

Abstract
Acetylcholine (ACh) is known to shape the adult neocortical activity related to behavioral states and processing of sensory information. However, the impact of cholinergic input on the neonatal neuronal activity remains widely unknown. Early during development, the principal activity pattern in the primary visual (V1) cortex is the intermittent self-organized spindle burst oscillation that can be driven by the retinal waves. Here, we assessed the relationship between this early activity pattern and the cholinergic drive by either blocking or augmenting the cholinergic input and investigating the resultant effects on the activity of the rat visual cortex during the first postnatal week in vivo. Blockade of the muscarinic receptors by intracerebroventricular, intracortical, or supracortical atropine application decreased the occurrence of V1 spindle bursts by 50%, both the retina-independent and the optic nerve-mediated spindle bursts being affected. In contrast, blockade of acetylcholine esterase with physostigmine augmented the occurrence, amplitude, and duration of V1 spindle bursts. Whereas direct stimulation of the cholinergic basal forebrain nuclei increased the occurrence probability of V1 spindle bursts, their chronic immunotoxic lesion using 192 IgG-saporin decreased the occurrence of neonatal V1 oscillatory activity by 87%. Thus, the cholinergic input facilitates the neonatal V1 spindle bursts and may prime the developing cortex to operate specifically on relevant early (retinal waves) and later (visual input) stimuli.
Keywords: development, in vivo, muscarinic, 192 IgG-saporin, oscillations, basal forebrain
Introduction
The cholinergic drive represents one of the major state-controlling systems of the neocortex. Acetylcholine (ACh) release correlated with EEG activation is responsible for behavioral arousal as first described by Moruzzi and Magoun (1949, 1995) in their principle of “neocortical readiness state.” The cholinergic system is involved in a large variety of cognitive processes (Damasio et al., 1985; Steriade et al., 1991; Everitt and Robbins, 1997) and gates sensory information processing (Singer, 1986; Juliano et al., 1991; Ma and Suga, 2005).
Anatomically, the cholinergic projections to the neocortex are well positioned for a modulatory role. These abundant cortical afferents mainly originate from the basal forebrain nuclei (BFn), including the nucleus basalis magnocellularis (nB) and medial septal nucleus, as well as horizontal limb nuclei of the diagonal band of Broca, which, despite their discrete localization, are often referred to as a continuum (Semba and Fibiger, 1989). Developmentally, cholinergic afferent ingrowth into the neocortex starts early, coinciding with neuronal differentiation, synapse formation, and circuit shaping (Mesulam et al., 1992). The cholinergic system modulates neuronal proliferation and differentiation and interferes with neonatal apoptosis and survival processes (Lipton and Kater, 1989; Role and Berg, 1996; Brimijoin and Koenigsberger, 1999; Pugh and Margiotta, 2000). Manipulation of the cortical ACh level leads to major morphogenetic alterations (Byers et al., 2005). In addition to its trophic role, the cholinergic input contributes to the maturation of functional synaptic contacts and the wiring of developing circuits (Maggi et al., 2003; Kuczewski et al., 2005; Myers et al., 2005). ACh plays a crucial role during the so-called critical period of the postnatal development, during which the cortex is highly susceptible to modulation by sensory input. In the visual cortex, for example, the cholinergic drive facilitates neuronal plasticity (Bear and Singer, 1986; Gu and Singer, 1993), whereas lesion of the nB transiently alters the formation of ocular dominance columns (Siciliano et al., 1997). However, already by birth, ACh-induced excitation is likely to stabilize the immature synaptic circuits (Hanganu and Luhmann, 2004). Moreover, in vitro data suggested that muscarinic acetylcholine receptor (mAChR)-dependent synchronized oscillatory activity in the alpha–beta frequency range may act as a functional template for the development of early cortical columnar networks and architecture (Dupont et al., 2006). Previously, we identified similar oscillatory activity patterns in the neonatal [postnatal day 2 (P2)–P6] visual cortex in vivo (Hanganu et al., 2006). These self-organized oscillations, termed as spindle bursts, are driven by retinal waves (Khazipov et al., 2004; Hanganu et al., 2006) and are likely to be subject to intense modulation (Luhmann and Khazipov, 2006). However, to which extent the cholinergic drive interacts with the early network activity in vivo remains widely unknown. Here, we performed in vivo recordings from the neonatal rat primary visual cortex (V1) paired with acute pharmacological manipulation of cholinergic drive, stimulation of BFn, and long-term immunotoxic lesion of BFn using 192 IgG-saporin. We provide evidence that the early V1 spindle bursts are facilitated by the cholinergic input from BFn and that the chronic cholinergic deprivation critically impairs the oscillatory network activity of the neonatal visual cortex.
Materials and Methods
Surgical, recording, and stimulation procedures.
All experiments were conducted in accordance with the national laws for the use of animals in research and approved by the local ethical committee. As determined previously (Hanganu et al., 2006), neocortical recordings were performed from the region of maximal responsiveness to optic nerve (ON) stimulation of the V1 (0–1.5 mm anterior to lambda suture and 2–3.5 mm from the midline) of P5–P6 rats using the experimental setup and conditions described previously (Leinekugel et al., 2002; Khazipov et al., 2004). Briefly, following the Guidelines of the Care and Use of Mammals in Neuroscience and Behavioral Research of the National Research council, the pups were anesthetized by hypothermia (ice-cooling anesthesia), known to preserve the anesthetic-sensitive retinal waves (Mooney et al., 1996). Under these conditions, the heads of the pups were fixed into the stereotaxic apparatus using two metal bars fixed with dental cement on the nasal and occipital bones, respectively. In some pups, the eye region was additionally anesthetized by hypothermia, the vitreous was extracted, and both retinas were removed with a thin cotton-covered bar. The bodies of the pups were surrounded by a cotton nest and kept at a constant temperature of 37°C by a temperature controller (TC-344B; Warner Instruments, Hamden, CT). After a 30–60 min recovery period, single tungsten electrodes (50 μm diameter; A-M Systems, Carlsborg, WA) were inserted vertically into the visual cortex to obtain simultaneous recordings of field potential and multiple unit activity (MUA). One or two silver wires were inserted into the cerebellum and served as ground electrodes. Stimulation of the ON was performed after surgically opening the lid under local anesthesia. Electrical pulses (50–100 V; 50 μs; 0.1 Hz) were applied via a bipolar tungsten electrode (2 MΩ; 160 μm diameter; California Fine Wire, Grover Beach, CA) inserted 3–4 mm through the cornea to reach the head of ON. Low-frequency stimulation was used to prevent accommodation. Stimulation of the cholinergic BFn was performed at stereotaxic coordinates (0–0.4 mm posterior to bregma, 1.5 mm lateral to midline, and 3.5–5 mm ventral to the pial surface) determined previously after staining for the vesicular acetylcholine transporter (VAChT). These coordinates correspond to the rostral region of the nB (Paxinos et al., 1991), known to project to the visual cortex (Gaykema et al., 1990). Single electrical pulses (5 V; 50 μs; 0.1 Hz) or tetanic stimulation (1–10 V; 10 pulses at 10 Hz; 15 s) were applied by a bipolar tungsten electrode (160 μm diameter; California Fine Wire) with a maximum tip separation of 400 μm. The stimulated hemisphere was referred to as ipsilateral. In some experiments, the exact depth of extracellular recordings and BFn stimulation was assessed by using 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indocarbocyanine (DiI; Invitrogen, Carlsbad, CA)-labeled electrodes (see Fig. 6B).
Figure 6.

Modulation of the V1 spindle bursts by electrical stimulation of the BFn. A, Schematic drawing of the BFn-stimulation (red) and V1 recording (blue) paradigm. B, Digital photomontage of DiI-labeled stimulation electrode in a 400-μm-thick coronal brain section superimposed on a schematic drawing of a coronal section of P0 rat brain. The basal forebrain nuclei with a high density of cholinergic neurons projecting to the V1 are marked in red. B, Nucleus basalis; Cpu, caudate putamen; HDB, horizontal limb diagonal band, LV, lateral ventricle; Par, parietal cortex (following Paxinos et al., 1991). C, Photograph of a coronal section (50 μm thick) from a P6 rat after immunostaining for VAChT. Note the presence of cholinergic neurons (black dots) in different nuclei of the basal forebrain. D, Extracellular recordings of the ipsilateral (black trace) and contralateral (gray trace) cortical response to electrical tetanic stimulation of the BFn in a P6 rat. Note the spindle bursts elicited on the ipsilateral side to stimulation and the absence of such response on the contralateral side. The displayed traces correspond to 20 averaged responses to BFn stimulation. E, Power spectrum of the averaged BFn-induced V1 bursts from the trace shown in D. The side of stimulation was defined as ipsilateral.
Pharmacological procedures.
Pharmacological modification of the cholinergic input to V1 was performed on pups fixed on the stereotaxic apparatus. For this purpose, a 26GA needle (World Precision Instruments, Sarasota, FL) attached to a microsyringe pump controller (Micro4; World Precision Instruments) was used to inject either 1 μl of solution intracerebroventricularly at a rate of 0.25 μl/min, 20–50 nl of solution intracortically at a rate of 25–50 nl/min, or 20–50 nl on the V1 surface at a rate of 20 nl/min. In some experiments, the V1 surface was covered with atropine containing (1 mm) low-melting agar. The stereotaxic coordinates for intracerebroventricular injection in P5–P6 pups (1.5 mm posterior to bregma, 1.5 mm lateral to midline, and 2–3 mm ventral to the pial surface) were determined previously in our lab by dye application (H. Becq and M. Milh, personal communication) and correspond to literature data (Pappas et al., 1996; Robertson et al., 1998). The stereotaxic coordinates for intracortical injections (2.5 mm posterior to bregma, 2.5 mm lateral to midline, and 300–500 μm ventral to pial surface) were chosen empirically to avoid side effects attributable to damage of the recorded V1. The injected hemisphere was referred to as ipsilateral. The applied substances were purchased as followed: atropine sulfate from Sigma (Taufkirchen, Germany) and physostigmine hemisulfate from Tocris (Ellisville, MO). Solutions of these drugs were prepared on the day of experiment in artificial CSF (ACSF) containing (in mm) 124 NaCl, 26 NaHCO3, 1.3 NaH2PO4, 1.9 MgCl2, 1.6 CaCl2, and 3 KCl, pH 7.4, osmolarity 333 mOsm. Control recordings were performed in each investigated pup before and after insertion of the injection syringe, and after solvent (ACSF) injection. At the end of each injection, the needle was withdrawn and a 5–10 min recovery period was allowed before starting the recordings.
Immunotoxin injection.
Male P0 pups were anesthetized by hypothermia, placed on a performed mold, and immobilized with tapes. A 26GA needle (WPI) attached to a microsyringe pump controller (Micro4; WPI) was used to inject, at a slow rate (0.1 μl/min), 0.5 μl of 192 IgG-saporin (0.2 μg/μl, solved in 0.1 m PBS, pH 7.4) or 0.5 μl of vehicle (0.1 m PBS, pH 7.4) into both lateral ventricles (1 mm posterior to bregma, 1 mm lateral to midline, and 2–2.5 mm ventral to pial surface). After injection, the needle was left in place for additional 1–3 min to allow optimal diffusion of the solution. The scalp wound was closed with tissue adhesive. Pups were warmed up under a filament bulb and returned to the dam only after full recovery of body temperature and motility (30–60 min) to prevent maternal cannibalism. In each investigated litter, control (nontreated), PBS-, and immunotoxin-treated pups were observed daily (general and feeding behavior, weight). At P5–P6, the pups were deeply anesthetized by hypothermia, fixed in the stereotaxic apparatus as described above, and investigated for their V1 activity patterns.
Immunocytochemical analysis of the immunotoxin lesions.
To examine the effects of 192 IgG-saporin on the BFn cholinergic neurons, at the end of recordings, the pups were perfused with 4% of paraformaldehyde dissolved in 0.1 m phosphate buffer (PB), pH 7.4. The brains were removed and postfixed in the same solution for 22–24 h. Blocks of tissue containing the BFn were then rinsed several times with PB and sectioned coronally on a freezing microtome at 50 μm. Several series of sections were stained with an antibody against VAChT raised in goat (Millipore, Schwalbach, Germany), which is considered to by a highly specific marker for cholinergic neurons and, in addition, a robust antigen detectable with very high sensitivity (Schafer et al., 1995; Weihe et al., 1996). A previously published standard protocol was used for immunostaining (Staiger et al., 2004). Briefly, free-floating sections were rinsed with 0.1 m PB and 0.05 m Tris-buffered saline (with 0.3% Triton X-100; TBST), pH 7.6. All sera were diluted in TBST and, between all subsequent steps, the tissue was rinsed with TBST unless otherwise noted. Preincubation of the tissue in 10% normal serum (Vector Laboratories, Burlingame, CA) was followed by exposure to the primary antiserum (1:5.000) at 6°C for 36 h under gentle agitation. The tissue was afterward incubated with biotinylated rabbit anti-goat serum (1:200; Vector Laboratories) for 2 h at room temperature (RT) and subsequently treated with peroxidase coupled to an avidin-biotin complex (ABC, 1:400; Vector Laboratories) for 2 h at RT. The sections were then rinsed in TBST and 0.05 m Tris-buffer (TB, pH 7.6). On the next step, the buffer was exchanged with a chromogen solution consisting of 0.5% 3,3′-diaminobenzidine (Sigma). After a preincubation of 10 min, the reaction was started by adding hydrogen peroxide to a final concentration of 0.1%. The reaction was stopped by rinsing with TB when no additional increase in the staining intensity of neuronal processes could be visually detected (usually after 6 min). All tissue from all investigated animals was treated equally in the same experimental run. Because the specificity of the antisera was extensively tested and approved by the manufacturers, only immunocytochemical controls (replacement of the primary antiserum by the appropriately diluted normal serum) were performed and led to the absence of any staining. The sections were air dried and coverslipped through xylene with Entellan (Merck, Darmstadt, Germany).
Data analysis.
Data were acquired at a sampling rate of 10 kHz and analyzed off-line using ClampFit software (Molecular Devices, Union City, CA). Oscillatory events were detected by eye and confirmed after filtering the raw data between 5 and 50 Hz using a Butterworth filter. Only events lasting >100 ms and containing more than three cycles were considered for analysis. MUA was analyzed after filtering at 200 Hz high-pass filter. The VAChT-stained sections were analyzed using Neurolucida (MBF Bioscience, Magdeburg, Germany). The stained cells in the nB [as identified according to Paxinos et al. (1980) and Mesulam et al. (1983)] were counted and their density (stained cells per millimeter squared) calculated. Statistical analyses were performed with Systat (SPSS, Chicago, IL) and Origin 7 software (OriginLab, Northampton, MA) using Student's t test, ANOVA, and Wilcoxon test. Significance levels of p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***) were considered. Data are presented as mean ± SEM.
Results
Pharmacological manipulation of the cholinergic input modulates V1 spindle bursts
To assess the role of cholinergic innervation for the generation of spindle bursts in the visual cortex of the newborn rat, we first investigated the effects of mAChR blockade on V1 activity. Intracerebroventricular injection of the selective mAChR antagonist atropine (1 μl, 10 mg/kg body weight, in ACSF) was performed unilaterally in 7 P5–P6 pups. Although a very small volume was applied and the speed of injection (0.25 μl/min) was very slow, some of the atropine-induced effects could have been caused by pressure- or lesion-related action. To test this possibility, injections using only the vehicle (1 μl of ACSF) were performed before atropine application in all seven pups investigated (Fig. 1A,C). The occurrence of spindle bursts was modified neither on the contralateral (control, 2.3 ± 0.2 bursts/min; ACSF, 2.2 ± 0.3 bursts/min) nor on the ipsilateral hemisphere (2 ± 0.1 bursts/min; ACSF, 1.8 ± 0.2 bursts/min) regarding to the side of injection. In addition, the amplitude (contralateral control, 192.5 ± 23.8 μV; ACSF, 176.8 ± 21 μV; ipsilateral control, 221.4 ± 49.3 μV; ACSF, 160.2 ± 30 μV), frequency within burst (contralateral control, 13.6 ± 1.2 Hz; ACSF, 15 ± 1 Hz; ipsilateral control, 15 ± 1.2 Hz; ACSF, 15.2 ± 1.3 Hz), and duration (contralateral control, 3.8 ± 0.2 s; ACSF, 3.7 ± 0.2 s; ipsilateral control, 3.9 ± 0.1 s; ACSF, 3.2 ± 0.3 s) of the spindle bursts were not affected by the ACSF injection. Moreover, the MUA showed a similar frequency under control conditions (contralateral, 0.4 ± 0.1 Hz; ipsilateral, 0.36 ± 0.1 Hz) and after ACSF injection (contralateral, 0.56 ± 0.2 Hz; ipsilateral, 0.31 ± 0.08 Hz; n = 7 pups). In contrast, intracerebroventricular injection of atropine had a strong effect on the V1 activity in both hemispheres and especially on the ipsilateral side (n = 7 pups) (Fig. 1A,C). The occurrence of spindle bursts was significantly reduced on the contralateral (1.1 ± 0.1 bursts/min; p = 0.0001) and ipsilateral (0.65 ± 0.3 bursts/min; p = 0.004) hemispheres after atropine application. Similarly, the amplitude of spindle bursts was reduced on both the contralateral V1 to 145.1 ± 16.6 μV and the ipsilateral V1 to 132.8 ± 15 μV. In contrast, the mean frequency within burst (contralateral, 13.6 ± 1.4 Hz; ipsilateral, 15.1 ± 0.6 Hz), as well as the prevalence of alpha- and beta-band (relative power in control, 89.9 ± 1.1%; atropine, 91.1 ± 1.7%; n = 7 pups) versus gamma-band activity (relative power in control, 11.1 ± 1.2%; atropine, 8.9 ± 1.7%; n = 7 pups) were not significantly affected by atropine. Whereas the duration of spindle bursts on the contralateral side to injection remained unchanged (3.6 ± 0.3 s), a slight reduction in the duration of ipsilateral bursts to 2.1 ± 0.1 s was observed after intracerebroventricular injection of atropine. To characterize the dynamics of atropine action on cortical activity, its effect on the spindle bursts was investigated during 1 h. The atropine-induced reduction in the occurrence of spindle bursts on both hemispheres was generally constant over the investigated period (Fig. 1B); in only three of seven pups a slight recovery being observed. Pharmacokinetics of atropine in vivo is poorly understood; however, it may be suggested that the high-binding affinity of atropine as well as limited atropine washout through the blood–brain barrier into a small body volume in pups may account for the persistence of atropine-induced blockade. In addition, atropine application seems to interfere with the patterns of interhemispheric synchronization. Although the fraction of spindle bursts occurring within a time window ±1 s on both hemispheres was similar before (control, 24.3 ± 5.7%; ACSF, 24.3 ± 6.5%; n = 7 pups) and after atropine injection (20 ± 7.2%; n = 7 pups), the interhemispheric cross-correlation coefficients significantly (p = 0.02) decreased (control, 0.16 ± 0.03; atropine, 0.08 ± 0.03; n = 7 pups). These results suggest that the cholinergic input from the basal forebrain may reinforce the interhemispheric synchronization of spindle burst activity that is mainly achieved by the callosal and commissural connections (Innocenti, 1995; Bloom and Hynd, 2005). The ability of intracerebroventricularly injected atropine to diminish the V1 oscillatory activity supports the hypothesis that the cholinergic innervation acting on mAChR exerts a powerful modulation of cortical activity, facilitating the generation of V1 spindle bursts.
Figure 1.

Effects of mAChR blockade by intracerebroventricular injection of atropine on spindle bursts in the V1 cortex of newborn rat in vivo. A, Extracellular field potential recordings from the ipsilateral (black trace) and contralateral (gray trace) V1 of a P6 rat under control conditions, after injection of 1 μl of ACSF, and after injection of 1 μl of atropine (10 μg/g body weight in ACSF). Inset, Typical spindle bursts displayed at expanded time scale and averaged power spectrum of the field potential oscillations corresponding to the displayed traces. Note the reduced burst occurrence and the unchanged amplitude and frequency within burst in the presence of atropine. B, Time dependence of atropine effect on the spindle bursts recorded on the ipsilateral (black dots) and contralateral (gray dots) hemispheres in seven P5–P6 pups and compared with control. Each dot corresponds to the averaged burst occurrence per min. C, Bar diagram displaying the effects of intracerebroventricularly applied atropine on the occurrence of spindle bursts recorded on the ipsilateral (black bars) and contralateral (gray bars) V1 cortices of seven pups. The hemisphere of intracerebroventricular injection was defined as ipsilateral.
Because blockade of mAChR using intracerebroventricular injection of atropine may not only affect the cortical receptors, but also have a global effect on subcortical regions, we applied atropine directly to the cortex (n = 9 pups) using two approaches. First, atropine was injected intracortically at 1.5–3 mm distance from the recorded V1 site. The injected volume did not exceed 50 nl and a very low injection speed (usually 25 nl/min) was used. In addition to these precautions, we first tested the nonspecific effects of ACSF injection (n = 4 pups). ACSF had no significant effects on the spindle bursts or MUA of both injected and noninjected hemispheres (Fig. 2A,B). A different situation could be observed when atropine was intracortically injected into one hemisphere (n = 4 pups). On the contralateral side, neither the occurrence (control, 1.6 ± 0.2 bursts/min; atropine, 1.6 ± 0.2 bursts/min), nor the amplitude (control, 136.4 ± 5.5 μV; atropine, 157.4 ± 6.2 μV), the frequency within the burst (control, 16.6 ± 1.2 Hz; atropine, 17.8 ± 0.4 Hz), or the duration of the burst (control, 3.4 ± 0.5 s; atropine, 3 ± 0.2 s) was significantly modified by atropine. In contrast, a significant drop in the ipsilateral V1 activity was observed after cortical mAChR blockade. The occurrence of V1 spindle bursts significantly decreased from 1.9 ± 0.3 to 0.8 ± 0.3 bursts/min (p = 0.0002) (Fig. 2A,B) and the duration of single bursts was significantly reduced from 3.6 ± 0.3 to 2.6 ± 0.03 s (p = 0.045; n = 4 pups). The amplitude (control, 193.7 ± 15.2 μV; atropine, 169.8 ± 20.4 μV) and the frequency within bursts (control, 17.4 ± 1.1 Hz; atropine, 18.5 ± 0.4 Hz) of the ipsilateral spindle bursts were not modified by intracortical injection of atropine.
Figure 2.
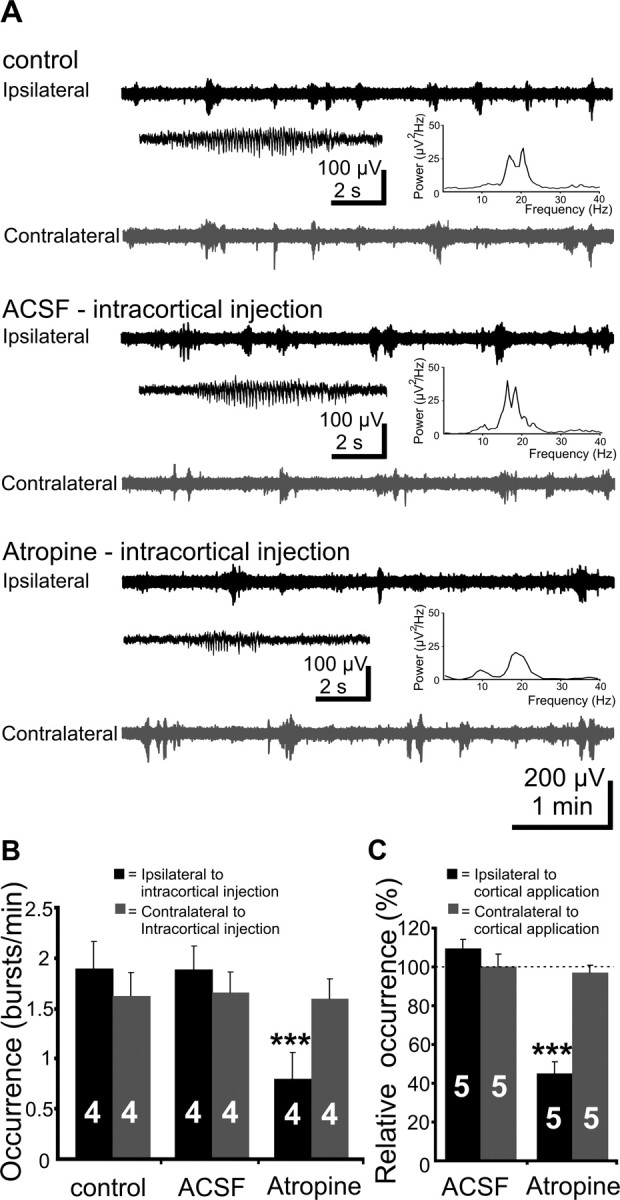
Effects of mAChR blockade by intracortical injection or by application of atropine to the cortical surface on V1 spindle bursts. A, Extracellular field potential recordings from ipsilateral (black trace) and contralateral (gray trace) V1 of a P5 rat under control conditions, after intracortical injection of 20 nl of ACSF, and after intracortical injection of 20 nl of atropine (10 μg/g body weight, in ACSF). Inset, Typical spindle bursts displayed at an expanded time scale and averaged power spectrum of the field potential oscillations corresponding to the displayed traces. Note the reduced burst occurrence on the side of atropine injection. B, Bar diagram displaying the effects of intracortical atropine injection on the occurrence of bursts in the ipsilateral (black bars) and contralateral (gray bars) V1 of four P5–P6 pups. C, Bar diagram displaying the effects of atropine application to the V1 surface on the occurrence of bursts in the ipsilateral (black bars) and contralateral (gray bars) V1 of five P5–P6 pups. The occurrence of spindle bursts before cortical application was considered as 100%. The hemisphere of intracortical injection or cortical application was defined as ipsilateral.
In the second approach, atropine was either unilaterally superfused on the V1 surface (n = 3 pups) or mixed with low-melting agar to unilaterally cover V1 (n = 2 pups). Because both methods led to similar results, the data were pooled together. Whereas direct delivery of ACSF on V1 had no effect on the ipsilateral and contralateral spindle bursts, direct atropine application diminished the V1 activity on the treated hemisphere (Fig. 2C). The occurrence of the ipsilateral spindle bursts significantly (p = 0.003) decreased from 1.14 ± 0.07 bursts/min in control and 1.24 ± 0.09 bursts/min after ACSF application to 0.5 ± 0.05 bursts/min after atropine application (n = 5 pups), whereas the contralateral side of application was unaffected (control, 0.9 ± 0.1 bursts/min; ACSF, 0.9 ± 0.1 bursts/min; atropine, 0.86 ± 0.1 bursts/min). Moreover, atropine significantly (p = 0.006) decreased the duration of ipsilateral spindle bursts (control, 4.6 ± 0.1 s; ACSF, 4.6 ± 0.4 s; atropine, 3 ± 0.3 s). Similar to the intracortically injected atropine, the direct application of the antagonist on the V1 did not modify the amplitude (control, 126.6 ± 21.2 μV; atropine, 96.1 ± 7.2 μV) and the frequency within bursts (control, 11.1 ± 1.4 Hz; atropine, 13.1 ± 1.4 Hz) of the ipsilateral spindle bursts. These data reinforce our hypothesis that the atropine-induced diminishment of cortical activity is attributable to blockade of cortical mAChR, but not to subcortical effects.
Our previous data indicate that the V1 spindle bursts are self-organized oscillations that can be selectively triggered by retinal waves (Hanganu et al., 2006), but not by cortical stimulation (R. Khazipov, unpublished observation). Therefore, we separately investigated the effects of cholinergic drive on the spontaneous, retina-independent spindle bursts as well as on the retina-driven oscillatory activity. To abolish the retinal input, both retinas were removed (n = 5 pups). According to our previous data (Hanganu et al., 2006), spontaneous spindle bursts occurred at a lower frequency in the retinas-ablated animals (0.7 ± 0.15 bursts/min; n = 5 pups) (Fig. 3A). Additionally, retina removal slightly shifted the power of the spindle bursts toward alpha-band frequency (8–12 Hz) and decreased the amount of beta-band (12–30 Hz) oscillations (compare Figs. 1A, 3A, power spectra), but these changes were not at a significant level (p = 0.138). Intracerebroventricular injection of ACSF modified neither their occurrence (contralateral control, 0.73 ± 0.13 bursts/min; ACSF, 0.64 ± 0.13 bursts/min; ipsilateral control, 0.65 ± 0.2 bursts/min; ACSF, 0.66 ± 0.2 bursts/min; n = 5 pups) nor the amplitude (contralateral control, 129.8 ± 15.1 μV; ACSF, 128 ± 20.2 μV; ipsilateral control, 150.3 ± 42 μV; ACSF, 214.7 ± 74 μV), duration (contralateral control, 1.9 ± 0.3 s; ACSF, 2.1 ± 0.3 s; ipsilateral control, 2 ± 0.4 s; ACSF, 1.9 ± 0.8 s), or frequency within burst (contralateral control, 10.3 ± 1 Hz; ACSF, 11 ± 1 Hz; ipsilateral control, 12.9 ± 1.7 Hz; ACSF, 14.8 ± 2.5 Hz) (Fig. 3A,B). In contrast, intracerebroventricular injection of 1 μl of atropine solution (10 mg/kg body weight in ACSF) significantly reduced the occurrence of spindle bursts to 0.43 ± 0.08 bursts/min (p = 0.018) on the contralateral side of injection and to 0.3 ± 0.16 bursts/min (p = 0.039) on the ipsilateral side (n = 5 pups) (Fig. 3A,B). The amplitude (contralateral, 103.3 ± 7.2 μV; ipsilateral, 131.4 ± 25.8 μV), frequency within the burst (contralateral, 11 ± 1 Hz; ipsilateral, 14.8 ± 2.5 Hz), and duration (contralateral, 2.1 ± 0.3 s; ipsilateral, 1.9 ± 0.3 s) of endogenous V1 spindle bursts were not significantly modified by atropine. These data indicate that the cholinergic input facilitates the endogenously generated retina-independent V1 activity.
Figure 3.
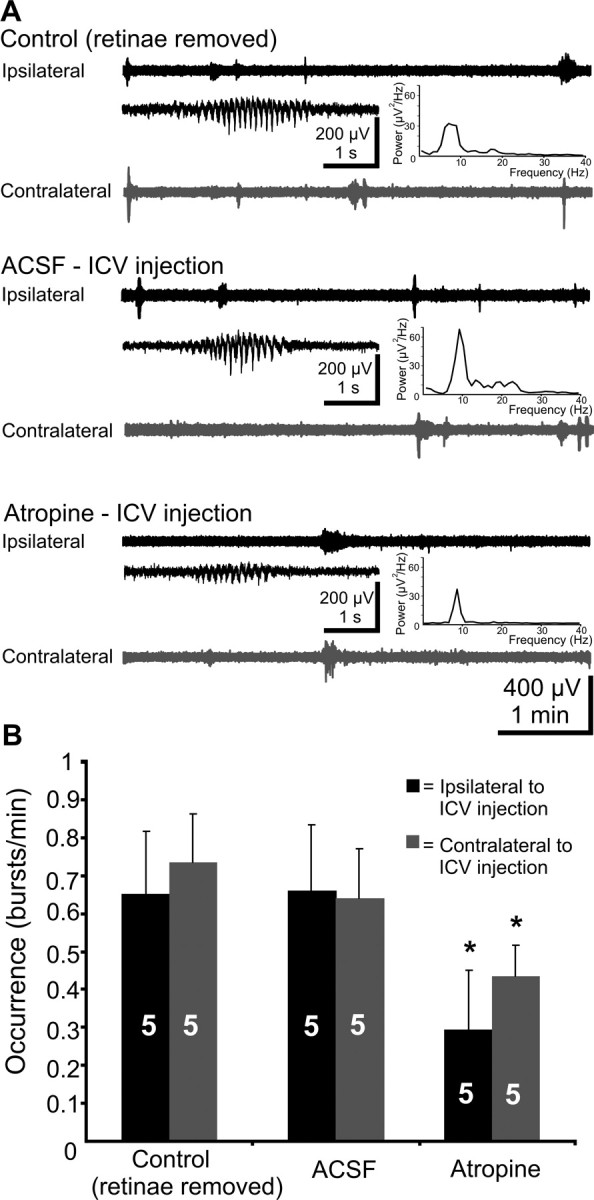
Effects of mAChR blockade by intracerebroventricular injection of atropine on the endogenous V1 spindle bursts in the retinas-ablated pups. A, Extracellular field potential recordings from ipsilateral (black trace) and contralateral (gray trace) V1 of a P6 rat under control conditions (both retinas removed), after injection of 1 μl of ACSF, and after injection of 1 μl of atropine (10 μg/g body weight, in ACSF). Inset, Typical spindle bursts displayed at expanded time scale and averaged power spectrum of the field potential oscillations corresponding to the displayed traces. Note that atropine reduced the occurrence of endogenous spindle bursts. B, Bar diagram displaying the effects of intracerebroventricularly applied atropine on the occurrence of bursts on the ipsilateral (black bars) and contralateral (gray bars) V1 of five P5–P6 pups. The hemisphere of intracerebroventricular injection was defined as ipsilateral.
To approach the role of cholinergic innervation for the generation of retina-driven spindle bursts, we recorded the V1 activity elicited by electrical stimulation of the ON before and after intracortical atropine injection. As reported previously (Hanganu et al. 2006), ON stimulation led to two types of responses on the contralateral V1, a direct response being often followed by ON-induced spindle bursts (Fig. 4Ai). The robust direct response with a mean amplitude of 154.7 ± 22.9 μV started 29 ± 3 ms from stimulation and lasted 0.7 ± 0.1 s (n = 5 pups). It was followed in 12.6 ± 2.3% of stimulations by spindle bursts with mean amplitude of 138.9 ± 17.7 μV, starting 0.9 ± 0.7 s from stimulation and lasting for 2.9 ± 0.4 s. The ON-induced V1 spindle bursts revealed a wide distribution of frequencies within burst ranging from 8 to 22 Hz with a mean of 12.4 ± 1.2 Hz (Fig. 4Aii,iii). Whereas the direct response was not affected by intracortical injection of atropine (onset, 31 ± 5 ms; amplitude, 161.4 ± 33 μV; duration, 0.8 ± 0.2 s; n = 5) (Fig. 4Ai,Bi,C), the ON-induced spindle bursts were diminished by blockade of cortical mAChR (Fig. 4Aii,Bii,D). The occurrence of ON-induced V1 spindle bursts significantly decreased to 2.8 ± 0.9% of stimulations (p = 0.004). The onset (0.95 ± 0.2 s), amplitude (106.8 ± 8.7 μV), duration (2.1 ± 0.6 s), and frequency within the bursts (12.9 ± 0.5 Hz) (Fig. 4Biii) were not modified by atropine. These data indicate that the cholinergic input controls not only the spontaneous, but also the retina-triggered V1 spindle bursts.
Figure 4.
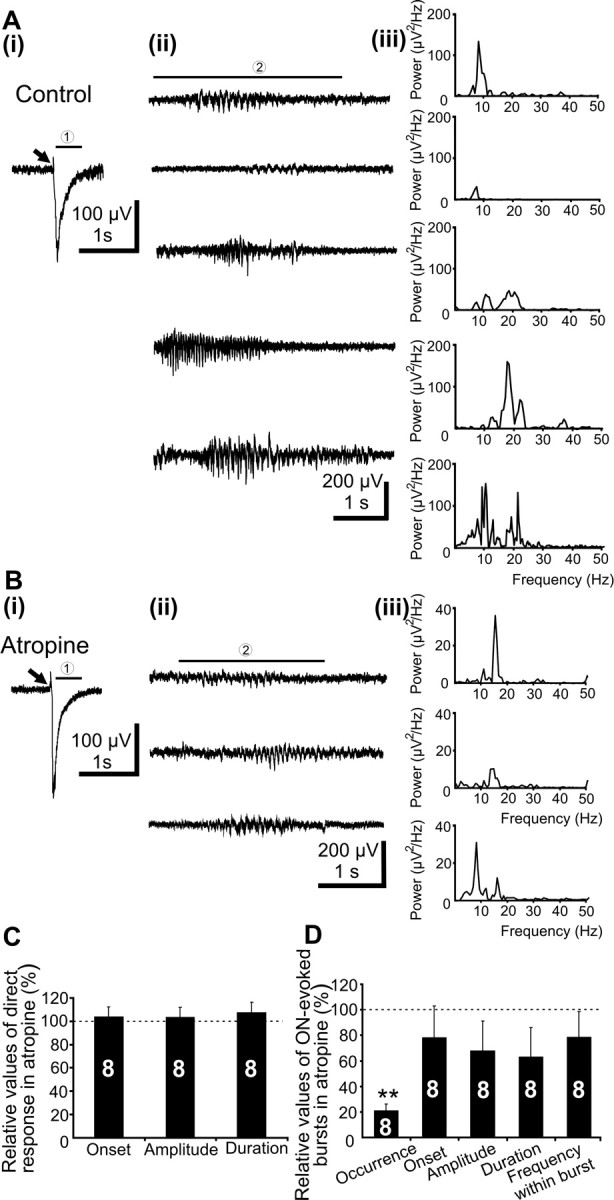
Effects of intracortical injection of atropine on the V1 spindle bursts elicited by ON stimulation. A, Averaged ON-evoked cortical responses (1; i) followed by individual spindle bursts (2; ii) in response to electrical stimulation (black arrow) of the contralateral ON in a P5 pup under control conditions. Note the various amplitude and duration of the ON-induced spindle bursts. Aiii, Power spectra of the single ON-induced V1 spindle bursts shown in ii. B, Averaged direct cortical responses (1; i) followed by individual spindle bursts (2; ii) in response to electrical stimulation (black arrow) of the contralateral ON in a P5 pup after intracortical injection of 50 nl of atropine (10 μg/g body weight, in ACSF; coordinates of injection on the recorded side, −2 mm anterior to bregma, 2.5 mm lateral to midline, and 400 μm below the pial surface). Note that atropine had no effect on the direct response, but diminished the ON-induced spindle bursts. Biii, Power spectra of the single ON-induced V1 spindle bursts shown in ii. C, Bar diagram displaying the relative onset, amplitude, and duration of the ON-induced direct response (1) in the presence of atropine when compared with controls (100%). D, Bar diagram displaying the properties of ON-induced spindle bursts in the V1 of five pups before and after intracortical injection of atropine on the recorded hemisphere.
We further investigated the effects of increased ACh level on V1 spindle bursts by blocking the ACh degrading enzyme acetylcholine esterase (AChE) with the specific inhibitor physostigmine (Grundfest et al., 1952). Physostigmine was intracortically injected (300–600 μm deep, at 1.5–3 mm anterior from the recoding site) in six P5–P6 pups or topically applied on the V1 surface in five P5–P6 pups. This local type of application was used because intracerebroventricularly injected physostigmine (n = 4 pups) caused a dramatic increase in the motor activity (n = 4 pups), seizure-like behavior (n = 4 pups), and even death (n = 1 pup). Control experiments were performed by injecting the vehicle (ACSF) at the same volume and speed as physostigmine. Neither the MUA frequency nor the properties of spindle bursts on both hemispheres were modified by intracortical injection or cortical application of ACSF (Fig. 5A). In contrast, physostigmine (20–50 nl; 130 μg/kg body weight, in ACSF) generally modified the bursting pattern on the side of injection (Fig. 5A,B). The occurrence of ipsilateral V1 spindle bursts was significantly increased from 1.2 ± 0.1 bursts/min under control conditions to 1.8 ± 0.1 bursts/min (p = 0.0002) in the presence of physostigmine. Their amplitude and duration were also significantly augmented from 165.1 ± 23.9 μV in control to 230.9 ± 28.6 μV in physostigmine (p = 0.03; n = 6 pups) and from 3.35 ± 0.3 s in control to 5.2 ± 0.5 s in physostigmine (p = 0.007; n = 6 pups), respectively. However, the frequency within burst remained constant (control, 13.9 ± 1.4 Hz; physostigmine, 12.7 ± 1 Hz; n = 6 pups) (Fig. 5A, insets, B). Associated with the augmentation of spindle bursts, the frequency of MUA on the ipsilateral V1 was significantly increased from 0.8 ± 0.3 to 3.4 ± 1 Hz (p = 0.026; n = 6 pups). The activity on the noninjected hemisphere was not significantly augmented by physostigmine, neither the occurrence (control, 1.5 ± 0.2 bursts/min; physostigmine, 1.6 ± 0.2 bursts/min), amplitude (control, 134.9 ± 30.7 μV; physostigmine, 129.2 ± 22.3 μV), or duration (control, 3.8 ± 0.4 s; physostigmine, 3.8 ± 0.4 s) of spindle bursts, nor the frequency within burst (control, 12.8 ± 1.4 Hz; physostigmine, 12.7 ± 1 Hz) or the MUA frequency (control, 0.26 ± 0.2 Hz; physostigmine, 0.37 ± 0.3 Hz) being modified (Fig. 5A,B). Similarly, the direct application of physostigmine (130 μg/kg body weight; 20–50 nl at 20 nl/min) on the V1 surface led to a significant (p = 0.02) increase in the occurrence of ipsilateral spindle bursts from 0.8 ± 0.1 bursts/min to 1.36 ± 0.1 bursts/min, but had no effects on the contralateral activity.
Figure 5.
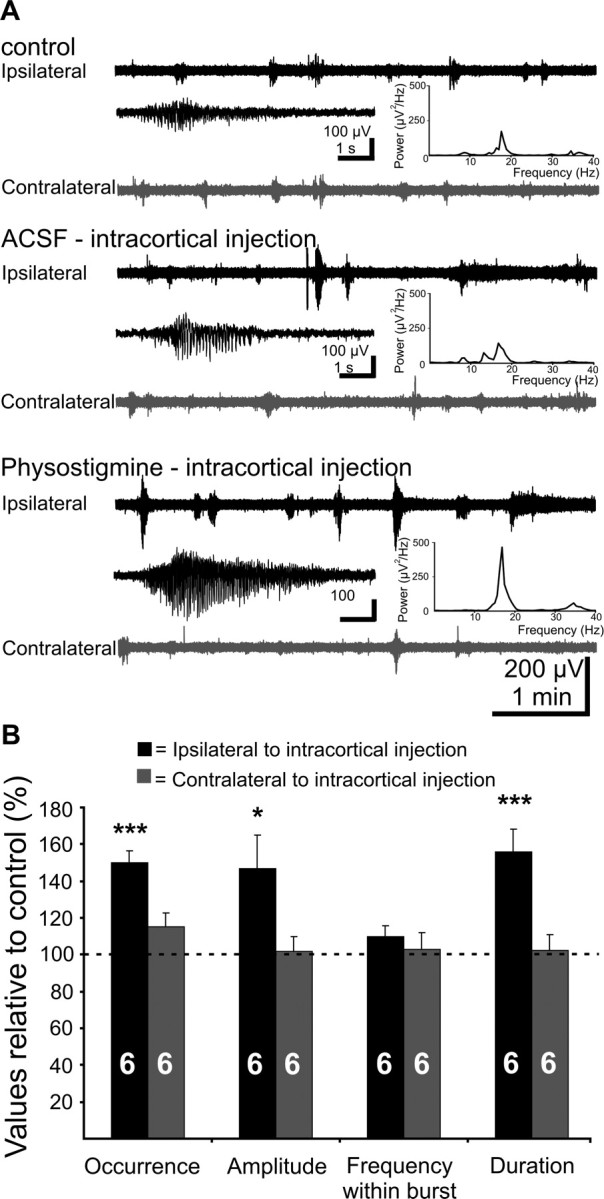
Effects of increased cortical ACh levels by blockade of the AChE with physostigmine on the V1 spindle burst activity of the newborn rat in vivo. A, Extracellular field potential recordings from ipsilateral (black trace) and contralateral (gray trace) V1 of a P6 rat under control conditions, after intracortical injection of 20 nl of ACSF, and after intracortical injection of 20 nl of physostigmine (130 μg/kg body weight, in ACSF). Note the increased occurrence, amplitude, and duration of the V1 bursts on the side of injection. Inset, Typical spindle bursts displayed at an expanded time scale and averaged power spectrum of the field potential oscillations corresponding to the displayed traces. Note the dramatic physostigmine-induced increase in the burst power. B, Bar diagram displaying the effects of physostigmine on the occurrence, amplitude, frequency within burst, and duration of ipsilateral (black bars) and contralateral (gray bars) V1 bursts of six pups. Control values recorded before physostigmine application were considered as 100%. The hemisphere of intracortical injection was defined as ipsilateral.
These results reinforce the hypothesis that the cholinergic input facilitates the generation of spindle bursts in the neonatal visual cortex.
Stimulation of the cholinergic basal forebrain nuclei induces V1 spindle bursts
Most of the cholinergic axons innervating the neocortex originate in the BFn (Metherate et al., 1992). They start to grow into the cortical layers by birth, maturating during the first postnatal weeks (Kiss and Patel, 1992). The contribution of BFn to the cholinergic control of V1 spindle bursts was assessed by simultaneously stimulating the BFn and recording the V1 cortical activity in three P0–P1 and eight P5–P6 rats (Fig. 6A). The electrode for BFn stimulation was inserted 0–0.4 mm posterior to bregma, 1.5 mm lateral to midline, and 3.5–5 mm ventral to the pial surface. These stereotaxic coordinates correspond to the location of the BFn described previously (Paxinos et al., 1991) (Fig. 6B) and were confirmed by DiI-labeling of electrodes (Fig. 6B) and immunocytochemical staining for the VAChT (Fig. 6C).
Stimulation of the BFn was performed taking several precautions. The deep insertion of the stimulation electrode required by the ventral localization of the BFn may have damaging effects on the brain structures with more dorsal positions and, therefore, a nonspecific action on the V1 activity. To decide on the consequences of this lesion, we investigated the V1 oscillatory activity before and after insertion of the stimulation electrode (n = 6 pups). The frequency of MUA was not significantly modified by the insertion of the electrode (1.1 ± 0.4 Hz in control and 0.6 ± 0.2 Hz after insertion). A similar lack of effects was observed when the properties of V1 spindle bursts were investigated. Their occurrence was completely comparable before (contralateral, 1.5 ± 0.2 bursts/min; ipsilateral, 1.9 ± 0.4 bursts/min) and after electrode insertion (contralateral, 1.1 ± 0.2 bursts/min; ipsilateral, 1.9 ± 0.3 bursts/min), and no significant changes could be observed in the amplitude (contralateral, 106 ± 10 μV in control and 106 ± 11 μV after insertion; ipsilateral, 197 ± 44 μV in control and 180.6 ± 31 μV after insertion) or the duration (contralateral, 3.1 ± 0.5 s in control and 2.7 ± 0.2 s after insertion; ipsilateral, 3.6 ± 0.5 s in control and 3.3 ± 0.5 s after insertion) of V1 spindle bursts. Thus, the insertion of the stimulation electrode into BFn does not affect the V1 activity.
Because a single stimulation of the BFn had no effect on the V1 activity (n = 3 pups), we used a previously described tetanic stimulation protocol (Metherate and Ashe, 1991; Rasmusson et al., 1994) to increase the efficiency of stimulation. In eight investigated P5–P6 pups, repetitive stimulation of the BFn at 10 Hz for 1 s performed with a bipolar electrode every 15 s induced spindle bursts on the ipsilateral side of stimulation in 15.6 ± 2.4% of stimulations (Fig. 6D, top, E). BFn-evoked spindle bursts started 1.5 ± 0.1 s after the tetanic train of pulses and showed similar amplitude (149.1 ± 19.5 μV; n = 8 pups), frequency within bursts (13.8 ± 0.9 Hz; n = 8 pups) (Fig. 6D inset), and duration (2.2 ± 0.3 s; n = 8 pups) as the spontaneously occurring V1 spindle bursts. The generation probability of spindle bursts in the contralateral V1 after BFn stimulation was significantly lower (6.9 ± 1.2%; p = 0.01; n = 8 pups) (Fig. 6D, bottom, E) when compared with the stimulated side. These data suggest that activation of BFn afferents projecting to the visual cortex significantly increases the generation probability of V1 spindle bursts. We also tested the effect of BFn stimulation in newborn rats. None of the three investigated P0–P1 pups displayed spindle bursts in control conditions (see also Hanganu et al., 2006) or in response to BFn-stimulation. Because mAChRs agonists evoke beta oscillations in the P0–P1 neocortex in vitro (Dupont et al., 2006), the lack in the effect of BFn stimulation may reflect the immaturity of the cholinergic BFn input to neocortex at birth.
Immunotoxic lesion of cholinergic basal forebrain nuclei dramatically diminishes the V1 spindle bursts
The basal forebrain cholinergic projections have been already described as a major factor in the maintenance of normal cortical neuronal structure and function (Hohmann, 2003). Therefore, in the next series of experiments, we studied the effects of long-lasting alteration of the cortical cholinergic input on the V1 spindle bursts. For this purpose, we used a selective method to permanently impair the basal forebrain cholinergic neurons. In contrast to the electrolytic and excitatory amino acid-induced lesions (Hohmann and Coyle, 1988), with an inherent lack of selectivity, the immunolesion technique using 192 IgG-saporin leads to exclusive destruction of the cholinergic neurons in the basal forebrain (Wiley et al., 1991; Book et al., 1992; Robertson et al., 1998; Pappas and Sherren, 2003). The immunotoxin termed 192 IgG-saporin has been obtained by connecting saporin, a plant-derived ribosome inactivating protein, with an antibody against the low-affinity neurotrophin receptor, p75 (Wiley et al., 1991).
The effects of a selective immunolesion of cholinergic neurons on the oscillatory activity of the V1 were investigated in 21 P5–P6 rats. For these investigations, only the male pups from three litters were used to avoid the influence of different maturation dynamic in males and females of the cholinergic system (Arters et al., 1998) in particular, and of the neocortex in general (Gregory, 1975; Munoz-Cueto et al., 1991). The pups were divided into three groups: (1) pups nontreated, serving as control (n = 8), (2) pups receiving a bilateral intracerebroventricular injection of a total of 1 μl of PBS (solvent for 192 IgG-saporin) at P0 (n = 5), and (3) pups receiving a bilateral injection of a total of 1 μl of 192 IgG-saporin (0.2 μg/μl) in PBS at P0 (n = 8). An extensive analysis of the histological changes of 192 IgG-saporin-treated pups is already available (Robertson et al., 1998). In accordance with these data, the VAChT staining performed on a total of 10 P6 pups revealed a dramatic reduction in the number, but not a complete destruction of the basal forebrain cholinergic neurons in the 192 IgG-saporin-injected pups (n = 4) when compared with the controls (Fig. 7Ai,iii), whereas no significant changes were observed in the PBS-treated pups (n = 4) (Fig. 7Ai,ii). We quantified the neuronal loss in the rostral parts of the BFn (including magnocellular medial preoptic nucleus, horizontal limb of the diagonal band, and rostral nucleus basalis), known to preferentially project to the occipital cortex (Mesulam et al., 1983; Gaykema et al., 1990). The density of cholinergic neurons significantly decreased from 168.3 ± 10.4 neurons/mm2 in controls to 102.3 ± 6.5 neurons/mm2 (p = 0.019) in saporin-treated pups, but was not affected by the PBS treatment (149.4 ± 14.6 neurons/mm2) (Fig. 7B).
Figure 7.
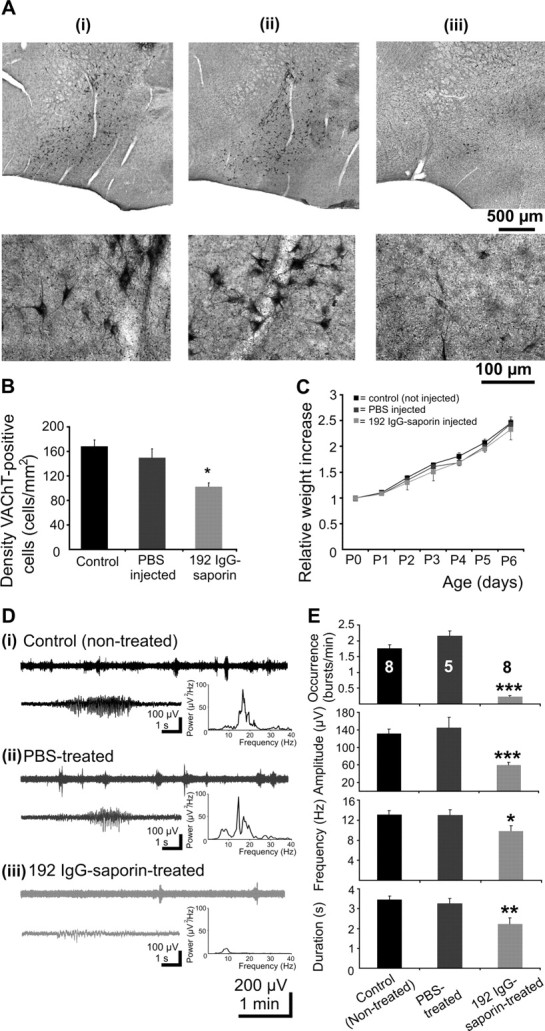
Long-term consequences of selective immunolesion of BFn cholinergic neurons using 192 IgG-saporin on the V1 oscillatory activity. A, Immunocytochemistry of BFn VAChT-positive neurons in control (nontreated; i), PBS-treated (ii), and 192 IgG-saporin-treated (iii) pups. The pups were injected at P0 and the brains fixed by perfusion at P6. Top, Note the dramatic decrease of the number of cholinergic neurons in saporin-treated pups when compared with controls and PBS-treated animals. Bottom, Higher magnification of the sections displaying a comparable portion of the rostral part of the basal nucleus to allow the recognition of the morphology of cholinergic neurons. B, Bar diagram displaying the density of VAChT-positive cells under control conditions and after PBS and 192 IgG-saporin treatment. C, Effects of 192 IgG-saporin treatment on the body weight during early postnatal development. The weight increase for controls, PBS-treated, and 192 IgG-saporin-treated animals is displayed as relative to the P0 weight. Note the similar increase in the body weight of all investigated pups. D, Extracellular field potential recordings from ipsilateral and contralateral V1 of a nontreated P6 pup (i; black traces), a PBS-treated P6 pup (ii; dark gray traces), and a 192 IgG-saporin-treated P6 pup (iii; light gray traces). Inset, Typical spindle bursts displayed at an enlarged scale and averaged power spectrum of the field potential oscillations corresponding to the displayed traces. Note the dramatic reduction in the occurrence and power of spindle bursts in 192 IgG-saporin-treated animals. E, Bar diagram displaying the mean occurrence, amplitude, frequency within burst and duration of V1 spindle bursts recorded in eight control (nontreated), five PBS-treated, and eight 192 IgG saporin-treated pups.
Because 192 IgG-saporin treatment may generally impair the development of animals by affecting their behavior and feeding abilities (Leanza et al., 1996; Pappas et al., 1996; Robertson et al., 1998), we also investigated the developmental increase in the body weight of control and treated animals. No significant difference was observed between the weight of control, PBS-, and 192 IgG-saporin-treated pups over the investigated time-window, P0–P6 (Fig. 7C). These results are in accordance with previous data, showing that the immunotoxin treatment leads to weight loss starting from P8 (Robertson et al., 1998). Additionally, without being the main focus of our study and therefore not systematically quantified, the observed general behavior (locomotor ability, sleep-awake cyclus, feeding ability, response to maternal behavior) of PBS- and 192 IgG-saporin-treated pups did not differ from that of the controls (nontreated).
The V1 activity of the control animals was characterized by the presence of spindle bursts as the main activity pattern (Fig. 7Di). The bursts recorded at the same depth in all investigated pups occurred at similar frequencies on both hemispheres (1.75 ± 0.2 and 1.75 ± 0.24 bursts/min; n = 8 pups) and had similar amplitudes (135.9 ± 14.3 and 125.5 ± 17.7 μV), frequency within burst (14 ± 1 and 12 ± 1 Hz), duration (3.5 ± 0.3 and 3.4 ± 0.3 s), and power (133.8 ± 42.2 and 190.8 ± 91.8 μV2/Hz). The spindle bursts were accompanied by MUA occurring at an average frequency of 0.23 ± 0.08 Hz (n = 8 pups). Before characterizing the action of 192 IgG-saporin on the cortical activity, we tested the effects of needle insertion and intracerebroventricular solvent (PBS) injection. Five to 6 d after 192 IgG-saporin injection, no evidence of major damage caused by the injection needle was recognizable. The V1 oscillatory activity recorded in PBS-treated pups (n = 5) was similar to that recorded in control animals (Fig. 7Dii,E). The spindle bursts of each of the two hemispheres occurred at frequencies of 2 ± 0.3 and 2.3 ± 0.2 bursts/min, respectively, and had amplitudes of 140.3 ± 28 and 150 ± 39.4 μV, respectively. The duration (3.5 ± 0.5 and 3 ± 0.3 s) and power (89.3 ± 26.3 and 295 ± 200 μV2/Hz) of spindle bursts as well as the frequency within burst (13.9 ± 0.6 and 12.1 ± 2 Hz) were not significantly different from the controls. The frequency of MUA (0.2 ± 0.08 Hz) remained unchanged in PBS-treated pups when compared with controls. In contrast, neonatal treatment with 192 IgG-saporin caused major changes in the V1 oscillatory activity of the eight pups investigated 5–6 d after immunotoxin injection. The activity on both hemispheres diminished (Fig. 7Diii,E). The occurrence of spindle bursts on each of the two hemispheres was significantly reduced to 0.2 ± 0.09 bursts/min (p = 2.5 × 10−6) and 0.3 ± 0.07 bursts/min (p = 3.8 × 10−5), respectively, and the amplitude significantly decreased (70.2 ± 7.9 μV, p = 0.003; 59.3 ± 4.4 μV, p = 0.005). The spindle bursts recorded in 192 IgG-saporin-treated pups were shorter (1.8 ± 0.3 and 2.8 ± 0.4 s; n = 8) than in controls and the frequency within bursts significantly lower (10.3 ± 1 Hz, p = 0.004; 10.7 ± 1 Hz, p = 0.035; n = 8). The power of spindle bursts on each of the two hemispheres decreased to 11.2 ± 4.7 μV2/Hz (p = 0.029) and 8.4 ± 1.5 μV2/Hz (p = 0.08), respectively, after treatment with 192 IgG-saporin. Thus, these results indicate that the selective lesion of cholinergic neurons in the basal forebrain using 192 IgG-saporin dramatically reduces the V1 oscillatory activity, supporting the role of cholinergic innervation for the normal development of cortical circuitry and function.
Discussion
In the present study, we investigated the impact of cholinergic input on the oscillatory activity of the visual cortex during the first postnatal week, which is a critical period for the fine-tuning of the geniculocortical connections (Cang et al., 2005; del Rio and Feller, 2006; Huberman et al., 2006). Using in vivo recordings from V1 of neonatal rats paired with short-term and long-term modification of the cholinergic input, we showed that (1) acute pharmacological blockade of the cortical mAChR or increase in the ACh level modulates both spontaneous and retina-triggered V1 spindle bursts, (2) electrical stimulation of the BFn responsible for the cortical cholinergic input elicits V1 spindle bursts, and (3) irreversible immunotoxic lesion of the BFn dramatically diminishes the oscillatory spindle bursts activity of the V1 cortex. Thus, these data indicate that the cholinergic drive to the visual cortex facilitates the generation of spontaneous and retina-driven V1 spindle bursts and reinforce the idea that ACh is one of the neuromodulatory inputs able to shape the immature synaptic circuits by optimally controlling their activity.
Cholinergic control of V1 spindle bursts
Several lines of evidence indicate that the cholinergic input from the BFn facilitates the generation of spindle bursts in the neonatal visual cortex. First, mAChR blockade by atropine applied via various routes reduced the occurrence of the V1 spindle bursts. Similar effects have been documented in the adult visual cortex, where iontophoretic blockade of the mAChR reduced the endogenous fast gamma oscillations in vivo (Rodriguez et al., 2004). In contrast, the literature data concerning the cholinergic influence on circuit activity in the neonatal cortex are less uniform. Whereas application of cholinergic agonists in vitro may occasionally inhibit the responses of developing circuits (Roerig et al., 1997), their potentiation seems to be the dominant function of cholinergic innervation. Application of cholinergic agonists on cortical slices induced periodic oscillatory activity (Kilb and Luhmann, 2003) (I. L. Hanganu, unpublished observations) capable of spreading over large distances (Peinado, 2000) or facilitated the cortical synaptic transmission in an age-dependent manner (Kuczewski et al., 2005). To the best of our knowledge, the present data are the first in vivo proof of the causality between the acute modification of the mAChR and the modulation of neonatal cortical activity and, thus, confirm the positive role of cholinergic input on the early V1 activity. Remarkably, both retina-independent and retina-triggered spindle bursts are modified by atropine, indicating a global action of the cholinergic input on V1 activity. However, atropine diminished more strongly the retina-driven activity than the endogenous V1 oscillations, both the occurrence and the amplitude of retina-triggered spindle bursts being significantly reduced. It remains to be elucidated whether this early selective potentiation is a prerequisite for the dependence of plasticity in the adult visual cortex on cholinergic innervation (Gu and Singer, 1993).
Second, the augmentation of the cortical activity by blockade of AChE is likely to reinforce the cholinergic modulation of V1 activity. Physostigmine as a potent AChE antagonist enhanced the V1 ability to generate spindle bursts. A similar increase in the frequency of the giant depolarizing potentials was observed after AChE blockade in the neonatal rat hippocampus in vitro (Avignone and Cherubini, 1999).
Third, stimulation of the BFn increased the generation probability of V1 spindle bursts. The reliable facilitation of the spindle bursts in P5–P6 pups, and the lack of BFn-induced spindle bursts at an early postnatal age (P0–P1) when spontaneous V1 bursts are also absent, supports the modulatory role of the BFn cholinergic input. The low incidence of BFn-induced spindle bursts could be partially attributable to the relative immaturity of the BFn-to-cortex pathway. The distribution of cholinergic fibers in cortical areas shows an adult-like pattern starting at P11 (Calarco and Robertson, 1995). However, we cannot rule out an ineffective stimulation paradigm as a cause for the low incidence of BF-induced oscillations. Despite the fact that BFn stimulation was performed according to the stereotaxic coordinates determined in the present study by VAChT staining and corresponding to the literature data (Gaykema et al., 1990), the deep and discrete localization, as well as the small size of BFn, precluded a more precise activation of the cholinergic input using bipolar stimulation electrodes. However, none of the nuclei located in the direct neighborhood of the BFn directly projects to V1 and, therefore, the probability of nonspecific activation remains relative low. Such technical difficulties concerning the access to BFn neurons in newborns may explain the lack of previous data concerning the effect of BFn stimulation on the neonatal cortical activity. Our data, describing for the first time the BFn modulation of neonatal spindle bursts, are similar to the effect observed in adult animals, where stimulation of BFn elicited EEG activation via cortical mAChR (Metherate et al., 1992).
Finally, the chronic effects of an immunotoxic lesion of the BFn represent a solid piece of evidence for the cholinergic modulation of V1 activity. According to anatomical studies, the destruction of cholinergic BFn neurons starts 24 h after injection of 192 IgG-saporin, reaches its maximum 10–20 d later, and lasts for at least 6 month (Robertson et al., 1998; Koh et al., 2005). Despite the fact that 5–6 d after intracerebroventricular injection of immunotoxin the lesion of BFn cholinergic neurons is not complete (see VAChT staining), a dramatic reduction in the occurrence and a significant modification of the properties of V1 spindle bursts could be observed. Profound impairment of V1 spindle bursts indicates abnormal cortical development in 192 IgG saporin-treated animals. This is in keeping with the results of a large number of studies that characterized the morphological and behavioral effects of the 192 IgG-saporin injections. Reduction of the cortical thickness and cholinergic innervation (Robertson et al., 1998; Koh et al., 2005) associated with more or less pronounced impairment of visual attention and working memory (Leanza et al., 1996; Pappas et al., 2005; McGaughy et al., 2005) have been described. Yet, in contrast to robust modification of activity in neonates, the adult cortical activity is less impaired by 192 IgG-saporin (Wenk et al., 1994; Holschneider et al., 1999). Therefore, the shaping ability of the cholinergic input from the BFn seems to be optimal during a defined phase of development and to decrease at adulthood.
In addition to the immunolesion of cholinergic BFn neurons, an additional mechanism may account for the significant effects of saporin treatment, performed directly at birth, on the cortical activity. 192 IgG-saporin selectively lesions the p75 expressing neurons, which cluster within the BFn, but are present in other brain regions, however, at lower density. In the neonatal cortex, the expression of p75 receptor is highly restricted to a transiently expressed layer of neurons, the subplate (Koh and Loy, 1989; Meinecke and Rakic, 1993). The subplate neurons are requested for the generation of oscillatory activity patterns in the neonatal cortex by birth (Dupont et al., 2006). To which extent lesion of the p75-expressing subplate neurons contributes to the 192 IgG saporin-induced impairment of spindle bursts remains to be elucidated.
The mechanisms by which the cholinergic input controls and enhances the synchronized cortical activity are not completely understood. The excitatory mAChR-mediated ACh action, inducing sustained increase in membrane resistance and depolarization by interfering with K+ conductance and IP3-dependent Ca2+ release (Krnjevic et al., 1971; McCormick and Prince, 1985), is necessary, but probably not sufficient to generate these effects. The oscillatory bursting pattern of cholinergic BFn projecting to cortical areas (Lee et al., 2005) may also contribute to the synchronization of activity in V1. The efficiency of 10 Hz stimulation (as reported here and in other studies) to induce optimal activation of the rhythmically discharging BFn supports the previously proposed gating function of the cholinergic input (Metherate et al., 1992; Bao et al., 2003). The activity of the cholinergic BFn allows the cortex to operate specifically on relevant stimuli. However, several other afferent systems (dopaminergic, noradrenergic, serotonergic, etc.) are present at this developmental stage (Lidov and Molliver, 1982; Latsari et al., 2002). Similar to cholinergic input, they may control the cortical activity and set the threshold for activity-dependent circuitry shaping (Gu, 2002). The existence of cholinergic-independent activity patterns, as shown here, supports this assumption. Moreover, these neuromodulatory inputs may synergistically interact with the abundant cholinergic afferents in facilitating the synchronization of cortical activity.
Physiological role of the cholinergic drive on the neonatal oscillatory activity
Previous data indicate that the cholinergic innervation critically interferes with the cortical development by finely tuning the sensory maps. Damage of the BFn impairs the barrel formation in the somatosensory cortex (Baskerville et al., 1997; Zhu and Waite, 1998) and ACh acting on mAChR modulates the tonotopic maps in the developing auditory cortex (Zhang et al., 2005). In the neonatal visual cortex, the formation of the visual feature maps, described as ocular dominance columns (ODCs), has been extensively investigated. Blockade of the mAChR disrupted the organization of ODCs (Gu et al., 1989; Gu and Singer, 1993) whereas iontophoretic application of ACh facilitated receptive-field modifications when paired with visual stimulation (Greuel et al., 1988). Despite the abundance of experimental evidence supporting the ability of cholinergic input to modify sensory maps, the underlying mechanisms are widely unknown. We propose that the cholinergic control of cortical activity, as reported here, may represent one possible way that the cholinergic innervation accomplishes the fine tuning of sensory maps. Our previous data showed that the V1 spindle bursts are generally spatially confined (Hanganu et al., 2006). The ACh-induced augmentation of this synchronized oscillatory activity in neonatal cortical networks may be necessary to facilitate incoming activity (retinal wave, driven during early development; visual stimulus, triggered later during development). Although the direct causality between early oscillatory activity and cortical maturation is still subject to debate, the here reported ability of the cholinergic drive from the BFn to facilitate early oscillatory activity may represent one of the key mechanisms by which acetylcholine controls cortical development.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Sta 431/2-4; I.L.H.), Institut National de la Santé et de la Recherche Médicale, Fondation pour la Recherche Médicale, and Agence Nationale de la Recherche (R.K., Y.B.A.). We thank Dr. Georg Haase for help with histology, and Monika Knoblich and Nadine Ferrand for technical assistance. We thank Drs. W. Kilb, T. Opatz, and M. Colonnese for helpful comments on this manuscript.
References
- Arters J, Hohmann CF, Mills J, Olaghere O, Berger-Sweeney J. Sexually dimorphic responses to neonatal basal forebrain lesions in mice: I. Behavior and neurochemistry. J Neurobiol. 1998;37:582–594. doi: 10.1002/(sici)1097-4695(199812)37:4<582::aid-neu7>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Avignone E, Cherubini E. Muscarinic receptor modulation of GABA-mediated giant depolarizing potentials in the neonatal rat hippocampus. J Physiol (Lond) 1999;518:97–107. doi: 10.1111/j.1469-7793.1999.0097r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Chang EF, Davis JD, Gobeske KT, Merzenich MM. Progressive degradation and subsequent refinement of acoustic representations in the adult auditory cortex. J Neurosci. 2003;23:10765–10775. doi: 10.1523/JNEUROSCI.23-34-10765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville KA, Schweitzer JP, Herron P. Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience. 1997;80:1159–1169. doi: 10.1016/s0306-4522(97)00064-x. [DOI] [PubMed] [Google Scholar]
- Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- Bloom JS, Hynd GW. The role of the corpus callosum in interhemispheric transfer of information: excitation or inhibition? Neuropsychol Rev. 2005;15:59–71. doi: 10.1007/s11065-005-6252-y. [DOI] [PubMed] [Google Scholar]
- Book AA, Wiley RG, Schweitzer JB. Specificity of 192 IgG-saporin for NGF receptor-positive cholinergic basal forebrain neurons in the rat. Brain Res. 1992;590:350–355. doi: 10.1016/0006-8993(92)91121-t. [DOI] [PubMed] [Google Scholar]
- Brimijoin S, Koenigsberger C. Cholinesterases in neural development: new findings and toxicologic implications. Environ Health Perspect. 1999;107(Suppl 1):59–64. doi: 10.1289/ehp.99107s159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers DM, Irwin LN, Moss DE, Sumaya IC, Hohmann CF. Prenatal exposure to the acetylcholinesterase inhibitor methanesulfonyl fluoride alters forebrain morphology and gene expression. Dev Brain Res. 2005;158:13–22. doi: 10.1016/j.devbrainres.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Calarco CA, Robertson RT. Development of basal forebrain projections to visual cortex: DiI studies in rat. J Comp Neurol. 1995;354:608–626. doi: 10.1002/cne.903540409. [DOI] [PubMed] [Google Scholar]
- Cang J, Renteria RC, Kaneko M, Liu X, Copenhagen DR, Stryker MP. Development of precise maps in visual cortex requires patterned spontaneous activity in the retina. Neuron. 2005;48:797–809. doi: 10.1016/j.neuron.2005.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio AR, Graff-Radford NR, Eslinger PJ, Damasio H, Kassell N. Amnesia following basal forebrain lesions. Arch Neurol. 1985;42:263–271. doi: 10.1001/archneur.1985.04060030081013. [DOI] [PubMed] [Google Scholar]
- del Rio T, Feller MB. Early retinal activity and visual circuit development. Neuron. 2006;52:221–222. doi: 10.1016/j.neuron.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Dupont E, Hanganu IL, Kilb W, Hirsch S, Luhmann HJ. Rapid developmental switch in the mechanisms driving early cortical columnar networks. Nature. 2006;439:79–83. doi: 10.1038/nature04264. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Gaykema RP, Luiten PG, Nyakas C, Traber J. Cortical projection patterns of the medial septum-diagonal band complex. J Comp Neurol. 1990;293:103–124. doi: 10.1002/cne.902930109. [DOI] [PubMed] [Google Scholar]
- Gregory E. Comparison of postnatal CNS development between male and female rats. Brain Res. 1975;99:152–156. doi: 10.1016/0006-8993(75)90618-6. [DOI] [PubMed] [Google Scholar]
- Greuel JM, Luhmann HJ, Singer W. Pharmacological induction of use-dependent receptive field modifications in the visual cortex. Science. 1988;242:74–77. doi: 10.1126/science.2902687. [DOI] [PubMed] [Google Scholar]
- Grundfest H, Nachmansohn D, Kao CY, Chambers R. Mode of blocking of axonal activity by curare and inhibitors of acetylcholinesterase. Nature. 1952;169:190. doi: 10.1038/169190a0. [DOI] [PubMed] [Google Scholar]
- Gu Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience. 2002;111:815–835. doi: 10.1016/s0306-4522(02)00026-x. [DOI] [PubMed] [Google Scholar]
- Gu Q, Singer W. Effects of intracortical infusion of anticholinergic drugs on neuronal plasticity in kitten striate cortex. Eur J Neurosci. 1993;5:475–485. doi: 10.1111/j.1460-9568.1993.tb00514.x. [DOI] [PubMed] [Google Scholar]
- Gu QA, Bear MF, Singer W. Blockade of NMDA-receptors prevents ocularity changes in kitten visual cortex after reversed monocular deprivation. Dev Brain Res. 1989;47:281–288. doi: 10.1016/0165-3806(89)90183-1. [DOI] [PubMed] [Google Scholar]
- Hanganu IL, Luhmann HJ. Functional nicotinic acetylcholine receptors on subplate neurons in neonatal rat somatosensory cortex. J Neurophysiol. 2004;92:189–198. doi: 10.1152/jn.00010.2004. [DOI] [PubMed] [Google Scholar]
- Hanganu IL, Ben Ari Y, Khazipov R. Retinal waves trigger spindle bursts in the neonatal rat visual cortex. J Neurosci. 2006;26:6728–6736. doi: 10.1523/JNEUROSCI.0752-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann CF. A morphogenetic role for acetylcholine in mouse cerebral neocortex. Neurosci Biobehav Rev. 2003;27:351–363. doi: 10.1016/s0149-7634(03)00066-6. [DOI] [PubMed] [Google Scholar]
- Hohmann CF, Coyle JT. Long-term effects of basal forebrain lesions on cholinergic, noradrenergic and serotonergic markers in mouse neocortex. Brain Res Bull. 1988;21:13–20. doi: 10.1016/0361-9230(88)90113-x. [DOI] [PubMed] [Google Scholar]
- Holschneider DP, Waite JJ, Leuchter AF, Walton NY, Scremin OU. Changes in electrocortical power and coherence in response to the selective cholinergic immunotoxin 192 IgG-saporin. Exp Brain Res. 1999;126:270–280. doi: 10.1007/s002210050736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Speer CM, Chapman B. Spontaneous retinal activity mediates development of ocular dominance columns and binocular receptive fields in V1. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti GM. Exuberant development of connections, and its possible permissive role in cortical evolution. Trends Neurosci. 1995;18(9):397–402. doi: 10.1016/0166-2236(95)93936-r. [DOI] [PubMed] [Google Scholar]
- Juliano SL, Ma W, Eslin D. Cholinergic depletion prevents expansion of topographic maps in somatosensory cortex. Proc Natl Acad Sci USA. 1991;88:780–784. doi: 10.1073/pnas.88.3.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazipov R, Sirota A, Leinekugel X, Holmes GL, Ben-Ari Y, Buzsáki G. Early motor activity drives spindle bursts in the developing somatosensory cortex. Nature. 2004;432:758–761. doi: 10.1038/nature03132. [DOI] [PubMed] [Google Scholar]
- Kilb W, Luhmann HJ. Carbachol-induced network oscillations in the intact cerebral cortex of the newborn rat. Cereb Cortex. 2003;13:409–421. doi: 10.1093/cercor/13.4.409. [DOI] [PubMed] [Google Scholar]
- Kiss J, Patel AJ. Development of the cholinergic fibres innervating the cerebral cortex of the rat. Int J Dev Neurosci. 1992;10:153–170. doi: 10.1016/0736-5748(92)90043-y. [DOI] [PubMed] [Google Scholar]
- Koh S, Loy R. Localization and development of nerve growth factor-sensitive rat basal forebrain neurons and their afferent projections to hippocampus and neocortex. J Neurosci. 1989;9:2999–3018. doi: 10.1523/JNEUROSCI.09-09-02999.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Santos TC, Cole AJ. Susceptibility to seizure-induced injury and acquired microencephaly following intraventricular injection of saporin-conjugated 192 IgG in developing rat brain. Exp Neurol. 2005;194:457–466. doi: 10.1016/j.expneurol.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Krnjevic K, Pumain R, Renaud L. The mechanism of excitation by acetylcholine in the cerebral cortex. J Physiol (Lond) 1971;215:247–268. doi: 10.1113/jphysiol.1971.sp009467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Aztiria E, Leanza G, Domenici L. Selective cholinergic immunolesioning affects synaptic plasticity in developing visual cortex. Eur J Neurosci. 2005;21:1807–1814. doi: 10.1111/j.1460-9568.2005.04014.x. [DOI] [PubMed] [Google Scholar]
- Latsari M, Dori I, Antonopoulos J, Chiotelli M, Dinopoulos A. Noradrenergic innervation of the developing and mature visual and motor cortex of the rat brain: a light and electron microscopic immunocytochemical analysis. J Comp Neurol. 2002;445:145–158. doi: 10.1002/cne.10156. [DOI] [PubMed] [Google Scholar]
- Leanza G, Nilsson OG, Nikkhah G, Wiley RG, Bjorklund A. Effects of neonatal lesions of the basal forebrain cholinergic system by 192 immunoglobulin G-saporin: biochemical, behavioural and morphological characterization. Neuroscience. 1996;74:119–141. doi: 10.1016/0306-4522(96)00095-4. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE. Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci. 2005;25:4365–4369. doi: 10.1523/JNEUROSCI.0178-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Khazipov R, Cannon R, Hirase H, Ben-Ari Y, Buzsaki G. Correlated bursts of activity in the neonatal hippocampus in vivo. Science. 2002;296:2049–2052. doi: 10.1126/science.1071111. [DOI] [PubMed] [Google Scholar]
- Lidov HG, Molliver ME. An immunohistochemical study of serotonin neuron development in the rat: ascending pathways and terminal fields. Brain Res Bull. 1982;8:389–430. doi: 10.1016/0361-9230(82)90077-6. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Kater SB. Neurotransmitter regulation of neuronal outgrowth, plasticity and survival. Trends Neurosci. 1989;12:265–270. doi: 10.1016/0166-2236(89)90026-x. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Khazipov R. Early patterns of electrical activity in the developing cerebral cortex of humans and rodents. Trends Neurosci. 2006;29:414–418. doi: 10.1016/j.tins.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Ma X, Suga N. Long-term cortical plasticity evoked by electric stimulation and acetylcholine applied to the auditory cortex. Proc Natl Acad Sci USA. 2005;102:9335–9340. doi: 10.1073/pnas.0503851102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci USA. 2003;100:2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci USA. 1985;82:6344–6348. doi: 10.1073/pnas.82.18.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughy J, Koene RA, Eichenbaum H, Hasselmo ME. Cholinergic deafferentation of the entorhinal cortex in rats impairs encoding of novel but not familiar stimuli in a delayed nonmatch-to-sample task. J Neurosci. 2005;25:10273–10281. doi: 10.1523/JNEUROSCI.2386-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinecke DL, Rakic P. Low-affinity p75 nerve growth factor receptor expression in the embryonic monkey telencephalon: timing and localization in diverse cellular elements. Neuroscience. 1993;54:105–116. doi: 10.1016/0306-4522(93)90386-t. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6) Neuroscience. 1983;10:1185–1201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Hersh LB, Mash DC, Geula C. Differential cholinergic innervation within functional subdivisions of the human cerebral cortex: a choline acetyltransferase study. J Comp Neurol. 1992;318:316–328. doi: 10.1002/cne.903180308. [DOI] [PubMed] [Google Scholar]
- Metherate R, Ashe JH. Basal forebrain stimulation modifies auditory cortex responsiveness by an action at muscarinic receptors. Brain Res. 1991;559:163–167. doi: 10.1016/0006-8993(91)90301-b. [DOI] [PubMed] [Google Scholar]
- Metherate R, Cox CL, Ashe JH. Cellular bases of neocortical activation: modulation of neural oscillations by the nucleus basalis and endogenous acetylcholine. J Neurosci. 1992;12:4701–4711. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney R, Penn AA, Gallego R, Shatz CJ. Thalamic relay of spontaneous retinal activity prior to vision. Neuron. 1996;17:863–874. doi: 10.1016/s0896-6273(00)80218-4. [DOI] [PubMed] [Google Scholar]
- Moruzzi G, Magoun W. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455–473. [PubMed] [Google Scholar]
- Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. 1949. J Neuropsychiatry Clin Neurosci. 1995;7:251–267. doi: 10.1176/jnp.7.2.251. [DOI] [PubMed] [Google Scholar]
- Munoz-Cueto JA, Garcia-Segura LM, Ruiz-Marcos A. Regional sex differences in spine density along the apical shaft of visual cortex pyramids during postnatal development. Brain Res. 1991;540:41–47. doi: 10.1016/0006-8993(91)90490-m. [DOI] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL. Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron. 2005;46:37–49. doi: 10.1016/j.neuron.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Pappas BA, Sherren N. Neonatal 192 IgG-saporin lesion of forebrain cholinergic neurons: focus on the life span? Neurosci Biobehav Rev. 2003;27:365–376. doi: 10.1016/s0149-7634(03)00067-8. [DOI] [PubMed] [Google Scholar]
- Pappas BA, Davidson CM, Fortin T, Nallathamby S, Park GA, Mohr E, Wiley RG. 192 IgG-saporin lesion of basal forebrain cholinergic neurons in neonatal rats. Brain Res Dev Brain Res. 1996;96:52–61. doi: 10.1016/0165-3806(96)00095-8. [DOI] [PubMed] [Google Scholar]
- Pappas BA, Payne KB, Fortin T, Sherren N. Neonatal lesion of forebrain cholinergic neurons: further characterization of behavioral effects and permanency. Neuroscience. 2005;133:485–492. doi: 10.1016/j.neuroscience.2005.02.040. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson CR, Emson PC. AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods. 1980;3:129–149. doi: 10.1016/0165-0270(80)90021-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Törk I, Tecott LH, Valentino KL. San Diego: Academic; 1991. Atlas of the developing rat brain. [Google Scholar]
- Peinado A. Traveling slow waves of neural activity: a novel form of network activity in developing neocortex. J Neurosci. 2000;20(RC54) doi: 10.1523/JNEUROSCI.20-02-j0002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh PC, Margiotta JF. Nicotinic acetylcholine receptor agonists promote survival and reduce apoptosis of chick ciliary ganglion neurons. Mol Cell Neurosci. 2000;15:113–122. doi: 10.1006/mcne.1999.0810. [DOI] [PubMed] [Google Scholar]
- Rasmusson DD, Clow K, Szerb JC. Modification of neocortical acetylcholine release and electroencephalogram desynchronization due to brainstem stimulation by drugs applied to the basal forebrain. Neuroscience. 1994;60:665–677. doi: 10.1016/0306-4522(94)90495-2. [DOI] [PubMed] [Google Scholar]
- Robertson RT, Gallardo KA, Claytor KJ, Ha DH, Ku KH, Yu BP, Lauterborn JC, Wiley RG, Yu J, Gall CM, Leslie FM. Neonatal treatment with 192 IgG-saporin produces long-term forebrain cholinergic deficits and reduces dendritic branching and spine density of neocortical pyramidal neurons. Cereb Cortex. 1998;8:142–155. doi: 10.1093/cercor/8.2.142. [DOI] [PubMed] [Google Scholar]
- Rodriguez R, Kallenbach U, Singer W, Munk MH. Short- and long-term effects of cholinergic modulation on gamma oscillations and response synchronization in the visual cortex. J Neurosci. 2004;24:10369–10378. doi: 10.1523/JNEUROSCI.1839-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17:8353–8362. doi: 10.1523/JNEUROSCI.17-21-08353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Weihe E, Erickson JD, Eiden LE. Human and monkey cholinergic neurons visualized in paraffin-embedded tissues by immunoreactivity for VAChT, the vesicular acetylcholine transporter. J Mol Neurosci. 1995;6:225–235. doi: 10.1007/BF02736782. [DOI] [PubMed] [Google Scholar]
- Semba K, Fibiger HC. Organization of central cholinergic systems. Prog Brain Res. 1989;79:37–63. doi: 10.1016/s0079-6123(08)62464-4. [DOI] [PubMed] [Google Scholar]
- Siciliano R, Fontanesi G, Casamenti F, Berardi N, Bagnoli P, Domenici L. Postnatal development of functional properties of visual cortical cells in rats with excitotoxic lesions of basal forebrain cholinergic neurons. Vis Neurosci. 1997;14:111–123. doi: 10.1017/s0952523800008816. [DOI] [PubMed] [Google Scholar]
- Singer W. The brain as a self-organizing system. Eur Arch Psychiatry Neurol Sci. 1986;236:4–9. doi: 10.1007/BF00641050. [DOI] [PubMed] [Google Scholar]
- Staiger JF, Masanneck C, Schleicher A, Zuschratter W. Calbindin-containing interneurons are a target for VIP-immunoreactive synapses in rat primary somatosensory cortex. J Comp Neurol. 2004;468:179–189. doi: 10.1002/cne.10953. [DOI] [PubMed] [Google Scholar]
- Steriade M, Dossi RC, Paré D, Oakson G. Fast oscillations (20–40 Hz) in thalamocortical systems and their potentiation by mesopontine cholinergic nuclei in the cat. Proc Natl Acad Sci USA. 1991;88:4396–4400. doi: 10.1073/pnas.88.10.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihe E, Tao-Cheng JH, Schafer MK, Erickson JD, Eiden LE. Visualization of the vesicular acetylcholine transporter in cholinergic nerve terminals and its targeting to a specific population of small synaptic vesicles. Proc Natl Acad Sci USA. 1996;93:3547–3552. doi: 10.1073/pnas.93.8.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL, Stoehr JD, Quintana G, Mobley S, Wiley RG. Behavioral, biochemical, histological, and electrophysiological effects of 192 IgG-saporin injections into the basal forebrain of rats. J Neurosci. 1994;14:5986–5995. doi: 10.1523/JNEUROSCI.14-10-05986.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley RG, Oeltmann TN, Lappi DA. Immunolesioning: selective destruction of neurons using immunotoxin to rat NGF receptor. Brain Res. 1991;562:149–153. doi: 10.1016/0006-8993(91)91199-b. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dyck RH, Hamilton SE, Nathanson NM, Yan J. Disrupted tonotopy of the auditory cortex in mice lacking M1 muscarinic acetylcholine receptor. Hear Res. 2005;201:145–155. doi: 10.1016/j.heares.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Zhu XO, Waite PM. Cholinergic depletion reduces plasticity of barrel field cortex. Cereb Cortex. 1998;8:63–72. doi: 10.1093/cercor/8.1.63. [DOI] [PubMed] [Google Scholar]