

Abstract
Basic fibroblast growth factor (FGF-2) is involved in the development and maintenance of the nervous system. Exogenous administration of FGF-2 increased dopaminergic (DA) graft survival in different animal models of Parkinson's disease. To study the physiological function of the endogenous FGF-2 system, we analyzed the nigrostriatal system of mice lacking FGF-2, mice overexpressing FGF-2, and FGF-receptor-3 (FGFR3)-deficient mice both after development and after 6-hydroxydopamine lesion. FGFR3-deficient mice (+/−) displayed a reduced number of DA neurons compared with the respective wild type. Whereas absence of FGF-2 led to significantly increased numbers of DA neurons, enhanced amount of the growth factor in mice overexpressing FGF-2 resulted in less tyrosine hydroxylase expression and a reduced DA cell density. The volumes of the substantia nigra were enlarged in both FGF-2−/− and in FGF-2 transgenic mice, suggesting an important role of FGF-2 for the establishment of the proper number of DA neurons and a normal sized substantia nigra during development. In a second set of experiments, the putative relevance of endogenous FGF-2 after neurotoxin application was investigated regarding the number of rescued DA neurons after partial 6-OHDA lesion. Interestingly, the results after lesion were directly opposed to the results after development: significantly less DA neurons survived in FGF-2−/− mice compared with wild-type mice. Together, the results indicate that FGFR3 is crucially involved in regulating the number of DA neurons. The lack of FGF-2 seems to be (over)compensated during development, but, after lesion, compensation mechanisms fail. The transgenic mice showed that endogenous FGF-2 protects DA neurons from 6-OHDA neurotoxicity.
Keywords: basic fibroblast growth factor, midbrain, mutant mice, Parkinson's disease, stereology, substantia nigra, terminal 6-OHDA lesion
Introduction
Parkinson's disease (PD) is the most common neurodegenerative disease affecting the nigrostriatal system. The disorder is characterized by the progressive degeneration of dopaminergic (DA) neurons in the pars compacta of the substantia nigra (SNpc), resulting in severe motor dysfunctions. The etiology of PD is associated with genetic defects (e.g., α-synuclein, parkin) in only ∼10% of the cases (Feany and Bender, 2000; Shimura et al., 2000; Giasson and Lee, 2001), whereas in the majority of patients its pathogenesis is unknown (Goldman and Tanner, 1998; Lotharius and Brundin, 2002). All clinical approaches for the therapy of PD, including drug treatment, deep brain stimulation, and striatal cell transplantation, are symptomatic and do not prevent the progression of the disease. To develop new causal therapeutic strategies and/or to prevent the progression of the disease, one has to identify the molecular mechanisms that regulate the generation, survival, and differentiation of nigral DA neurons, both during development and after lesion.
Basic fibroblast growth factor (FGF-2) is involved in the development, maintenance, and survival of the nervous system and, as one of the most potent survival factors, exerts neurotrophic activity on DA neurons in vitro and in vivo. In vitro, exogenous administration of FGF-2 or cocultivation with FGF-2-producing Schwann cells stimulated survival and neurite formation of DA neuronal cultures and protected them from 6-hydroxydopamine (6-OHDA)-induced neurotoxicity (Ferrari et al., 1989; Knusel et al., 1990; Engele and Bohn, 1991; Beck et al., 1993; Mayer et al., 1993a; Bouvier and Mytilineou, 1995; Grothe et al., 2000). With regard to the in vivo situation, FGF-2 has been efficiently used in different treatment protocols in mouse and rat PD models. Intrastriatal infusions of FGF-2 induced recovery of striatal DA fibers and DA content in the mouse 1-methyl-1,2,3,6-tetrahydopyridine (MPTP)-model and increased the protection of the neurotoxicity-induced lesioned nigrostriatal DA system (Date et al., 1993). In addition, survival of grafted DA neurons was also enhanced after repeated intracerebral infusions or pretreatment of transplanted cells with FGF-2 (Mayer et al., 1993b). Furthermore, cografting of FGF-2-producing fibroblasts (Takayama et al., 1995) or Schwann cells (Timmer et al., 2004) with DA neurons mediated growth-promoting effects on the transplants and improved the behavioral deficits of parkinsonian rats.
Interestingly, in the SNpc of control brains, ∼82% of the melanin containing nigral cells are FGF-2 positive, whereas in the SNpc of patients suffering from PD only, ∼13% of the remaining DA neurons are positive for FGF-2, suggesting a profound depletion of FGF-2 in the remaining DA neurons, and this depletion may be related to the disease process (Tooyama et al., 1993, 1994).
The FGF-2 signal is mediated via four high-affinity tyrosine kinase receptors (R) that are crucial for development and regeneration of the CNS and PNS. In the dopaminergic nigrostriatal system, FGF-2 and three of its respective receptors (FGFR1–FGFR3) are endogenously expressed (Claus et al., 2004; Timmer et al., 2004).
The physiological roles of FGF-2 and its receptors can be investigated because mutant mice (1) lacking FGF-2 (Dono et al., 1998; Zhou et al., 1998), (2) overexpressing FGF-2 (Coffin et al., 1995), and (3) with a reduced level of FGFR3 (Colvin et al., 1996) have been generated.
Using these mice, it has been shown previously that different parts of the CNS and PNS are affected. FGF-2 null mutants are viable and the phenotype looked quite normal (Dono et al., 1998; Zhou et al., 1998), but more detailed evaluation revealed that FGF-2−/− displayed cerebral neocortex defects at birth (Dono et al., 1998).
In addition, we have shown that targeted disruption of the FGF-2 gene affects the response to peripheral nerve injury, whereas the PNS development showed no defects (Jungnickel et al., 2004b). In mice overexpressing FGF-2, we found faster nerve regeneration after sciatic nerve injury (Jungnickel et al., 2006) and a regulation of injury-related effects in the peripheral nervous system via FGFR3 signaling (Jungnickel et al., 2004a). Moreover, FGF-2 and FGFR3 play an important role in the regulation of neuronal death after peripheral nerve injury (Jungnickel et al., 2005; for review, see Grothe et al., 2006).
In the present study, we quantified the nigrostriatal system of the above mentioned mice strains after both development and lesion to analyze the physiological role of FGF-2 in the CNS. Our results show an increase of the DA neuron number in FGF-2−/− mice after development but an increased cell death after lesion, suggesting different functions or, more probably, failing compensation mechanisms within the adult nigrostriatal system. Moreover, mice overexpressing FGF-2 protected DA neurons from 6-OHDA toxicity. Reduced DA neuron numbers in mice deleted for FGFR3 (genotype +/−) during development indicate a major role of released FGF-2 compared with intracellular one. These alterations remain during adulthood.
Materials and Methods
Mutant mice
All experimental protocols were done in accordance with the German law for the protection of animals with a permit by the local authority (Bezirksregierung Hannover; guidelines of the Tierschutzgesetz i.d.F.v. December 25, 1998). Three mutant mice of the FGF-2 system were used for the present study, including (1) receptor-deficient mice (FGFR3+/−), (2) knock-out (ko) mice (FGF-2−/−), and (3) transgene mice (overexpressing FGF-2; TgFGF-2+/−) (Table 1):
Table 1.
Mouse strains analyzed in the present study
| Mouse strain | Abbreviation | Genotype | Background |
|---|---|---|---|
| FGFR3 koa | FGFR3 = FGFR3+/− | Heterozygous | C57BL/6J |
| FGFR3 ko (wild type)a | FGFR3 wt = FGFR3+/+ | Homozygous | C57BL/6J |
| FGF-2 kob | FGF-2 ko = FGF-2−/− | Homozygous | 129P2/OlaHsd:Black Swiss |
| FGF-2 ko (wild type)b | FGF-2 ko-wt = FGF-2+/+ | Homozygous | 129P2/OlaHsd:Black Swiss |
| Transgenic FGF-2c | TgFGF-2 = TgFGF-2+/− | Heterozygous | FVB/N |
| Transgenic FGF-2 (wild type)c | TgFGF-2 wt | Homozygous | FVB/N |
aFor additional details, see Colvin et al., 1996.
bFor additional details, see Dono et al., 1998, Zhou et al., 1998.
cFor additional details, see Coffin et al., 1995.
FGFR3-deficient mice.
FGFR3 heterozygous mice (Colvin et al., 1996) (background in this study, C57BL/6) were obtained from heterozygous crosses of breeders received from Dr. A. Vortkamp (Max-Planck-Institute of Molecular Genetics, Berlin, Germany). The original colony created in the laboratory of Dr. D. Ornitz (Washington University, St. Louis, MO) was founded in a mixed C57BL/6 × 129 background and maintained via successive C57BL/6 backcrosses. In the present study, FGFR3 heterozygotes were used, because the viability of FGFR3-deleted homozygous animals is low. The heterozygous mice exhibit nearly the same characteristic phenotype as FGFR3 null mice, including kyphotic and scoliotic deformations of the vertebral column at different extent (Colvin et al., 1996; Oh et al., 2003; Jungnickel et al., 2004a). FGFR3 is expressed before embryonic day 16.5 (E16.5). At this stage, the first phenotypical alterations were found in FGFR3+/− mice (Colvin et al., 1996). Genotypes were determined by PCR of the tail DNA.
FGF-2-deleted mice.
FGF-2 ko and wild-type (wt) mice (Zhou et al., 1998) were obtained from The Jackson Laboratory (Bar Harbor, ME). Wild-type (FGF-2+/+; 129P2/OlaHsd:Black Swiss) and homozygous knock-out (FGF-2−/−) mice were studied, and genotypes were determined by PCR of the tail DNA. The lack of the protein was proven by Western blot (Jungnickel et al., 2004b). FGF-2+/+ littermates served as the control group.
Mice overexpressing FGF-2.
Heterozygous TgFGF-2 mice (Coffin et al., 1995) were received from Dr. S. Wolfram (Max-Planck-Institute, Bad Nauheim, Germany) and maintained on a FVB/N background. Wild-type and transgenic littermates were studied, and genotypes were determined by PCR of the tail DNA (Jungnickel et al., 2006). Overexpression of the different FGF-2 isoforms was verified in brain and lung tissue from transgenic mice by Western blot (data not shown).
Experimental design.
For the first part of the experiments (nonlesioned), 9- to 12- week-old male wild-type (FGFR3 wt, n = 5; FGF-2 ko wt, n = 6; TgFGF-2 wt, n = 6) and male mutant mice (FGFR3, n = 6; FGF-2 ko, n = 6; TgFGF-2, n = 6) were studied. Male mice older than 3 months were used for the lesion studies, because the cranial bone must be thick enough for putting the mice into the stereotactical frame (n > 8 per group). Female wild-type and mutant mice were taken for Western blot analysis (n = 3). The genetically altered animals represent wild-type littermates (animals born together from the same female). Wild-type and knock-out animals were obtained by crossbreeding with heterozygous genetically altered animals.
Tyrosine hydroxylase immunohistochemistry
The mice were deeply anesthetized with ketamine and Rompun and perfused transcardially with 20 ml of 0.9% saline, followed by 40 ml of 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were postfixed overnight and dehydrated in 20% sucrose. Serial coronal sections were cut on a freezing microtome at 30 μm thickness. To quantify the substantia nigra morphologically, every third section was processed for tyrosine hydroxylase (TH) immunohistochemistry (mouse anti-rat TH monoclonal antibody, 1:2500; catalog #T1299; Sigma, St. Louis, MO) using the avidin–biotin complex kit and 3′,3-diaminobenzidine for visualization, as described previously (Nikkhah et al., 1994). Before putting the first antibody, the sections were preincubated using null serum.
Morphological evaluation of the substantia nigra
All morphological procedures were performed using an Olympus Optical (Tokyo, Japan) microscope (BX 60). We quantified the substantia nigra with regard to the number of DA neurons, the volumes, and the cell density (Fig. 1).
Figure 1.

Overview of coronal sections through the midbrain. The photomicrographs show five levels of the substantia nigra (and the VTA) from rostral to occipital after tyrosine hydroxylase staining. In the present study, every third section of the midbrain was analyzed and not only the five sections shown here. Scale bar, 2000 μm.
Neuron counts.
Stereological methods were used to evaluate the number of neurons in the SNpc. Total numbers of TH-positive (TH+)cells were estimated with the optical disector method (Gundersen, 1977, 1986; Gundersen and Jensen, 1987; Gundersen et al., 1988; West and Gundersen, 1990; West et al., 1991) according to Oorschot (1996). Sections used for counting covered the entire substantia nigra. The borders of the SNpc at all levels in the rostrocaudal axis were defined at 4× magnification. The medial border was defined by a vertical line passing through the medial tip of the cerebral peduncle and by the medial terminal nucleus of the accessory nucleus of the optic tract, when present in sections, thereby excluding the TH-positive cells in the ventral tegmental area (VTA). Ventral border followed the dorsal border of the cerebral peduncle, thereby including the TH-positive cells in pars reticularis (SNpr), and the area extended laterally to include the pars lateralis in addition to pars compacta (Kirik et al., 2001). One-third of the sections through the substantia nigra stained with TH immunohistochemistry was evaluated under 40× magnification (objective: numerical aperture, 0.85). The measurements in the z-axis were done using the electric microcator (ND 281; Heidenhain, Traunreut, Germany) of the Olympus Optical CAST-Grid system. The counting frame was placed randomly on the first counting area and systematically moved through all fields. Only the profiles that came into focus within the counting volume were counted. The estimate of the total number of neurons was calculated according to the optical disector formula (for more details, see Oorschot, 1996). The coefficient of error was calculated according to Gundersen and Jensen (1987), and values <0.10 were accepted. TH-positive cells were counted only when the nucleus could be identified.
The x-step length of 320 μm and a y-step length of 210 μm between the samples were chosen, and a rectangular counting frame of 26.3% (exact size of the counting frame, 0.01728 mm2) of the total visual field was used. All neurons that were found inside of the counting frame or touching the inclusion lines (upper and right sides) were counted, and neurons touching the exclusion lines (bottom and left sides) or located outside the counting frame were excluded. All of the results (number of counted neurons, the number of samples, and the area of the counting frame) were collected in a table for additional analysis. The formula for the calculation of the total number of neurons in a selected region was based on stereological methods. The formula used was V(ref) × NV = N (unbiased estimate of the total number of neurons) (for details, see Oorschot, 1996). Our mathematical approach was N = I × II × III × IV, where I is total number of counted neurons in the selected area. Thus, I = NV. Hence, II × III × IV = Vref: I, the total number of counted neurons in the selected area was multiplied with II. II, The total number of histological series in which the nucleus was divided was multiplied with III. III, Optical disector is the result from the division between the real physical thickness of the evaluated slice, which is given by the electronic microcator from the actual evaluated slice thickness (real physical slice thickness − excluded bottom and top areas) (the guard zones were 5 μm from the bottom and 3 μm from the top) was multiplied with IV. IV, The area evaluated was estimated as the product of the x-step length and y-step length divided by the area of the counting frame. This resulted in the total estimated DA neuron number within this basal ganglia.
Volume measurement.
The volume of the SNpc was measured by outlining the areas of the SNpc on each section (magnification 10×). The SN was outlined on the computer screen using the software AnalySIS Pro (version 3.00; Soft Imaging System, Münster, Germany). Afterward, the total volume was calculated by multiplying the area of each section with three (because we only used every third section) and multiplying the result with 18 μm (thickness of the slice after shrinkage; before, 30 μm). Both the results and figures show the volume after shrinkage, and, therefore, to get the physiological volumes, one has to multiply all shown values with 1.7. In addition to the volume measurement, the area of both SNpc and SNpr were measured using the same system but only at three different coronal levels with regard to the bregma. The latter approach was performed to compare both parts of the SN.
Cell density.
The cell density was calculated by dividing the counted cell number by the measured volume.
TH Western blot
Western blotting (n = 3) was done after homogenization of the tissue (one hemisphere per probe, excluding the rostral part of the brain and the cerebellum) and separation of it using an 8% SDS-PAGE. The same amount of protein was loaded on each gel. Western blot analysis was performed as described previously (Meisinger et al., 1996). Detection of TH was done using a monoclonal anti-TH antibody (1:25,000; Sigma) and the enhanced chemiluminescence system from Amersham Biosciences (Braunschweig, Germany). Coomassie gels and β-III-tubulin lanes (on the same blot as TH; blot was cut before primary antibody) were used as a loading control (each lane, 100 μg of total protein). Blot bands were photographed and quantified using Scion (Frederick, MD) Image by density measurements.
Terminal 6-OHDA lesion
For the second part of the experiment, FGF-2 ko and TgFGF-2 mice received terminal unilateral 6-OHDA lesions to quantify the number of DA neurons, which survive this kind of intrastriatal lesion comparing mutant and wild-type mice. All surgical operations were performed under intraperitoneal anesthesia by using 10% ketamine/2% Rompun (0.1 ml/10 g). The animals received two stereotaxic injections of 6-OHDA at a concentration of 3 μg/μl (in 0.2 mg/ml l-ascorbate saline) using a 10 μl Hamilton syringe plus glass capillary into the right striatum (for coordinates, see Table 2) (Lundblad et al., 2004). The injection rate was 0.5 μl/min, and the cannula was left in place for an additional 3 min before slowly retracting it. The amount and concentration of the toxin was evaluated before the main experiment to fulfill the following two criteria: (1) reduce the number of DA neurons in wt mice to ∼50%; (2) get the lowest possible mortality rate at the same time. The mice were killed 2.5 months after lesion.
Table 2.
Stereotactic coordinates for the intrastriatal partial 6-OHDA lesion (Lundblad et al., 2004)
| Injection Tract | TB | AP | LAT | VERT | Volume |
|---|---|---|---|---|---|
| I | * | +1.0 | −2.1 | −2.9 | 2.5 μl |
| II | * | +0.3 | −2.3 | −2.9 | 2.5 μl |
The coordinates are given in millimeters with reference to bregma and dura. The lesion was performed unilaterally (right hemisphere). The neonatal rat/mouse adaptor was used [Cunningham hypothermic miniaturized stereotaxic device (Cunningham and McKay, 1993); Stoelting, Keil, WI]. * indicates tooth bar (∼2.4) and ear bar (∼8) were set that bregma and lambda were within the same horizontal level. The postoperative mortality was ∼35%. TB, Tooth bar; AP, anteroposterior to bregma; LAT, lateral from the midline; VERT, vertical (ventral to dura mater).
Statistics
Data are expressed as means ± SEM. Statistical analysis was performed using StatView 5.0 for Windows (SAS, Cary, NC). Comparisons between wild-type and mutant animals were performed with an unpaired Student's t test after testing normality. A p value of <0.05 was defined as significant.
Results
DA neuron numbers differ between mouse strains
We used three different mouse mutants to analyze the influence of FGF-2 on the nigrostriatal system during development: (1) FGF-2 knock-out mice (FGF-2−/−); (2) FGF-2 overexpressing mice (TgFGF-2), and (3) mice with a reduced level of FGFR3. Because the different mouse mutants of the FGF-2 system displayed different genetic backgrounds, the morphometric parameters of the SNpc were compared between the different strains (Table 1).
The number of DA neurons was quantified in ∼2.5-month-old adult mice. Comparing the wild types of these three strains, the results displayed different DA cell numbers depending on the mouse strain analyzed (Table 3). FVB/N mice (TgFGF-2 wt) have 8032.25 ± 414.49 nigral DA neurons, whereas 129P2 mice (FGF-2 ko wt) only have 6057.2 ± 283.50 DA neurons. This difference is significant (p < 0.001). C57BL/6 mice (FGFR3 wt) showed the highest number of nigral neurons with 8350.5 ± 448.86 neurons per SNpc, which was significantly higher than FGF-2 ko wt (p < 0.0005). TgFGF-2 wt mice and FGFR3 wt ones do not differ and show similar DA cell numbers (difference of 3.9%; p = 0.6; F = 5.684).
Table 3.
Morphological properties of the SN between wild-type mice of different mouse strains
| Mouse strain | DA neurons | Volume of SNpc (mm3) | Cell density (cells/mm3) |
|---|---|---|---|
| C57BL/6J | 8350.50 ± 448.86b*** | 0.12 ± 0.005 | 65,451.33 ± 2,078.2b,c*** |
| 129P2 | 6057.20 ± 283.50c***,a*** | 0.12 ± 0.006 | 51,088.51 ± 1,987.5a*** |
| FVB/N | 8032.25 ± 414.49b*** | 0.13 ± 0.004 | 54,286.74 ± 1,743.6a*** |
aSignificant to C57BL/6J.
bSignificant to 129P2.
cSignificant to FVB/N.
***p < 0.001.
The volumes of the SNpc were nearly equal between the different strains (p > 0.05). The exact values are depicted in Table 3. These data suggest that the volume of the SNpc is subject to stronger adjustments than the neuron number. Consequently, the cell density also shows significant differences between the three strains. The lowest density was found in FGF-2 ko wt mice. TgFGF-2 wt mice DA neuron density was 1.06-fold greater (p > 0.05, not significant), and the density in FGFR3 wt mice was 1.28-fold greater than in FGF-2 ko wt mice (p < 0.0001; F = 13.756). The DA densitometric difference between FGFR3 wt and TgFGF-2 wt was significant (p = 0.0005) (absolute values, see below).
Number of DA neurons reduced in FGFR3-deficient mice
Recent studies showed an important functional relevance of FGFR3 in the PNS. After peripheral nerve injury, FGFR3 was upregulated in contrast to the other FGF-2 receptors (Ji et al., 1995; Grothe and Nikkhah, 2001). Furthermore, this receptor is crucially involved in the regulation of survival of sensory neurons after lesion but not during peripheral nerve development (Jungnickel et al., 2004a, 2005). Regarding the CNS, it has been shown that the high-affinity tyrosine kinase receptor FGFR3 is expressed in the substantia nigra (Claus et al., 2004). In contrast to the PNS, FGFR3 expression displayed no alterations after 6-OHDA medial forebrain bundle lesion (Claus et al., 2004).
Therefore, we wanted to analyze the role of FGFR3 concerning the development of the nigrostriatal system. The SNpc was evaluated in terms of (1) DA neuron number, (2) SNpc volume, and (3) the density of DA neurons within the SNpc. Interestingly, FGFR3+/− mice (n = 6) displayed a significant lower number of DA neurons compared with their wild-type littermates [+/−, 6692.94 ± 307.3 (n = 5) vs wt, 8350.50 ± 448.9 (n = 6); p < 0.01; F = 8.541] (Fig. 2A,C,E). This decrease in DA neuron number after development in mice with a reduced level of FGFR3 suggests a physiological function for this receptor during development. The volume of the SNpc was not altered in FGFR3 mutant mice: the nigral volume of FGFR3+/− mice was 0.12 ± 0.005 mm3, and the volume of FGFR3wt was 0.12 ± 0.007 mm3 (p > 0.05; not significant; F = 0.001) (Fig. 2A,C,F). Consequently, the neuronal cell density was significantly decreased in FGFR3+/− (56,568.17 ± 2,764.9 DA neurons/mm3) compared with the corresponding wild type (FGFR3wt, 65,451.33 ± 2078.2 DA neurons/mm3; p < 0.05; F = 6.842) (Fig. 2B,D,G).
Figure 2.

FGFR3-deficient mice (+/−) display a reduced number of DA neurons. A–D, TH-immunostained coronal sections illustrating morphological parameters of the SN. The number of DA neurons and the size of the SN can be seen in an FGFR3 depleted (A) and a wild-type (wt) mouse (C). Note the reduced DA neuron number in FGFR3-deleted mice (E). The volume of the SNpc was not altered in FGFR3-deficient mice (F). The DA neuron density is depicted in the higher-magnification photomicrographs (FGFR3, B; wt, D; note the frames in A and C). No difference was found concerning the DA cell density (G). Scale bars: 500 μm; higher magnifications, 100 μm. *p < 0.01 (DA cell number); *p < 0.05 (DA cell intensity).
FGF-2 level is reciprocal to number of DA neurons
FGF-2 is a potent survival factor and is, for example, used as mitogen for the expansion of DA progenitor cells in vitro (Studer et al., 1998; Timmer et al., 2006) and promotes the survival of grafted embryonic DA neurons in vivo (Mayer et al., 1993b; Timmer et al. 2004). However, only little is known about the physiological role of FGF-2 on DA neurons and the nigrostriatal system. To study the influence of FGF-2 on the development of the nigrostriatal system, we quantified the number of DA neurons. Mice lacking FGF-2 (FGF-2 ko; n = 6) resulted in 7170.60 ± 228.9 DA neurons, whereas wild-type mice (FGF-2 ko wt; n = 6) only resulted in 6057.20 ± 283.5 (Fig. 3A,C,E). This result suggests that mice without the important mitogen and survival factor FGF-2 show a significant increase in the number of nigral neurons during development, which remains during adulthood (p < 0.01; F = 9.336). Moreover, mice overexpressing FGF-2 displayed a reduction of dopaminergic neurons after development [TgFGF-2 (n = 6), 6902.23 ± 414.2 DA neurons compared with TgFGF-2 wt (n = 6), 8032.25 ± 414.5] (Fig. 4A,C,E). However, the latter difference was not significant (p = 0.06; F = 3.719). Summarizing these results, it appears that an inverse correlation exists between the number of DA neurons and the level of FGF-2 during development.
Figure 3.

More DA neurons and enlarged SNpc volume in FGF−/− mice. A, C, The overview photomicrographs show examples of an FGF-2 ko (A) and a wild-type (C) midbrain section illustrating the volume of the SN and the DA neuron number. Scale bars: 500 μm; higher magnifications, 100 μm. B, D, The frames are mapped in more detail indicating the DA cell density. Note the significantly higher DA neuron number and the bigger volume of the SNpc in FGF-2 ko mice (E, F). Consequently, no difference appeared with regard to the DA neuron density (G). H, The TH protein level was evaluated by Western blot and densitometrically analyzed. A Coomassie gel verifies similar loaded protein amounts. *p < 0.01 (DA cell number); *p < 0.05 (SNpc volume).
Figure 4.

Substantia nigra in adult mice overexpressing FGF-2. A–D, TH immunohistochemistry of the SN [TgFGF-2 (A, B) vs WT (C, D)]. The frames within the overviews (A, C) are enlarged in B and D indicating the DA cell density. E, Quantification of the DA neuron number. F, G, Note the nigral volume enlargement (F) and the reduced TH+ cell density in TgFGF-2 mice. H, The TH protein amount is shown for two animals (n = 3) by Western blot and the corresponding Coomassie gel. I, Quantification of the Western blot revealed a lower TH level in TgFGF-2 mice. Scale bars: 500 μm; higher magnifications, 100 μm. *p < 0.05 (TH expression); *p < 0.001 (SNpc volume); *p < 0.0001 (DA cell density).
Volume of SNpc enlarged in both FGF-2 ko and Tg mice
The volume of the SNpc was relatively constant between the different mouse strains (see above). Therefore, the question whether endogenous FGF-2 can change the nigral volume is of importance. The volume of the SNpc in wild-type FGF-2-deleted mice was 0.120 ± 0.009 mm3 (Fig. 3C,F). The volume in the corresponding mutant strain (FGF-2 ko) was significantly enlarged (0.143 ± 0.006 mm3; p < 0.05; F = 4.628) (Fig. 3A,F). These data suggest that FGF-2 is crucially involved in the development of a proper sized SN. Moreover, the nigral volume was also significantly bigger in mice overexpressing FGF-2. The volume in TgFGF-2 mice was 0.177 ± 0.007 (wt, 0.134 ± 0.005; p < 0.001; F = 24.438) (Fig. 4A,C,F). An alteration of the endogenous FGF-2 concentration seems to result in an enlargement of the SNpc regardless of whether more or less FGF-2 is present.
TH-positive cell densities in the SNpc in FGF-2 mouse mutants
The DA neuron density was unchanged in FGF-2 knock-out mice. The cell density within the SN in FGF-2 ko mice was 50,733.97 ± 2038.9 DA neurons/mm3 and 51,088.51 ± 1987.5 cells/mm3 in the corresponding wild type (p = 0.9; F = 0.016) (Fig. 3B,D,G). In contrast, transgenic mice displayed a highly significant difference (p < 0.0001; F = 23.407). TgFGF-2 wt mice showed a DA neuron density of 54,286.74 ± 1743.6 (Fig. 4D,G). TgFGF-2 have a reduced DA cell density of 39,536.72 ± 2500.9 (Fig. 4B,G). This difference in TH-positive cell density further supports the hypothesis that FGF-2 is important for the development of the substantia nigra.
TH expression in FGF-2 mutant mice
The expression of the DA rate-limiting enzyme TH was evaluated using Western blot analysis. The densities of the bands were measured (n = 3 animals per group) compared with the respective wild-type animals (set as 100%). The same amount of protein was loaded on each lane. This was determined before and confirmed by a Coomassie gel. FGF-2−/− mice displayed increased TH expression levels (112 ± 8.8%) compared with wild-type mice (100 ± 5.9%; p > 0.05) (Fig. 3H). Two examples of TH Western blot of transgenic mice are depicted in Figure 4H (of n = 3). Transgenic mice show decreased TH expression levels of 73 ± 10.9% compared with wt animals (100 ± 8.6%). This difference was significant (p < 0.05) (Fig. 4I). These data confirm the results of DA neuron counting and also suggest an inverse relationship between the TH and the FGF-2 levels in adult mice.
Substantia nigra: compacta and reticularis
In addition to the measurement of the volume of the SNpc of the whole midbrain, we measured the area of the SNpc and of the SNpr at three different coronal levels of the midbrain in FGF-2 ko mice (Fig. 5A). The question was whether the increased nigral size was attributable to a change in the rostral, medial, or occipital part of the SN and whether the pars reticularis was also affected. Compared with wt mice (wt was set as 100%), FGF-2 ko mice displayed a 16% increase of the SNpc area at −2.9 mm, a 26.9% increase at −3.3 mm, and a 23.7% increase at −3.8 mm from the bregma. All three levels showed a significant area increase of the SNpc (p < 0.05), indicating that the SNpc is enlarged throughout the midbrain (Fig. 5B). In contrast, the pars reticularis was not affected. FGF-2 ko mice displayed the following area values of the SNpr: rostral, 108.3 ± 9%; medial, 104.2 ± 11%; and occipital, 110.9 ± 9% (p > 0.05).
Figure 5.

SNpc and SNpr in FGF-2 ko mice. A, In addition to the volume measurement of the complete SN, the area of both parts of the SN (pars compacta and pars reticularis) was analyzed at three different midbrain levels (−2.9, −3.3, and −3.8 occipital from the bregma; see also the middle three levels of Fig. 1) (Paxinos and Franklin, 2001). B, Quantitative results of the SN area of the three coronal levels. The area of wt mice was set as 100% in all levels. The SNpc was enlarged throughout the midbrain, whereas the pars reticularis was not changed in FGF-2 ko mice. *p < 0.05.
Less surviving DA neurons in FGF-2 ko mice after lesion
To understand the physiological role of FGF-2 after CNS lesion could have impact on the development of new therapeutic strategies. Here, we addressed the question of whether endogenous FGF-2 could protect DA neurons from 6-OHDA neurotoxicity. For this purpose, the number of DA nigral neurons was quantified after striatal 6-OHDA lesion.
It has been shown recently that FGF-2 immunoreactivity increases in reactive astrocytes after a striatal 6-OHDA-induced partial lesion of the nigrostriatal system (Chadi and Gomide, 2004). In addition, FGF-2-producing fibroblasts protect the nigrostriatal DA system from 6-OHDA-lesioned rats (Shults et al., 2000). In the present study, the lesion parameters (6-OHDA concentration used and coordinates) were chosen to destroy ∼50% of DA neurons in wild-type mice. FGF-2 ko wt mice result in 2749.6 ± 151.1 surviving DA neurons, whereas in FGF-2−/−, only 2144.3 ± 137.0 survived the terminal lesion (Fig. 6A–C). This significant difference (p < 0.05) suggests that other trophic factors are not able to compensate for the missing endogenous FGF-2 after lesion. The difference becomes even more significant when the lesioned hemisphere is compared with the nonlesioned side, because unlesioned wild-type mice display significantly less DA neurons than FGF-deleted mice (see above): 45.3% of the DA neurons survived in FGF-2 ko wild-type mice, whereas only 29.9% survived in FGF-2 ko mice lacking the protein (Fig. 6C).
Figure 6.

DA neuron number after 6-OHDA lesion in FGF-2 ko mice. A, Representative coronal TH-stained section of an FGF-2−/− mice after unilateral striatal 6-OHDA lesion. The right hemisphere (H.) was lesioned. The two insets show higher magnifications of the two frames in the overview. B, Representative photomicrograph analog to A of wild type. Scale bars, 500 μm. C, Quantification of TH+ cells at the lesioned side (left) and the relative cell survival compared with the unlesioned side (right). D, TH Western blot comparing knock-out and wt mice before and after lesion. β-III-Tubulin (Tub.) was used as standard. *p < 0.05.
DA neurons rescued in TgFGF-2 mice after 6-OHDA lesion
Mice overexpressing FGF-2 were able to protect DA neurons from 6-OHDA neurotoxicity, because 3657.1 ± 128.4 DA neurons survived the 6-OHDA lesion in wild-type mice, whereas in TgFGF-2 mice, 4023.9 ± 166.7 DA neurons survived the lesion (Fig. 7A–C). This significant difference (p < 0.05) also becomes more significant when the lesioned hemisphere is compared with the nonlesioned side, because unlesioned wild-type mice displayed more DA neurons than transgenic mice (see above). A total of 45.5% of the DA neurons survived in TgFGF-2 wt mice, whereas 58.3% survived in TgFGF-2 overexpressing the protein (Fig. 7C). These results suggest that endogenous FGF-2 can protect DA neurons from lesion-induced cell death.
Figure 7.
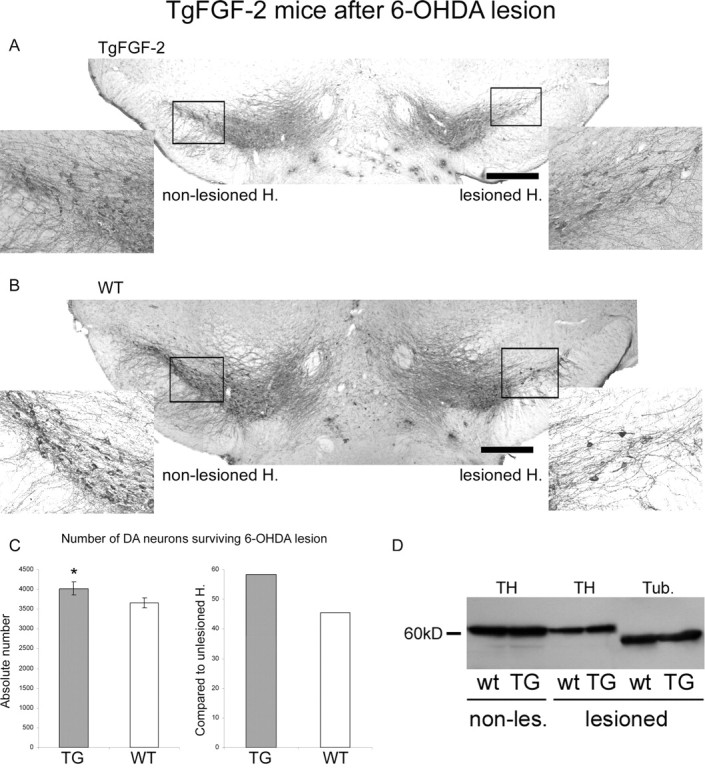
TgFGF-2 mice after terminal 6-OHDA lesion. A, Representative coronal TH-stained section of mice overexpressing FGF-2 after unilateral 6-OHDA lesion. Lesioned hemisphere (H.), Right. The two inserts show higher magnifications of the two frames in the overview. B, Representative photomicrograph analog to A of wild type. Scale bars, 500 μm. C, Quantification of TH+ cells at the lesioned side (left) and the relative cell survival compared with the unlesioned side (right). Significantly more DA neurons were rescued from 6-OHDA neurotoxicity in mice overexpressing FGF-2. D, TH Western blot comparing transgenic and wt mice before and after lesion. β-III-Tubulin (Tub.) was used as standard. Abbreviations: see Figure 6. *p < 0.005.
TH expression levels after striatal 6-OHDA lesion
The TH Western blots confirmed the quantification of DA neurons after 6-OHDA lesion. A profound reduction of TH was found after terminal lesion (Figs. 6D, 7D). In addition, TH expression after 6-OHDA lesion was lowered in FGF-2 ko animals compared with wt mice (Fig. 6D). Furthermore, FGF-2 transgenic mice showed higher TH expression after lesion than the corresponding wt mice (Fig. 7D).
Discussion
To study the relevance of the FGF-2 system in SNpc development, we used mice overexpressing FGF-2, without FGF-2 (ko) and lacking FGFR3, one of the high-affinity receptors of FGF-2. The present study revealed crucial functions of the endogenous FGF-2 system in the developing SNpc and after lesion, including four main findings: (1) an increase in DA neuron number in mice lacking FGF-2 and, moreover, a TH decrease in mice overexpressing the factor; (2) a vice versa situation under lesioning conditions in which more DA neurons were rescued in TgFGF-2 mice and less in FGF-2 ko ones; (3) an enlargement of the volume of the SNpc when the FGF-2 level was altered regardless whether this change was an increase or a decrease; and (4) a significantly lower number of DA neurons in FGFR3+/− deficient mice, which suggests an involvement of FGFR3 signaling during development (Table 4).
Table 4.
Semiquantitative summary of results
| Parameter | FGFR3 | FGF-2 ko | TgFGF-2 |
|---|---|---|---|
| Amount of FGF-2 proteina | Ø | — | + + + |
| Number of DA neurons (adult mice) | < wt | > wt | < wt |
| Volume of SNpc (adult mice) | = wt | > wt | > wt |
| DA cell density (adult mice) | < wt | = wt | < wt |
| Number of DA neurons after lesion | n.d. | < wt | > wt |
| TH Western blot density (intact) | n.d. | > wt | < wt |
| TH Western blot density (lesioned) | n.d. | < wt | > wt |
n.d., Not done; ø, normal level; —, nil; +++, increased level.
a Determined by Western blot (data not shown).
Striatal 6-OHDA lesion provides a useful tool
Inducing PD in animal models can be achieved by different methods such as 6-OHDA, rotenone, paraquat, and maneb (Betarbet et al., 2002; Soderstrom et al., 2006). The most common one in mice is MPTP treatment. Most studies analyzing mutant mice regarding the nigrostriatal system use this approach (Sriram et al., 2002; Zechel et al., 2006). This technique is easily applicable but has several disadvantages, including a recovery of motor impairments after some days and enhanced de novo DA neurogenesis after MPTP lesion (Shan et al., 2006). Furthermore, mice are less sensitive to MPTP treatment compared with primates, and different mouse strains are differently sensitive to MPTP toxicity, with the highest sensitivity in C57BL/6. The degree of the damage is also age related (Gerlach et al., 1991; Schober, 2004). To overcome these limitations, we used the 6-OHDA lesion, which has to be performed stereotactically. Normally, this method is only used in rats, because they are insensitive against systemic MPTP applications attributable to a high endothelial monoamine oxidase-B activity. The common 6-OHDA medial forebrain bundle lesion completely destroys all DA projections (Ungerstedt and Arbuthnott, 1970). More recently, terminal lesions of the rat nigrostriatal pathway induced by intrastriatally injected 6-OHDA have been developed (Sauer and Oertel, 1994; Lee et al., 1996; Kirik et al., 1998; Rodter et al., 2000). This animal model of PD has the advantages that (1) one can directly influence the extent of the lesion by altering the dose given, and (2) this kind of lesion mimics early stages of the disease and is therefore useful for the development of neuroprotective therapies.
FGF-2 as a key factor: influence of the FGF-2 level on DA neurons
FGF-2 and its high-affinity tyrosine kinase receptors (FGFR1–FGFR3) are expressed in the developing and adult mesencephalon and especially in the SN (Bean et al., 1991; Belluardo et al., 1997; Bansal et al., 2003). FGF-2 displays different isoforms with different subcellular distributions and functions (Stachowiak et al., 1994; Timmer et al., 2004). In the mutant mice analyzed (transgenic and knock-out), all three FGF-2 isoforms (18, 21, and 23 kDa) are deleted or overexpressed, respectively.
FGF-2 mainly acts as a survival factor and mitogen in many tissues, including the PNS and CNS (Xu et al., 2005). On the one hand, it is upregulated in several carcinogenic cells (Ueba et al., 1994), and, on the other hand, it is used to stimulate the survival of grafted (stem) cells in regenerative medicine. In PD, it was successfully applied to enhance the survival and differentiation of transplanted embryonic DA cells (Takayama et al., 1995; Timmer et al., 2004), and it was taken as mitogen to expand neural progenitor cells before differentiation (Studer et al., 1998; Timmer et al., 2006). Moreover, FGF-2 promotes neurogenesis and neuroprotection (Jin et al., 2005). On the basis of these facts, an increase in DA neuron number in mice overexpressing FGF-2 and a decrease in FGF-2-deleted mice could be expected. Interestingly, the opposite was found in this study. Significantly more DA neurons were found in FGF-2 ko mice. A possible mechanism explaining this finding could be that the lack of FGF-2 is (over) compensated during development by other trophic factors, which could be upregulated. Such compensatory regulations are reported from many knock-out mice displaying only some deficits and nearly normal phenotypes.
The compensation of FGF-2 in FGF-2−/− mice could theoretically occur through (1) other members of the FGF family or (2) other trophic factors [for example, brain-derived neurotrophic factor (BDNF)]. The latter possibility is unlikely, because no compensation takes place when the FGF receptor is deleted, suggesting that other FGFs are responsible for the compensation (see below). To date, the FGF proteins are encoded by 22 distinct genes known as Fgf1–Fgf18 and Fgf20–Fgf23 in the mouse species (Dono, 2003). Human and chick Fgf19 is the ortholog of mouse Fgf15 (Wright et al., 2004). To evaluate a putative candidate for the compensation of FGF-2, first all FGF members have to be excluded that are not expressed in the SN. Most FGFs are expressed in the brain, whereas FGF-21 is preferentially expressed in the liver (Nishimura et al., 2000) and FGF-22 in the inner root sheath of the hair follicle (Nakatake et al., 2001). Other FGF family members are only expressed in distinct parts of the brain, such as FGF-23, which is expressed in the ventrolateral thalamic nucleus (Yamashita et al., 2000). Many studies showed that FGF-4 is not expressed in the brain, but a recent study revealed localization of mouse Fgf4 mRNA in the hippocampus, subventricular zone, and the rostral migratory stream in which new neurons are generated and migrate (Kosaka et al., 2006). The Fgf8/Fgf17/Fgf18 subgroup of Fgfs mainly have midbrain and cerebellum regulatory properties in very early stages of development (Reifers et al., 2000; Liu et al., 2003), but there is some evidence that they are also expressed during later stages in the SN. In addition, FGF-1, FGF-9, FGF-11, FGF-12, FGF-13, FGF-14, FGF-15, and FGF-20 are expressed in the SN and could be responsible for the compensation when FGF-2 is deleted (Todo et al., 1998; Ohmachi et al., 2000; Dono, 2003). FGF-3, FGF-5, FGF-6, FGF-7, FGF-10, and FGF-16 could theoretically also compensate FGF-2, but, so far, no evidence has been published that these FGF members are expressed in the SN.
Concerning the volume of the SN, it seems that overcompensation of the deleted FGF-2 by other FGFs leads to a similar effect as overexpressed FGF-2 in transgenic FGF-2 mice. That both FGF-2 and other FGFs (in FGF-2 knock-out mice) play a crucial role on the size (in terms of an enlargement) of the SN is very likely.
In mice overexpressing FGF-2, we found a nonsignificant decreased DA cell number and, in addition, a significantly lower TH expression level, suggesting that too much FGF-2 may be harmful for DA neurons during development. A recent finding supports this hypothesis because it was shown in vitro that increased concentration of exogenous FGF-2 leads to reduced outgrowth of human embryonic stem cell (hESC) colonies with time in culture (Dvorak et al., 2005). In addition, we have shown recently that FGF-2 can exhibit toxic effects on transfected DA progenitor cells overexpressing FGF-2 (Cesnulevicius et al., 2006). Moreover, continuous exposure of glial cell line-derived neurotrophic factor (GDNF) to mature DA transplants impairs the ability of the graft to improve spontaneous motor behavior in parkinsonian rats (Winkler et al., 2006).
The SNpc and its trophic factors
Several growth factors promote the survival of mesencephalic dopaminergic neurons such as GDNF, a member of the transforming growth factors β superfamily, neurotrophins including brain-derived neurotrophic factor (BDNF), and low molecular compounds mimicking growth factor activities (Krieglstein, 2004). Currently, the available data support GDNF to be a highly promising candidate for the treatment of PD in terms of exogenous application [e.g., viral (Kirik et al., 2004)]. However, GDNF-null mutants lack a nigral DA phenotype (Granholm et al., 1997). In another experimental approach by transplanting fetal neural tissues from GDNF−/− into wild-type mice, it was demonstrated that the continued postnatal development of these grafted DA neurons was severely disturbed in terms of reduced DA neuron numbers and fiber outgrowth (Granholm et al., 2000).
Wnt-BDNFko mice revealed a persistent 23% reduction of TH+ cells at postnatal day 21 (P21) compared with controls. The reduced number of TH-expressing neurons was present at birth and continued through P120 (Baquet et al., 2005). Heterozygous mice (BDNF+/−) show no reduction in TH+ cell numbers, and BDNF−/− mice are postnatal lethal.
It was reported recently that FGF-2 deficiency alters neither the DA cell density nor the vulnerability of DA neurons toward MPTP intoxication, which was evaluated on the basis of TH+ cell density (Zechel et al., 2006). In accordance with our results, they found no difference between unlesioned FGF-2 null mutants and wild-type mice. As we could show in this study, the unaltered cell density in FGF−/− is attributable to an increased overall DA neuron number and, simultaneously, an enlarged volume. After lesion, Zechel et al. (2006) found no difference of the TH+ cell density, whereas we found a significant lower number of TH+ cells afterward. This apparent discrepancy could be attributable to (1) different lesion models (Zechel et al., 2006, MPTP; this study, unilateral, terminal 6-OHDA) and (2) different kinds of analysis (Zechel et al., 2006, TH+ cell density; present study, stereologically evaluated absolute TH+ cell number). Our results suggest that endogenous FGF-2 is a physiological key player within the nigrostriatal system important for the development and protection/repair during adulthood. This finding is supported by previous studies in which the most striking difference in patients suffering from PD was found in FGF-2 expression: a profound depletion of FGF-2 [from 82% (control brains) down to 12.7% (PD brains)] in the remaining DA neurons was found (Tooyama et al., 1994), whereas BDNF was only reduced from 65 to 43% (Parain et al., 1999). In situ hybridization failed to show any detectable levels of GDNF in PD or in control cases, suggesting that GDNF is not a major physiological factor during adulthood (Hunot et al., 1996). These postmortem studies are in good correlation with the present null mutant studies. FGF-2 seems to be one of the first growth factors that affects the nigrostriatal system during development, with an effect throughout adulthood.
It is not very likely that a single neurotrophic factor supports survival of a specific cell population. Neuron losses in BDNF/GDNF double mutants are not additive compared with the losses in single null mutants, indicating that in vivo survival in many cells, including DA neurons, is supported by more than one factor (Erickson et al., 2001). However, additional studies are needed to understand the interplay between the different growth factors and to exactly evaluate which factor is able to compensate or support each other. Double-knock-out mice could help to address this question.
FGFR3 involved in postnatal DA neuron survival
The present study shows a significant decrease of DA midbrain neurons in unlesioned FGFR3 knock-out mice (+/−), suggesting an involvement of FGFR3 during DA neuron development. This finding could mean that FGFR3 is involved in DA neuron development. Expression analyses of FGFR3 during embryonic development revealed that FGFR3 displayed a diffuse expression throughout the brain at E14 (Claus and Grothe, 2001). Postpartum, there is a wave of FGFR3 mRNA expression in the normal postnatal brain that spreads from the hindbrain to the forebrain. At P1, FGFR3 was expressed diffusely and localized in cells (Peters et al., 1993). At P2, FGFR3 showed little expression in the midbrain. By P4, the FGFR3 mRNA expression increased in the midbrain (Oh et al., 2003), where it remained during adulthood (Belluardo et al., 1997; Oh et al., 2003; Claus et al., 2004; Timmer et al., 2004). This FGFR3 expression pattern suggests that FGFR3 does not play an important role during embryonic midbrain development but could be an important player after birth. A similar result was shown recently in which the postnatal expression pattern of FGFR3 parallels the appearance of differentiated oligodendrocytes (OLs), and the absence of FGFR3 expression in FGFR3 null mutants resulted in reduced numbers of differentiated OLs (Oh et al., 2003). Interestingly, significant neurophysiological and neuropharmacological changes, including DA neuron cell death, take place within the SN in the first postnatal month (Tepper et al., 1994). Tepper et al. could show that the number of TH+ neurons decreased significantly in the SN from P1 to P14, with the most striking effect between P1 and P7. This time frame correlates with the increase of FGFR3 expression in the SN (see above), indicating that FGFR3 could have the highest impact on the development of DA neurons during the first week after birth as survival-promoting factor. Nevertheless, the alterations must also have an effect during embryonic stages because the effect is too big and can therefore not only be explained by postnatal changes. The ∼20% reduced DA neuron number in FGFR3+/− knock-out mice demonstrates that FGFR3 can, in contrast to FGF-2 (see above), not be compensated (e.g., by FGFR1 or FGFR2).
One major question is whether this dramatic effect is mediated via FGF-2 or via another ligand. FGF-1–FGF-3, FGF-5–FGF-18, and FGF-20 are possible candidates because of their expression pattern (see above; this list includes all possible candidates). The intracellular FGF-11 to FGF-14 do not bind to FGFRs and can therefore be excluded (Dono, 2003). FGF-19, the ortholog of FGF-15, displays unique specificity for FGFR4 (Xie et al., 1999). FGF-6 is only expressed at very low levels in the brain, making this candidate very unlikely (Ozawa et al., 1996). The FGF ligands bind to the different FGF receptors with different affinities. FGF-3, FGF-7, FGF-10, and FGF-18 do not bind to FGFR3 or only with a low affinity (Ford-Perriss et al., 2001). However, a more recent publication showed that FGF-18 can bind to FGFR3 (Zhang et al., 2006). One of the most promising protein is FGF-20, because it is preferentially expressed in the substantia nigra pars compacta (Ohmachi et al., 2000), and it promotes the differentiation of Nurr1-overexpressing neural stem cells into TH+ neurons (Grothe et al., 2004). Up to now, it was thought that FGF-20 preferentially acts via FGFR1c (Ohmachi et al., 2003), but a new binding study revealed that the binding affinity between FGF-20 and FGFR3 is ∼90% (Zhang et al., 2006), making it a “high potential” candidate for compensating FGF-2 in FGF-2 ko mice. Thus, FGF-1, FGF-2, FGF-5, FGF-8, FGF-9, FGF-16, FGF-17, FGF-18, and FGF-20 could act via FGFR3 in the SN during postnatal development. However, to date, no published data showed FGF-5 and FGF-16 in the SN, making both candidates unlikely. FGF1 would be a good candidate for compensating FGF2, but it has been shown previously that FGF1 does not compensate FGF2 during development because FGF1–FGF2 double-knock-out mice were viable and fertile and did not display any gross phenotypic defects other than seen in FGF2 knock-out mice (Miller et al., 2000). Taking all of these results from literature and this study together suggest that the FGF-8/FGF-17/FGF-18 and the FGF-9/FGF-20 subfamilies might be the candidates that (over)compensate FGF-2 in FGF-2 ko mice (see above).
FGFR3 is a transmembrane tyrosine kinase receptor mediating the extracellular FGF signal. The lacking receptor prevents secreted FGFs (2, 8, 17, 18, 9, 20) from entering the cytoplasm and acting in an autocrine/paracrine manner. Thus, deleted FGFR3 knocks down most FGFs at the same time. This would explain why no compensation, like in FGF-2 ko mice, is possible and at the same time confirms the hypothesis that, in FGF-2 ko mice, other FGFs come into play. The results also suggest that the other two FGF receptors [FGFR1 and FGFR2; FGFR4 is not present in the nigrostriatal system (Timmer et al., 2004)] cannot fully compensate the lack of FGFR3 or may fulfill different functions (Corso et al., 2005; Klejbor et al., 2006). This idea is supported by an oligodendrocyte study showing that distinct FGF/FGFR signaling pairs initiate diverse cellular responses in cell lineage (Fortin et al., 2005).
FGFR3c is preferentially found in glia but also in DA neurons (Yazaki et al., 1994), suggesting an indirect effect. We have shown previously that FGFR3 was not reduced after 6-OHDA lesion (Claus et al., 2004), suggesting that the majority of the FGFR3 population might be located on cells other than DA neurons. This finding further supports the hypothesis of an indirect (i.e., glia-mediated) effect. Moreover, released FGF (e.g., FGF-2) must be crucially involved in DA neuron development and survival, because intracellular FGF-2 is not affected in FGFR3 knock-out mice but the extracellular cannot act as ligand without its receptor. This consideration is supported by a recent study in which FGFRs were inhibited. The resultant inhibition of FGFR suppresses activation of downstream protein kinases and causes rapid cell differentiation, suggesting an involvement of autocrine FGF signals in the maintenance of proliferating hESCs in the undifferentiated state (Dvorak et al., 2005).
The finding of the present study, that released FGF is important, is supported by a recent publication showing that autocrine FGF-2 signaling is critical for self-renewal and differentiation of mesenchymal stem cells (Zaragosi et al., 2006). Moreover, extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway is involved in FGF-2-mediated self-renewal but not in differentiation. Another study showed that rotenone-induced apoptosis in DA cells was attenuated by pretreatment with FGF-2 via activation of ERK1/2 and phosphatidylinositol-3 kinase (PI3-kinase) pathways (Hsuan et al., 2006). Ectopic activation of ERK1/2 or PI3-kinase protected against rotenone, whereas inhibition of either pathway attenuated FGF-2 protection. These data indicate that second-messenger systems play a crucial role in this scenario. FGFR3 is expressed as two main splice isoforms (IIIb or IIIc). We have shown previously that FGFR3 mediates neuronal death in the PNS (Jungnickel et al., 2004a), whereas this study suggests that FGFR3 supports survival and inhibits naturally occurring apoptosis during development. How signaling through FGFR3 can lead to apoptosis, differentiation, or cell proliferation is not fully understood. It seems that STAT1 (signal transducer and activator of transcription 1) phosphorylation is involved in inhibition of proliferation, whereas activation of the ERK pathway inhibits differentiation (L'Hote and Knowles, 2005). Some of the cell specificity may arise via modulation of signaling by crosstalk with other signaling pathways. Together, FGFR3 seems to be essential for (at least) a ∼20% subpopulation of DA neurons during development.
Different mechanisms during development and after lesion?
Our data after terminal 6-OHDA lesion clearly indicate a physiological neuroprotective function of FGF-2 because significantly more DA neurons survived after lesion in mice overexpressing FGF-2 compared with wild-type mice. This result together with the finding that released FGF-2 seems to be important in this context (see above) confirms the physiological trophic effects of FGF-2 on DA neurons and justifies the exogenous application of FGF-2 to (1) prevent the degeneration of the remaining DA neurons in PD patients or (2) enhance DA graft survival and benefit. Moreover, these findings encourage the development of new FGF-2-mediated neuroprotective therapies for neurodegenerative disorders. The observed effects, at least in FGF-2 ko mice, seem to be specific to DA neurons and not systemic/general because other studies showed different results for other brain regions/neuronal populations [e.g., impaired cerebral cortex development (Dono et al., 1998)].
After lesion, significant more DA neurons died in FGF-2 ko mice than in wild-type mice. This further underlies the survival-promoting and neuroprotective effect of FGF-2. In contrast to the situation during development, the compensation mechanisms (e.g., upregulation of other members of the FGF family) seem to fail or at least are not as sufficient as in fetal/neonatal animals. These results verify that plasticity and repair mechanisms are much more powerful in developing systems. Furthermore, this is a fundamental finding for neuroprotective and restorative therapies because it shows restrictions in terms of limited endogenous repair functions and could help to overcome these limitations.
Conclusion
The present findings not only depict the function and relevance of FGF-2 but also support the hypothesis that most biological systems are redundant, and even one of the most important survival factors seems to be (over)compensated. However, this compensation is by far not perfect and fails when lesioning adult mice.
Thus, FGFR3 seems to be crucial during (postnatal) development of DA neurons, and FGF-2 seems to fulfill an unique physiological function within the nigrostriatal system after lesion. It has to be further analyzed whether FGF-2 is supportive or essential. This gain in knowledge could help for a better understanding of how FGF-2 regulates DA survival and differentiation and how compensation mechanisms come into play or fail. In addition to development, the neuronal changes observed in adult mice could also reflect an altered cell homeostasis.
Moreover, these findings are also relevant to exogenous FGF-2 applications [e.g., transplantation experiments (Cesnulevicius et al., 2006)] because detailed information about the growth factors used is necessary for a proper application. Together, these data could help to develop new therapeutic approaches for PD, including protective ones. Moreover, these results exemplify how narrow the range between toxic and beneficial effects of e.g., growth factors is.
Footnotes
We are very grateful to Dr. J. D. Coffin (University of Montana, Missoula, MN) and Dr. S. Wolfram (Max-Planck-Institute, Bad Nauheim, Germany) for providing the TgFGF-2 breeders. We acknowledge the excellent technical assistance of G. Braun, H. Brinkmann, N. Heidrich, K. Kuhlemann, and J. Wittek. We thank Dr. K. Haastert for critical reading and comments on this manuscript.
References
- Bansal R, Lakhina V, Remedios R, Tole S. Expression of FGF receptors 1, 2, 3 in the embryonic and postnatal mouse brain compared with Pdgfralpha, Olig2 and Plp/dm20: implications for oligodendrocyte development. Dev Neurosci. 2003;25:83–95. doi: 10.1159/000072258. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean AJ, Elde R, Cao YH, Oellig C, Tamminga C, Goldstein M, Pettersson RF, Hokfelt T. Expression of acidic and basic fibroblast growth factors in the substantia nigra of rat, monkey, and human. Proc Natl Acad Sci USA. 1991;88:10237–10241. doi: 10.1073/pnas.88.22.10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KD, Knusel B, Hefti F. The nature of the trophic action of brain-derived neurotrophic factor, des(1–3)-insulin-like growth factor-1, and basic fibroblast growth factor on mesencephalic dopaminergic neurons developing in culture. Neuroscience. 1993;52:855–866. doi: 10.1016/0306-4522(93)90534-m. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Wu G, Mudo G, Hansson AC, Pettersson R, Fuxe K. Comparative localization of fibroblast growth factor receptor-1, -2, and -3 mRNAs in the rat brain: in situ hybridization analysis. J Comp Neurol. 1997;379:226–246. [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson's disease. BioEssays. 2002;24:308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- Bouvier MM, Mytilineou C. Basic fibroblast growth factor increases division and delays differentiation of dopamine precursors in vitro. J Neurosci. 1995;15:7141–7149. doi: 10.1523/JNEUROSCI.15-11-07141.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesnulevicius K, Timmer M, Wesemann M, Thomas T, Barkhausen T, Grothe C. Nucleofection is the most efficient nonviral transfection method for neuronal stem cells derived from ventral mesencephali with no changes in cell composition or dopaminergic fate. Stem Cells. 2006;24:2776–2791. doi: 10.1634/stemcells.2006-0176. [DOI] [PubMed] [Google Scholar]
- Chadi G, Gomide VC. FGF-2 and S100beta immunoreactivities increase in reactive astrocytes, but not in microglia, in ascending dopamine pathways following a striatal 6-OHDA-induced partial lesion of the nigrostriatal system. Cell Biol Int. 2004;28:849–861. doi: 10.1016/j.cellbi.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Claus P, Grothe C. Molecular cloning and developmental expression of rat fibroblast growth factor receptor 3. Histochem Cell Biol. 2001;115:147–155. doi: 10.1007/s004180000215. [DOI] [PubMed] [Google Scholar]
- Claus P, Werner S, Timmer M, Grothe C. Expression of the fibroblast growth factor-2 isoforms and the FGF receptor 1–4 transcripts in the rat model system of Parkinson's disease. Neurosci Lett. 2004;360:117–120. doi: 10.1016/j.neulet.2004.01.046. [DOI] [PubMed] [Google Scholar]
- Coffin JD, Florkiewicz RZ, Neumann J, Mort-Hopkins T, Dorn GW, II, Lightfoot P, German R, Howles PN, Kier A, O'Toole BA, Sasse J, Gonzalez AM, Baird A, Doetschman T. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol Biol Cell. 1995;6:1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Corso TD, Torres G, Goulah C, Roy I, Gambino AS, Nayda J, Buckley T, Stachowiak EK, Bergey EJ, Pudavar H, Dutta P, Bloom DC, Bowers WJ, Stachowiak MK. Transfection of tyrosine kinase deleted FGF receptor-1 into rat brain substantia nigra reduces the number of tyrosine hydroxylase expressing neurons and decreases concentration levels of striatal dopamine. Brain Res Mol Brain Res. 2005;139:361–366. doi: 10.1016/j.molbrainres.2005.05.032. [DOI] [PubMed] [Google Scholar]
- Cunningham MG, McKay RD. A hypothermic miniaturized stereotaxic instrument for surgery in newborn rats. J Neurosci Methods. 1993;47:105–114. doi: 10.1016/0165-0270(93)90026-n. [DOI] [PubMed] [Google Scholar]
- Date I, Yoshimoto Y, Imaoka T, Miyoshi Y, Gohda Y, Furuta T, Asari S, Ohmoto T. Enhanced recovery of the nigrostriatal dopaminergic system in MPTP-treated mice following intrastriatal injection of basic fibroblast growth factor in relation to aging. Brain Res. 1993;621:150–154. doi: 10.1016/0006-8993(93)90312-b. [DOI] [PubMed] [Google Scholar]
- Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dono R. Fibroblast growth factors as regulators of central nervous system development and function. Am J Physiol Regul Integr Comp Physiol. 2003;284:R867–R881. doi: 10.1152/ajpregu.00533.2002. [DOI] [PubMed] [Google Scholar]
- Dvorak P, Dvorakova D, Koskova S, Vodinska M, Najvirtova M, Krekac D, Hampl A. Expression and potential role of fibroblast growth factor 2 and its receptors in human embryonic stem cells. Stem Cells. 2005;23:1200–1211. doi: 10.1634/stemcells.2004-0303. [DOI] [PubMed] [Google Scholar]
- Engele J, Bohn MC. The neurotrophic effects of fibroblast growth factors on dopaminergic neurons in vitro are mediated by mesencephalic gliak. J Neurosci. 1991;11:3070–3078. doi: 10.1523/JNEUROSCI.11-10-03070.1991. [Erratum (1992) 12:685] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Brosenitsch TA, Katz DM. Brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor are required simultaneously for survival of dopaminergic primary sensory neurons in vivo. J Neurosci. 2001;21:581–589. doi: 10.1523/JNEUROSCI.21-02-00581.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Minozzi MC, Toffano G, Leon A, Skaper SD. Basic fibroblast growth factor promotes the survival and development of mesencephalic neurons in culture. Dev Biol. 1989;133:140–147. doi: 10.1016/0012-1606(89)90305-9. [DOI] [PubMed] [Google Scholar]
- Ford-Perriss M, Abud H, Murphy M. Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol. 2001;28:493–503. doi: 10.1046/j.1440-1681.2001.03477.x. [DOI] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach M, Riederer P, Przuntek H, Youdim MB. MPTP mechanisms of neurotoxicity and their implications for Parkinson's disease. Eur J Pharmacol. 1991;208:273–286. doi: 10.1016/0922-4106(91)90073-q. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM. Parkin and the molecular pathways of Parkinson's disease. Neuron. 2001;31:885–888. doi: 10.1016/s0896-6273(01)00439-1. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Tanner C. Etiology of Parkinson's disease. In: Jankovic J, Tolosa E, editors. Parkinson's disease and movement disorder. Baltimore: Williams and Wilkins; 1998. pp. 1333–1358. [Google Scholar]
- Granholm AC, Srivastava N, Mott JL, Henry S, Henry M, Westphal H, Pichel JG, Shen L, Hoffer BJ. Morphological alterations in the peripheral and central nervous systems of mice lacking glial cell line-derived neurotrophic factor (GDNF): immunohistochemical studies. J Neurosci. 1997;17:1168–1178. doi: 10.1523/JNEUROSCI.17-03-01168.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G, Shen L, Westphal H, Hoffer B. Glial cell line-derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J Neurosci. 2000;20:3182–3190. doi: 10.1523/JNEUROSCI.20-09-03182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe C, Nikkhah G. The role of basic fibroblast growth factor in peripheral nerve regeneration. Anat Embryol (Berl) 2001;204:171–177. doi: 10.1007/s004290100205. [DOI] [PubMed] [Google Scholar]
- Grothe C, Schulze A, Semkova I, Muller-Ostermeyer F, Rege A, Wewetzer K. The high molecular weight fibroblast growth factor-2 isoforms (21,000 mol. wt and 23,000 mol. wt) mediate neurotrophic activity on rat embryonic mesencephalic dopaminergic neurons in vitro. Neuroscience. 2000;100:73–86. doi: 10.1016/s0306-4522(00)00247-5. [DOI] [PubMed] [Google Scholar]
- Grothe C, Timmer M, Scholz T, Winkler C, Nikkhah G, Claus P, Itoh N, Arenas E. Fibroblast growth factor-20 promotes the differentiation of Nurr1-overexpressing neural stem cells into tyrosine hydroxylase-positive neurons. Neurobiol Dis. 2004;17:163–170. doi: 10.1016/j.nbd.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Grothe C, Haastert K, Jungnickel J. Physiological function and putative therapeutic impact of the FGF-2 system in peripheral nerve regeneration: lessons from in vivo studies in mice and rats. Brain Res Brain Res Rev. 2006;51:293–299. doi: 10.1016/j.brainresrev.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ. Notes on the estimation of numerical density of arbitrary profiles: the edge effect. J Microsc. 1977;111:219–223. [Google Scholar]
- Gundersen HJ. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bagger P, Bendtsen TF, Evans SM, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorenson FB, Vesterby A, West MJ. The new stereological tools: disector, fractionator, nucleator and point-sampled intercepts and their use in pathological research and diagnosis. Acta Pathol Microbiol Immunol Scand. 1988;96:857–881. doi: 10.1111/j.1699-0463.1988.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Hsuan SL, Klintworth HM, Xia Z. Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. J Neurosci. 2006;26:4481–4491. doi: 10.1523/JNEUROSCI.4922-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S, Bernard V, Faucheux B, Boissiere F, Leguern E, Brana C, Gautris PP, Guerin J, Bloch B, Agid Y, Hirsch EC. Glial cell line-derived neurotrophic factor (GDNF) gene expression in the human brain: a post mortem in situ hybridization study with special reference to Parkinson's disease. J Neural Transm. 1996;103:1043–1052. doi: 10.1007/BF01291789. [DOI] [PubMed] [Google Scholar]
- Ji RR, Zhang Q, Zhang X, Piehl F, Reilly T, Pettersson RF, Hokfelt T. Prominent expression of bFGF in dorsal root ganglia after axotomy. Eur J Neurosci. 1995;7:2458–2468. doi: 10.1111/j.1460-9568.1995.tb01044.x. [DOI] [PubMed] [Google Scholar]
- Jin K, LaFevre-Bernt M, Sun Y, Chen S, Gafni J, Crippen D, Logvinova A, Ross CA, Greenberg DA, Ellerby LM. FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2005;102:18189–18194. doi: 10.1073/pnas.0506375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungnickel J, Gransalke K, Timmer M, Grothe C. Fibroblast growth factor receptor 3 signaling regulates injury-related effects in the peripheral nervous system. Mol Cell Neurosci. 2004a;25:21–29. doi: 10.1016/j.mcn.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Jungnickel J, Claus P, Gransalke K, Timmer M, Grothe C. Targeted disruption of the FGF-2 gene affects the response to peripheral nerve injury. Mol Cell Neurosci. 2004b;25:444–452. doi: 10.1016/j.mcn.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Jungnickel J, Klutzny A, Guhr S, Meyer K, Grothe C. Regulation of neuronal death and calcitonin gene-related peptide by fibroblast growth factor-2 and FGFR3 after peripheral nerve injury: evidence from mouse mutants. Neuroscience. 2005;134:1343–1350. doi: 10.1016/j.neuroscience.2005.04.066. [DOI] [PubMed] [Google Scholar]
- Jungnickel J, Haase K, Konitzer J, Timmer M, Grothe C. Faster nerve regeneration after sciatic nerve injury in mice over-expressing basic fibroblast growth factor. J Neurobiol. 2006;66:940–948. doi: 10.1002/neu.20265. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Bjorklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- Kirik D, Georgievska B, Rosenblad C, Bjorklund A. Delayed infusion of GDNF promotes recovery of motor function in the partial lesion model of Parkinson's disease. Eur J Neurosci. 2001;13:1589–1599. doi: 10.1046/j.0953-816x.2001.01534.x. [DOI] [PubMed] [Google Scholar]
- Kirik D, Georgievska B, Bjorklund A. Localized striatal delivery of GDNF as a treatment for Parkinson disease. Nat Neurosci. 2004;7:105–110. doi: 10.1038/nn1175. [DOI] [PubMed] [Google Scholar]
- Klejbor I, Myers JM, Hausknecht K, Corso TD, Gambino AS, Morys J, Maher PA, Hard R, Richards J, Stachowiak EK, Stachowiak MK. Fibroblast growth factor receptor signaling affects development and function of dopamine neurons: inhibition results in a schizophrenia-like syndrome in transgenic mice. J Neurochem. 2006;97:1243–1258. doi: 10.1111/j.1471-4159.2006.03754.x. [DOI] [PubMed] [Google Scholar]
- Knusel B, Michel PP, Schwaber JS, Hefti F. Selective and nonselective stimulation of central cholinergic and dopaminergic development in vitro by nerve growth factor, basic fibroblast growth factor, epidermal growth factor, insulin and the insulin-like growth factors I and II. J Neurosci. 1990;10:558–570. doi: 10.1523/JNEUROSCI.10-02-00558.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, Kodama M, Sasaki H, Yamamoto Y, Takeshita F, Takahama Y, Sakamoto H, Kato T, Terada M, Ochiya T. FGF-4 regulates neural progenitor cell proliferation and neuronal differentiation. FASEB J. 2006;20:1484–1485. doi: 10.1096/fj.05-5293fje. [DOI] [PubMed] [Google Scholar]
- Krieglstein K. Factors promoting survival of mesencephalic dopaminergic neurons. Cell Tissue Res. 2004;318:73–80. doi: 10.1007/s00441-004-0920-8. [DOI] [PubMed] [Google Scholar]
- Lee CS, Sauer H, Bjorklund A. Dopaminergic neuronal degeneration and motor impairments following axon terminal lesion by instrastriatal 6-hydroxydopamine in the rat. Neuroscience. 1996;72:641–653. doi: 10.1016/0306-4522(95)00571-4. [DOI] [PubMed] [Google Scholar]
- L'Hote CG, Knowles MA. Cell responses to FGFR3 signalling: growth, differentiation and apoptosis. Exp Cell Res. 2005;304:417–431. doi: 10.1016/j.yexcr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Liu A, Li JY, Bromleigh C, Lao Z, Niswander LA, Joyner AL. FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development. 2003;130:6175–6185. doi: 10.1242/dev.00845. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of l-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2004;16:110–123. doi: 10.1016/j.nbd.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Mayer E, Dunnett SB, Pellitteri R, Fawcett JW. Basic fibroblast growth factor promotes the survival of embryonic ventral mesencephalic dopaminergic neurons. I. Effects in vitro. Neuroscience. 1993a;56:379–388. doi: 10.1016/0306-4522(93)90339-h. [DOI] [PubMed] [Google Scholar]
- Mayer E, Fawcett JW, Dunnett SB. Basic fibroblast growth factor promotes the survival of embryonic ventral mesencephalic dopaminergic neurons. II. Effects on nigral transplants in vivo. Neuroscience. 1993b;56:389–398. doi: 10.1016/0306-4522(93)90340-l. [DOI] [PubMed] [Google Scholar]
- Meisinger C, Zeschnigk C, Grothe C. In vivo and in vitro effect of glucocorticoids on fibroblast growth factor (FGF)-2 and FGF receptor 1 expression. J Biol Chem. 1996;271:16520–16525. doi: 10.1074/jbc.271.28.16520. [DOI] [PubMed] [Google Scholar]
- Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260–2268. doi: 10.1128/mcb.20.6.2260-2268.2000. [Erratum (2000) 20:3752] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatake Y, Hoshikawa M, Asaki T, Kassai Y, Itoh N. Identification of a novel fibroblast growth factor, FGF-22, preferentially expressed in the inner root sheath of the hair follicle. Biochim Biophys Acta. 2001;1517:460–463. doi: 10.1016/s0167-4781(00)00302-x. [DOI] [PubMed] [Google Scholar]
- Nikkhah G, Cunningham MG, Jodicke A, Knappe U, Bjorklund A. Improved graft survival and striatal reinnervation by microtransplantation of fetal nigral cell suspensions in the rat Parkinson model. Brain Res. 1994;633:133–143. doi: 10.1016/0006-8993(94)91532-6. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492:203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci. 2003;23:883–894. doi: 10.1523/JNEUROSCI.23-03-00883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmachi S, Watanabe Y, Mikami T, Kusu N, Ibi T, Akaike A, Itoh N. FGF-20, a novel neurotrophic factor, preferentially expressed in the substantia nigra pars compacta of rat brain. Biochem Biophys Res Commun. 2000;277:355–360. doi: 10.1006/bbrc.2000.3675. [DOI] [PubMed] [Google Scholar]
- Ohmachi S, Mikami T, Konishi M, Miyake A, Itoh N. Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J Neurosci Res. 2003;72:436–443. doi: 10.1002/jnr.10592. [DOI] [PubMed] [Google Scholar]
- Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996;366:580–599. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Uruno T, Miyakawa K, Seo M, Imamura T. Expression of the fibroblast growth factor family and their receptor family genes during mouse brain development. Brain Res Mol Brain Res. 1996;41:279–288. doi: 10.1016/0169-328x(96)00108-8. [DOI] [PubMed] [Google Scholar]
- Parain K, Murer MG, Yan Q, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R. Reduced expression of brain-derived neurotrophic factor protein in Parkinson's disease substantia nigra. NeuroReport. 1999;10:557–561. doi: 10.1097/00001756-199902250-00021. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- Reifers F, Adams J, Mason IJ, Schulte-Merker S, Brand M. Overlapping and distinct functions provided by fgf17, a new zebrafish member of the Fgf8/17/18 subgroup of Fgfs. Mech Dev. 2000;99:39–49. doi: 10.1016/s0925-4773(00)00475-5. [DOI] [PubMed] [Google Scholar]
- Rodter A, Winkler C, Samii M, Nikkhah G. Complex sensorimotor behavioral changes after terminal striatal 6-OHDA lesion and transplantation of dopaminergic embryonic micrografts. Cell Transplant. 2000;9:197–214. doi: 10.1177/096368970000900206. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6-hydroxydopamine: a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Schober A. Classic toxin-induced animal models of Parkinson's disease: 6-OHDA and MPTP. Cell Tissue Res. 2004;318:215–224. doi: 10.1007/s00441-004-0938-y. [DOI] [PubMed] [Google Scholar]
- Shan X, Chi L, Bishop M, Luo C, Lien L, Zhang Z, Liu R. Enhanced de novo neurogenesis and dopaminergic neurogenesis in the substantia nigra of 1-methyl-4-phyenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease-like mice. Stem Cells. 2006;24:1280–1287. doi: 10.1634/stemcells.2005-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shults CW, Ray J, Tsuboi K, Gage FH. Fibroblast growth factor-2-producing fibroblasts protect the nigrostriatal dopaminergic system from 6-hydroxydopamine. Brain Res. 2000;883:192–204. doi: 10.1016/s0006-8993(00)02900-0. [DOI] [PubMed] [Google Scholar]
- Soderstrom K, O'Malley J, Steece-Collier K, Kordower JH. Neural repair strategies for Parkinson's disease: insights from primate models. Cell Transplant. 2006;15:251–265. doi: 10.3727/000000006783982025. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: implications for Parkinson's disease. FASEB J. 2002;16:1474–1476. doi: 10.1096/fj.02-0216fje. [DOI] [PubMed] [Google Scholar]
- Stachowiak MK, Moffett J, Joy A, Puchacz E, Florkiewicz R, Stachowiak EK. Regulation of bFGF gene expression and subcellular distribution of bFGF protein in adrenal medullary cells. J Cell Biol. 1994;127:203–223. doi: 10.1083/jcb.127.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L, Tabar V, McKay RD. Transplantation of expanded mesencephalic precursors leads to recovery in parkinsonian rats. Nat Neurosci. 1998;1:290–295. doi: 10.1038/1105. [DOI] [PubMed] [Google Scholar]
- Takayama H, Ray J, Raymon HK, Baird A, Hogg J, Fisher LJ, Gage FH. Basic fibroblast growth factor increases dopaminergic graft survival and function in a rat model of Parkinson's disease. Nat Med. 1995;1:53–58. doi: 10.1038/nm0195-53. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Damlama M, Trent F. Postnatal changes in the distribution and morphology of rat substantia nigra dopaminergic neurons. Neuroscience. 1994;60:469–477. doi: 10.1016/0306-4522(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Timmer M, Muller-Ostermeyer F, Kloth V, Winkler C, Grothe C, Nikkhah G. Enhanced survival, reinnervation, and functional recovery of intrastriatal dopamine grafts co-transplanted with Schwann cells overexpressing high molecular weight FGF-2 isoforms. Exp Neurol. 2004;187:118–136. doi: 10.1016/j.expneurol.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Timmer M, Grosskreutz J, Schlesinger F, Krampfl K, Wesemann M, Just L, Bufler J, Grothe C. Dopaminergic properties and function after grafting of attached neural precursor cultures. Neurobiol Dis. 2006;21:587–606. doi: 10.1016/j.nbd.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Todo T, Kondo T, Nakamura S, Kirino T, Kurokawa T, Ikeda K. Neuronal localization of fibroblast growth factor-9 immunoreactivity in human and rat brain. Brain Res. 1998;783:179–187. doi: 10.1016/s0006-8993(97)01340-1. [DOI] [PubMed] [Google Scholar]
- Tooyama I, Kawamata T, Walker D, Yamada T, Hanai K, Kimura H, Iwane M, Igarashi K, McGeer EG, McGeer PL. Loss of basic fibroblast growth factor in substantia nigra neurons in Parkinson's disease. Neurology. 1993;43:372–376. doi: 10.1212/wnl.43.2.372. [Erratum 43:815–816] [DOI] [PubMed] [Google Scholar]
- Tooyama I, McGeer EG, Kawamata T, Kimura H, McGeer PL. Retention of basic fibroblast growth factor immunoreactivity in dopaminergic neurons of the substantia nigra during normal aging in humans contrasts with loss in Parkinson's disease. Brain Res. 1994;656:165–168. doi: 10.1016/0006-8993(94)91378-1. [DOI] [PubMed] [Google Scholar]
- Ueba T, Nosaka T, Takahashi JA, Shibata F, Florkiewicz RZ, Vogelstein B, Oda Y, Kikuchi H, Hatanaka M. Transcriptional regulation of basic fibroblast growth factor gene by p53 in human glioblastoma and hepatocellular carcinoma cells. Proc Natl Acad Sci USA. 1994;91:9009–9013. doi: 10.1073/pnas.91.19.9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24:485–493. doi: 10.1016/0006-8993(70)90187-3. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:l-22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Winkler C, Georgievska B, Carlsson T, Lacar B, Kirik D. Continuous exposure to glial cell line-derived neurotrophic factor to mature dopaminergic transplants impairs the graft's ability to improve spontaneous motor behavior in parkinsonian rats. Neuroscience. 2006;141:521–531. doi: 10.1016/j.neuroscience.2006.03.068. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Ladher R, McWhirter J, Murre C, Schoenwolf GC, Mansour SL. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev Biol. 2004;269:264–275. doi: 10.1016/j.ydbio.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Xie MH, Holcomb I, Deuel B, Dowd P, Huang A, Vagts A, Foster J, Liang J, Brush J, Gu Q, Hillan K, Goddard A, Gurney AL. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine. 1999;11:729–735. doi: 10.1006/cyto.1999.0485. [DOI] [PubMed] [Google Scholar]
- Xu C, Rosler E, Jiang J, Lebkowski JS, Gold JD, O'Sullivan C, Delavan-Boorsma K, Mok M, Bronstein A, Carpenter MK. Basic fibroblast growth factor supports undifferentiated human embryonic stem cell growth without conditioned medium. Stem Cells. 2005;23:315–323. doi: 10.1634/stemcells.2004-0211. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- Yazaki N, Hosoi Y, Kawabata K, Miyake A, Minami M, Satoh M, Ohta M, Kawasaki T, Itoh N. Differential expression patterns of mRNAs for members of the fibroblast growth factor receptor family, FGFR-1-FGFR-4, in rat brain. J Neurosci Res. 1994;37:445–452. doi: 10.1002/jnr.490370403. [DOI] [PubMed] [Google Scholar]
- Zaragosi LE, Ailhaud G, Dani C. Autocrine FGF2 signaling is critical for self-renewal of human multipotent adipose-derived stem cells. Stem Cells. 2006;24:2412–2419. doi: 10.1634/stemcells.2006-0006. [DOI] [PubMed] [Google Scholar]
- Zechel S, Jarosik J, Kiprianova I, Schober A, Unsicker K, von Bohlen und Halbach O. FGF-2 deficiency does not alter vulnerability of the dopaminergic nigrostriatal system towards MPTP intoxication in mice. Eur J Neurosci. 2006;23:1671–1675. doi: 10.1111/j.1460-9568.2006.04700.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 control of vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]