in accordance with the European Communities Council, directive 86/609/EEC. Rats (n ≥ 5 per group) were anesthetized by intraperitoneal injection of ketamine (60 mg/kg) and Domtor (5 μg/kg) and placed into a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). KA (1 μg in 2.5 μl PBS) alone or in combination with NP031112 (2 ng in 2.5 μl PBS) was injected into the hippocampus [coordinates from bregma: posterior, −3.0 mm; lateral, −2.0 mm; depth, 3.5 mm; according to the atlas of Paxinos and Watson (1998)]. Control animals of the same age were injected with vehicle. Two groups of animals also received 0.7 μg of the PPARγ antagonist GW9662 (2-chloro-5-nitrobenzanilide), either alone or in combination with KA. Each injection was performed for >2.5 min using a micropump (KD Scientific, Holliston, MA). The amounts of NP031112 and GW9662 used were calculated based on the in vitro results to reach active concentrations within the hippocampus. Lithium chloride (LiCl), a potent inhibitor of GSK-3β activity, was administered (40 mg/kg/d) by intraperitoneal injection to a further two groups of animals, either alone or in combination with KA. The rats were then housed individually to recover.
Seizures were induced by intraperitoneal administration of rats with KA (10 mg/kg) in PBS. Control animals received saline only. Behavioral analysis was monitored for a period of 3 h by trained observers blind to the treatment of the rats. The convulsive behavior was classified according to Racine (1972) and Sperk et al. (1985) as follows: stage 0, no changes; stage 0.5, wet dog shakes (WDS); stage 1, mouth and facial movements; stage 2, head nodding; stage 3, forelimbs clonus; stage 4, rearing; stage 5, rearing and falling; stage 6, death. Status epilepticus (SE) was defined as continuous behavioral seizure activity (stage 5) for ≥5 min. The number of WDS before SE was also examined. In trials using NP031112, the TDZD was administered intragastrically (50 mg/kg) 1 h before KA injection.
Magnetic resonance imaging.
Magnetic resonance imaging (MRI) was performed using a 7.0 tesla horizontal bore (16 cm) magnet interfaced with a Bruker Pharmascan console (Bruker Medical, Ettlingen, Germany). Rat brain MRI was performed with a 90 mm gradient insert and a concentrical 38 mm birdcage resonator, using Paravision version 3.1 software (Bruker Medical), as implemented in a Hewlett-Packard (Palo Alto, CA) Linux platform. MRI examinations used adult male Wistar rats (n = 5; 250 g) anesthetized through a plastic mask with 2% isoflurane in 99.9% O2. Animals were allowed to breath spontaneously during the experiment and were placed in a heated cradle to maintain the core body temperature at ∼37°C. The physiological state of the animal was monitored throughout MRI acquisition through the respiratory rate and body temperature, as monitored by a rectal probe. T2-weighted spin-echo images were acquired at 1, 2, 3, and 9 d after KA injection with a rapid acquisition with relaxation enhancement (Hennig et al., 1986) sequence in axial orientations [repetition time (TR), 2500 ms; echo time (TE), 60 ms; averages, 3; field of view, 2.65 cm; acquisition matrix, 256 × 256; slice thickness, 1.50 mm; number of slices, 15]. The in vivo spectroscopy protocol acquired two 4 × 4 × 4 mm voxels in the hippocampal area, using a point-resolved spatially spectroscopy (Bottomley, 1987) protocol, combined with VAPOR water suppression (Tkac et al., 1999) (TR, 3000 ms; TE, 35 ms; averages, 128). The lesion area was calculated from T2-weighted images using image analySIS software (Soft Imaging System). Lesion volume was estimated from the summation of areas of hyperintensity on each slice, multiplied by slice thickness. Average lesion volume was calculated for each treatment.
Immunohistochemistry.
At different times after stereotaxic injection, the animals were anesthetized and perfused transcardially with 4% paraformaldehyde solution. The brains were removed, postfixed in the same solution at 4°C overnight, cryoprotected in the paraformaldehyde solution containing 30% sucrose, frozen, and 30 μm coronal sections were obtained in a cryostat. Free-floating sections were processed for cresyl violet (Nissl stain) or immunohistochemistry using the diaminobenzidine method or double-immunofluorescence analysis. For the diaminobenzidine method, floating sections were immersed for 15 min in 3% H2O2 to inactivate endogenous peroxidase, and then blocked for 2 h at room temperature (RT) in 5% normal goat serum (NGS; Vector Laboratories Burlingame, CA) in PBS, containing 4% bovine serum albumin, 0.1 m lysine, and 0.1% Triton X-100. Afterward, the sections were incubated overnight with the corresponding primary antibodies. After several rinses, sections were incubated for 1 h with a biotinylated secondary antibody. Finally, the sections were processed after the avidin-biotin protocol (Vectastain ABC kit; Vector Laboratories). Tissues were mounted onto gelatin-coated slides and were let to dry. Finally, the slides were dehydrated, cleared in xylene, and mounted with DePeX (Serva, Heidelberg, Germany). The slides were examined with a Zeiss (Oberkochen, Germany) Axiophot microscope, equipped with an Olympus Optical (Tokyo, Japan) DP-50 digital camera, and a Leica (Nussloch, Germany) MZ6 modular stereomicroscope. Neuronal integrity was assessed by counting the percentage of Nissl-positive cells in the CA3 region of the hippocampus in five independent well defined high-magnification (400×) fields per animal using a computer-assisted image analySIS software (Soft Imaging System). The extent of microgliosis was quantified by counting the number of OX-42-positive cells in five independent well defined high-magnification (400×) fields per animal, as described above. To evaluate astrogliosis, two different parameters were quantified: number of activated cells, based on the calculation of highly immunostained cell body profiles, and immunosignal intensity, based on the measurement of optical density. Individual cell bodies were manually traced, and their mean staining intensity was normalized against the background of the respective section, defined as tissue devoid of specific immunostaining. The procedure resulted in arbitrary values on a scale from 1 (background staining) to 256.
For the double-immunofluorescence, the protocol was similar to the one described above with some modifications. Briefly, the floating sections were blocked for 1 h in PBS containing 0.25% Triton X-100 and 3% NGS and incubated overnight with the corresponding primary antibodies. Then, AlexaFluor 647 and AlexaFluor 488 secondary antibodies were added for 1 h at RT. Finally, the tissue was mounted with mowiol and the sections were examined as described for immunocytochemistry. The sequential mode was used to acquire fluorescence images to avoid any interference from overlapping fluorescence. The images were obtained using a series of 0.5-μm (depth)-spaced cell fluorescent slices (z-axis).
Statistical determinations.
The data shown are the means ± SD of at least three independent experiments. Statistical comparisons for significance between cells with different treatments were performed using the Student's test, with p ≤ 0.05. ANOVA was used to analyze the data of Figures 2–6.
Figure 2.

Effect of NP031112 on KA-induced brain edema as detected by MRI. T2-weighted imaging was performed at 7 tesla as described in Materials and Methods at different times after KA injection. a, Representative coronal images of rat brain injected with vehicle (top), KA (middle), and KA plus NP031112 (bottom). Hyperintensity areas in T2-weighted MRIs reveal regions of edema after KA injection. Hyperintense areas were reduced in the NP031112-treated rats compared with the KA-injected rats, demonstrating a decrease in the injured area. No hyperintensity was found in the vehicle-injected rats. Arrows indicate the injection site. An anatomic diagram (Paxinos, 1998) showing (arrow) the precise localization of the microinjection in the rat hippocampus is shown. b, Quantitative analysis of total lesion volumes of KA- and KA plus NP031112-injected rats. The volume of the edemas was significantly lower in NP031112-treated rats, compared with KA-treated group at all of the times studied. Values represent the mean ± SD from five different animals and five independent sections per animal. ***p ≤ 0.001. c, Representative point-resolved spatially spectra acquired from the hippocampal region (white box) with resonances from NAA, lactate (Lac), and creatine (Cr) 9 d after treatment. d, Time course of variation of NAA or lactate values (mean ± SD) in the ipsilateral hemisphere of control-, KA-, and KA plus NP031112-treated animals during 9 d, normalized to the creatine peak. V, Vehicle; NP, NP031112. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001, versus vehicle-injected animals at each time point.
Figure 3.
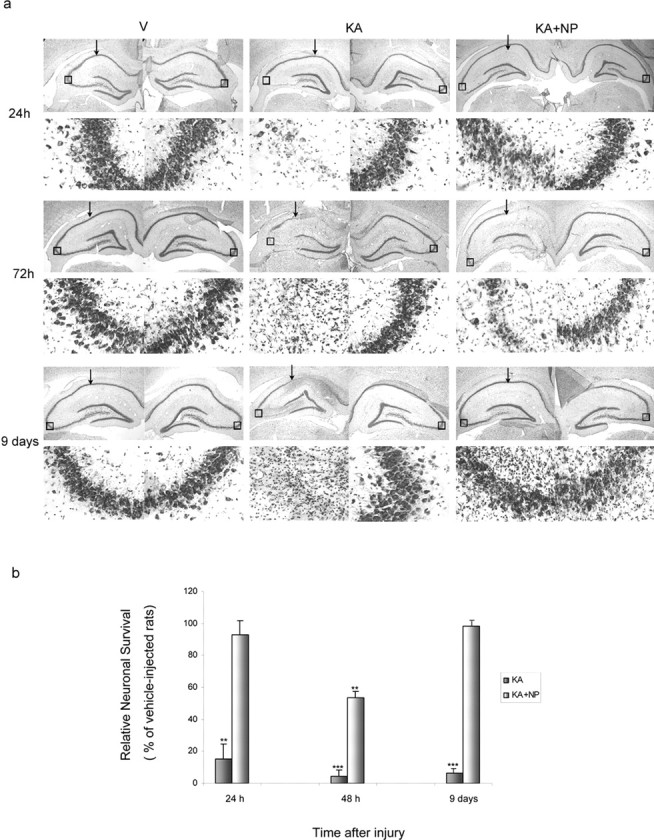
Neuroprotective effects of NP031112 on KA-induced excitotoxicity. a, Rats were injected with saline, KA, or KA plus NP031112 and killed at different times after injection. Neuronal cell loss was assessed in coronal brain sections, using a Nissl stain. Arrows indicate the injection site. Insets, 400× magnifications of Nissl-stained CA3 cells (boxed areas). No apparent cell loss was observed in control animals. Three days after KA, cell loss was apparent on the CA3 and CA1 subfields of the hippocampus in both KA and KA plus NP031112-injected rats. By day 9, cell loss was more prominent in KA-injected animals, whereas animals treated with NP031112 showed no cell loss. V, Vehicle; NP, NP031112. b, The extent of neuronal damage in the CA3 area of the hippocampus was quantified as described in Materials and Methods. Data were normalized against the mean values given by vehicle-injected rats. Values represent the mean ± SD from five different animals and five independent sections per animal. **p ≤ 0.01; ***p ≤ 0.001, versus vehicle-injected animals at each time point.
Figure 4.

Effect of NP031112 administration on KA-induced glial activation. a–d, GFAP- and OX-42-positive cells in the hippocampus. Rats were injected with saline, KA, or KA plus NP031112 and killed 9 d after injection. Arrows indicate the injection site. Coronal sections (30 μm) were stained with mouse monoclonal antibodies directed against either GFAP (a) or cd11b (OX-42; c) to detect astrocytes or microglial cells, respectively, as described in Materials and Methods. Insets, 400× magnifications of GFAP- and OX-42-stained cells (boxed regions). A significant increase in gliosis is observed after KA injection, which is prevented in the animals treated with NP031112. b, Quantification of the number of reactive astrocytes and their mean immunostaining intensity evaluated in the molecular layer of the hippocampus. Values represent the mean ± SD from five different animals and five independent sections per animal. ***p ≤ 0.001. d, Quantification of the number of reactive microglial cells analyzed in the molecular layer of the hippocampus. Values represent the mean ± SD from five different animals and five independent sections per animal. ***p ≤ 0.001. e, Induction of TNF-α in astrocytes, microglia, and neurons after KA injection is abrogated by NP031112. Coronal sections were double stained with a polyclonal antibody directed against TNF-α and with mouse monoclonal antibodies directed against NeuN, GFAP, or cd11b (OX-42) and examined by confocal microscopy. Photomicrographs showing representative CA3 fields for each group are shown. Scale bars, 10 μm. DAPI was used as a counterstain.
Figure 5.

The PPARγ antagonist GW9662 abolishes NP031112 neuroprotection after KA injection. Rats were injected with saline (V), KA, KA plus NP031112 (NP), or KA plus LiCl and killed 24 h after injection. Some animals were also injected with 0.7 μg GW9662 (GW). Arrows indicate the injection site. a, Neuronal cell loss was assessed in coronal brain sections using a Nissl stain. NP031112 treatment affords robust neuroprotection in the CA3 regions of the hippocampus 24 h after KA injection, whereas LiCl administration only had a small effect on the decrease in CA3 neurons elicited by KA injection. Injection of the PPARγ antagonist GW9662 after NP031112 and KA treatment abolished this neuroprotection. Treatment with the PPARγ antagonist did not affect the number of surviving neurons in any of the other treatment groups. b, Gliosis was assessed in similar brain sections by staining with a mouse monoclonal antibody directed against cd11b (OX-42) to detect microglial cells. Treatment with GW9662 blocked the anti-inflammatory effect of NP031112. c, Quantification of neuronal damage in the CA3 area of the hippocampus. d, Quantification of the number of reactive microglial cells analyzed in the molecular layer of the hippocampus. Values represent the mean ± SD from five different animals and five independent sections per animal. ***p ≤ 0.001 versus vehicle-injected animals; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001 versus KA-injected animals.
Figure 6.

Effect of NP031112 administered orally on behavioral activity and neuronal loss after injection of KA. a, b, NP031112 (50 mg/kg) was administered intragastrically 1 h before intraperitoneal injection of KA (10 mg/kg), and the behavioral activity was analyzed. NP031112 has no significant effects on behavioral seizures induced by KA. Each value represents the mean ± SEM from at least nine animals. c, The data correspond to animals that entered SE. Animals were treated as in a, and 72 h after KA injections, animals were killed and coronal hippocampal sections were stained with Nissl. NP031112-treated rats exhibited a diminished loss of neurons in the CA1 and CA3 regions, when compared with KA-injected rats. V, Vehicle; NP, NP031112. Values represent the mean ± SD from five different animals and five independent sections per animal. ***p ≤ 0.001 versus vehicle-injected animals.
Results
NP031112 inhibits glutamate-induced glial activation and protects cortical neurons from cell death in vitro
We first analyzed whether NP031112 affected the glutamate-induced expression of TNF-α and COX-2, two well known proinflammatory agents, in primary glial cultures. As shown in Figure 1a, incubation of both astrocyte and microglial cultures with NP031112 completely abrogated the induction of TNF-α and COX-2 expression after glutamate treatment. These effects of NP031112 were not caused by a loss of cell viability, because the 24 h exposure of astrocyte and microglial cells to this TDZD did not modify cell viability (data not shown).
Figure 1.
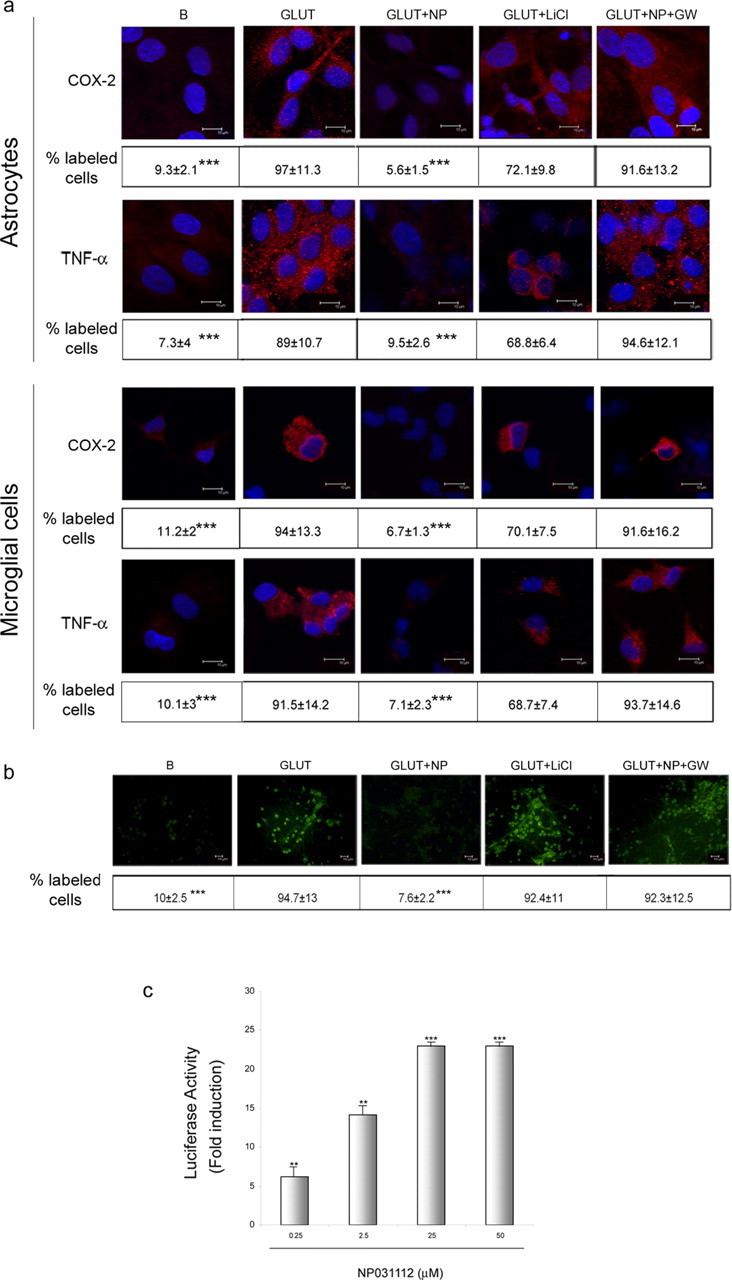
Effects of NP031112 on excitotoxic neural injury in vitro. a, Rat primary astrocyte or microglial cultures were treated for 24 h with glutamate (500 μm) in the absence or presence of NP031112 (2.5 μm), and the expression of TNF-α and COX-2 was evaluated by immunofluorescence analysis and confocal microscopy using specific antibodies, as described in Materials and Methods. Some cultures were preincubated 1 h with 30 μm GW9662 before the addition of NP031112 or with 20 mm LiCl before the addition of glutamate. Representative results of three different experiments are shown. Scale bars, 10 μm. Nuclei were counterstained with DAPI (blue). b, Rat primary neuronal cultures were treated for 24 h with glutamate (100 μm) in the absence or presence of NP031112 (2.5 μm). Some cultures were preincubated 1 h with 30 μm GW9662 before the addition of NP031112 or with 20 mm LiCl before the addition of glutamate, and apoptosis was assessed by Annexin-V-FITC staining as described in Materials and Methods. Representative confocal images of three independent experiments are shown. Scale bars, 10 μm. Values in a and b represent the mean ± SD from three different experiments and five independent fields (≥ 50 cells/field) per culture. ***p ≤ 0.001. c, Activation of a PPRE (peroxisome proliferator response element) reporter gene by NP031112. Rat primary astrocytes were transfected with 0.2 μg of the PPRE-tk-luc reporter plasmid, cells were harvested 24 h after treatment with NP031112 at the indicated concentrations, and luciferase activity of cell lysates was determined. Data are expressed relative to the basal values and represent the mean ± SD luciferase activity determined in triplicate in three independent experiments. **p ≤ 0.01; ***p ≤ 0.001, versus control nontreated cultures. GLUT, Glutamate; B, basal; GW, GW9662; NP, NP031112.
TDZD compounds were originally described as GSK-3β inhibitors (Martinez et al., 2002a,b) and, more recently, we have shown that their effects on neural cells in vitro seems to be mediated through activation of the PPARγ nuclear receptor (Luna-Medina et al., 2005). Therefore, we next tested whether LiCl (an inhibitor of GSK-3β) mimicked the effect of NP031112 and whether the specific PPARγ antagonist GW9662 impaired the action of this TDZD. As shown in Figure 1a, treatment of astrocyte and microglial cultures with LiCl did not have a major effect on the increase in TNF-α and COX-2 expression elicited by treatment with glutamate. On the contrary, GW9662 significantly inhibited the blocking effects of NP031112 on TNF-α and COX-2 expression after glutamate treatment of glial cultures, suggesting an involvement of PPARγ in the protective effects of NP031112 against the response of glial cultures to glutamate treatment.
In addition to its blocking effect in glial activation after an excitotoxic insult triggered by glutamate treatment, NP031112 was also able to exert a potent neuroprotective effect on cortical neurons from glutamate-induced excitotoxicity (Fig. 1b). Treatment of neuronal cultures with NP031112 resulted in a significant reduction in the number of Annexin-V-positive cells compared with untreated cells. As observed with glial cultures, the PPARγ antagonist GW9662 attenuated the neuroprotective effects of NP031112, whereas the GSK-3β inhibitor LiCl did not have any effect on glutamate-induced cell death or inflammatory response.
To further analyze the possible involvement of PPARγ on NP031112 actions, we next examined whether this compound could activate a reporter construct containing three consensus PPARγ response elements (PPRE-tk-luc) in primary cultures of astrocytes. Figure 1c shows a dose–response of the induction by NP031112 of PPRE-tk-luc reporter construct. Half-maximal stimulation was reached at 1.16 μm and maximal stimulation (23-fold) at 25 μm NP031112. No bigger induction was observed with higher doses of the compound. These results further suggest that NP031112 can be acting through a PPARγ-dependent mechanism.
Neuroprotective role of NP031112 after excitotoxic brain injury
Given the in vitro anti-inflammatory and neuroprotective effects described above, we then assessed the efficacy of NP031112 in an established focal excitotoxic model in vivo. To this end, adult rats received intrahippocampal injections of vehicle, KA, or KA plus NP031112 and were killed at different times after injection. Because brain edema is a common pathological trait of brain injury, we first used MRI to determine the neuroprotective efficacy of NP031112 in vivo. T2-weighted MRI allows the visualization of areas of brain edema as hyperintense regions. Indeed, areas of hyperintensity in T2-weighted images, reflecting cerebral edema or neurodegeneration, were found in the ipsilateral hemisphere of KA-injected rats 24 h after KA injection, their hyperintensity becoming further enhanced after 2, 3, and 9 d (Fig. 2a). Coinjection of NP031112 resulted in a significant decrease in the hyperintensity of T2-weighted images, compared with the untreated KA-injected animals. Lesion volume was significantly reduced at all of the times studied after KA injection in the NP031112-treated group. By day 9, the damage in NP031112-treated animals was almost completely absent (98% reduction relative to KA-injected animals) (Fig. 2b). Single-voxel proton nuclear magnetic resonance spectra from the hippocampal region of vehicle-, KA, and KA plus NP031112-injected animals are shown in Figure 2c. The spectral patterns between the vehicle- and the KA-injected rats were considerably different (Fig. 2d). In particular, KA administration was associated with a progressive and significant decrease of N-acetyl-aspartate (NAA)/creatine ratio and an increase in lactate/creatine ratio in the ipsilateral hippocampus, suggesting neuronal loss. On the contrary, the spectra observed in NP031112-injected rats were very similar to those found in vehicle-treated animals. These findings indicate that NP031112 protects the brain against KA-induced damage.
The MRI results were further confirmed by histological analysis of the hippocampus. Nissl staining was used to evaluate the extent of neuronal loss in the hippocampus of vehicle-, KA-, and KA plus NP031112-injected rats. A significant preservation of hippocampal cells was found in NP031112-injected rats compared with abundant neuronal loss in the CA1, CA3, and hilus after injection with KA (Fig. 3a). Three days after the injection, cell loss in CA1 and CA3 layers was apparent in both groups of animals, although it became more intense in the KA-injected group. Remarkably, by 9 d, both CA1 and CA3 regions of the hippocampus of NP031112-injected rats were almost completely normal, compared with KA-injected cells, which still presented a significant neuronal loss and a disorganization of both regions of the hippocampus. Quantitative studies (Fig. 3b) showed a decrease of 85, 96, and 94%, compared with the vehicle-injected rats, in the number of neurons in the CA3 subfield of the hippocampus 1, 3, and 9 d, respectively, after KA injection. In contrast, in the NP031112-treated group, only a moderate decrease (47%) in cell number was observed 3 d after KA injection. These results extend the observations made in vitro and suggest that treatment of KA-injected animals with NP031112 results in an almost complete recovery of the regions more affected by the injury.
One of the events that take place in the hippocampus after excitotoxic injury is the sequential activation of microglia and astroglia. Concerning astrogliosis, resting astrocytes were distributed mostly in the stratum lacunosum-moleculare layer of the hippocampus (LMol) of vehicle-injected animals (Fig. 4a). Twenty-four hours after KA injection, the highest GFAP immunoreactive signal intensity was not only observed in the LMol but also in the hilus of the dentate gyrus (data not shown). Nine days after injury, a dramatic increased in GFAP immunoreactivity was detected in all regions of the hippocampus (Fig. 4a). This strong astrogliosis was completely absent in the animals treated with NP031112, and the pattern of GFAP immunostaining in these animals was indistinguishable from that of control animals. Quantification of the data revealed a 6.7- and 2.2-fold decrease in both the number of strongly GFAP+ astrocytes and staining intensity, respectively, in the NP031112-treated animals, compared with the KA-injected group (Fig. 4b). Microglial activation in the contralateral hippocampus of kainic-injected rats, as well as in the hippocampus of vehicle-injected animals, was at almost undetectable levels in all regions of the hippocampus (Fig. 4c). After KA injection, numerous reactive microglial cells appeared in the ipsilateral part of all regions of the hippocampus. KA treatment resulted in morphological changes in microglial cells, which included swelling of the cell somas and thickening and retraction of the cell processes, two characteristic features of reactive microglia (Jorgensen et al., 1993; Ladeby et al., 2005). Coadministration of NP031112 together with KA completely blocked microglial activation. Quantitative analysis of OX-42+ cells from KA- and KA plus NP031112-injected animals shows a 40-fold decrease in the number of microglial cells 9 d after KA injection in the NP031112 group (Fig. 4d).
Double immunofluorescence staining of the rat hippocampus 9 d after injection of KA revealed an induction of the TNF-α cytokine, both in astrocytes and microglial cells (Fig. 4e), whereas the expression of this cytokine was almost completely absent in the neuronal population. Injection of KA in the presence of NP031112 significantly decreased TNF-α-positive staining both in astrocytes and microglia.
Subsequently, in view of the in vitro results described above, which suggested an implication of the nuclear receptor PPARγ in the action of NP031112, we next examined the possible involvement of this receptor on NP031112 actions in vivo. To this end, some rats were injected with the PPARγ antagonist GW9662 together with KA and NP031112 and killed 24 h later for histological evaluation. As expected, KA injection induced a substantial loss of hippocampal CA3 neurons and, once more, NP031112 treatment afforded robust neuroprotection (Fig. 5a,c). The PPARγ antagonist GW9662 alone did not modify the number of surviving neurons in vehicle-injected rats but abrogated NP031112-elicited neuroprotection. These findings suggest that NP031112 acts via activation of PPARγ to elicit protection of hippocampal neurons. Treatment of rats with GW9662 also inhibited the blocking effects of NP031112 on activation of microglia in the hippocampus, as shown in Figure 5, b and d. Overall, these findings indicate that NP031112 acts via PPARγ to protect hippocampal neurons from KA-induced cell death and inflammation. Next, we analyzed the neuroprotective effect of the GSK-3β inhibitor LiCl. For this purpose, two additional groups of rats were injected either with KA or with KA plus LiCl, and the number of CA3 neurons and activated microglial cells were determined in the hippocampus 24 h later. As expected, LiCl alone does not alter any of the parameters analyzed (data not shown). In contrast, LiCl administration caused a moderate neuroprotective effect against KA-induced injury (Fig. 5a,c) that was much lower than the one observed in NP031112-treated animals. Correspondingly, the number of OX-42-positive cells was slightly reduced in animals treated with LiCl, compared with the KA-injected group (Fig. 5b,d). These data further suggest that the effects of NP031112 are mediated mainly through PPARγ activation, although some effects because of its inhibition of GSK-3β cannot be discarded.
Susceptibility of NP031112-treated rats to KA-induced seizures
KA administered intraperitoneally at a dose of 10 mg/kg caused the entrance in SE in 80% of rats (Fig. 6a). Intragastrical administration of NP031112 1 h before KA injection had no effect on the percentage of rats reaching SE (80%), and only a small delay in the entrance of SE could be observed [126 ± 8.9 min in the NP031112-treated rats, compared with 104 ± 5.5 min (p < 0.01) in the control animals]. During the latency period, KA-injected rats displayed approximately the same number of WDS than NP031112-treated rats (Fig. 6b). We next evaluated neuronal damage in coronal brain sections of surviving rats 72 h after KA administration (Fig. 6c). Low-power images of the Nissl-stained hippocampus revealed extensive damage, evident in the CA1 and CA3 regions, which was substantially attenuated by orally administered NP031112. Together, these results suggest that the neuroprotective effects of NP031112 cannot be attributed to an anticonvulsant action.
Discussion
Collectively, our data clearly show that administration of the TDZD NP031112 in a rat model of excitotoxicity markedly induces neuroprotection and attenuates the production of proin flammatory cytokines and activation of astrocytes and microglial cells. Additionally, NP031112 actions appear to be mediated mainly through activation of the nuclear receptor PPARγ within the hippocampal cells, although part of its effects could be attributable to its inhibitory action of GSK-3β activity, because a modest neuroprotective effect of LiCl was observed in vivo. These results suggest that NP031112 can be a therapeutic agent in neurodegenerative disorders in which excitotoxic neuronal cell death and inflammation processes are involved.
We initially analyzed the anti-inflammatory and neuroprotective effects of NP031112 in an in vitro model of excitotoxicity, using primary cultures of astrocytes, microglia, and neurons. We show that NP031112 attenuates glutamate-induced excitotoxic glial activation and neuronal damage. NP031112 appears to be an extremely potent agent, because its effects were achieved at much lower concentrations than the two other TDZD compounds previously examined (Luna-Medina et al., 2005). The mechanism of action of all of these TDZD compounds in the neuroprotection experiments in vitro seems to be the activation of the nuclear receptor PPARγ, as suggested by the activation of a reporter gene containing a PPARγ response element and the blocking effect of the PPARγ antagonist GW9662 of their anti-inflammatory and neuroprotective actions. Although the TDZD compounds have been identified previously as ATP-noncompetitive inhibitors of GSK-3β activity (Martinez et al., 2002a,b), and it has been shown that inactivation of GSK-3β protects against KA-induced neurotoxicity in vivo (Goodenough et al., 2004), it is unlikely that a direct inhibition of this enzyme by TDZDs represents the major mechanism for their anti-inflammatory and neuroprotective effects in vitro, because these effects were not entirely mimicked by LiCl, a selective inhibitor of GSK-3β activity.
In the present study, we focused on the study of the anti-inflammatory and neuroprotective effects of NP031112, in an in vivo experimental model of excitotoxicity. Excitotoxicity is a concept of neuronal cell death caused by overactivation of excitatory amino-acid receptors and it is thought to represent a common biochemical pathway, which plays an important role in the pathogenesis of many acute and chronic neurodegenerative disorders such as stroke, traumatic brain injury, amyotrophic lateral sclerosis, and Parkinson's, Huntington's, and Alzheimer's diseases (Dirnagl et al., 1999; Doble, 1999; Nicotera et al., 1999). Also, and as pointed out in the Introduction, one of the events associated to many brain neurodegenerative diseases is an inflammation response, which has long been linked to neuronal cell death (Barone and Feuerstein, 1999; del Zoppo et al., 2000). Inhibition of this process could then protect against neurodegeneration and expansion of brain injury. This view is further supported by epidemiological data showing that long-term treatment with NSAIDs may protect against Alzheimer's and Parkinson's diseases (Chen et al., 2003; Hald and Lotharius, 2005; Townsend and Pratico, 2005).
Here, we show that NP031112 has a potent anti-inflammatory effect in vivo after KA-induced excitotoxicity. In the MRI studies, we analyzed the effect of NP031112 on the inflammatory process induced by KA at different times after injection. NP031112 treatment reduced the extension and intensity of the inflammation in the hippocampus and completely abrogated the reduction in NAA, which has been widely used as an indicator of brain pathology and of disease progression (Demougeot et al., 2004). NP031112 also reduced the increase in lactate observed in the KA-injected animals. Increases of lactate concentration are often related to pathologies, which enhance the anaerobic pathway (Choi et al., 2005; Moller et al., 2005; Williams et al., 2005). In agreement with these findings, our results also demonstrate that NP031112 significantly reduces other inflammatory parameters, such as the accumulation of reactive astrocytes and microglia in the hippocampus. The underlying mechanisms of this anti-inflammatory effect of NP031112 may involve the suppression of certain cytokines [e.g., TNF-α, because it has been shown that the production of this cytokine can predispose to local inflammation and contribute to brain injury initiation and progression (Chao et al., 1995; Rizzi et al., 2003)]. Indeed, our results show that administration of NP031112 significantly decreased TNF-α levels in vitro and in astrocytes and microglial cells in vivo after KA injection. Together, these data clearly indicate that NP031112 plays an important role in neuroinflammation and it is able to efficiently block this process when locally injected at very low concentrations in the damaged hippocampus.
In addition to this potent anti-inflammatory action of NP031112, the administration of this compound also causes a significant delay of neuronal cell loss in the CA1 and CA3 subregions of the hippocampus. Twenty-four hours after intrahippocampal injection, KA causes a prominent decrease in the number of neurons in the CA3 field of the hippocampus, which is blocked by NP031112 administration (Fig. 3). These in vivo data, together with the observed neuroprotective action in vitro against glutamate-induced neurotoxicity, suggest that, in addition to its anti-inflammatory effects, NP031112 could exert a more direct neuroprotective action. Notably, 9 d after NP031112 injection, both areas of the hippocampus were almost completely recovered. As expected, intraperitoneal injection of KA induced typical behavioral activity and histopathological changes in the hippocampus (loss of pyramidal cells in CA1 and CA3 areas). NP031112 acute administration had no effect on the behavioral seizures; however, it inhibited hippocampal damage, suggesting that the neuroprotective effects of this compound are not exerted through an inhibition of seizure activity.
Regarding the molecular mechanisms underlying the effects of NP031112, the present data are consistent with PPARγ playing a major role on its action. Our results demonstrate that NP031112 is able to strongly activate a consensus PPARγ response element in primary astrocytes in culture and that blocking this activation with a known PPARγ antagonist results in a prevention of the NP031112 neuroprotective effects in vitro as well as in vivo. These results, together with the in vitro and in vivo experiments using the GSK-3β inhibitor LiCl, suggest that, although TDZDs were originally identified as GSK-3β inhibitors, the mechanisms by which NP031112 exerts its neuroprotective and anti-inflammatory effects in excitotoxic injury involve mainly the activation of the nuclear receptor PPARγ. In agreement with these results, an anti-inflammatory and neuroprotective effect has been suggested for other PPARγ agonists (for review, see Landreth and Heneka, 2001; Townsend and Pratico, 2005). Epidemiological studies have shown that the long-term use of NSAIDs reduces the risk of developing and delays the onset of both Alzheimer's and Parkinson's diseases (Chen et al., 2003). Ibuprofen, an NSAID, reduces microglial activation induced by β-amyloid peptide (Combs et al., 2000) and amyloid plaque load in an animal model of Alzheimer's disease (Yan et al., 2003). PPARγ agonists of the TZD family, including rosiglitazone, pioglitazone, and troglitazone, have also been shown to have anti-inflammatory and neuroprotective effects (Diab et al., 2002; Camacho et al., 2004; Kiaei et al., 2005). Additionally, 15-deoxy-Δ-12,14-prostaglandin J2, the most potent natural ligand of PPARγ, reduces in vitro microglial activation (Bernardo et al., 2000) and T cell activity in an animal model of human multiple sclerosis (Diab et al., 2002). More recently, a protective role of PPARγ agonists against ischemic brain injury has been demonstrated (Shimazu et al., 2005; Sundararajan et al., 2005; Zhao et al., 2006).
In conclusion, we show that one single intrahippocampal injection of the TDZD compound NP031112 affords robust neuroprotection of hippocampal neurons against excitotoxic injury in adult rats. Our findings also suggest that the nuclear receptor PPARγ participates in this NP031112-induced neuroprotection. Hence, NP031112 may hold promise for preventing permanent neurological impairment in adults after a brain injury.
Footnotes
This work was supported by Ministerio de Educacion y Ciencia Grants SAF2004-06263-CO2-01 and 95-0764.OP (A.P.-C.) and SAF2004-06263-CO2-02 (A.S.) and Comunidad de Madrid Grant GR/SAL/0033/2004 (A.P.-C.). R.L.-M. is a fellow from the Ministerio de Educación y Ciencia. M.C.-C. is a postdoctoral fellow of the Consejo Superior de Investigaciones Científicas. We thank Marina Sanz-SanCristobal for excellent technical assistance and Ricardo Uña for helping with the quantification analysis. We are indebted to Dr. Sebastián Cerdán for helpful comments and critical reading of this manuscript and to Dr. Pilar López for expert handling of the nuclear magnetic resonance (NMR) instrumentation, as provided by Servicio de Imagen y Espectroscopía por Resonancia Magnética de Alto Campo, the core NMR Facility for small animal imaging of the Institute of Biomedical Research.
References
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Tremblay E, Ottersen OP. Injections of kainic acid into the amygdaloid complex of the rat: an electrographic, clinical and histological study in relation to the pathology of epilepsy. Neuroscience. 1980;5:515–528. doi: 10.1016/0306-4522(80)90049-4. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-Delta12, 14-prostaglandin J2 in the regulation of microglial functions. Eur J Neurosci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann NY Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- Camacho IE, Serneels L, Spittaels K, Merchiers P, Dominguez D, De Strooper B. Peroxisome-proliferator-activated receptor γ induces a clearance mechanism for the amyloid-β peptide. J Neurosci. 2004;24:10908–10917. doi: 10.1523/JNEUROSCI.3987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-d-aspartate receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol. 2003;60:1059–1064. doi: 10.1001/archneur.60.8.1059. [DOI] [PubMed] [Google Scholar]
- Choi CB, Kim HY, Han DY, Kang YW, Han YM, Jeun SS, Choe BY. In vivo 1H MR spectroscopic findings in traumatic contusion of ICR mouse brain induced by fluid percussion injury. Eur J Radiol. 2005;55:96–101. doi: 10.1016/j.ejrad.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT. Neurotoxic action of kainic acid. J Neurochem. 1983;41:1–11. doi: 10.1111/j.1471-4159.1983.tb11808.x. [DOI] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demougeot C, Marie C, Giroud M, Beley A. N-acetylaspartate: a literature review of animal research on brain ischaemia. J Neurochem. 2004;90:776–783. doi: 10.1111/j.1471-4159.2004.02583.x. [DOI] [PubMed] [Google Scholar]
- Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-δ(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- Goodenough S, Conrad S, Skutella T, Behl C. Inactivation of glycogen synthase kinase-3beta protects against kainic acid-induced neurotoxicity in vivo. Brain Res. 2004;1026:116–125. doi: 10.1016/j.brainres.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson's disease: is there a causal link? Exp Neurol. 2005;193:279–290. doi: 10.1016/j.expneurol.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Hennig J, Nauerth A, Friedburg H. RARE imaging: a fast imaging method for clinical MR. Magn Reson Med. 1986;3:823–833. doi: 10.1002/mrm.1910030602. [DOI] [PubMed] [Google Scholar]
- Jorgensen MB, Finsen BR, Jensen MB, Castellano B, Diemer NH, Zimmer J. Microglial and astroglial reactions to ischemic and kainic acid-induced lesions of the adult rat hippocampus. Exp Neurol. 1993;120:70–88. doi: 10.1006/exnr.1993.1041. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191:331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res. 2003;71:315–325. doi: 10.1002/jnr.10501. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer's disease. Neurobiol Aging. 2001;22:937–944. doi: 10.1016/s0197-4580(01)00296-2. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Nicotera P. Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium. 1998;23:165–171. doi: 10.1016/s0143-4160(98)90115-4. [DOI] [PubMed] [Google Scholar]
- Luna-Medina R, Cortes-Canteli M, Alonso M, Santos A, Martinez A, Perez-Castillo A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor {gamma} activation. J Biol Chem. 2005;280:21453–21462. doi: 10.1074/jbc.M414390200. [DOI] [PubMed] [Google Scholar]
- Martinez A, Castro A, Dorronsoro I, Alonso M. GSK-3 inhibitors as promising drugs for the treatment of Alzheimer disease, cancer, diabetes, and inflammation. Med Res Rev. 2002a;22:373–384. doi: 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J Med Chem. 2002b;45:1292–1299. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- Moller HE, Kurlemann G, Putzler M, Wiedermann D, Hilbich T, Fiedler B. Magnetic resonance spectroscopy in patients with MELAS. J Neurol Sci. 2005;229–230:131–139. doi: 10.1016/j.jns.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Nicotera P, Leist M, Manzo L. Neuronal cell death: a demise with different shapes. Trends Pharmacol Sci. 1999;20:46–51. doi: 10.1016/s0165-6147(99)01304-8. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Ed 4. San Diego: Academic; 1998. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang JT, Welch JS, Glass CK. The peroxisome proliferator-activated receptor (PPARgamma) as a regulator of monocyte/macrophage function. J Leukoc Biol. 1999;66:733–739. doi: 10.1002/jlb.66.5.733. [DOI] [PubMed] [Google Scholar]
- Rizzi M, Perego C, Aliprandi M, Richichi C, Ravizza T, Colella D, Veliskova J, Moshe SL, De Simoni MG, Vezzani A. Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol Dis. 2003;14:494–503. doi: 10.1016/j.nbd.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Inoue I, Araki N, Asano Y, Sawada M, Furuya D, Nagoya H, Greenberg JH. A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke. 2005;36:353–359. doi: 10.1161/01.STR.0000152271.21943.a2. [DOI] [PubMed] [Google Scholar]
- Sperk G. Kainic acid seizures in the rat. Prog Neurobiol. 1994;42:1–32. doi: 10.1016/0301-0082(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 1985;338:289–295. doi: 10.1016/0006-8993(85)90159-3. [DOI] [PubMed] [Google Scholar]
- Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE. Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130:685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkac I, Starcuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med. 1999;41:649–656. doi: 10.1002/(sici)1522-2594(199904)41:4<649::aid-mrm2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Pratico D. Novel therapeutic opportunities for Alzheimer's disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19:1592–1601. doi: 10.1096/fj.04-3620rev. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31:3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- Williams K, Westmoreland S, Greco J, Ratai E, Lentz M, Kim WK, Fuller RA, Kim JP, Autissier P, Sehgal PK, Schinazi RF, Bischofberger N, Piatak M, Lifson JD, Masliah E, Gonzalez RG. Magnetic resonance spectroscopy reveals that activated monocytes contribute to neuronal injury in SIV neuroAIDS. J Clin Invest. 2005;115:2534–2545. doi: 10.1172/JCI22953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters β-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Patzer A, Herdegen T, Gohlke P, Culman J. Activation of cerebral peroxisome proliferator-activated receptors gamma promotes neuroprotection by attenuation of neuronal cyclooxygenase-2 overexpression after focal cerebral ischemia in rats. FASEB J. 2006;20:1162–1175. doi: 10.1096/fj.05-5007com. [DOI] [PubMed] [Google Scholar]