

Abstract
A pathological hallmark of Alzheimer's disease is accumulation of amyloid-β peptide (Aβ) in senile plaques. Aβ has also been implicated in vascular degeneration in cerebral amyloid angiopathy because of its cytotoxic effects on non-neuronal cells, including cerebral endothelial cells (CECs). We explore the role of apoptosis signal-regulating kinase 1 (ASK1) in Aβ-induced death in primary cultures of murine CECs. Aβ induced ASK1 dephosphorylation, which could be prevented by selective inhibition of protein phosphatase 2A (PP2A) but not PP2B. ASK1 dephosphorylation resulted in its dissociation from 14-3-3. ASK1, released from 14-3-3 inhibition, activated p38 mitogen-activated protein kinase (p38MAPK), leading to p53 phosphorylation. p53, a proapoptotic transcription factor, in turn transactivated the expression of Bax, a proapoptotic protein. Transfection with various dominant-negative mutants (DNs), including ASK1 DN and p38MAPK DN, suppressed Aβ-induced p38MAPK activation, p53 phosphorylation, and Bax upregulation and partially prevented CEC death. Bax knockdown using a bax small interfering RNA strategy also reduced Bax expression and subsequent CEC death. These results suggest that Aβ activates the ASK1–p38MAPK–p53–Bax cascade to cause CEC death in a PP2A-dependent manner.
Keywords: angiopathy, ASK1, Bax, cerebrovascular diseases, p38 mitogen-activated protein kinase, p38MAPK, p53
Introduction
Amyloid-β peptide (Aβ) is a primary mediator of neuronal degeneration in Alzheimer's disease (AD) (Yankner et al., 1989). Prevailing evidence suggests that Aβ induces apoptosis not only in neurons (Behl et al., 1994) but also in cerebral endothelial cells (CECs) (Xu et al., 2001a; Yin et al., 2002, 2006), cerebrovascular smooth muscle cells (Davis-Salinas and Van Nostrand, 1995), oligodendrocytes (Xu et al., 2001b; Lee et al., 2004), and astrocytes (Yang et al., 2004). CECs and astrocytes constitute the blood–brain barrier to shield the brain from damage by harmful circulating toxins or deleterious cellular elements. As a result, CEC apoptosis appears to be a contributing factor in Aβ-induced cerebrovascular degeneration (Yang et al., 2004) characterized by cerebral amyloid angiopathy. However, the molecular mechanism of Aβ-induced CEC apoptosis has not been fully delineated.
Reversible protein phosphorylation catalyzed by protein kinases and protein phosphatases regulates various cellular processes, including apoptosis (Hunter, 2000). Recent studies have highlighted a major role of serine/threonine protein phosphatases, including protein phosphatase 2A (PP2A) in apoptosis (Ray et al., 2005; Yin et al., 2006). PP2A, a member of the ceramide-activated protein phosphatases (CAPPs), has been implicated in Aβ-induced apoptosis through the ceramide–PP2A–Bim cascade (Yin et al., 2006). Bim is a member in the “BH3-only proteins,” a subgroup of Bcl-2 apoptotic regulators that contain only one of the bcl-2 homology regions (BH3). In response to apoptotic stimuli, BH3-only proteins translocate to the mitochondrial membrane from other cellular compartments to interfere with the function of antiapoptotic Bcl-2 family members, leading to apoptotic cell death (Huang and Strasser, 2000).
Kinases may be involved in Aβ-induced apoptosis as well but independent of the BH3-only death paradigm. In the present study, we explored apoptosis signal-regulating kinase 1 (ASK1) and mitogen-activated protein kinase (MAPK), serine/threonine-specific protein kinases, in Aβ-induced CEC death. The rationale for targeting ASK1 and MAPK was based on the findings that oxidative stress is causally related to Aβ-induced apoptotic neuronal (Behl et al., 1994; Hensley et al., 1994) as well as CEC (Xu et al., 2001a; Yin et al., 2002) death and apoptosis associated with the activation of ASK1 (Kanamoto et al., 2000; Tobiume et al., 2001; Matsuzawa et al., 2002; Nishitoh et al., 2002; Noguchi et al., 2005) and MAPK (Tamagno et al., 2003; Okuno et al., 2004; Kamada et al., 2007) is characterized by a heightened oxidative tension in several death paradigms.
We aimed to determine whether activation of the ASK1–MAPK pathway contributes to Aβ-induced CEC death. Results from the present study provide experimental evidence to support the contention that activation of the ASK1–MAPK kinase 3/6 (MKK3/6)–p38MAPK–p53–Bax pathway contributes to Aβ-induced CEC apoptosis in a PP2A-dependent manner. These findings suggest that more than one apoptotic pathway may be in operation downstream of PP2A in Aβ-induced CEC death.
Materials and Methods
Materials.
DMEM, optiMEM, fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA); the enhanced chemiluminescence detection kit was from GE Healthcare (Little Chalfont, UK); amyloid peptide (Aβ1–40) and a cytotoxic fragment (Aβ25–35), protein A/G beads, anti-mouse and anti-rabbit IgG-conjugated horseradish peroxidase antibodies, and rabbit polyclonal antibodies specific for ASK1, p53, p38MAPK, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), α-tubulin, Bax, 14-3-, and hemagglutinin (HA) were from Santa Cruz Biotechnology (Santa Cruz, CA); antibodies against p53 phosphorylated at Ser 15, ASK1 phosphorylated at Ser 967, or p38MAPK phosphorylated at Ser/Thr residues were from New England Biolabs (Beverly, MA); all reagents for SDS-PAGE were from Bio-Rad (Richmond, CA); okadaic acid was from Upstate Biotechnology (Lake Placid, NY); SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole] was from Calbiochem (San Diego, CA); [γ-32P]ATP (6000 Ci/mmol) was from GE Healthcare; Lipofectamine Plus was from Invitrogen; and all other chemicals were from Sigma (St. Louis, MO).
The HA-tagged expression constructs for catalytically inactive ASK1–K709E [ASK1 dominant-negative mutant (DN)] and pcDNA were derived as described previously (Chen et al., 2003). MKK3 DN, MKK6 DN, and p38MAPK DN were kindly provided by Dr. C. M. Teng (National Taiwan University, Taipei, Taiwan). Enhanced green fluorescent protein expression construct (pEGFP) was kindly provided by Dr. M. L. Kuo (National Taiwan University, Taipei, Taiwan).
Aβ preparation.
Aβ was aggregated before experiments in the present study. For aggregation, amyloid peptide was dissolved in sterile double-distilled H2O to a concentration of 1 mm and then maintained for 3 d at 37°C to allow polymerization.
Mouse CEC primary culture.
Mouse CECs were prepared as described previously (Xu et al., 1992; Yin et al., 2002). Briefly, mouse CECs migrating from isolated microvessel preparations were pooled to form a proliferating cell culture that was maintained in DMEM, with high glucose and l-glutamine supplemented with 10% FBS, 0.5 mg/ml heparin, and 75 μg/ml endothelial cell growth supplements. Mouse CECs (between passages 4 and 15) uniformly positive for factor VIII, vimentin, and characteristic bradykinin receptors (>95% endothelial cell purity) were grown to 85–95% confluence before use.
Flow cytometric analysis.
CECs were cultured in 6 cm dishes. After reaching confluence, cells were treated with vehicle or Aβ with or without additional interventions (e.g., addition of a specific inhibitor of p38MAPK) or previous transfection with ASK1 DN, MKK3 DN, MKK6 DN, or p38MAPK DN. At the end of the experiments, CECs were washed twice with PBS (in mm: 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, and 1.5 KH2PO4, pH 7.4) and resuspended in ice-cold 70% ethanol at 0°C overnight. CECs were washed for 5 min with 0.4 ml phosphate-citric acid buffer, pH 7.8, containing 50 mm Na2HPO4, 25 mm citric acid, and 0.1% Triton X-100 and subsequently stained with 1.5 ml of propidium iodide (PI) staining buffer containing 0.1% Triton X-100, 10 mm PIPES, 100 mm NaCl, 2 mm MgCl2, 100 μg/ml RNase A, and 50 μg/ml PI for 30 min in the dark before flow cytometric analysis. Cells were filtered on a nylon mesh filter. The samples were analyzed by the FACScan and Cellquest program (BD Biosciences, San Jose, CA). Each experiment was repeated at least three times.
Plasmid DNA transfection.
For transfection, 105 CECs were seeded onto 12-well plates and transfected with Lipofectamine Plus with 1 μg of pcDNA, ASK1 DN, MKK3 DN, MKK6 DN, p38MAPK DN, or pEGFP in optiMEM for 24 h. The transfection medium was then replaced by fresh DMEM, and CECs were exposed to Aβ (20 μm) for various time periods.
Transfection efficiency analysis.
CECs were cultured in 6 cm dishes. After reaching confluence, cells were transfected with pEGFP, a green fluorescence (GF) protein expression vector. After transfection, CECs were washed twice and resuspended in ice-cold PBS in the dark before flow cytometric analysis. Cells were filtered on a nylon mesh filter, and green fluorescence derived from successful transfected cells was analyzed using the FACScan and Cellquest program (BD Biosciences). Transfection efficiency was defined as the percentage of cells expressing green fluorescence.
Suppression of bax and pp2a expression.
Several protocols for target gene suppression were used in the present study. For bax suppression, cells were transfected with pSiREN–bax, a plasmid that generates small interfering RNAs (siRNA) targeting bax mRNA for degradation. To produce pSiREN–bax, the following complementary oligonucleotides were annealed and cloned into BamHI/EcoRI-digested pSiREN (BD Biosciences): bax sense, 5′-gatccgactggtgctcaaggccctgttcaagagacagggccttgagcaccagcttttttg-3′; bax antisense, 3′-gctgaccacgagttccgggacaag-ttctctgtcccggaactcgtggtcagaaaaaacttaag-5′. A control RNA interference (RNAi) was also constructed by cloning custom-synthesized oligonucleotides (BD Biosciences) into BamHI/EcoRI-digested pSiREN. We also use predesigned siRNAs to suppress bax expression. The siRNAs targeting the mouse bax gene was purchased from Ambion (Austin, TX). The siRNA oligonucleotides targeting the coding regions of mouse bax mRNA were as follows: bax siRNA-I sense, 5′-ggaugauugcugacguggatt-3′; bax siRNA-II sense, 5′-ggcccugugcacuaaagugtt-3′. For pp2a suppression, predesigned siRNAs targeting the mouse pp2a gene was also purchased from Ambion. The siRNA oligonucleotides targeting the coding regions of mouse PP2A catalytic subunit (PP2A-C) mRNA were as follows: pp2a siRNA-1 sense, 5′-ccauacuccgagggaaucatt-3′; pp2a siRNA-2 sense, 5′-ccguauauugaccuaauggtt-3′. The negative control siRNA comprising a 19 bp scrambled sequence with 3′ dT overhangs was also purchased from Ambion.
Western blot analysis.
To determine the expressions of ASK1, ASK1 phosphorylated at Ser967,14-3-3, p38MAPK, p38MAPK phosphorylated at Ser/Thr residues, p53, p53 phosphorylated at Ser15, and Bax in CECs using α-tubulin and GAPDH, as the internal controls, proteins were extracted and analyzed by Western blotting as described previously (Yin et al., 2002; Chen et al., 2004). Briefly, CECs were cultured in 6 cm dishes. After reaching confluence, cells were treated with vehicle or specific inhibitors followed by Aβ for various time intervals. After incubation, cells were washed twice in ice-cold PBS and solubilized in extraction buffer containing 10 mm Tris, pH 7.0, 140 mm NaCl, 2 mm phenylmethylsulfonyl fluoride, 5 mm dithiothreitol, 0.5% Nonidet P-40, 0.05 mm pepstatin A, and 0.2 mm leupeptin. Samples of equal amounts of protein (60 μg) were subjected to SDS-PAGE and then transferred onto a polyvinylidene difluoride membrane that was later incubated in TBST buffer (150 mm NaCl, 20 mm Tris-HCl, and 0.02% Tween 20, pH 7.4) containing 5% nonfat milk. Proteins were incubated with first specific primary antibodies and then horseradish peroxidase-conjugated secondary antibodies. Specific bands were detected based on enhanced chemiluminescence per the instructions of the manufacturer. Quantitative data were obtained using a computing densitometer with scientific imaging systems (Eastman Kodak, Rochester, NY).
Immunoprecipitation and protein kinase assays.
CECs were grown in 6 cm dishes. After reaching confluence, cells were treated with 20 μm Aβ for the indicated time intervals. After incubation, cells were washed twice with ice-cold PBS, lysed in 1 ml of lysis buffer containing 20 mm Tris-HCl, pH 7.5, 1 mm MgCl2, 125 mm NaCl, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 25 mm β-glycerophosphate, 50 mm NaF, and 100 μm sodium orthovanadate and were centrifuged. The supernatant was immunoprecipitated overnight with polyclonal antibodies against ASK1 or p38MAPK in the presence of protein A/G-agarose beads. The beads were washed three times with lysis buffer and two times with kinase buffer containing 20 mm HEPES, pH 7.4, 20 mm MgCl2, and 2 mm dithiothreitol. The kinase reactions were performed by incubating immunoprecipitated complex with 20 μl of kinase buffer supplemented with 20 μm ATP and 3 μCi of [γ-32P]ATP at 30°C for 30 min. To assess ASK1 and p38MAPK activities, 50 μg/ml myelin basic protein (MBP) was added to serve as the substrate. The reaction mixtures were analyzed by 15% SDS-PAGE, followed by autoradiography.
Coimmunoprecipitation.
CECs were grown in 6 cm dishes. After reaching confluence, cells were treated with 20 μm Aβ for the indicated time intervals. The cells were harvested, lysed in 1 ml of PD buffer (40 mm Tris-HCl, pH 8.0, 500 mm NaCl, 0.1% Nonidet P-40, 6 mm EGTA, 10 mm β-glycerophosphate, 10 mm NaF, 300 μm sodium orthovanadate, 2 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm dithiothreitol), and centrifuged. The supernatant was immunoprecipitated overnight with specific antibodies against ASK1 in the presence of protein A/G-agarose beads at 4°C. The immunoprecipitated complex was washed three times with PD buffer. The samples were fractionated on 15% SDS-PAGE, transferred to a polyvinylidene difluoride membrane, and subjected to immunoblotting with antibodies specific for 14-3-3.
Preparation of nuclear extracts and electrophoretic mobility shift assays.
CECs were grown in 6 cm dishes. After reaching confluence, cells were treated with 20 μm Aβ for the indicated time intervals. The nuclear protein fractions were then prepared as described previously (Xu et al., 2001b; Chen et al., 2004). Briefly, cells were washed with ice-cold PBS and then centrifuged. The cell pellet was resuspended in hypotonic buffer (10 mm HEPES, 10 mm KCl, 0.5 mm DTT, 10 mm aprotinin, 10 mm leupeptin, and 20 mm PMSF) for 15 min on ice and vortexed for 10 s. The nuclei were pelleted by centrifugation at 15,000 × g for 1 min. The pellet containing nuclei was resuspended in hypertonic buffer (20 mm HEPES, pH 7.6, 25% glycerol, 1.5 mm MgCl2, 4 mm EDTA, 0.05 mm DTT, 20 mm PMSF, 10 mm aprotinin, and 10 mm leupeptin) for 30 min on ice. The supernatants containing the nuclear proteins were collected by centrifugation at 15,000 × g for 2 min and stored at −70°C. A double-stranded oligonucleotide probe containing the p53 sequence (5′-GAACATGTCTAAGCATGCTG-3′; Santa Cruz Biotechnology) was end labeled with [γ-32P]ATP using T4 polynucleotide kinase. The nuclear extract (2.5–5 μg) was incubated with 1 ng of a 32P-labeled p53 probe (50,000–75,000 cpm) in 10 μl of binding buffer containing 1 μg of poly(dI-dc), 15 mm HEPES, pH 7.6, 80 mm NaCl, 1 mm EDTA, 1 mm DTT, and 10% glycerol at 30°C for 25 min. DNA/nuclear protein complexes were separated from the DNA probe by electrophoresis on 6% polyacrylamide gels. The gels were vacuum dried and subjected to autoradiography with an intensifying screen at −80°C.
Statistical analysis.
Results are presented as mean ± SEM from at least three independent experiments. One-way ANOVA, followed by Bonferroni's multiple range tests when appropriate, was used to determine the statistical significance of the difference between the means. A p value <0.05 was considered statistically significant.
Results
ASK1 activation in Aβ-induced CEC death
As reported previously, Aβ1–40 and Aβ25–35 have equal potency for inducing apoptosis in CECs (Xu et al., 2001a). In the present study, we also demonstrated that the potency of Aβ1–40 in causing CEC death was similar to that of Aβ25–35 as determined by the MTT assay (data not shown). Thus, this study was conducted using mainly Aβ25–35 with key experiments confirmed with Aβ1–40. ASK1 activation is a pivotal mechanism in a broad range of cell death paradigms (Nishitoh et al., 2002). To explore whether ASK1 activation contributes to Aβ-induced CEC death, CECs were transiently transfected with HA epitope-tagged ASK1 DN before Aβ treatment. We first confirmed that the protein encoded by ASK1 DN plasmid was expressed in transfected CECs. Based on immunoblotting using specific antibodies to HA or ASK1, HA-tagged ASK1 DN protein was highly expressed in ASK1 DN-transfected cells compared with the control group (transfected with pcDNA) (Fig. 1A). Using the same transfection protocol, we also assessed the transfection efficiency based on pEGFP, a green fluorescence protein expression vector. After transfection, green fluorescence derived from successful transfected cells was analyzed using flow cytometry. Transfection efficiency was defined as the percentage of cells expressing GF. The compiled results show a transfection rate of 54.7 ± 2.9% (n = 3) in the bottom in Figure 1B. To elucidate whether Aβ-induced CEC death is attributable to apoptosis, flow cytometric analysis was then used. As shown in Figure 1C, the percentage of PI-stained cells in the apoptotic region (sub-G0/G1 peak, subdiploid peak) was significantly increased after 20 μm Aβ treatment (Fig. 1Cc) compared with the vehicle-treated group (Fig. 1Ca). The compiled data are shown in the bottom in Figure 1C (Aβ, 64.0 ± 5.9%; vehicle, 15.0 ± 2.1%; p < 0.05). Aβ-induced CEC apoptosis was attenuated by transfection with ASK1 DN (Fig. 1Cd and bottom: Aβ plus ASK1 DN, 28.1 ± 2.6%; p < 0.05 compared with Aβ plus pcDNA). To further elucidate whether ASK1 activation is involved in the signaling cascade of Aβ-induced cell death, ASK1 kinase activity was measured after Aβ exposure. Treatment with Aβ rapidly increased ASK1 kinase activity in CECs as early as 5 min and returned to basal level within 1 h (Fig. 1D). We next explored the mechanism by which Aβ induces ASK1 activation. ASK1 binds to 14-3-3, an inhibitory protein, to stay inactive. Dissociation of ASK1 from 14-3-3 leads to ASK1 activation. Phosphorylation of the ASK1 Ser967 residue is required for ASK1 binding to 14-3-3 (Zhang et al., 1999). We examined whether the extent of ASK1 Ser967 phosphorylation is altered after Aβ exposure. Aβ had no effects on ASK1 Ser967 dephosphorylation at 1 min. However, Aβ caused a significant decrease in ASK1 Ser967 phosphorylation after exposure to Aβ for 2 min or longer, whereas vehicle control was without effects (Fig. 1E). Coimmunoprecipitation was then used to confirm the hypothesis that Aβ-induced ASK1 dephosphorylation was accompanied by the dissociation of the ASK1–14-3-3 complex. As shown in Figure 1F, Aβ rapidly caused ASK1 dissociation from 14-3-3. This response began as early as 2 min after Aβ exposure. These findings together suggest that ASK1 Ser967 dephosphorylation and subsequent ASK1 dissociation from 14-3-3 were rapid in response to Aβ-induced stress in CECs. However, the mechanism that regulates ASK1 dephosphorylation at the Ser967 residue remains to be identified. It is conceivable that Aβ may activate a protein phosphatase that dephosphorylates Ser967, leading to ASK1 activation. Activation of PP2A was shown recently to be causally related to Aβ-induced CEC death (Yin et al., 2006). We, therefore, explore whether PP2A is involved in ASK1 dephosphorylation. Okadaic acid, a selective PP2A inhibitor, reduced ASK1 dephosphorylation in CECs treated with Aβ. Cyclosporin A, a specific PP2B/calcineurin inhibitor, was without effect (Fig. 2A). To further confirm more specifically that Aβ-induced ASK1 dephosphorylation was mediated by PP2A, two pp2a siRNA oligonucleotides (pp2a siRNA-1 and pp2a siRNA-2) were used. As shown in Figure 2B, transfection of CECs with pp2a siRNA-1 or pp2a siRNA-2 significantly reduced Aβ-induced ASK1 dephosphorylation (Fig. 2B). Furthermore, siRNA experiments revealed that pp2a siRNA-1 or pp2a siRNA-2 suppressed the basal level of PP2A-C (Fig. 2C). These results suggest that PP2A may be specifically responsible for Aβ-induced dephosphorylation of ASK1 Ser967 in CECs.
Figure 1.

ASK1 in Aβ-induced CEC death. A, CECs were transiently transfected with pcDNA (mock transfection) or HA–ASK1 DN for 24 h. After transfection, cells were harvested and the level of HA–ASK1 DN was determined by immunoblotting using anti-HA (top) and anti-ASK1 (bottom) antibody. B, CECs were transiently transfected with pEGFP for 24 h. After transfection, cells were harvested, and green fluorescence derived from successful transfected cells was measured using flow cytometry as described in Materials and Methods. Transfection efficiency is defined as the percentage of cells expressing GF. Compiled results are shown at the bottom. Each column represents the mean ± SEM of three independent experiments. C, After transfection as described above in A, cells were treated with vehicle or 20 μm Aβ for another 48 h. The percentage of apoptotic cells was then analyzed by flow cytometric analysis of PI-stained cells as described in Materials and Methods. Compiled results are shown at the bottom. Each column represents the mean ± SEM of at least three independent experiments. *p < 0.05 by comparing the Aβ plus ASK1DN and Aβ plus pcDNA groups. D, CECs were treated with 20 μm Aβ for the indicated time intervals, and ASK1 kinase activity was assessed using MBP as a substrate. After SDS-PAGE, γ-32P-labeled MBP was visualized by autoradiography. Immunoblotting confirming equal amount of immunoprecipitated ASK1 for each sample is shown at the bottom. Data shown are representative of three separate experiments with similar results. KA, Kinase assay; IB, immunoblotting. E, CECs were treated with vehicle or 20 μm Aβ for various time intervals as indicated. Cell lysates were then prepared and subjected to immunoblotting with anti-pSer967–ASK1 antibody. Equal loading in each lane is reflected by similar intensities of ASK1 at the bottom. *p < 0.05 compared with the control group. F, Cells were treated with 20 μm Aβ for 0–30 min and then immunoprecipitated with the anti-ASK1 antibody. The immunoprecipitated complex was then subjected to immunoblotting with an anti-14-3-3 antibody. Typical bands representative of three separate experiments with similar results are shown. Immunoblotting confirming equal amount of immunoprecipitated ASK1 for each sample is shown at the bottom. IP, Immunoprecipitation; IB, immunoblotting.
Figure 2.

PP2A in Aβ-induced ASK1 Ser967 dephosphorylation. A, CECs were pretreated with vehicle, 1–10 nm okadaic acid (OA), or 100 nm cyclosporine A (CA) for 30 min before treatment with 20 μm Aβ for another 10 min. Cell lysates were then prepared and subjected to immunoblotting with anti-pSer967–ASK1 antibody. Equal loading in each lane is reflected by similar intensities of ASK1 at the bottom. *p < 0.05 compared with the vehicle-treated group in the presence of Aβ. B, CECs were transiently transfected with control siRNA, pp2a siRNA-1, or pp2a siRNA-2 for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 10 min. Cell lysates were then prepared and subjected to immunoblotting with an anti-pSer967–ASK1 antibody. Equal loading in each lane is reflected by similar intensities of ASK1 at the bottom. *p < 0.05 compared with the control siRNA group in the presence of Aβ. C, After transfection as described above in B, the expression of PP2A-C was then determined by immunoblotting with an anti-PP2A-C antibody. Data shown are representative of three independent experiments with similar results.
p38MAPK activation in Aβ-induced CEC apoptosis
ASK1 belongs to the MAPK kinase kinase family and activates the c-Jun N-terminal kinase (JNK) and p38MAPK pathways via MKK4/7 and MKK3/6, respectively (Ichijo et al., 1997). JNK-mediated signaling cascade has been shown previously to participate in Aβ-induced CEC death (Yin et al., 2002). In the present study, we focused on the role of p38MAPK signaling cascade in Aβ-induced CEC death. We examined whether p38MAPK signaling events are involved in Aβ-induced CEC apoptosis using flow cytometry. As shown in Figure 3A, the percentage of PI-stained cells in the apoptotic region was significantly increased after 20 μm Aβ treatment (Fig. 3Ac) compared with the vehicle-treated group (Fig. 3Aa) The compiled data are shown in the bottom of Figure 3A (Aβ, 64.2 ± 4.6%; vehicle, 7.2 ± 2.3%; p < 0.05). Aβ-induced CEC apoptosis was attenuated by SB203580, a p38MAPK inhibitor (Fig. 3Ad and bottom: Aβ plus SB203580, 45.2 ± 2.6%; Aβ, 64.2 ± 4.6%; p < 0.05). The time course of Aβ-induced p38MAPK phosphorylation is shown in Figure 3B. Aβ increased p38MAPK phosphorylation in a time-dependent manner. In parallel, using MBP as a p38MAPK substrate, a time-dependent increase in p38MAPK activity was also observed in Aβ-treated CECs (Fig. 3C). To further ascertain the linkage between ASK1 and p38MAPK signaling cascade downstream of Aβ, we determined the effect of ASK1 DN on Aβ-induced p38MAPK activation. As shown in Figure 4A, transfection with ASK1 DN significantly reduced Aβ-induced p38MAPK activation. ASK1 activation of p38MAPK is via the MKK3/6 pathway (Ichijo et al., 1997). MKK3 DN and MKK6 DN also attenuated Aβ-induced p38MAPK activation (Fig. 4B).
Figure 3.
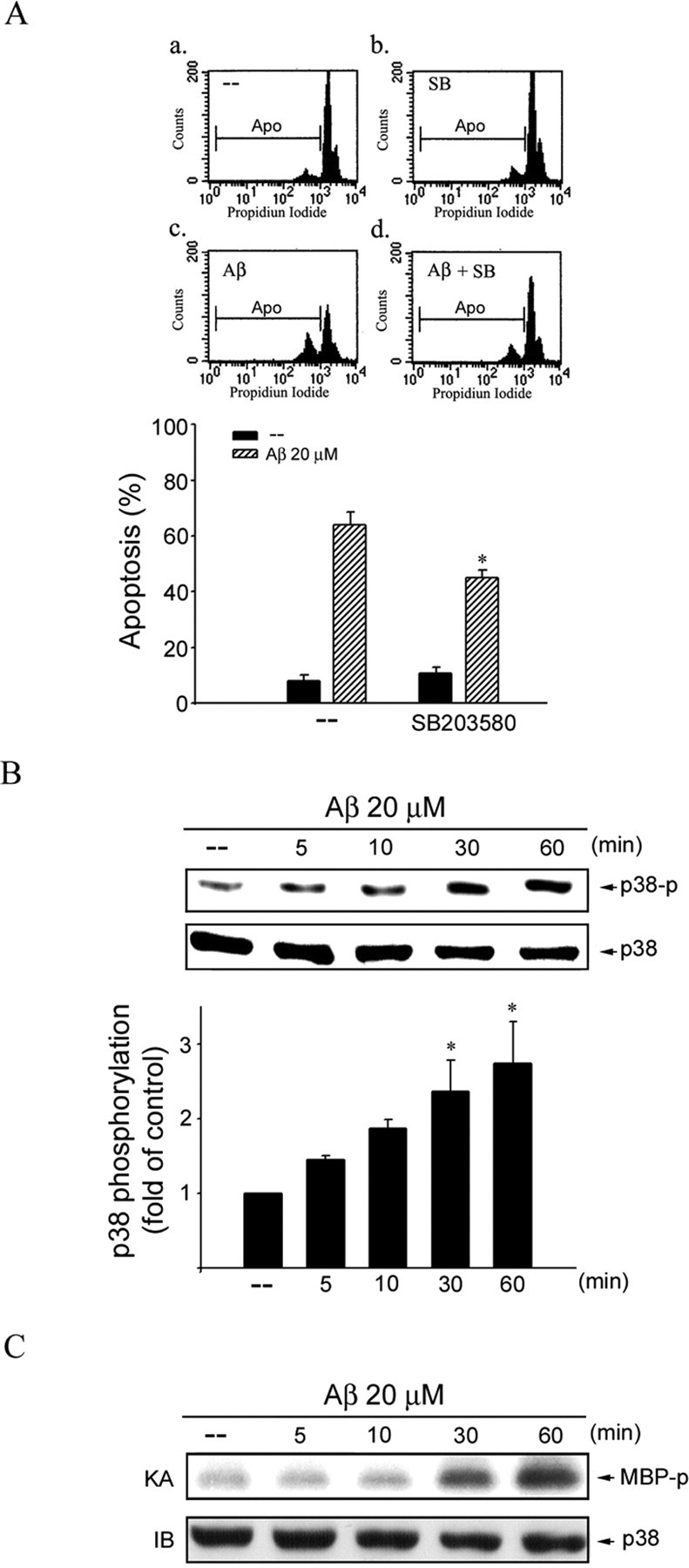
p38MAPK in Aβ-induced CEC apoptosis. A, CECs were pretreated with vehicle or 10 μm SB203580 (SB), a specific p38MAPK inhibitor, for 30 min before treatment with 20 μm Aβ for another 48 h. The percentage of apoptotic cells was then analyzed by flow cytometric analysis of PI-stained cells as described in Materials and Methods. Compiled results are shown at the bottom. Each column represents the mean ± SEM of at least three independent experiments. *p < 0.05 compared with the group treated with Aβ alone. B, CECs were treated with 20 μm Aβ for 0–60 min, and p38MAPK phosphorylation was determined by immunoblotting with anti-phospho-p38MAPK antibody. Equal loading in each lane is reflected by approximately similar intensities of p38MAPK at the bottom. *p < 0.05 compared with the control group. C, Cells were treated with 20 μm Aβ for 0–60 min, and p38MAPK activity was assessed using MBP as a substrate. After SDS-PAGE, γ-32P-labeled MBP was visualized by autoradiography. Typical bands representative of three independent experiments with similar results are shown. KA, Kinase assay; IB, immunoblotting; AP, apoptotic region.
Figure 4.

ASK1, MKK3, and MKK6 in Aβ-induced p38MAPK activation and CEC death. A, CECs were transiently transfected with pcDNA or ASK1 DN for 24 h. After transfection, CECs were treated with vehicle or 20 μm Aβ for indicated time intervals and harvested for assessing the level of p38MAPK phosphorylation by immunoblotting with an anti-phospho-p38MAPK antibody. Equal loading in each lane is reflected by approximately similar intensities of the GAPDH bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ. B, CECs were transiently transfected with pcDNA, MKK3 DN, or MKK6 DN for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for 30 min and then harvested, and the level of p38MAPK phosphorylation was determined as described above in A. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ. C, CECs were transiently transfected with pcDNA, MKK3 DN, MKK6 DN, or p38MAPKα DN for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h. The percentage of apoptotic cells was then analyzed by flow cytometric analysis of PI-stained cells as described in Materials and Methods. Each column represents the mean ± SEM of at least three independent experiments. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ.
We also used MKK3 DN, MKK6 DN, and p38MAPKα DN to block MKK3/6–p38MAPK signaling pathway to confirm whether the MKK3/6–p38MAPK cascade participates in Aβ-induced CEC death. Flow cytometric analysis demonstrated that transfection with MKK3 DN, MKK6 DN, or p38MAPKα DN attenuated Aβ-induced CEC apoptosis with the extent of cell apoptosis reduced from 53.4 ± 5.2 to 31.8 ± 5.9, 31.2 ± 8.5, and 26.3 ± 8.4%, respectively (Fig. 4C). Together, these findings suggest that the ASK1–MKK3/6–p38MAPK cascade is in operation in Aβ-induced CEC death.
p53 activation in Aβ-induced CEC death
p53 is a transcription factor that plays a key role in the regulation of cell viability downstream of MAP kinase. Activation of p53 entails phosphorylation of its serine residues, primarily Ser15 (Dumaz and Meek, 1999; Meek, 1999). To explore the role of p53 in Aβ death signaling downstream of MAP kinase, we examined the effect of Aβ on p53 phosphorylation at Ser15. Figure 5A shows that Aβ caused an increase in p53 phosphorylation at Ser15 in a time-dependent manner. Phosphorylation began at 10 min, peaked at 2 h, and declined toward the basal level 6 h after Aβ treatment. Phosphorylation at Ser15 is responsible for p53 binding to its cognate DNA binding sequence (Dumaz and Meek, 1999). The nuclear extracts of CECs with and without Aβ treatment were subjected to an electrophoretic mobility shift assay (EMSA) using p53-specific oligonucleotides as the probe. As shown in Figure 5B, p53-specific DNA–protein binding activity, low in vehicle-treated CECs, was markedly increased 1–2 h after Aβ treatment (Fig. 5B). Pretreatment of nuclear extracts with specific antibody against p53 reduced p53-specific DNA–protein binding activity, demonstrating the specificity of p53–DNA binding activity (Fig. 5C).
Figure 5.
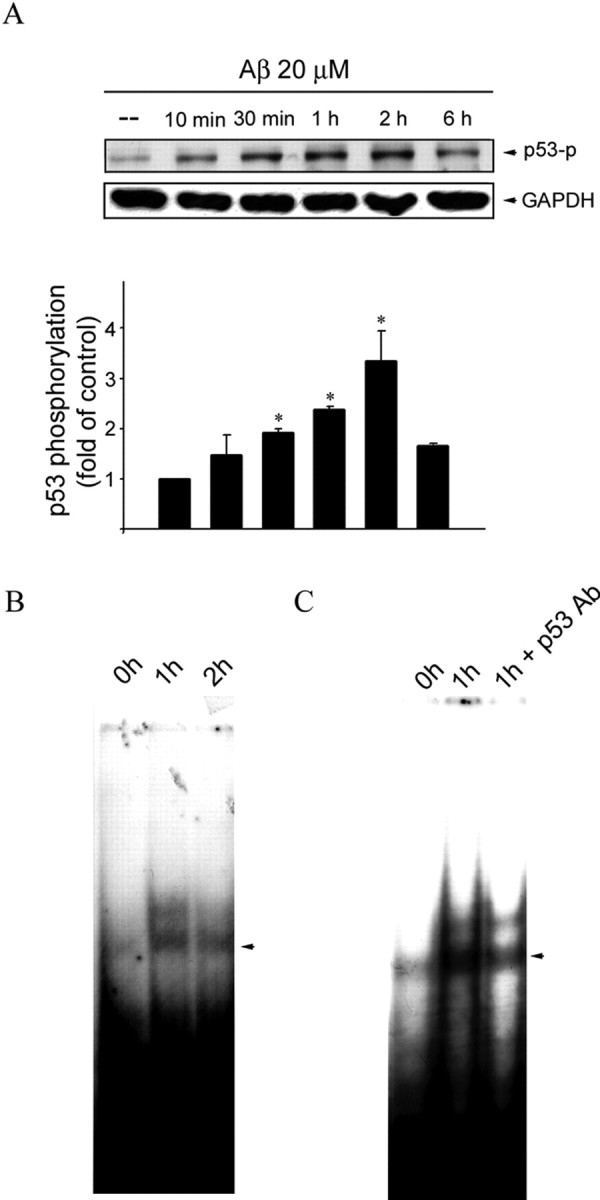
Aβ-induced p53 phosphorylation and increase in p53 binding activity in CECs. A, CECs were treated with 20 μm Aβ for indicated time periods. Cells were then harvested, and p53 phosphorylation at Ser15 was determined by immunoblotting with an anti-pSer15–p53 antibody. Equal loading in each lane is shown by the similar intensities of GAPDH. Compiled results are shown at the bottom. *p < 0.05 compared with the control group. B, CECs were incubated with 20 μm Aβ for 1 and 2 h. After incubation, the nuclear protein fraction was prepared for EMSA as described in Materials and Methods. C, An anti-p53 antibody (Ab) was included before EMSA to detect the specificity of p53 binding activity. Data shown are representative of three independent experiments with similar results.
ASK1 DN was then used to block ASK1 signaling to determine whether ASK1 mediates Aβ-induced p53 activation. As shown in Figure 6A, ASK1 DN significantly suppressed Aβ-induced p53 phosphorylation at Ser15 residue. Similarly, both MKK3 DN and MKK6 DN were effective in attenuating Aβ-induced p53 phosphorylation at Ser15 (Fig. 6B). Furthermore, SB203580, a p38MAPK inhibitor, also inhibited Aβ-induced p53 activation (Fig. 6C). These results support a causal role of the ASK1–MKK3/6–p38MAPK signaling cascade in Aβ-induced p53 activation.
Figure 6.
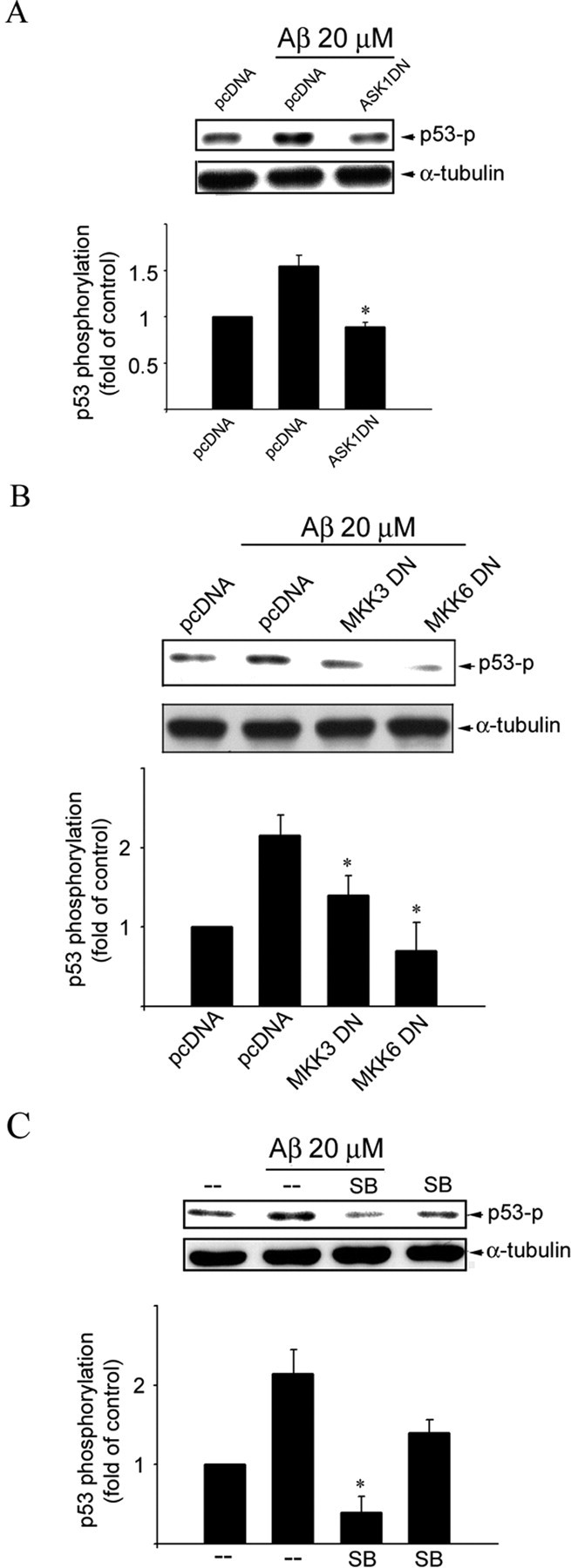
Involvement of ASK1, MKK3, MKK6, and p38MAPK in Aβ-induced p53 phosphorylation in CECs. A, CECs were transiently transfected with pcDNA or ASK1 DN for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for 1 h and then harvested for immunoblotting to assess the level of p53 phosphorylation at Ser15 using an anti-pSer15–p53 anti-body. Equal loading in each lane is reflected by approximately similar intensities of the α-tubulin bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ. B, CECs were transiently transfected with pcDNA, MKK3 DN, or MKK6 DN for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for 1 h and then harvested, and the level of p53 phosphorylation at Ser15 was determined as described in A. Equal loading in each lane is reflected by approximately similar intensities of the α-tubulin bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ. C, CECs were treated with 10 μm SB203580 (SB), specific p38MAPK inhibitor, for 30 min, followed by vehicle or 20 μm Aβ for another 1 h and were then harvested for assessing the level of p53 phosphorylation at Ser15 as described in A. Equal loading in each lane is reflected by approximately similar intensities of the α-tubulin bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ.
Bax upregulation in Aβ-induced CEC apoptosis
p53 has been shown to induce apoptosis by causing mitochondrial dysfunction via transactivation of Bax expression (Jeffers et al., 2003; Hastak et al., 2005). We, therefore, examined whether Aβ is capable of inducing Bax expression in CECs. As shown in Figure 7A, Aβ elevated cellular level of Bax in a time-dependent manner. To further confirm that Aβ-induced cell death was mediated by Bax, Bax expression was silenced using RNAi strategy. RNAi experiments revealed that the basal Bax level in CECs was only slightly affected by bax RNAi. In contrast, bax RNAi suppressed Aβ-induced Bax expression (Fig. 7B). To further confirm the results of Bax RNAi experiments. We also used siRNAs to suppress Bax expression. As shown in Figure 7, C and D, bax siRNA-I and bax siRNA-II significantly suppressed Aβ-induced Bax expression. Similar to RNAi experiments, the basal Bax level was only slightly affected by bax siRNA-I (Fig. 7C). bax siRNA-II appears to suppress the basal Bax level (Fig. 7D). Transfection of CECs with bax siRNA-II attenuated Aβ-induced CEC apoptosis with the extent of apoptosis reduced from 55.1 ± 7.0 to 29.0 ± 7.7% (Fig. 7E).
Figure 7.
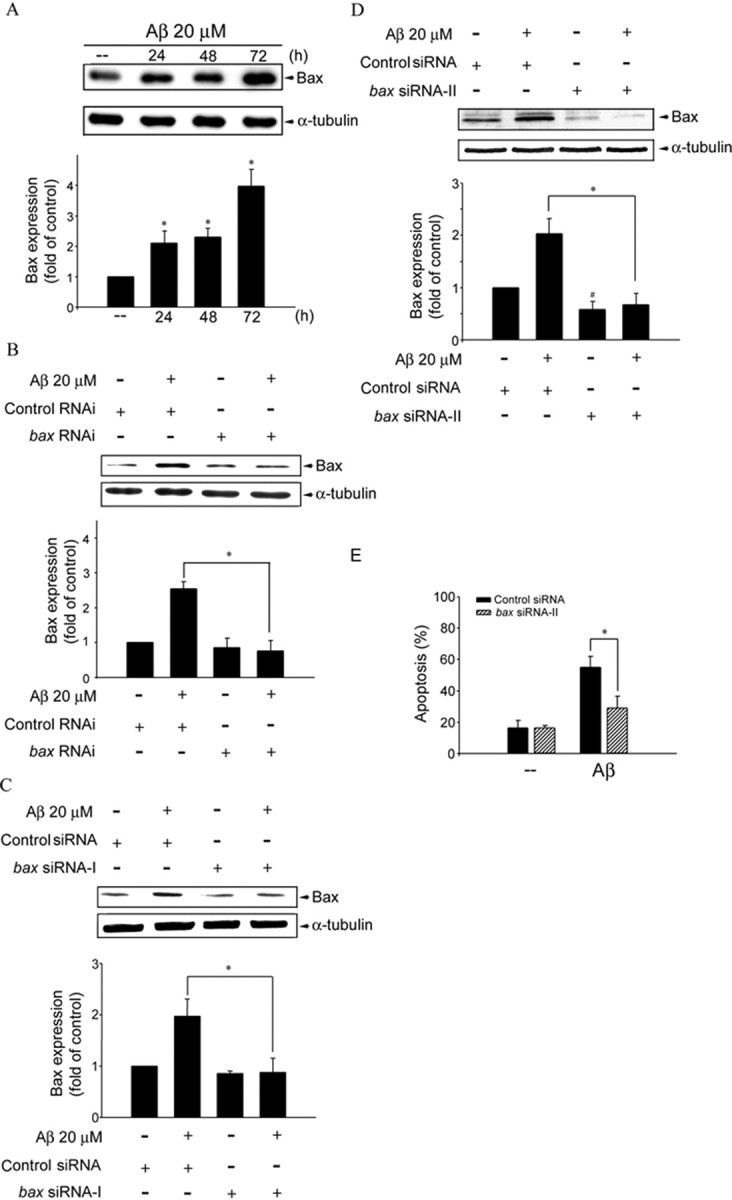
Bax expression in Aβ-induced CEC death. A, CECs were treated with 20 μm Aβ for 24–72 h and then harvested for assessing the extent of Bax expression by immunoblotting with an anti-Bax antibody. Compiled results are shown at the bottom. *p < 0.05 compared with the control group. B, CECs were transfected with control RNAi or bax RNAi for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h. The expression of Bax was then determined by immunoblotting with an anti-Bax antibody. Compiled results are shown at the bottom. *p < 0.05 compared with the control RNAi group in the presence of Aβ. C, CECs were transfected with control siRNA or bax siRNA-I for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h. The expression of Bax was then determined by immunoblotting with an anti-Bax antibody. Compiled results are shown at the bottom. *p < 0.05 compared with the control siRNA group in the presence of Aβ. D, CECs were transfected with control siRNA or bax siRNA-II for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h. The expression of Bax was then determined by immunoblotting with an anti-Bax antibody. Compiled results are shown at the bottom. *p < 0.05 compared with the control siRNA group in the presence of Aβ. #p < 0.05 compared with the control siRNA group. E, CECs were transiently transfected with control siRNA or bax siRNA-II for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h, and cell apoptosis was determined by flow cytometry. *p < 0.05 compared with the control siRNA group in the presence of Aβ.
We then tested the hypothesis that Aβ may induce Bax expression through the ASK1–MKK3/6–p38MAPK–p53 signaling cascade. As shown in Figure 8A, ASK1 DN noticeably inhibited Aβ upregulation of Bax expression in CECs. Moreover, MKK3 DN, MKK6 DN (Fig. 8B), and the p38MAPK inhibitor SB203580 (Fig. 8C) also attenuated Aβ-induced Bax expression, respectively. These findings support the contention that Aβ activates the ASK1–MKK3/6–p38MAPK–p53 signaling cascade to induce Bax expression, resulting in CEC apoptosis.
Figure 8.
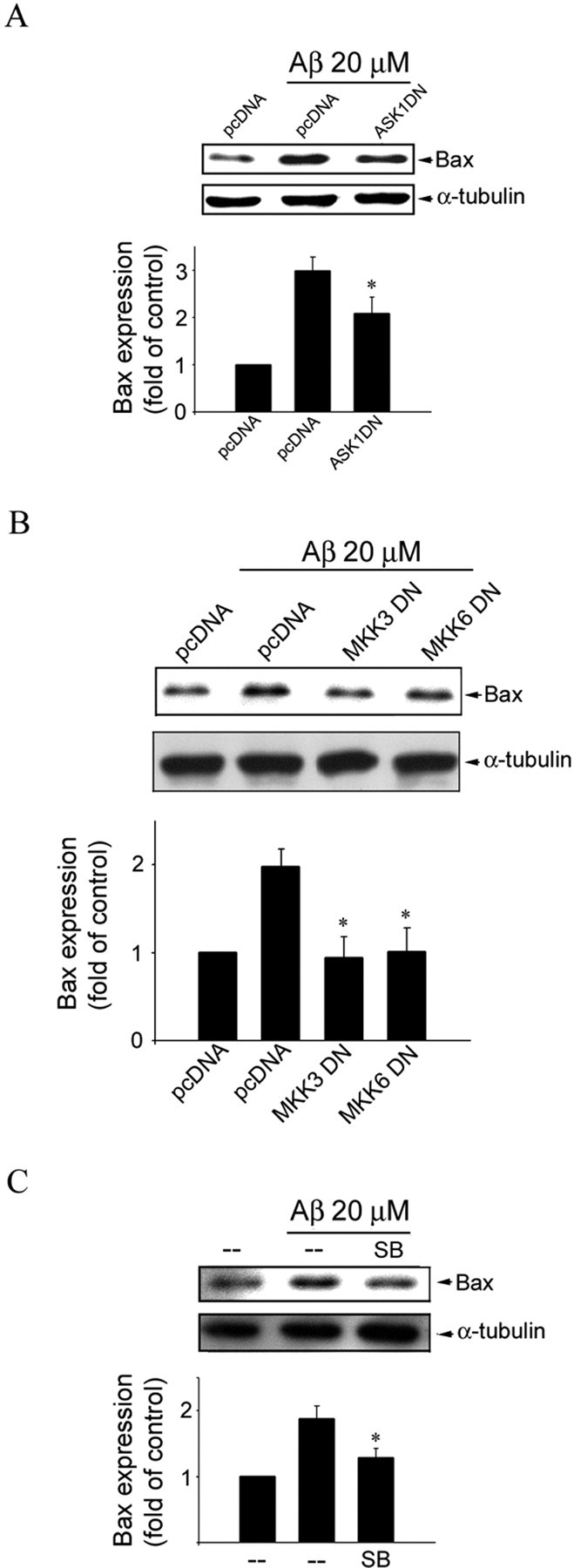
ASK1, MKK3, MKK6, and p38MAPK in Aβ-induced Bax expression in CECs. A, Cells were transiently transfected with pcDNA or ASK1 DN for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h. Cells were then harvested, and the extent of Bax expression was determined by immunoblotting with an anti-Bax antibody. Equal loading in each lane is shown by approximately similar intensities of the α-tubulin bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ. B, CECs were transiently transfected with pcDNA, MKK3 DN, or MKK6 DN for 24 h. After transfection, cells were treated with vehicle or 20 μm Aβ for another 48 h and then harvested for assessing the extent of Bax expression as described in A. Equal loading in each lane is shown by approximately similar intensities of the α-tubulin bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ. C, CECs were pretreated with vehicle or 10 μm SB203580 (SB), a specific p38MAPK inhibitor, for 30 min, followed by vehicle or 20 μm Aβ for another 48 h, and then harvested for assessing the extent of Bax expression as described in A. Equal loading in each lane is shown by approximately similar intensities of the α-tubulin bands. Compiled results are shown at the bottom. *p < 0.05 compared with the pcDNA (mock transfection) group in the presence of Aβ.
Discussion
Aβ has been implicated as the primary neurotoxic factor in the pathogenesis of AD. Results from the present study, similar to those reported previously (Xu et al., 2001a; Yin et al., 2002, 2005; Yang et al., 2004), show that Aβ is also cytotoxic to CECs. Aβ-induced apoptosis is a multifactorial process involving excessive formation of reactive oxygen species (Schapira, 1996), alteration of intracellular calcium homeostasis (Kriem et al., 2005), and mitochondrial dysfunction and caspases activation (Hitomi et al., 2004). However, the precise molecular mechanism responsible for the apoptotic action of Aβ remains to be fully characterized. We demonstrated that the activation of the ASK1–MKK3/6–p38MAPK signaling cascade followed by p53 activation and Bax expression contributes to Aβ-induced CEC death.
Phosphorylation of the ASK1 Ser967 residue is required for the formation of the ASK1–14-3-3 complex to keep ASK1 inactive (Zhang et al., 1999). However, the signaling pathways that control ASK1 function through Ser967 remain unresolved. Activation of an unknown protein phosphatase is required for tumor necrosis factor-α-induced ASK1 activation by dephosphorylating ASK1 Ser967 (Zhang et al., 2003). Goldman et al. (2004) further demonstrated that an okadaic acid-sensitive phosphatase is required for H2O2 dephosphorylation of ASK1 Ser967 in COS7 cells. We have shown that Aβ-induced CEC death was causally related to PP2A activation (Yin et al., 2006). In the present study, we demonstrated that okadaic acid, a specific inhibitor of PP2A, inhibited Aβ dephosphorylation of the ASK1 Ser967 residue. Cyclosporin A, a specific inhibitor of PP2B, however, failed to block Aβ-induced Ser967 dephosphorylation. Furthermore, pp2a siRNAs, which silenced PP2A-C, also attenuated Aβ dephosphorylation of Ser967. These findings suggest that PP2A may play a pivotal role in ASK1 dephosphorylation and the subsequent signaling events.
Activated ASK1 plays a critical role in apoptosis by stimulating the downstream signaling events, including the activation of MKK3/6 and p38MAPK in sequence (Yamaguchi et al., 2004; Holasek et al., 2005). Whether the p38MAPK signaling pathway participates in Aβ-induced CEC death has not been demonstrated previously. We show in the present study that p38MAPK was activated and causally related to Aβ-induced CEC death. In addition, we noted that Aβ-induced p38MAPK activation and subsequent CEC death was mediated by ASK1 and MKK3/6. These findings are consistent with the observation that SB203580, a selective inhibitor of p38MAPK, and dominant-negative mutants of ASK1, MKK3, and MKK6 diminished Aβ-induced p38MAPK activation and CEC death. Moreover, increased oxidative stress has been proposed to play an important role in Aβ-induced cell death in neurons (Behl et al., 1994) and CECs (Xu et al., 2001a). It has been demonstrated recently that oxidative stress signals such as H2O2 may activate PP2A-like protein phosphatase to cause ASK1 Ser967 dephosphorylation and subsequent cell death (Goldman et al., 2004). Studies with ASK1 knock-out mice demonstrated that ASK1–p38MAPK pathway is required for oxidative stress-induced mouse embryo fibroblast apoptosis (Tobiume et al., 2001). These findings together with results presented in this communication suggest that Aβ-induced oxidative stress may be critical for activating PP2A–ASK1–MKK3/6–p38MAPK apoptotic signaling cascade in CECs.
A number of studies have indicated that p53 plays an important role in promoting cell apoptosis by regulating the transcription of proapoptotic genes (el-Deiry et al., 1995). Aβ was shown recently to promote phosphorylation-mediated stabilization of p53 and subsequent cortical neuron death (Fogarty et al., 2003). p53 phosphorylation by p38MAPK contributed to nitric oxide-induced apoptosis (Kim et al., 2002). In agreement with these observations, we noted that the dominant-negative mutants of ASK1, MKK3 and MKK6, or SB203580, a selective p38MAPK inhibitor, prevented Aβ-induced p53 phosphorylation and CEC death. Thus, it is plausible that Aβ activates the ASK1–MKK3/6–p38MAPK cascade to cause p53 phosphorylation and subsequent cell death. In addition, the finding in this study that the transfection efficiencies do not match the effects of the dominant-negative mutants in immunoblotting experiments has also been noted by others. For instance, in a study of ASK1 dominant-negative strategy to prevent apoptosis, Chen et al. (1999) noted the transfection of a dominant-negative ASK1 mutant resulted in similar findings with the biological effects greater than transfection efficiency. Similar findings have been reported by others (Huang et al., 2003; Li et al., 2005).
The BclII family proteins regulate mitochondria-dependent apoptosis, with the balance of the antiapoptotic and proapoptotic members arbitrating the life-or-death decisions. Bax, a proapoptotic member of the Bcl-2 family, causes apoptosis by disrupting mitochondrial integrity. bax expression is induced by p53 in response to selected stress signals (Zhang et al., 2005). In the present study, bax siRNA attenuated Aβ-induced CEC apoptosis, suggesting that Bax expression is causally related to Aβ- induced CEC apoptosis. In addition to Bax, recent reports have indicated that BH3-only members of BclII family, such as Bim (Yin et al., 2002) and Bad (Yin et al., 2005), also participate in Aβ-induced CEC death. The link between these proapoptotic Bcl-2 family protein-mediated cell death pathways remains to be established. Moreover, transcription factors other than p53 may also contribute to Aβ-induced CEC apoptosis. These include AP-1 (activator protein-1) (Yin et al., 2002) and FKHRL1 (forkhead in rhabdomyosarcoma-like 1) (Yin et al., 2006). These observations explain, at least in part, why blocking the ASK1 signaling cascade could not completely abolish Aβ-induced CEC death.
As described above, PP2A appears to play an important role in activating ASK1. PP2A is a member in the CAPP family. We established previously that Aβ-induced death of non-neuronal cells, including CECs, astrocytes (Yang et al., 2004), and oligodendrocytes (Lee et al., 2004), involved activation of neutral sphingomyelinase to release ceramide from membrane sphingomyelin. Results from the present studies suggest that ceramide activation of PP2A may be involved in Aβ-induced death paradigms in CECs by dephosphorylating both Akt (Yin et al., 2006) as well as ASK1. Together, these results suggest that Aβ activation of PP2A may activate at least two separate pathways: one on the Akt–FKHRL1–Bim cascade (Yin et al., 2006) and another on the ASK1–MAP kinase-p53–Bax cascade as reported here. The differential mechanisms of Aβ actions in driving these two separate death signaling pathways downstream of PP2A remain to be elucidated. It is likely that two different pathways culminating in Bim and Bax expression, respectively, are in cooperation. Both Bim and Bax are Bcl-2 family proteins. However, Bim is a proapoptotic protein belonging to the BH3-only subfamily, whereas Bax is not. Whether Bim and Bax act in sequence or in synergy remains controversial (Willis and Adams, 2005). Additional works are needed to characterize the interrelationship between Bim and Bax in Aβ-induced CEC death.
The signaling events before PP2A–ASK1 activation have not been delineated but are likely to involve Aβ interaction with cell membrane proteins. Cytotoxic actions of Aβ have been ascribed to various types of receptor mechanisms. Among the postulated Aβ receptors, p75 neurotrophin receptor (p75NTR) is the most likely candidate to mediate Aβ cytotoxic effect. The first link between Aβ neurotoxicity and p75NTR was noted by Ye et al. (1999), who found that expression of p75NTR enhances Aβ toxicity in PC12 cells. Yaar et al. (2002) found that Aβ binds to p75NTR directly and causes JNK activation and apoptotic cell death in p75NTR-expressing cells (Yaar et al., 2002). Kuner and Hertel (1998) confirmed that Aβ binds to p75NTR in neuroblastoma cells (Kuner et al., 1998). In our preliminary studies in CECs, p75NTR is also an attractive candidate. We found that p75NTR blockade by anti-p75NTR antibody significantly inhibit Aβ-induced CEC death (our unpublished data). These findings raise the possibility of a receptor-mediated event in Aβ apoptotic action. It may explain, at least in part, why ASK1 Ser967 dephosphorylation induced by Aβ was a rapid event within minutes in the present study.
In conclusion, results from the present study demonstrated for the first time that Aβ-induced CEC death involves at least in part the activation of the PP2A–ASK1–MKK3/6–p38MAPK signaling cascade to induce p53 activation and Bax expression (Fig. 9).
Figure 9.
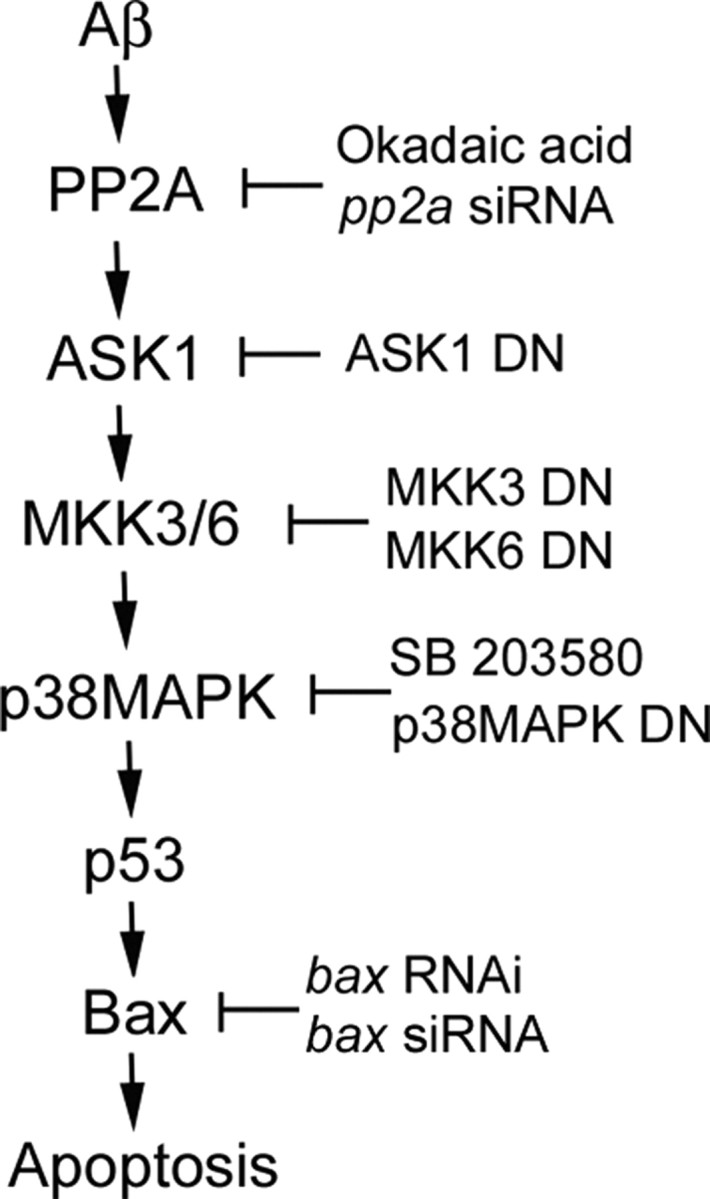
Schematic summary of ASK1-mediated apoptotic pathway in Aβ-induced CEC death. Aβ activates the PP2A–ASK1–MKK3/6–p38MAPK signaling cascade, leading to p53 activation and Bax expression in Aβ-induced CEC death. Approaches applied in the present studies are shown to support the causal role of each step in the cascade.
Footnotes
This work was supported by National Science Council of Taiwan Grants NSC 92-2321-B-038-002 and NSC 93-2321-B-038-002, by Taipei Medical University Topnotch Stroke Research Center grant from the Ministry of Education, by Department of Health Clinical Research Center of Excellence Grant DOH-TD-B-111-002, and by Chi-Chin Huang Stroke Research Center.
References
- Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Chen BC, Yu CC, Lei HC, Chang MS, Hsu MJ, Huang CL, Chen MC, Sheu JR, Chen TF, Chen TL, Inoue H, Lin CH. Bradykinin B2 receptor mediates NF-kappaB activation and cyclooxygenase-2 expression via the Ras/Raf-1/ERK pathway in human airway epithelial cells. J Immunol. 2004;173:5219–5228. doi: 10.4049/jimmunol.173.8.5219. [DOI] [PubMed] [Google Scholar]
- Chen MC, Hwang MJ, Chou YC, Chen WH, Cheng G, Nakano H, Luh TY, Mai SC, Hsieh SL. The role of apoptosis signal-regulating kinase 1 in lymphotoxin-beta receptor-mediated cell death. J Biol Chem. 2003;278:16073–16081. doi: 10.1074/jbc.M208661200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Seimiya H, Naito M, Mashima T, Kizaki A, Dan S, Imaizumi M, Ichijo H, Miyazono K, Tsuruo T. ASK1 mediates apoptotic cell death induced by genotoxic stress. Oncogene. 1999;18:173–180. doi: 10.1038/sj.onc.1202276. [DOI] [PubMed] [Google Scholar]
- Davis-Salinas J, Van Nostrand WE. Amyloid beta-protein aggregation nullifies its pathologic properties in cultured cerebrovascular smooth muscle cells. J Biol Chem. 1995;270:20887–20890. doi: 10.1074/jbc.270.36.20887. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18:7002–7010. doi: 10.1093/emboj/18.24.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Waldman T, Oliner JD, Velculescu VE, Burrell M, Hill DE, Healy E, Rees JL, Hamilton SR, Kinzler KW, Vogelstein B. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–2919. [PubMed] [Google Scholar]
- Fogarty MP, Downer EJ, Campbell V. A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the beta-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem J. 2003;371:789–798. doi: 10.1042/BJ20021660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman EH, Chen L, Fu H. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3- dissociation. J Biol Chem. 2004;279:10442–10449. doi: 10.1074/jbc.M311129200. [DOI] [PubMed] [Google Scholar]
- Hastak K, Agarwal MK, Mukhtar H, Agarwal ML. Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J. 2005;19:789–791. doi: 10.1096/fj.04-2226fje. [DOI] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holasek SS, Wengenack TM, Kandimalla KK, Montano C, Gregor DM, Curran GL, Poduslo JF. Activation of the stress-activated MAP kinase, p38, but not JNK in cortical motor neurons during early presymptomatic stages of amyotrophic lateral sclerosis in transgenic mice. Brain Res. 2005;1045:185–198. doi: 10.1016/j.brainres.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, Houghton PJ. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol Cell. 2003;11:1491–1501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Kamada H, Nito C, Endo H, Chan PH. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2007;27:521–533. doi: 10.1038/sj.jcbfm.9600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamoto T, Mota M, Takeda K, Rubin LL, Miyazono K, Ichijo H, Bazenet CE. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol. 2000;20:196–204. doi: 10.1128/mcb.20.1.196-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Hwang SG, Shin DY, Kang SS, Chun JS. p38 kinase regulates nitric oxide-induced apoptosis of articular chondrocytes by accumulating p53 via NFkappa B-dependent transcription and stabilization by serine 15 phosphorylation. J Biol Chem. 2002;277:33501–33508. doi: 10.1074/jbc.M202862200. [DOI] [PubMed] [Google Scholar]
- Kriem B, Sponne I, Fifre A, Malaplate-Armand C, Lozac'h-Pillot K, Koziel V, Yen-Potin FT, Bihain B, Oster T, Olivier JL, Pillot T. Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-beta peptide. FASEB J. 2005;19:85–87. doi: 10.1096/fj.04-1807fje. [DOI] [PubMed] [Google Scholar]
- Kuner P, Schubenel R, Hertel C. Beta-amyloid binds to p57NTR and activates NFkappaB in human neuroblastoma cells. J Neurosci Res. 1998;54:798–804. doi: 10.1002/(SICI)1097-4547(19981215)54:6<798::AID-JNR7>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, Chen S, Hsu CY. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–131. doi: 10.1083/jcb.200307017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang R, Luo D, Park SJ, Wang Q, Kim Y, Min W. Tumor necrosis factor alpha-induced desumoylation and cytoplasmic translocation of homeodomain-interacting protein kinase 1 are critical for apoptosis signal-regulating kinase 1-JNK/p38 activation. J Biol Chem. 2005;280:15061–15070. doi: 10.1074/jbc.M414262200. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Nishitoh H, Tobiume K, Takeda K, Ichijo H. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid Redox Signal. 2002;4:415–425. doi: 10.1089/15230860260196218. [DOI] [PubMed] [Google Scholar]
- Meek DW. Mechanisms of switching on p53: a role for covalent modification? Oncogene. 1999;18:7666–7675. doi: 10.1038/sj.onc.1202951. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi T, Takeda K, Matsuzawa A, Saegusa K, Nakano H, Gohda J, Inoue JI, Ichijo H. Recruitment of TRAF family proteins to the ASK1 signalosome is essential for oxidative stress-induced cell death. J Biol Chem. 2005;280:37033–37040. doi: 10.1074/jbc.M506771200. [DOI] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RM, Bhattacharya S, Johnson LR. Protein phosphatase 2A regulates apoptosis in intestinal epithelial cells. J Biol Chem. 2005;280:31091–31100. doi: 10.1074/jbc.M503041200. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Oxidative stress and mitochondrial dysfunction in neurodegeneration. Curr Opin Neurol. 1996;9:260–264. doi: 10.1097/00019052-199608000-00003. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Robino G, Obbili A, Bardini P, Aragno M, Parola M, Danni O. H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPK. Exp Neurol. 2003;180:144–155. doi: 10.1016/s0014-4886(02)00059-6. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Qu ZX, Moore SA, Hsu CY, Hogan EL. Receptor-linked hydrolysis of phosphoinositides and production of prostacyclin in cerebral endothelial cells. J Neurochem. 1992;58:1930–1935. doi: 10.1111/j.1471-4159.1992.tb10071.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen S, Ku G, Ahmed SH, Chen H, Hsu CY. Amyloid beta peptide-induced cerebral endothelial cell death involves mitochondrial dysfunction and caspase activation. J Cereb Blood Flow Metab. 2001a;21:702–710. doi: 10.1097/00004647-200106000-00008. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen S, Ahmed SH, Chen H, Ku G, Goldberg MP, Hsu CY. Amyloid-β peptides are cytotoxic to oligodendrocytes. J Neurosci. 2001b;21(RC118):1–5. doi: 10.1523/JNEUROSCI.21-01-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Fine RE, Eisenhauer PB, Arble BL, Stewart KB, Gilchrest BA. Amyloid beta binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J Biol Chem. 2002;277:7720–7725. doi: 10.1074/jbc.M110929200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi O, Watanabe T, Nishida K, Kashiwase K, Higuchi Y, Takeda T, Hikoso S, Hirotani S, Asahi M, Taniike M, Nakai A, Tsujimoto I, Matsumura Y, Miyazaki J, Chien KR, Matsuzawa A, Sadamitsu C, Ichijo H, Baccarini M, Hori M, Otsu K. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J Clin Invest. 2004;114:937–943. doi: 10.1172/JCI20317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DI, Yeh CH, Chen S, Xu J, Hsu CY. Neutral sphingomyelinase activation in endothelial and glial cell death induced by amyloid beta-peptide. Neurobiol Dis. 2004;17:99–107. doi: 10.1016/j.nbd.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, Wang JJ, Leo E, Zapata J, Hauser CA, Reed JC, Bredesen DE. TRAF family proteins interact with the common neurotrophin receptor and modulate apoptosis induction. J Biol Chem. 1999;274:30202–30208. doi: 10.1074/jbc.274.42.30202. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Lee JM, Chen SD, Xu J, Hsu CY. Amyloid-β induces Smac release via AP-1/Bim activation in cerebral endothelial cells. J Neurosci. 2002;22:9764–9770. doi: 10.1523/JNEUROSCI.22-22-09764.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Lee JM, Chen H, Xu J, Hsu CY. Abeta(25–35) alters Akt activity, resulting in Bad translocation and mitochondrial dysfunction in cerebrovascular endothelial cells. J Cereb Blood Flow Metab. 2005;25:1445–1455. doi: 10.1038/sj.jcbfm.9600139. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Hsu CY, Hu XY, Chen H, Chen SW, Xu J, Lee JM. Protein phosphatase 2A regulates bim expression via the Akt/FKHRL1 signaling pathway in amyloid-β peptide-induced cerebrovascular endothelial cell death. J Neurosci. 2006;26:2290–2299. doi: 10.1523/JNEUROSCI.5103-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HM, Yuan J, Cheung P, Chau D, Wong BW, McManus BM, Yang D. Gamma interferon-inducible protein 10 induces HeLa cell apoptosis through a p53-dependent pathway initiated by suppression of human papillomavirus type 18 E6 and E7 expression. Mol Cell Biol. 2005;25:6247–6258. doi: 10.1128/MCB.25.14.6247-6258.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen J, Fu H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci USA. 1999;96:8511–8515. doi: 10.1073/pnas.96.15.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2003;111:1933–1943. doi: 10.1172/JCI17790. [DOI] [PMC free article] [PubMed] [Google Scholar]