

Abstract
The glial cell line-derived neurotrophic factor (GDNF) and neurturin (NTN) belong to a structurally related family of neurotrophic factors. NTN exerts its effect through a multicomponent receptor system consisting of the GDNF family receptor α2 (GFRα2), RET, and/or NCAM (neural cell adhesion molecule). GFRα2 is alternatively spliced into at least three isoforms (GFRα2a, GFRα2b, and GFRα2c). It is currently unknown whether these isoforms share similar functional and biochemical properties. Using highly specific and sensitive quantitative real-time PCR, these isoforms were found to be expressed at comparable levels in various regions of the human brain. When stimulated with GDNF and NTN, both GFRα2a and GFRα2c, but not GFRα2b, promoted neurite outgrowth in transfected Neuro2A cells. These isoforms showed ligand selectivity in MAPK (mitogen-activated protein kinase) [ERK1/2 (extracellular signal-regulated kinase 1/2)] and Akt signaling. In addition, the GFRα2 isoforms regulated different early-response genes when stimulated with GDNF or NTN. In coexpression studies, GFRα2b was found to inhibit ligand-induced neurite outgrowth by GFRα2a and GFRα2c. Stimulation of GFRα2b also inhibited the neurite outgrowth induced by GFRα1a, another member of the GFRα. Furthermore, activation of GFRα2b inhibited neurite outgrowth induced by retinoic acid and activated RhoA. Together, these data suggest a novel paradigm for the regulation of growth factor signaling and neurite outgrowth via an inhibitory splice variant of the receptor. Thus, depending on the expressions of specific GFRα2 receptor spliced isoforms, GDNF and NTN may promote or inhibit neurite outgrowth through the multicomponent receptor complex.
Keywords: GDNF, NTN, GFRα2, RhoA, inhibitory splice isoforms, neuroblastoma
Introduction
Neurturin (NTN), glial cell line-derived neurotrophic factor (GDNF), Artemin, and Persephin are cysteine knot proteins and are members of the GDNF family ligands (GFLs) (Kotzbauer et al., 1996; Airaksinen and Saarma, 2002). These GFLs have been shown to support the growth, maintenance, and differentiation of a wide variety of neuronal and extraneuronal systems (Saarma and Sariola, 1999). Each GFL is known to bind preferentially to one GDNF family receptor α (GFRα) in vitro, and the activation of the multicomponent receptor system shows some degree of promiscuity in their ligand specificities (Horger et al., 1998; Airaksinen et al., 1999; Cik et al., 2000; Wang et al., 2000; Scott and Ibanez, 2001). NTN is thought to signal through its preferred receptor complex consisting of GFRα2, RET, and/or neural cell adhesion molecule (NCAM) (Baloh et al., 1997; Buj-Bello et al., 1997; Widenfalk et al., 1997; Paratcha et al., 2003).
Alternative splicing is prevalent in many mammalian genomes, as a means of producing functionally diverse polypeptides from a single gene (Blencowe, 2006). It has been estimated that >50% of human multi-exon genes are alternatively spliced (Modrek and Lee, 2002). Multiple alternatively spliced variants of GFRα1 (Sanicola et al., 1997; Dey et al., 1998; Shefelbine et al., 1998), GFRα2 (Wong and Too, 1998; Dolatshad et al., 2002), and GFRα4 (Lindahl et al., 2000, 2001; Masure et al., 2000) have been reported. Similarly, alternative spliced isoforms of the coreceptors RET (Lorenzo et al., 1997; de Graaff et al., 2001; Lee et al., 2002) and NCAM (Povlsen et al., 2003; Buttner et al., 2004) have been reported. The alternatively spliced isoforms of GFRα1 have recently been shown to exhibit distinct biochemical functions (Charlet-Berguerand et al., 2004; Yoong et al., 2005). These observations are consistent with the emerging view that the combinatorial interactions of the spliced isoforms of GFRα, RET, and NCAM may contribute to the multicomponent signaling system to produce the myriad of observed biological responses.
We have previously shown that all three isoforms of GFRα2 are expressed at significant levels in the murine whole brain and embryo (Too, 2003). It is, however, unknown whether these isoforms serve distinct or redundant functions. To gain a better insight into their biological relevance in the CNS, the expression levels of the isoforms in different regions of the human brain were quantified by highly specific real-time PCR assays. The biological functions of the isoforms were then examined in a neuronal differentiation model using Neuro2A cells and in BE(2)-C cells, which express the spliced isoforms endogenously.
Here, we showed that ligand activation of the isoforms differentially activated mitogen-activated protein kinase (MAPK) [extracellular signal-regulated kinase 1/2 (ERK1/2)] and AKT signaling and regulated distinct early-response genes. Furthermore, both GDNF and NTN induced neurite outgrowth through GFRα2a and GFRα2c, but not GFRα2b. Activation of GFRα2b inhibited neurite outgrowth induced by the other two GFRα2 isoforms as well as GFRα1a and retinoic acid. RhoA was also found to be activated by GDNF and NTN through GFRα2b. This study thus provides the first piece of evidence of a dominant inhibitory activity of GFRα2b on neurite outgrowth and distinct signaling mechanisms underlying the activation of spliced isoforms.
Materials and Methods
Cell culture.
Neuro2A (catalog #CCL-131; American Type Culture Collection, Manassas, VA) and BE(2)-C (catalog #CRL-2268; American Type Culture Collection) cells were grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone, Logan, UT). All cultures were maintained in a 5% CO2 humidified atmosphere at 37°C.
Reverse transcription reaction.
Total RNA for different human brain regions was purchased from Clontech (Palo Alto, CA). Total RNA from Neuro2A cells was prepared as described previously (Too and Maggio, 1995). The integrity of isolated total RNA was validated by denaturing agarose gel electrophoresis. Five micrograms of total RNA were reverse transcribed using 400 U of ImpromII and 0.5 μg of random hexamer (Promega, Madison, WI) for 60 min at 42°C according to the manufacturer's instructions. The reaction was terminated by heating at 70°C for 5 min, and the cDNA was used directly for quantitative real-time PCR. Three independent preparations of cDNA were used for the study. All measurements were performed in triplicate.
Plasmids constructions.
To prepare plasmid standards for quantitative real-time PCR, open reading frames of human GFRα2 isoforms and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were subcloned into p-GEMT (Promega). For early-response genes and transcriptional factors, partial sequences were subcloned using the same primers used for real-time PCR quantification. XbaI or XmnI (Promega) was used to linearize plasmids to be used as templates for real-time PCR amplifications.
Sequence-independent real-time PCR.
Real-time PCR was performed on the iCycler iQ (Bio-Rad, Hercules, CA) using SYBR Green I. The threshold cycles (Ct) were calculated using the Optical interface version 3.0B. Real-time PCR was performed after an initial denaturation for 3 min at 95°C, followed by 40–60 cycles of 60 s denaturation at 95°C, 30 s annealing at 60°C, and 60 s extension at 72°C. Fluorescent detection was performed at the annealing phase. The reaction was performed in a total volume of 50 μl in 1× XtensaMix-SG (BioWORKS, Singapore), containing 2.5 mm MgCl2, 10 pmol of primer, and 1.25 U of Platinum DNA polymerase (Invitrogen, Carlsbad, CA). Melt-curve analyses were performed at the end of PCR to verify the identity of the products. A specific exon-overlapping forward primer used for amplification of human GFRα2a was designated as “2a 15 + 9F” (5′-TCTTCTTCTTTCTAGACGAGACCC-3′), for human GFRα2b as “2b 17 + 7F” (5′-CCTCTTCTTCTTTCTAGGTGAGGA-3′), and for human GFRα2c as “2c 18 + 5F” (5′-GCCTCTTCTTCTTTCTAGGGACA-3′). A common reverse primer, designated as “553R” (5′-GCAGATGGAGATGTAGGAGGAG-3′), was used for all three isoforms. The primer pair 5′-GATCATCAGCAATGCCTCCT-3′ and 5′-GCCATCACGCCACAGTTT-3′ was used to amplify human GAPDH. All real-time PCR quantification was performed simultaneously with linearized plasmid standards and a nontemplate control. The gene expression levels were interpolated from standard curves and normalized to the expressions of GAPDH in the same samples. Differences in the expression levels of GFRα2 isoforms were analyzed using the paired Student's t test with a level of significance of p < 0.05.
Generation of Neuro2A cells expressing GFRα2 isoforms.
The murine neuroblastoma cell line Neuro2A, which express endogenous RET and NCAM, was stably transfected with murine GFRα2a, GFRα2b, GFRα2c, or vector control pIRESneo (Clontech) using Fugene-6 (Roche, Mannheim, Germany) and selected with 0.8 mg/ml G418 (Promega), over a period of 2 months. Primers used for measuring GFRα2 isoforms, RET, and NCAM expression were as described previously (Too, 2003; Yoong et al., 2005). For coexpression studies, GFRα2a, GFRα2c, or GFRα1a was cloned into the proximal 5′ multiple cloning site, whereas GFRα2b was cloned into a distal 3′ multiple cloning site of the bicistronic pIRES vector (Clontech). All studies were performed with three independent clones.
Assessment of neurite outgrowth in GFRα2-transfected Neuro2A cells.
Twenty thousand to 50,000 cells per well were seeded on six-well plates overnight, in DMEM supplemented with 10% FBS. Cells were then incubated with medium containing 0.5% FBS, with or without 50 ng/ml recombinant human GDNF (Biosource, Camarillo, CA) or NTN (ProSpec-Tany TechnoGene, Rehovot, Israel). Cells were incubated for 3 more days. All-trans retinoic acid (5 μm; Sigma, St. Louis, MO) was used as a positive control for inducing neurite outgrowth. Cells bearing at least one neurite twice the length of the cell bodies were scored. More than 600 cells from three different fields were counted per well. Statistical significance between ligand-stimulated and control samples was calculated using the paired Student's t test. A value of p < 0.05 was considered significant.
Immunocytochemistry and confocal microscopy.
Cells were seeded on chamber slides, fixed with 4% paraformaldehyde in 1× PBS for 15 min at 37°C, and subsequently fixed in methanol at −20°C for an additional 15 min. After three washes with 1× PBS, cells were permeabilized and blocked with serum (1:10; Dako, Glostrup, Denmark) and 0.5% Triton X-100 in 1× PBS for 30 min at room temperature. The cells were then incubated with F-actin (phalloidin-conjugated tetramethylrhodamine isothiocyanate) and high-molecular-weight neurofilament protein (NF-200) antibody (Sigma) in 0.1% Triton X-100, 0.1% BSA, and 1× PBS for 1 h at 37°C and washed three times in 1× PBS. A secondary antibody (Alexa Fluor 488; Invitrogen, Eugene, OR) was then added at a dilution of 1:200 and incubated for 1 h. The cells were then washed in 1× PBS and mounted for visualization. Image acquisition was obtained using a Zeiss (Oberkochen, Germany) 510 meta confocal microscope equipped with fluorescence detection.
Immunoblotting.
Phosphorylation of MAPK (ERK1/2) or Akt was analyzed as follows. Cells were initially seeded in DMEM with 10% FBS for 24 h, and serum was depleted (0.5% FBS) for 16 h. The cells were then treated with 50 ng/ml GDNF, NTN, Artemin, or Persephin (PreproTech, London, UK) in serum-free medium for different periods of time at 37°C. For dose–response studies, cells were stimulated with different concentrations of ligands for 10 min at 37°C. Control treatment with 1 m Sorbitol (Sigma) was performed simultaneously. The supernatants were then removed, and cells were washed once with 1× PBS and subsequently lysed in 2% SDS. Protein concentrations were estimated using the BCA assay (Pierce, Rockford, IL). ERK1/2 or Akt phosphorylation was analyzed by Western blot using phospho-specific antibodies according to the manufacturer's instructions (Cell Signaling Technology, Danvers, MA). Blots were stripped with Restore Western Stripping Buffer (Pierce) and reprobed with pan antibodies to verify equal loading of protein.
For studying kinetics and dose–response of ligand-induced ERK1/2 activation, dot blot analysis was performed using the BIO Dot Apparatus (Bio-Rad). Five micrograms of protein were loaded per well in triplicates. Blots were then detected by anti-phospho ERK1/2 antibodies (Cell Signaling Technology) according to the manufacturer's instructions. Densities of blots were imaged and measured by Quantity One 4.0 (Bio-Rad).
Binding of [125I]GDNF to GFRα2 isoforms transfected Neuro2A cells.
[125I] GDNF (∼1000 mCi/mmol) was prepared using Bolton and Hunter reagent (Amersham Biosciences, Piscataway, NJ). Briefly, 10 μg of recombinant human GDNF (Biosource) was labeled with 1 mCi of Bolton and Hunter reagent for 1 h at room temperature according to the manufacturer's instructions. The reaction was then terminated by adding 10 μl of 0.1% tyrosine. Radiolabeled GDNF was then purified on a Sephadex G-10 column.
Binding studies were performed as described previously (Jing et al., 1997). Briefly, 0.1 million cells were seeded per well on 24-well Costar (Cambridge, MA) tissue culture plates for 2 d before the assay. Before the experiment, cells were placed on ice for 15–20 min and washed once with ice-cold DMEM buffer and 25 mm HEPES, pH 7.0. Cells were then incubated at 4°C for 3 h with 0.2 ml of binding buffer [DMEM, 25 mm HEPES, 2 mg/ml bovine albumin serum, and Complete Inhibitor Cocktail (Roche), pH 7.0] containing 50 pm [125I]GDNF and various concentrations of unlabeled GDNF. At the end of incubation, cells were washed three times with 0.3 ml of ice-cold washing buffer and lysed in 0.1% SDS containing 1 m NaOH. The radioactivity in lysates was measured using the auto gamma counter (PerkinElmer Packard, Wellesley, MA).
Measurements of early-response genes regulated by GDNF and NTN.
Cells were seeded in DMEM with 10% FBS for 24 h, followed by serum depletion (0.5% FBS) for 18–24 h. The cells were then treated with GDNF (50 ng/ml) or NTN (50 ng/ml) in serum-free medium for varying periods of time at 37°C. Total RNA was then isolated and reverse transcribed as described above. The gene expression levels were then quantified by real-time PCR using gene-specific primers. Primers used for amplification of early response genes were as follows: EGR-1-328F/EGR-1-459R (5′-GAGAAGGCGATGGTGGAGACGA-3′/5′-GCTGAAAAGGGGTTCAGGCCA-3′) for egr-I; EGR-2-1F/EGR-2-179R (5′-ATGAACGGAGTGGCGGGAGAT-3′/5′-TCTGGATAGCAGCTGGCACCAG-3′) for egr-2; mcfos(B)651F/mcfos(B)901R (5′-TGTGGCCTCCCTGGATTT-3′/5′-CTGCATAGAAGGAACCGGAC-3′) for c-fos; and mFosB(A)1926F/mFosB(A)2107R (5′-CAGGGTCAACATCCGCTAA-3′/5′-GGAAGTGTACGAAGGGCTAACA-3′) for fosB. Expression of target genes and GAPDH was interpolated from standard curves. The fold change of each target gene was calculated as a change in gene expression of the stimulated sample normalized to GAPDH compared with gene expression of the control sample normalized to GAPDH.
Silencing of GFRα2b in BE(2)-C.
Small interfering RNA (siRNA) duplexes (Invitrogen) were designed across specific exon (exons 1 and 3) boundaries of GFRα2b (siGFRα2b-15+5: TCTTCTTCTTTCTAGGTGAG; siGFRα2b-13+7: TCTTCTTCTTTCTAGGTGAGGA; siGFRα2b-10+10: TTCTTTCTAGGTGAGGAGTT; siGFRα2b-7+13: TTTCTAGGTGAGGAGTTCTA; siGFRα2b-5+15: TCTAGGTGAGGAGTTCTACG). Subconfluent cells (50–80%) were seeded on six-well plates, in 10% FBS DMEM. Cells were transfected with siRNA duplexes (20 pmol) using Transfectin (Bio-Rad) in 400 μl of 0.5% FBS DMEM per well. Total RNA was isolated 6 h after transfection, and gene expression was measured by real-time PCR. For differentiation studies using BE(2)-C, 6 h after silencing of GFRα2b, 2 ml of differentiation medium containing retinoic acid (5 μm), GDNF (50 ng/ml), or NTN (50 ng/ml) in 0.5% FBS DMEM was added to the medium. Analyses of morphological differences were performed after 3 d.
RhoA assay.
Neuro2A cells were seeded in 10% FBS DMEM and incubated for 18–24 h. Subsequently, the serum was reduced to 0.5% in DMEM, and the cells were incubated for an additional 18–24 h. Cells were then treated with 10 μm lysophosphatidic acid (LPA; Sigma), GDNF (50 ng/ml), or NTN (50 ng/ml) in serum-free DMEM for 10 min. Cells were lysed and used directly for the GTP-RhoA pull-down assay according to the manufacturer's instructions (Pierce). RhoA inhibitor exoenzyme C3 transferase and Rho kinase (ROCK) inhibitor Y27632 were purchased from Calbiochem (La Jolla, CA). Exoenzyme C3 transferase was transfected into cells using the lipotransfecting agent Transfectin (Bio-Rad), at 1 μl of Transfectin/1 μg of C3 transferase per well of a six-well plate, 4 h before start of the experiment. Cells were then treated with RhoA inhibitor exoenzyme C3 transferase (1 μg/ml) or ROCK inhibitor Y27632 (10 μm), in the presence or absence of differentiating stimuli. LPA (10 μm; Sigma) was used as a positive control for activities of Rho and ROCK inhibitor.
Results
Differential expression profiles of GFRα2 spliced variants
Currently, the expression levels of GFRα2 spliced variants in specific regions of the brain are unknown. To address this issue, we have developed sequence-independent real-time PCR assays to quantify each of the spliced variants with high specificity and sensitivity.
To discriminate between the three spliced variants of human GFRα2, overlapping exon primers were designed across exons 1 and 2, 1 and 3, or 1 and 4 to enable the specific detection and quantification of GFRα2a, GFRα2b, and GFRα2c, respectively (Fig. 1A). Because the amplification products of GFRα2a (545 bp), GFRα2b (233 bp), and GFRα2c (172 bp) were different in sizes, it was critical to determine the optimal cycling parameters for the selective amplification of each of the transcripts. A dwell time of 30 s for annealing, 60 s for denaturation at 95°C, and 60 s for extension at 72°C were found to be optimal for the amplifications of all three isoforms. The slopes of the plots of Ct versus log10 mole of the human GFRα2a, GFRα2b, and GFRα2c standards were 3.37 ± 0.30 (r2 = 0.98), 4.12 ± 0.41 (r2 = 0.99), and 3.82 ± 0.54 (r2 = 0.99), respectively. The samples diluted in parallel with the standards (data not shown). The specificity of amplifying a particular isoform compared with the other variants was >106-fold (data not shown). Hence, the amplifications of GFRα2b and GFRα2c were at least 106-fold less efficient than amplifying GFRα2a, when using GFRα2a exon-overlapping primers. The detection limits of the assays were estimated to be <100 copies of transcripts per reaction.
Figure 1.

Real-time PCR quantification of the expressions of GFRα2 isoforms in the human brain. A, A schematic diagram showing the protein coding exons and the positions of the primers used for quantitative real-time PCR. Exons 1–9 encode the full-length protein sequence of GFRα2a. Specific forward primers (2a 15 + 9F for GFRα2a, 2b 17 + 7F for GFRα2b, and 2c 18 + 5F for GFRα2c) were designed across exon junctions, whereas a common reverse primer (553R) was used for the amplification of all the three isoforms. B, The expression levels of GFRα2 isoforms in a different human brain region normalized to the levels of GAPDH in the same tissue. The results were expressed as mean ± SEM (n = 3). Significant differences between the expressions of the isoforms were calculated using paired Student's t test. A value of p < 0.05 was considered significant (**p = 0.001).
Using these highly sensitive and specific assays, the expression levels of the GFRα2 alternatively spliced isoforms were quantified in caudate nucleus, cortex, putamen, substantia nigra, subthalamic nucleus, and thalamus of the human brain (Fig. 1B). The three GFRα2 isoforms were detected at significant levels (>104 copies per reaction) in all areas of the brain, with expression levels highest in the cortex. In cortex, all three isoforms were expressed at comparable levels, with GFRα2b expression significantly lower than GFRα2c (p < 0.01).
GFRα2 isoforms differentially activated ERK1/2 and Akt
To investigate the biological significance of alternatively spliced GFRα2 isoforms, stable transfectants were generated in Neuro2a cells. We have shown previously that Neuro2a cells express RET and NCAM, but not GFRα2 receptors, endogenously (Yoong et al., 2005). The expression levels of GFRα2 isoforms in stably transfected Neuro2a cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) were comparable to that expressed in the human cortex (Fig. 1B).
When stimulated with NTN, all the three isoforms induced the rapid phosphorylation of ERK1/2 (Fig. 2A). However, when stimulated with GDNF, GFRα2a (Fig. 2A,C) and GFRα2c (Fig. 2A,E), but not GFRα2b (Fig. 2A,D), induced significant ERK1/2 phosphorylation (more than twofold). The extent of ERK1/2 phosphorylation was similar when GFRα2a (Fig. 2C) and GFRα2c (Fig. 2E) activated with either GDNF or NTN. However, GFRα2b showed rapid and significant phosphorylation of ERK1/2 only with NTN stimulation but not by GDNF (Fig. 2A,D). Both GDNF and NTN induced ERK1/2 phosphorylation in a dose–response manner in GFRα2a (Fig. 2F) and GFRα2c (Fig. 2H) transfectants. GDNF appeared to be slightly more potent than NTN in inducing ERK1/2 phosphorylation in both transfectants (Fig. 2F,H). Compared with the stimulation with NTN, GFRα2b when stimulated with GDNF showed no significant increase in ERK1/2 phosphorylation even at the highest dose (Fig. 2G). No significant increase in the phosphorylation of ERK1/2 was observed in vector (pIRESneo) control transfected Neuro2A cells when stimulated with either GDNF or NTN (data not shown).
Figure 2.
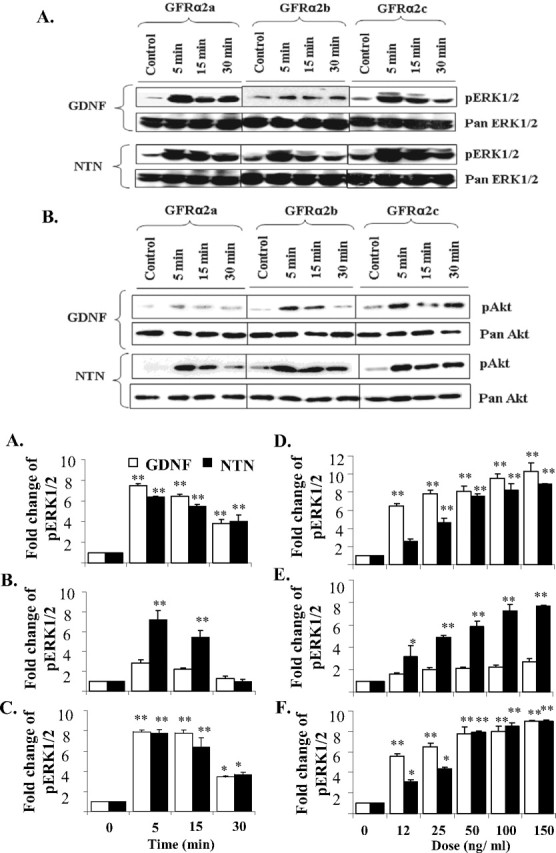
Activations of ERK1/2 and Akt in GFRα2 isoforms transfected Neuro2A cells when stimulated by either GDNF or NTN. Cells were stimulated in serum-free medium, with or without GDNF or NTN (50 ng/ml), for the period of time indicated. A, B, Five micrograms of protein were loaded and separated by SDS electrophoresis, and phosphorylated ERK1/2 (pERK1/2; A) or Akt (pAkt; B) was then detected by Western blot. Blots were stripped and reprobed with pan antibody as loading controls. C–E, Kinetics of GDNF and NTN induced ERK1/2 activations in GFRα2a (C), GFRα2b (D), and GFRα2c (E). Cells were treated with 50 ng/ml GDNF or NTN for 5, 15, and 30 min. F–H, Dose responses of the activation of ERK1/2 when stimulated with GDNF or NTN in GFRα2a (F), GFRα2b (G), and GFRα2c (H) isoforms. Cells were stimulated for 10 min with ligand at various doses. For kinetic and dose–response studies, 5 μg of protein was loaded per well for dot blot quantification of phospho-ERK1/2 (pERK1/2). The means ± SD were calculated from results obtained in triplicates. Significant differences in fold change of pERK1/2 between ligand stimulated and control were calculated using the paired Student's t test. A value of p < 0.05 was considered significant (**p < 0.001; *p < 0.05). Experiments were repeated three times with two independent clones with similar results.
We next investigated the ligand-regulated phosphorylation of Akt using GDNF or NTN (Fig. 2B). NTN induced rapid and significant phosphorylations of Akt in all three isoform transfectants. However, GDNF induced the rapid and significant phosphorylations of Akt in cells expressing GFRα2b and GFRα2c.
The other GFLs, Artemin and Persephin, did not induce significant phosphorylation of ERK1/2 or Akt in any of the GFRα2 isoform transfectants (data not shown). In addition, neither GDNF nor NTN was found to activate p38 and c-Jun N-terminal kinase (JNK) in any of the GFRα2 isoform transfectants, even at concentrations as high as 100 ng/ml and over a period of 1 h of ligand stimulation (data not shown).
[125I]GDNF bound equally well to all three GFRα2 isoforms
NTN has been shown to bind with similar affinities to the GFRα2 isoforms (Scott and Ibanez, 2001). Because GDNF failed to induce a significant increase in the phosphorylation of ERK1/2 in GFRα2b transfectants (Fig. 2A,D,G), it is possible that GDNF may not bind to this isoform. To address this possibility, we next performed a ligand displacement study using [125I]GDNF (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). GDNF displaced the binding of [125I]GDNF to the three GFRα2 isoforms with similar potencies. The IC50 for the displacements of cells transfected with GFRα2a, GFRα2b, and GFRα2c were 3.27 ± 0.02, 2.79 ± 0.16, and 2.31 ± 0.09 nm (mean ± SD), respectively. Parental Neuro2A or cells transfected with pIRESneo showed no significant binding to [125I]GDNF. This result indicates that GDNF binds to all three isoforms with similar affinities.
GFRα2 isoforms activated different transcriptional genes
The differential activation of ERK1/2 and Akt (Fig. 2) suggests the possibility that downstream biochemical mechanisms may differ. To explore this issue, we measured the changes in gene expression of the fos family (c-fos, fosB), jun family (c-jun, jun-b), egr family (egr1–4), and GDNF-inducible transcription factors mGIF and mGZF1 in response to GDNF and NTN (supplemental Table 1, available at www.jneurosci.org as supplemental material). These factors have previously been shown to be activated with GDNF or NTN (Yajima et al., 1997; Trupp et al., 1999; Kozlowski et al., 2000; Fukuda et al., 2003; Pezeshki et al., 2003). The kinetics of gene activations over a period of 6 h was quantified by real-time PCR (Fig. 3). Distinct ligand-induced early-response gene expressions were observed with the activation of the different GFRα2 isoforms. GFRα2a, when stimulated by GDNF (Fig. 3A) or NTN (Fig. 3B), upregulated egr-1 by as much as fourfold to fivefold. GFRα2b, when stimulated by GDNF (Fig. 3C) or NTN (Fig. 3D), upregulated fosB by >10-fold compared with control. When stimulated with GDNF (Fig. 3E) or NTN (Fig. 3F), GFRα2c upregulated the expressions of egr-1 and egr-2. With the other genes, no significant changes were observed with GDNF or NTN stimulations. These results showed that the activation of GFRα2b isoform regulates the transcription of specific sets of early-response genes.
Figure 3.

Kinetic analyses of the regulations of early-response genes by GDNF and NTN in GFRα2 isoform transfectants. The fold change of mRNA expressions of early-response genes in cells expressing GFRα2a (A), GFRα2b (C), and GFRα2c (E) when stimulated with GDNF and GFRα2a (B), GFRα2b (D), and GFRα2c (F) when stimulated with NTN at the designated period of time is shown. The expression levels were measured by quantitative real-time PCR. Similar results were obtained from more than three separate experiments. Error bars indicate SDs of triplicate measurements from one study. Significant differences in expression of genes between ligand stimulated and control were calculated using the paired Student's t test. A value of p < 0.05 was considered significant (**p < 0.001; *p < 0.05).
Neurite outgrowths were induced by GFRα2a and GFRα2c, but not GFRα2b
Neuro2a cells serve as an excellent in vitro model system for studying signaling pathways mediating neurite outgrowth. Under normal growth conditions, most Neuro2a cells spontaneously sprout a basal level of neurites. However, treatment with a variety of stimuli cause these cells to develop extensive neurites similar to changes observed in hippocampal and cortical cultures (Ahmari et al., 2000; Washbourne et al., 2002).
To investigate possible morphological changes induced by the activation of the GFRα2 isoforms, the transfectants were stimulated with either GDNF or NTN. Both GFRα2a and GFRα2c transfectants showed extensive neurite outgrowths when stimulated with either ligand, comparable to the effects of retinoic acid (Fig. 4). Unexpectedly, neither NTN nor GDNF induced neurite outgrowth in cells expressing GFRα2b (Fig. 4). Immunocytochemical staining for β-III tubulin confirmed these observations. (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Cells expressing GFRα2b extended neurite-like structures when treated with retinoic acid, indicative of the potential for neurite outgrowth (Fig. 4A). GDNF and NTN have no neuritogenic effect on control vector-transfected Neuro2A cells (Fig. 4).
Figure 4.
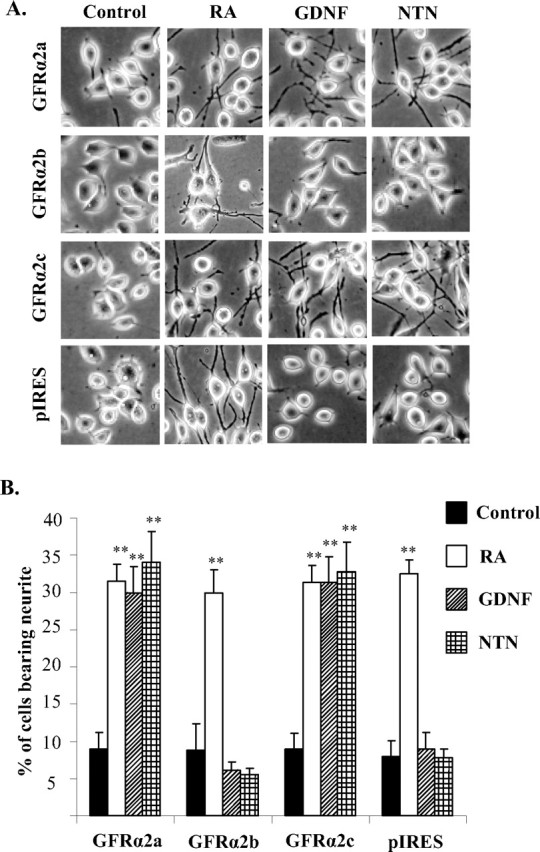
Differential neuritogenic activities of ligand-activated GFRα2 isoforms. Cells were seeded on six-well plates and incubated for 16–18 h in medium containing 10% serum. The cells were then exposed to GDNF or NTN (50 ng/ml) for 3 more days in 0.5% serum-containing medium. Retinoic acid (5 μm) was used as a positive control for cell differentiation. A, Digital phase-contrast images of Neuro2A cells stably expressing GFRα2a, GFRα2b, GFRα2c, or pIRES vector control when treated with retinoic acid, GDNF, or NTN. B, Percentages of cells bearing neurites that were at least twice the length of the cells bodies. More than 600 cells were counted per well, on at least three different fields. Experiments were repeated twice with three individual clones, with similar results. Significant differences in the percentage of cells bearing neurites between ligand stimulated and control were calculated using the paired Student's t test (*p < 0.002). Error bars indicate mean ± SD of triplicate measurements. RA, Retinoic acid.
To further examine the morphological changes in these cells, two major cytoskeletal components, F-actin and high-molecular-weight neurofilament protein (NF-H), which are involved in neurite outgrowth dynamics, were visualized by fluorescent staining (Myers et al., 2006). With ligand (GDNF or NTN)-stimulated GFRα2a and GFRα2c transfectants, NF-H-positive filopodia (axon-like processes) were relatively long and formed thick threads. Protrusions with F-actin staining were observed at the edges of the thick NF-H-positive axon-like elements and cell bodies (supplemental Fig. 4C, arrowheads, available at www.jneurosci.org as supplemental material). Engorgements were seen at some terminal structures that were both NF-H and F-actin positive (supplemental Fig. 4C, arrow, available at www.jneurosci.org as supplemental material). Long extensions were not obvious with cells expressing GFRα2b when stimulated with either ligand. Instead, F-actin-positive staining was found at the periphery of these cells where NF-H was not found to colocalize extensively (supplemental Fig. 4F, available at www.jneurosci.org as supplemental material). These observations provide additional evidence of the lack of neurite outgrowth in ligand-stimulated cells expressing GFRα2b and the neuritogenic activities of the other two isoforms.
GFRα2b inhibited neurite outgrowth mediated by GFRα2a, GFRα2c, and GFRα1a isoform
Because GFRα2b transfectants did not induce neurite outgrowth when stimulated with ligands, we explored the possibility that this isoform may affect the morphological changes in cells coexpressing GFRα2b and GFRα2a or GFRα2c. We first established stably transfected Neuro2A cells coexpressing GFRα2a and GFRα2b (GFRα2a + GFRα2b) using a bicistronic vector. Expression and membrane targeting of GFRα2a were not affected when coexpressed with GFRα2b (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). As shown previously, ligand-induced stimulation of GFRα2a but not GFRα2b induced neurite outgrowth (Fig. 4B). Ligand-induced stimulation of cells coexpressing GFRα2a + GFRα2b showed significantly less neurite outgrowth (Fig. 5A). However, these cells extended neurite when treated with retinoic acid. Similarly, ligand stimulation of cells coexpressing GFRα2c and GFRα2b (GFRα2c + GFRα2b) showed significantly less neurite outgrowth (Fig. 5A).
Figure 5.

Ligand-induced neurite outgrowth in Neuro2A coexpressing GFRα2b with other GFRα isoforms. GFRα2b was stably coexpressed with GFRα2a (GFRα2a + GFRα2b) or GFRα2c (GFRα2c + GFRα2b) (A) or GFRα1a (GFRα1a + GFRα2b) (B). Cells were treated with or without GDNF or NTN (50 ng/ml) for 3 d in 0.5% serum-containing medium. Retinoic acid (RA; 5 μm) differentiated all the transfectants efficiently. Experiments have been repeated twice with three independent clones, with similar results. Significant differences in the percentage of cells bearing neurites between ligand stimulated and control were calculated using the paired Student's t test (**p < 0.002; *p = 0.05). Error bars indicate mean ± SD of triplicate measurements.
Extending this finding, we next explored the possible inhibitory effect of the activation of GFRα2b on the neurite outgrowth induced by ligands in cells coexpressing GFRα1a. Cells expressing only GFRα1a showed significant neurite outgrowth when stimulated by GDNF, NTN, or retinoic acid (Fig. 5B). Interestingly, when stimulated by either GDNF or NTN, cells coexpressing GFRα1a and GFRα2b (GFRα1a + GFRα2b) showed significantly less neurite outgrowth. These observations indicate that the activation of GFRα2b inhibits neurite outgrowth induced by the activation of GFRα2a, GFRα2c, and even the structurally related GFRα1a.
Knock-down of GFRα2b resulted in an increase in neurite outgrowth
We next extended the above observation of the GFRα2b-induced inhibition of neurite outgrowth by investigating BE(2)-C cells that have previously been shown to endogenously express GFRα2 receptors (Kobori et al., 2004). These cells express a comparable level of GFRα2a and GFRα2b (Fig. 6A). GFRα2c was found to be expressed at a level close to the detection limit of the assay (data not shown). The presence of GFRα2 but not GFRα1 in BE(2)-C cells agrees with previous observations (Kobori et al., 2004; Yoong et al., 2006) but not with a recent report using semiquantitative PCR (Hansford and Marshall, 2005).
Figure 6.
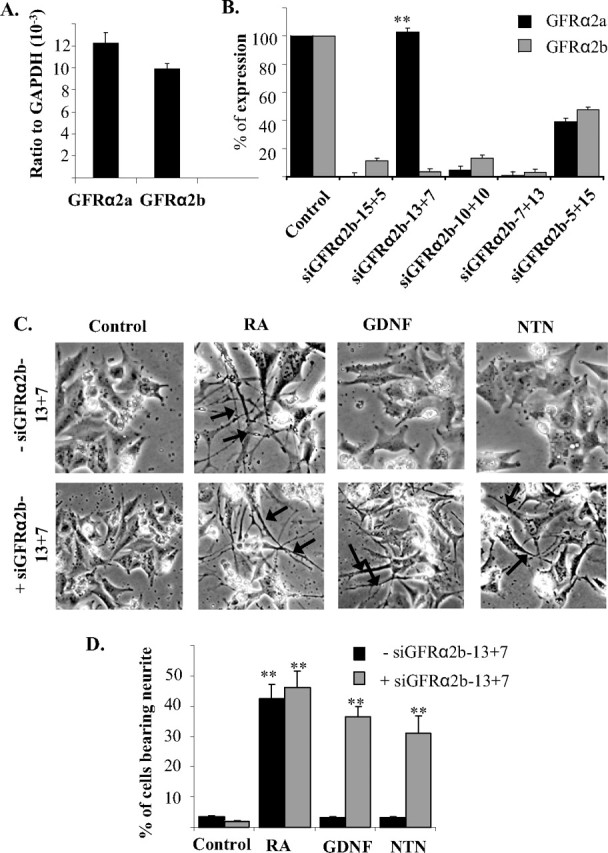
Silencing of GFRα2b expression in human BE(2)-C cells. A, The expression levels of GFRα2a and GFRα2b in BE(2)-C cells were determined using quantitative real-time PCR. B, Effects of various designs of siRNA sequences on the expressions of GFRα2a and GFRα2b in BE(2)-C. siRNA duplex (20 pmol) was transfected into cells, and total RNA was harvested 6 h later. The expressions of GFRα2a and GFRα2b were then measured by quantitative real-time PCR. Significant differences between the expression of the two isoforms after silencing with each of the siRNA designs were calculated using the paired Student's t test (**p = 0.001). C, D, Neurite outgrowth of BE(2)-C cells after silencing of GFRα2b. C, Top row, Cells were stimulated with retinoic acid (5 μm), GDNF, or NTN (50 ng/ml) in the absence of siRNA. Bottom row, Cells were transfected with siGFRα2b-13+7 for 6 h and subsequently stimulated with retinoic acid (5 μm), GDNF, or NTN (50 ng/ml). Pretreament of cells with siGFRα2b-13+7 and subsequent stimulation with GDNF or NTN resulted in the formation of neurite-like structures (arrows). D, Percentages of cells bearing neurites that were at least twice the length of the cells bodies were scored in the presence (+) or absence (−) of the siRNA siGFRα2b-13+7. Similar results were obtained from replicates of three individual experiments. Significant differences in the percentage of cells bearing neurites between ligand stimulated and control were calculated using the paired Student's t test (**p < 0.002; *p = 0.05). Error bars indicate mean ± SD of triplicate measurements. RA, Retinoic acid.
Similar to the above observations with the coexpression of GFRα2b with GFRα2a, GFRα2c, or GFRα1a, both GDNF and NTN failed to induce neurite outgrowth in BE(2)-C cells (Fig. 6C, control). Neurite outgrowth was, however, observed when these cells were treated with retinoic acid, an indication that BE(2)-C cells have the capability of forming neurite-like structures. To test the hypothesis that the activation of GFRα2b may inhibit neurite outgrowth induced by GFRα2a or GFRα2c in BE(2)-C cells, we attempted to silence the expression of GFRα2b using siRNA. Because GFRα2b has no unique sequences compared with GFRα2a, the design of a GFRα2b isoform-specific siRNA poses a significant challenge. A series of siRNA duplexes were then designed with sequences overlapping exons 1 and 3 of GFRα2b (Fig. 6B). Of the five designs tested, only the siGFRα2b-13+7 showed significant discrimination in silencing GFRα2a and GFRα2b (Fig. 6B). This particular siRNA design, siGFRα2b-13+7, inhibited the expression of GFRα2b to <10% of the control, with no significant reduction in the expression of GFRα2a.
When GFRα2b expression was silenced, the BE(2)-C cells extended neurite-like structures when stimulated with either GDNF and NTN (Fig. 6C). This observation supports the notion that the activation of GFRα2b inhibits neurite outgrowth induced by ligand stimulation of GFRα2a.
Signaling and biochemical activities of GFRα2 isoforms in the co-expression model
To further investigate the signaling and biochemical events underlying ligand activation of GFRα2b in the coexpression model, we first examined the stimulation of MAPK (ERK1/2). GDNF stimulated ERK1/2 phosphorylation in GFRα2a or GFRα2c, but not GFRα2b, transfectants (Fig. 2A). In the coexpression models, both GDNF and NTN induced rapid and transient phosphorylation of ERK1/2 (Fig. 7A).
Figure 7.

Ligand-regulated ERK1/2 signaling and expressions of immediate-early-response genes in Neuro2A coexpressing GFRα2b and the other GFRα2 isoforms. A, Western blot analyses of the activation of ERK1/2. Neuro2A cells stably coexpressing the isoforms GFRα2a and GFRα2b (GFRα2a + GFRα2b) or GFRα2c and GFRα2b (GFRα2c + GFRα2b) were treated with GDNF, NTN, or Sorbitol for the period of time indicated. Phospho-specific antibodies to ERK1/2 were used for detection, and the blots were reprobed with pan antibody serving as controls for protein loadings. B–E, Kinetic analyses of GDNF- or NTN-regulated expressions of early-response genes in the coexpression models. Expressions of fosB, egr-1, and egr-2 were measured with quantitative real-time PCR in cells stably coexpressing GFRα2a with GFRα2b (GFRα2a + GFRα2b) when stimulated with GDNF (B) or NTN (C); cells stably coexpressing GFRα2c with GFRα2b (GFRα2c + GFRα2b) were stimulated with GDNF (D) or NTN (E). Significant differences in expression of genes between ligand stimulated and control were calculated using the paired Student's t test. A value of p < 0.05 was considered significant (**p < 0.001).
Interestingly, when stimulated with either GDNF or NTN, no change in the expression of either egr-1 or egr-2 was observed (Fig. 7B–E). However, significant upregulation of the expression of fosB was observed in the coexpression of GFRα2b with GFRα2a (GFRα2a + GFRα2b) or with GFRα2c (GFRα2b + GFRα2c). This observation showed that the activation of coexpressed GFRα2b with either GFRα2a or GFRα2c results in the activation of fosB, an early-response gene, reminiscent of that observed in GFRα2b transfected alone (Fig. 3).
GFRα2b also inhibited retinoic acid-induced neurite outgrowth and activated RhoA
We next addressed the possibility that GFRα2b may affect neurite outgrowth induced by retinoic acid, a non-GFL stimulus. Using Neuro2A-expressing GFRα2b, retinoic acid treatment resulted in extensive neurite outgrowth. Both GDNF and NTN dramatically reduced the number of cells bearing neurite-like structures in retinoic acid-treated GFRα2b transfectant (Fig. 8).
Figure 8.

Ligand-activated GFRα2b antagonizes the neurite outgrowth induced by retinoic acid (RA). RA (5 μm) induced neurite outgrowth in GFRα2b-expressing Neuro2A cells. When treated together with GDNF or NTN (50 ng/ml), neurite outgrowth induced by RA was significantly attenuated. A, Phase-contrast images of Neuro2A cells stably expressing GFRα2b, treated with RA, GDNF, or NTN for 3 d. B, Graph of the percentage of cells bearing neurite with at least two times the length of the cell bodies and the effects of RA, GDNF, and NTN. Similar results were obtained from three independent clones. Significant differences in the percentage of cells bearing neurites between ligand stimulated and control were calculated using the paired Student's t test (*p < 0.002). Error bars indicate mean ± SD of triplicate measurements.
The Rho family of small GTPases and the associated regulators have been implicated in the modulation of neurite formation, axonal pathfinding, and dendritic arborization (Mackay et al., 1997; Van Aelst and Cline, 2004). Thus, it was of interest to examine the possibility that GFRα2b may activate the Rho family of GTPases. When stimulated with either GDNF or NTN, Neuro2a coexpressing GFRα2a and GFRα2b (GFRα2a + GFRα2b) or GFRα2c and GFRα2b (GFRα2c + GFRα2b) did not extend neurite-like structures (Fig. 5A). However, a significant number of these cells extended neurite-like structures in the presence of C3 transferase, suggesting the involvement of the Rho family of GTPases in the inhibitory effects of GFRα2b (Fig. 9A,B). Because Neuro2A cells have previously been shown to respond to LPA, resulting in the inhibition of neurite outgrowths through the RhoA-dependent mechanism (Sayas et al., 2002), it was not surprising that C3 transferase was found to inhibit LPA effects on retinoic acid-induced neurite outgrowth. At the concentration of C3 transferase used in this study, no significant cell death was observed (data not shown).
Figure 9.

Effects of RhoA and ROCK inhibitors in ligand-induced neurite outgrowth of GFRα2 isoform coexpression models and the ligand-induced activation of RhoA in GFRα2 isoform transfectants. A, B, Effects of RhoA inhibitor exoenzyme C3 transferase (1 μg/ml) and ROCK inhibitor Y27632 (10 μm) on ligand-induced neurite outgrowth in coexpression models of GFRα2a and GFRα2b (A) or GFRα2c and GFRα2b (B). LPA was used as a positive control in this study. LPA (10 μm) antagonizes neurite outgrowth induced by 5 μm retinoic acid (RA); such neurite inhibition of LPA was attenuated by C3 (1 μg/ml) and Y27632 (10 μm). The means ± SD were calculated from results obtained in triplicates. The effects of RhoA and ROCK inhibitors were compared with the effects of the inhibitors alone. With the concentrations of inhibitors used, no significant cell deaths were observed. Significant differences in the percentage of cells bearing neurites were calculated between ligand stimulated and control, using the paired Student's t test (*p ≤ 0.01). C, Analyses of RhoA activation in Neuro2A cells transfected with GFRα2 isoforms or pIRES control. After a 10 min pretreatment of LPA (10 μm), GDNF, or NTN (50 ng/ml), GTP-bound RhoA was pulled down from cell lysates using GST–Rhotekin and immunoblotted for RhoA. LPA served as a positive control for RhoA activation. Blotting of total RhoA in cell lysates showed similar loading of cell lysates.
To gain a better understanding of the mechanisms underlying the inhibitory effects of GFRα2b, we next examined the possible involvement of ROCK, which is known to be an effector of RhoA in the negative regulation of neurite outgrowth (Dickson, 2001; Sayas et al., 2002). Using the ROCK inhibitor Y27632, the inhibitory activity of LPA on retinoic acid-induced neurite outgrowth was significantly attenuated (Fig. 9A,B). However, the same concentration of Y27632 (10 μm) did not attenuate the inhibitory activity of GFRα2b (Fig. 9A,B). Higher concentrations of Y27632 (20 μm) resulted in significantly higher background neurite outgrowth and therefore complicated the interpretation of the study.
To investigate the possible involvement of RhoA in the inhibitory effects of GFRα2b, an attempt was made to pull down activated RhoA from cells lysates using glutathione S-transferase (GST)–Rhotekin and subsequently immunoblotted for RhoA. Similar to the effects of LPA, GFRα2b, when stimulated with either NTN or GDNF, was found to activate RhoA significantly (Fig. 9C). However, Neuro2A expressing GFRα2a, GFRα2c, or pIRES vector control did not activate RhoA significantly when stimulated with these ligands. This observation is consistent with the suggestion that RhoA and/or other Rho GTPases may be involved in the inhibition of neurite outgrowth mediated through GFRα2b.
The involvement of Rho in the activation of GFRα2b is not restricted to inhibiting GFRα1a-, GFRα2a-, or GFRα2c-induced neurite outgrowth but also to that induced by retinoic acid (Fig. 10A). Similar to the above observations, the inhibitory effects of GFRα2b on retinoic acid-induced neurite outgrowth appeared to be mediated through a Rho-dependent manner. Furthermore, the inhibition of ROCK may be sufficient to oppose the effects of LPA but not that of GFRα2b on retinoic acid-induced neurite outgrowth (Fig. 10B).
Figure 10.

Effects of RhoA and ROCK inhibitors on GFRα2b inhibition of retinoic acid (RA)-induced neurite outgrowth. A, RhoA inhibitor exoenzyme C3 transferase (1 μg/ml) inhibited the ligand-activated GFRα2b attenuation of neurite extension induced by RA (5 μm). The same concentration of exoenzyme C3 transferase also attenuated LPA (10 μm) inhibition of RA-induced neurite extension. B, Lack of effect of ROCK inhibitor Y27632 on the ligand-activated GFRα2b inhibition of RA induced neurite extension. The same concentration of Y27632 (10 μm) significantly attenuated the neurite outgrowth inhibition induced by LPA. The means ± SD were calculated from results obtained in triplicates. The effects of RhoA and ROCK inhibitors were compared with the effects of the inhibitors alone. With the concentrations of inhibitors used, no significant cell deaths were observed. Significant differences in the percentage of cells bearing neurites were calculated between ligand stimulated and control, using the paired Student's t test (*p ≤ 0.01).
Discussion
This study demonstrates a novel function of GFRα2b, an alternatively spliced isoform of GFRα2. When activated by ligands (GDNF or NTN), GFRα2b inhibited neurite outgrowth induced by GFRα1a, GFRα2a, and GFRα2c isoforms. Furthermore, GFRα2b was found to inhibit a non-GFRα stimulus, retinoic acid-induced neurite outgrowth, and to activate RhoA.
Alternative splicing is prevalent in many mammalian genomes and is a means of producing functionally diverse polypeptides from a single gene (Blencowe, 2006). Recently, genome-wide microarray and large-scale computational analyses of expressed-sequence tag and cDNA sequences have estimated that >50% of human multi-exon genes are alternatively spliced (Modrek and Lee, 2002). Comparative genomic analyses also demonstrated that the greatest amount of conserved alternative splicing occurs in the CNS (Kan et al., 2005). In many systems, alternative splicing events have been shown to produce isoforms with distinct activities and biochemical properties, as a means for diverse biological functions (Lee and Irizarry, 2003).
In the cortex of human, mouse, and rat brain, the expression of GFRα2 mRNA has been reported (Sanicola et al., 1997; Widenfalk et al., 1997; Golden et al., 1998, 1999; Trupp et al., 1998). However, the probes used in these studies cannot distinguish the expressions of the isoforms. In the present study, we were able to specifically amplify all three isoforms in the human brain regions using exon-overlapping primers (Too, 2003). In the human brain, all three GFRα2 isoforms are expressed at comparable levels, with GFRα2c significantly higher than the other two isoforms. Compared with the other regions of the human brain, the cortex expressed the highest levels of the isoforms. The functional significance of these isoforms in the cortex has yet to be defined. Interestingly, the high expressions of the GFRα2 isoforms in the cortex, a region of the brain involved in learning complex tasks, and the observation that GFRα2 knock-out mice show significant impairment in several memory tasks (Voikar et al., 2004) may suggest a possible role of GFRα2 signaling in the development and/or maintenance of cognitive abilities that help in solving complex learning tasks.
GDNF and NTN are known to similarly activate a number of signaling pathways, including ERK, phosphatidylinositol 3-kinase/AKT, p38 MAPK, and JNK (Trupp et al., 1999; Takahashi, 2001; Pezeshki et al., 2003; Ichihara et al., 2004), and regulate the expressions of various immediate-early-response genes (Fukuda et al., 2003; Pezeshki et al., 2003). In this study, it is intriguing to note that the activation of specific signaling pathways but not the early-response genes is dependent on the ligands used. For instance, GDNF was found to potently activate ERK1/2 through GFRα2a and GFRα2c in a dose- and time-dependent manner but did not activate GFRα2b significantly. This was not attributable to the failure of GDNF to interact with GFRα2b because GDNF displaced bound [125I]GDNF equally well with all three isoform transfectants. Similarly, GDNF activated AKT through GFRα2b and GFRα2c but not through GFRα2a. However, NTN showed similar activations of ERK1/2 and AKT through all of the three isoforms. GDNF at lower concentrations appeared to be slightly more potent than NTN in the activation of ERK1/2 but not at higher concentrations in GFRα2a and GFRα2c transfectants. The significance of this, however, is unclear presently.
Both GDNF and NTN have previously been shown to have similar properties in activating the multicomponent receptor complex (Baloh et al., 1997; Airaksinen et al., 1999; Wang et al., 2000; Scott and Ibanez, 2001; Coulpier et al., 2002; Charlet-Berguerand et al., 2004). In addition, midbrain dopaminergic neurons that only express GFRα1 appear to survive equally well with both GDNF and NTN in vitro and in vivo (Horger et al., 1998). However, there are observations of distinct functional differences with the use of specific ligands. Although GDNF and NTN promote the survival of dopaminergic neurons through GFRα1 (Cacalano et al., 1998; Akerud et al., 1999), only GDNF possess neuritogenic and hypertrophic effects (Akerud et al., 1999). In cultured sympathetic neurons, GDNF was able to promote the survival of culture sympathetic neurons through GFRα2, but NTN could not promote survival through GFRα1 (Buj-Bello et al., 1997). Furthermore, GDNF but not NTN could promote the axonal growth of DRG neurons through GFRα1 (Paveliev et al., 2004). Consistent with these studies, recent observations show differential ligand signaling through the activation of GFRα1 (Lee et al., 2006) and distinct activation of microRNAs by specific ligands through the GFRα2 receptor complexes (Yoong et al., 2006), supporting the emerging view that cross talk of exogenously applied GDNF and NTN with a specific receptor may, in some instances, result in distinct functions.
It is well documented that GDNF and NTN are potent trophic factors that have potent effects on neuronal differentiation and promote survival and sprouting of ventral mesencephalic dopaminergic neurons in primary cultures and other neuronal cultures (Lin et al., 1993; Akerud et al., 1999; Baloh et al., 2000; Yan et al., 2003; Wanigasekara and Keast, 2005; Zihlmann et al., 2005). The finding in this study of a particular alternatively spliced variant of GFRα2 inhibiting neurite outgrowth was unexpected. Unlike GFRα2a and GFRα2c, GFRα2b transfectants did not induce neurite outgrowth when activated by either GDNF or NTN. Both GFRα2a and GFRα2c (but not GFRα2b) activated the early-response gene egr1 (also known as NGFI-A, krox-24, zif-268, and TIS-8), consistent with a role of egr1 in neuronal differentiation (Pignatelli et al., 1999; Knapska and Kaczmarek, 2004). In coexpression studies, GFRα2b was found to inhibit ligand-induced neurite outgrowth by GFRα2a and GFRα2c. Similarly, in BE(2)-C cells endogenously expressing GFRα2b isoform, both GDNF and NTN did not significantly alter the morphology of the cells. However, the silencing of GFRα2b and subsequent treatment with either GDNF or NTN caused the cells to extend neurite-like structures. Interestingly, in coexpression studies, fosB was upregulated and paralleled the upregulation of the immediate-response gene observed in GFRα2b transfectants by GDNF or NTN. It is not known whether the coexpression of GFRα2b with GFRα2a or GFRα2c may have affected the protein expression levels of the latter two spliced variants resulting in the attenuation of neurite extension on ligand stimulation. The possibility of GFRα2b affecting the expression of the other spliced variants and the effects of expressions levels is currently investigated.
The inhibition of neurite outgrowth by GFRα2b is not restricted to the GFRα2 family of isoforms. Ligand-activated GFRα2b also inhibited the neurite outgrowth induced by GFRα1a, another member of GFR. Intriguingly, the activation of GFRα2b inhibited neurite outgrowth induced by retinoic acid. The underlying GFRα2b inhibitory mechanism appears to involve the Rho family of GTPases. RhoA is a member of the Rho GTPase family, which includes RhoA, Rac, and Cdc42 (Luo, 2000; Van Aelst and Cline, 2004). Although Rac and Cdc42 have been shown to be involved in promoting neurite and axonal outgrowth, RhoA has been the focus in studies of molecular mechanisms for some glia-derived neurite outgrowth inhibitory factors such as Nogo-A, myelin-associated glycoprotein (Niederost et al., 2002), and LPA (Sayas et al., 2002). More recent findings have revealed that RhoA mediates neurite outgrowth inhibition by reorganization of actin and the microtubular network (Dickson, 2001; Leung et al., 2002). Consistent with these findings is that GDNF and NTN increased the active form of RhoA in GFRα2b but not GFRα2a or GFRα2c transfectants. Furthermore, the Clostridium botulinum C3 exoenzyme specifically ADP-ribosylates and inactivates Rho, thereby increasing neurite outgrowths in GFRα2a/GFRα2b and GFRα2c/GFRα2b coexpression models. It is interesting to note that GDNF induced RET-mediated phosphorylation of focal adhesion kinase, paxillin, and p130C through the activation of the Rho family of GTPase and inhibited the outgrowth of neurites in TGW-I-nu cells (Murakami et al., 1999). It is, however, unclear whether this observation is mediated through GFRα2b.
Compared with GFRα2a, both GFRα2b and GFRα2c showed deletion of eight cysteine residues and N-glycosylation sites at the N terminus (Wong and Too, 1998). GFRα2 is thought to be structurally organized into three distinct domains. The N-terminal domain has previously been shown to be dispensable for ligand binding specificity and RET phosphorylation of GFRα receptors (Scott and Ibanez, 2001). Extending this observation, the N-terminal domain encoding the unique sequences of GFRα2a, GFRα2b, and GFRα2c may serve to regulate distinct biochemical and cellular processes. It is tempting to speculate that the expression and interactions of specific GFRα2 receptor spliced isoforms may play an important role in neuronal differentiation involving GDNF and NTN. The recent observation in which the expressions of GFRα2 isoforms are differentially regulated in Nurr1-induced dorpaminergic differentiation of embryonic stem cells is consistent with this suggestion (Sonntag et al., 2004).
In summary, this study provides the first evidence that GDNF and NTN have distinct neuritogenic effects mediated through specific GFRα2 isoforms. GFRα2b inhibited GFRα1 and GFRα2, and retinoic acid mediated neuritogenesis through the Rho family of GTPases. The emerging view is that the combinatorial interactions of the spliced isoforms of GFRα2, RET, and NCAM may contribute to the complexity of a multicomponent signaling system and may produce the myriad of observed biological responses.
Footnotes
This work was partially supported by a grant from the Singapore–Massachusetts Institute of Technology Alliance. We thank Wan Guoqiang for his efforts in the development of the real-time PCR assays.
References
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–394. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- Airaksinen MS, Titievsky A, Saarma M. GDNF family neurotrophic factor signaling: four masters, one servant? Mol Cell Neurosci. 1999;13:313–325. doi: 10.1006/mcne.1999.0754. [DOI] [PubMed] [Google Scholar]
- Akerud P, Alberch J, Eketjall S, Wagner J, Arenas E. Differential effects of glial cell line-derived neurotrophic factor and neurturin on developing and adult substantia nigra dopaminergic neurons. J Neurochem. 1999;73:70–78. doi: 10.1046/j.1471-4159.1999.0730070.x. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Tansey MG, Golden JP, Creedon DJ, Heuckeroth RO, Keck CL, Zimonjic DB, Popescu NC, Johnson EM, Jr, Milbrandt J. TrnR2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron. 1997;18:793–802. doi: 10.1016/s0896-6273(00)80318-9. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM, Jr, Milbrandt J. The GDNF family ligands and receptors—implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Buj-Bello A, Adu J, Pinon LG, Horton A, Thompson J, Rosenthal A, Chinchetru M, Buchman VL, Davies AM. Neurturin responsiveness requires a GPI-linked receptor and the Ret receptor tyrosine kinase. Nature. 1997;387:721–724. doi: 10.1038/42729. [DOI] [PubMed] [Google Scholar]
- Buttner B, Reutter W, Horstkorte R. Cytoplasmic domain of NCAM 180 reduces NCAM-mediated neurite outgrowth. J Neurosci Res. 2004;75:854–860. doi: 10.1002/jnr.20049. [DOI] [PubMed] [Google Scholar]
- Cacalano G, Farinas I, Wang LC, Hagler K, Forgie A, Moore M, Armanini M, Phillips H, Ryan AM, Reichardt LF, Hynes M, Davies A, Rosenthal A. GFRalpha1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron. 1998;21:53–62. doi: 10.1016/s0896-6273(00)80514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet-Berguerand N, Le Hir H, Incoronato M, di Porzio U, Yu Y, Jing S, de Franciscis V, Thermes C. Expression of GFRalpha1 receptor splicing variants with different biochemical properties is modulated during kidney development. Cell Signal. 2004;16:1425–1434. doi: 10.1016/j.cellsig.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Cik M, Masure S, Lesage AS, Van Der Linden I, Van Gompel P, Pangalos MN, Gordon RD, Leysen JE. Binding of GDNF and neurturin to human GDNF family receptor alpha 1 and 2. Influence of cRET and cooperative interactions. J Biol Chem. 2000;275:27505–27512. doi: 10.1074/jbc.M000306200. [DOI] [PubMed] [Google Scholar]
- Coulpier M, Anders J, Ibanez CF. Coordinated activation of autophosphorylation sites in the RET receptor tyrosine kinase: importance of tyrosine 1062 for GDNF mediated neuronal differentiation and survival. J Biol Chem. 2002;277:1991–1999. doi: 10.1074/jbc.M107992200. [DOI] [PubMed] [Google Scholar]
- de Graaff E, Srinivas S, Kilkenny C, D'Agati V, Mankoo BS, Costantini F, Pachnis V. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 2001;15:2433–2444. doi: 10.1101/gad.205001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey BK, Wong YW, Too HP. Cloning of a novel murine isoform of the glial cell line-derived neurotrophic factor receptor. NeuroReport. 1998;9:37–42. doi: 10.1097/00001756-199801050-00008. [DOI] [PubMed] [Google Scholar]
- Dickson BJ. Rho GTPases in growth cone guidance. Curr Opin Neurobiol. 2001;11:103–110. doi: 10.1016/s0959-4388(00)00180-x. [DOI] [PubMed] [Google Scholar]
- Dolatshad NF, Silva AT, Saffrey MJ. Identification of GFR alpha-2 isoforms in myenteric plexus of postnatal and adult rat intestine. Brain Res Mol Brain Res. 2002;107:32–38. doi: 10.1016/s0169-328x(02)00446-1. [DOI] [PubMed] [Google Scholar]
- Fukuda N, Ichihara M, Morinaga T, Kawai K, Hayashi H, Murakumo Y, Matsuo S, Takahashi M. Identification of a novel glial cell line-derived neurotrophic factor-inducible gene required for renal branching morphogenesis. J Biol Chem. 2003;278:50386–50392. doi: 10.1074/jbc.M309629200. [DOI] [PubMed] [Google Scholar]
- Golden JP, Baloh RH, Kotzbauer PT, Lampe PA, Osborne PA, Milbrandt J, Johnson EM., Jr Expression of neurturin, GDNF, and their receptors in the adult mouse CNS. J Comp Neurol. 1998;398:139–150. doi: 10.1002/(sici)1096-9861(19980817)398:1<139::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Golden JP, DeMaro JA, Osborne PA, Milbrandt J, Johnson EM., Jr Expression of neurturin, GDNF, and GDNF family-receptor mRNA in the developing and mature mouse. Exp Neurol. 1999;158:504–528. doi: 10.1006/exnr.1999.7127. [DOI] [PubMed] [Google Scholar]
- Hansford LM, Marshall GM. Glial cell line-derived neurotrophic factor (GDNF) family ligands reduce the sensitivity of neuroblastoma cells to pharmacologically induced cell death, growth arrest and differentiation. Neurosci Lett. 2005;389:77–82. doi: 10.1016/j.neulet.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Horger BA, Nishimura MC, Armanini MP, Wang LC, Poulsen KT, Rosenblad C, Kirik D, Moffat B, Simmons L, Johnson E, Jr, Milbrandt J, Rosenthal A, Bjorklund A, Vandlen RA, Hynes MA, Phillips HS. Neurturin exerts potent actions on survival and function of midbrain dopaminergic neurons. J Neurosci. 1998;18:4929–4937. doi: 10.1523/JNEUROSCI.18-13-04929.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett. 2004;204:197–211. doi: 10.1016/S0304-3835(03)00456-7. [DOI] [PubMed] [Google Scholar]
- Jing S, Yu Y, Fang M, Hu Z, Holst PL, Boone T, Delaney J, Schultz H, Zhou R, Fox GM. GFRalpha-2 and GFRalpha-3 are two new receptors for ligands of the GDNF family. J Biol Chem. 1997;272:33111–33117. doi: 10.1074/jbc.272.52.33111. [DOI] [PubMed] [Google Scholar]
- Kan Z, Garrett-Engele PW, Johnson JM, Castle JC. Evolutionarily conserved and diverged alternative splicing events show different expression and functional profiles. Nucleic Acids Res. 2005;33:5659–5666. doi: 10.1093/nar/gki834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapska E, Kaczmarek L. A gene for neuronal plasticity in the mammalian brain: Zif268/Egr-1/NGFI-A/Krox-24/TIS8/ZENK? Prog Neurobiol. 2004;74:183–211. doi: 10.1016/j.pneurobio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Kobori N, Waymire JC, Haycock JW, Clifton GL, Dash PK. Enhancement of tyrosine hydroxylase phosphorylation and activity by glial cell line-derived neurotrophic factor. J Biol Chem. 2004;279:2182–2191. doi: 10.1074/jbc.M310734200. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EM, Jr, Milbrandt J. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- Kozlowski DA, Connor B, Tillerson JL, Schallert T, Bohn MC. Delivery of a GDNF gene into the substantia nigra after a progressive 6-OHDA lesion maintains functional nigrostriatal connections. Exp Neurol. 2000;166:1–15. doi: 10.1006/exnr.2000.7463. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Irizarry K. Alternative splicing in the nervous system: an emerging source of diversity and regulation. Biol Psychiatry. 2003;54:771–776. doi: 10.1016/s0006-3223(03)00375-5. [DOI] [PubMed] [Google Scholar]
- Lee DC, Chan KW, Chan SY. RET receptor tyrosine kinase isoforms in kidney function and disease. Oncogene. 2002;21:5582–5592. doi: 10.1038/sj.onc.1205741. [DOI] [PubMed] [Google Scholar]
- Lee RH, Wong WL, Chan CH, Chan SY. Differential effects of glial cell line-derived neurotrophic factor and neurturin in RET/GFRalpha1-expressing cells. J Neurosci Res. 2006;83:80–90. doi: 10.1002/jnr.20701. [DOI] [PubMed] [Google Scholar]
- Leung T, Ng Y, Cheong A, Ng CH, Tan I, Hall C, Lim L. p80 ROKalpha binding protein is a novel splice variant of CRMP-1 which associates with CRMP-2 and modulates RhoA-induced neuronal morphology. FEBS Lett. 2002;532:445–449. doi: 10.1016/s0014-5793(02)03736-5. [DOI] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Lindahl M, Timmusk T, Rossi J, Saarma M, Airaksinen MS. Expression and alternative splicing of mouse Gfra4 suggest roles in endocrine cell development. Mol Cell Neurosci. 2000;15:522–533. doi: 10.1006/mcne.2000.0845. [DOI] [PubMed] [Google Scholar]
- Lindahl M, Poteryaev D, Yu L, Arumae U, Timmusk T, Bongarzone I, Aiello A, Pierotti MA, Airaksinen MS, Saarma M. Human glial cell line-derived neurotrophic factor receptor alpha 4 is the receptor for persephin and is predominantly expressed in normal and malignant thyroid medullary cells. J Biol Chem. 2001;276:9344–9351. doi: 10.1074/jbc.M008279200. [DOI] [PubMed] [Google Scholar]
- Lorenzo MJ, Gish GD, Houghton C, Stonehouse TJ, Pawson T, Ponder BA, Smith DP. RET alternate splicing influences the interaction of activated RET with the SH2 and PTB domains of Shc, and the SH2 domain of Grb2. Oncogene. 1997;14:763–771. doi: 10.1038/sj.onc.1200894. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Esch F, Furthmayr H, Hall A. Rho- and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: an essential role for ezrin/radixin/moesin proteins. J Cell Biol. 1997;138:927–938. doi: 10.1083/jcb.138.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masure S, Cik M, Hoefnagel E, Nosrat CA, Van der Linden I, Scott R, Van Gompel P, Lesage AS, Verhasselt P, Ibanez CF, Gordon RD. Mammalian GFRalpha-4, a divergent member of the GFRalpha family of coreceptors for glial cell line-derived neurotrophic factor family ligands, is a receptor for the neurotrophic factor persephin. J Biol Chem. 2000;275:39427–39434. doi: 10.1074/jbc.M003867200. [DOI] [PubMed] [Google Scholar]
- Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- Murakami H, Iwashita T, Asai N, Iwata Y, Narumiya S, Takahashi M. Rho-dependent and -independent tyrosine phosphorylation of focal adhesion kinase, paxillin and p130Cas mediated by Ret kinase. Oncogene. 1999;18:1975–1982. doi: 10.1038/sj.onc.1202514. [DOI] [PubMed] [Google Scholar]
- Myers KA, He Y, Hasaka TP, Baas PW. Microtubule transport in the axon: Re-thinking a potential role for the actin cytoskeleton. Neuroscientist. 2006;12:107–118. doi: 10.1177/1073858405283428. [DOI] [PubMed] [Google Scholar]
- Niederost B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE. Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J Neurosci. 2002;22:10368–10376. doi: 10.1523/JNEUROSCI.22-23-10368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibanez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/s0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Paveliev M, Airaksinen MS, Saarma M. GDNF family ligands activate multiple events during axonal growth in mature sensory neurons. Mol Cell Neurosci. 2004;25:453–459. doi: 10.1016/j.mcn.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Pezeshki G, Franke B, Engele J. GDNF elicits distinct immediate-early gene responses in cultured cortical and mesencephalic neurons. J Neurosci Res. 2003;71:478–484. doi: 10.1002/jnr.10513. [DOI] [PubMed] [Google Scholar]
- Pignatelli M, Cortes-Canteli M, Santos A, Perez-Castillo A. Involvement of the NGFI-A gene in the differentiation of neuroblastoma cells. FEBS Lett. 1999;461:37–42. doi: 10.1016/s0014-5793(99)01420-9. [DOI] [PubMed] [Google Scholar]
- Povlsen GK, Ditlevsen DK, Berezin V, Bock E. Intracellular signaling by the neural cell adhesion molecule. Neurochem Res. 2003;28:127–141. doi: 10.1023/a:1021660531484. [DOI] [PubMed] [Google Scholar]
- Saarma M, Sariola H. Other neurotrophic factors: glial cell line-derived neurotrophic factor (GDNF) Microsc Res Tech. 1999;45:292–302. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<292::AID-JEMT13>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Sanicola M, Hession C, Worley D, Carmillo P, Ehrenfels C, Walus L, Robinson S, Jaworski G, Wei H, Tizard R, Whitty A, Pepinsky RB, Cate RL. Glial cell line-derived neurotrophic factor-dependent RET activation can be mediated by two different cell-surface accessory proteins. Proc Natl Acad Sci USA. 1997;94:6238–6243. doi: 10.1073/pnas.94.12.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayas CL, Avila J, Wandosell F. Regulation of neuronal cytoskeleton by lysophosphatidic acid: role of GSK-3. Biochim Biophys Acta. 2002;1582:144–153. doi: 10.1016/s1388-1981(02)00149-x. [DOI] [PubMed] [Google Scholar]
- Scott RP, Ibanez CF. Determinants of ligand binding specificity in the glial cell line-derived neurotrophic factor family receptor alpha S. J Biol Chem. 2001;276:1450–1458. doi: 10.1074/jbc.M006157200. [DOI] [PubMed] [Google Scholar]
- Shefelbine SE, Khorana S, Schultz PN, Huang E, Thobe N, Hu ZJ, Fox GM, Jing S, Cote GJ, Gagel RF. Mutational analysis of the GDNF/RET-GDNFR alpha signaling complex in a kindred with vesicoureteral reflux. Hum Genet. 1998;102:474–478. doi: 10.1007/s004390050724. [DOI] [PubMed] [Google Scholar]
- Sonntag KC, Simantov R, Kim KS, Isacson O. Temporally induced Nurr1 can induce a non-neuronal dopaminergic cell type in embryonic stem cell differentiation. Eur J Neurosci. 2004;19:1141–1152. doi: 10.1111/j.1460-9568.2004.03204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12:361–373. doi: 10.1016/s1359-6101(01)00012-0. [DOI] [PubMed] [Google Scholar]
- Too HP. Real time PCR quantification of GFRalpha-2 alternatively spliced isoforms in murine brain and peripheral tissues. Brain Res Mol Brain Res. 2003;114:146–153. doi: 10.1016/s0169-328x(03)00169-4. [DOI] [PubMed] [Google Scholar]
- Too HP, Maggio JE. Simultaneous extraction of total RNA and peptides from tissues: application to tachykinins. Peptides. 1995;16:45–53. doi: 10.1016/0196-9781(94)00151-u. [DOI] [PubMed] [Google Scholar]
- Trupp M, Raynoschek C, Belluardo N, Ibanez CF. Multiple GPI-anchored receptors control GDNF-dependent and independent activation of the c-Ret receptor tyrosine kinase. Mol Cell Neurosci. 1998;11:47–63. doi: 10.1006/mcne.1998.0667. [DOI] [PubMed] [Google Scholar]
- Trupp M, Scott R, Whittemore SR, Ibanez CF. Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J Biol Chem. 1999;274:20885–20894. doi: 10.1074/jbc.274.30.20885. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Cline HT. Rho GTPases and activity-dependent dendrite development. Curr Opin Neurobiol. 2004;14:297–304. doi: 10.1016/j.conb.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Voikar V, Rossi J, Rauvala H, Airaksinen MS. Impaired behavioural flexibility and memory in mice lacking GDNF family receptor alpha2. Eur J Neurosci. 2004;20:308–312. doi: 10.1111/j.1460-9568.2004.03475.x. [DOI] [PubMed] [Google Scholar]
- Wang LC, Shih A, Hongo J, Devaux B, Hynes M. Broad specificity of GDNF family receptors GFRalpha1 and GFRalpha2 for GDNF and NTN in neurons and transfected cells. J Neurosci Res. 2000;61:1–9. doi: 10.1002/1097-4547(20000701)61:1<1::AID-JNR1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Wanigasekara Y, Keast JR. Neurturin has multiple neurotrophic effects on adult rat sacral parasympathetic ganglion neurons. Eur J Neurosci. 2005;22:595–604. doi: 10.1111/j.1460-9568.2005.04260.x. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK. Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci. 2002;5:751–759. doi: 10.1038/nn883. [DOI] [PubMed] [Google Scholar]
- Widenfalk J, Nosrat C, Tomac A, Westphal H, Hoffer B, Olson L. Neurturin and glial cell line-derived neurotrophic factor receptor-β (GDNFR-β), novel proteins related to GDNF and GDNFR-α with specific cellular patterns of expression suggesting roles in the developing and adult nervous system and in peripheral organs. J Neurosci. 1997;17:8506–8519. doi: 10.1523/JNEUROSCI.17-21-08506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YW, Too HP. Identification of mammalian GFRalpha-2 splice isoforms. NeuroReport. 1998;9:3767–3773. doi: 10.1097/00001756-199812010-00002. [DOI] [PubMed] [Google Scholar]
- Yajima S, Lammers CH, Lee SH, Hara Y, Mizuno K, Mouradian MM. Cloning and characterization of murine glial cell-derived neurotrophic factor inducible transcription factor (MGIF) J Neurosci. 1997;17:8657–8666. doi: 10.1523/JNEUROSCI.17-22-08657.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Newgreen DF, Young HM. Developmental changes in neurite outgrowth responses of dorsal root and sympathetic ganglia to GDNF, neurturin, and artemin. Dev Dyn. 2003;227:395–401. doi: 10.1002/dvdy.10294. [DOI] [PubMed] [Google Scholar]
- Yoong LF, Peng ZN, Wan G, Too HP. Tissue expression of alternatively spliced GFRalpha1, NCAM and RET isoforms and the distinct functional consequence of ligand-induced activation of GFRalpha1 isoforms. Brain Res Mol Brain Res. 2005;139:1–12. doi: 10.1016/j.molbrainres.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Yoong LF, Wan G, Too HP. Glial cell-line derived neurotrophic factor and neurturin regulate the expressions of distinct miRNA precursors through the activation of GFRa2. J Neurochem. 2006;98:1149–1158. doi: 10.1111/j.1471-4159.2006.03959.x. [DOI] [PubMed] [Google Scholar]
- Zihlmann KB, Ducray AD, Schaller B, Huber AW, Krebs SH, Andres RH, Seiler RW, Meyer M, Widmer HR. The GDNF family members neurturin, artemin and persephin promote the morphological differentiation of cultured ventral mesencephalic dopaminergic neurons. Brain Res Bull. 2005;68:42–53. doi: 10.1016/j.brainresbull.2004.10.012. [DOI] [PubMed] [Google Scholar]