

Abstract
Cellular cholesterol levels alter the processing of the amyloid precursor protein (APP) to produce Aβ. Activation of liver X receptors (LXRs), one cellular mechanism to regulate cholesterol homeostasis, has been found to alter Aβ levels in vitro and in vivo. To identify genes regulated by LXR, we treated human neuroblastoma cells with an LXR agonist (TO-901317) and examined gene expression by microarray. As expected, TO-901317 upregulated several cholesterol metabolism genes, but it also decreased expression of a metalloprotease inhibitor, TIMP-3. We confirmed this finding using real-time PCR and by measuring TIMP-3 protein in glia, SY5Y cells, and COS7 cells. TIMP-3 is a member of a family of metalloproteinase inhibitors and blocks A disintegrin and metalloproteinase-10 (ADAM-10) and ADAM-17, two APP α-secretases. We found that TIMP-3 inhibited α-secretase cleavage of APP and an apolipoprotein E (apoE) receptor, ApoER2. TIMP-3 decreased surface levels of ADAM-10, APP, and ApoER2. These changes were accompanied by increased APP β-C-terminal fragment and Aβ production. These data suggest that TIMP-3 preferentially routes APP and ApoER2 away from the cell surface and α-secretase cleavage and encourages endocytosis and β-secretase cleavage. In vivo, TO-901317 decreased brain TIMP-3 levels. TIMP-3 protein levels were increased in human Alzheimer's disease (AD) brain and in APP transgenic mice, suggesting that increased levels of TIMP-3 in AD may contribute to higher levels of Aβ.
Keywords: Alzheimer's disease, secretase, LXR, apolipoprotein, amyloid, neurodegeneration
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder of old age, characterized partially by the presence in the brain of senile plaques. These plaques are composed of the Aβ peptide, a 40 or 42 aa cleavage product of the amyloid precursor protein (APP). APP, a type I transmembrane protein, undergoes extracellular cleavage by one of two proteolytic activities, α- or β-secretase, resulting in the formation of a large N-terminal extracellular fragment [secreted APP (sAPP)] and smaller, membrane-bound C-terminal fragments (CTFs). Cleavage of APP by α-secretase precludes the formation of intact Aβ peptide, and it has been shown that the large ectodomain of APP released from the cell surface (sAPPα) has both neuroprotective and memory-enhancing effects (Furukawa et al., 1996). If the initial cleavage event occurs via a β-secretase activity, then cleavage of the CTF by γ-secretase results in the formation of Aβ (Walsh and Selkoe, 2004).
Little is known about the regulatory mechanisms of the enzymes responsible for the proteolysis of APP. TIMP-3 is a member of a family of four secreted proteins (TIMP-1 to TIMP-4) that were originally identified as inhibitors of the matrix metalloproteinases (MMPs) (Murphy et al., 1994). By inhibiting MMP activity, they participate in tissue remodeling of the extracellular matrix (ECM). TIMP-3 has recently been found to inhibit other zinc-dependent proteases, including members of the A disintegrin and metalloproteinase (ADAM) family (Loechel et al., 2000; Hashimoto et al., 2001). TIMP-3 has specifically been shown to inhibit the actions of ADAM-10 and ADAM-17 (Amour et al., 1998, 2000; Zou et al., 2004), two APP α-secretases (Allinson et al., 2004).
The APP secretases consist of membrane-spanning proteins, whose activities are affected by cholesterol levels in the plasma membrane. Cells have several mechanisms for maintaining tight control over cellular cholesterol levels (Weber et al., 2004). One mechanism involves the nuclear hormone receptor, LXR, which acts as a transcription factor (Lu et al., 2001). LXR induces expression of ABC-A1 and ABC-G1, which pump cholesterol and phospholipids from the membrane to the extracellular space, and apolipoprotein E (apoE), which acts as an acceptor for these lipids. Changes in cholesterol levels have been associated with changes in APP processing and Aβ production (Bodovitz and Klein, 1996; Simons et al., 1998).
In this work, we find that LXR activation reduced the expression of TIMP-3 in vitro and in vivo. TIMP-3 altered the intracellular trafficking of APP, reducing levels on the cell surface (and the associated α-secretase cleavage of APP at this site) and enhancing the β-secretase cleavage of APP. Similar effects were seen on another membrane-bound protein, apoE receptor 2 (ApoER2), which undergoes similar proteolytic processing to APP. Levels of TIMP-3 were increased in an animal model of AD and in human AD brain. TIMP-3 modulation could define a new mechanism by which APP processing is regulated.
Materials and Methods
Vectors.
Human TIMP-3 constructs were generated with a C-terminal myc tag in the pSecTag2/HygroB expression vector. Human APP constructs were generated with an N-terminal green fluorescent protein (GFP) tag in the enhanced GFP (EGFP)-C1 and C-terminal myc tag in the pSecTag2/HygroB. Recombinant DNA was confirmed by sequencing, and expression of correctly sized proteins was confirmed by Western blot. Dr. Stefan F. Lichtenthaler provided the hemagglutinin (HA)-tagged ADAM-10 cDNA.
RNA interference.
Small interfering RNA (siRNA) was obtained from Ambion (Austin, TX) against mouse TIMP-3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (as control) [catalog #4624 for GAPDH; identification (ID) #187322 and ID #187324 for TIMP-3]. siRNAs were transfected into N2a cells by using electroporation (AMEXA, Gaithersburg, MD). Cells were treated with AMEXA electroporation transfection solution and 10–40 nm siRNA using the C-9 program. After an overnight exposure, cells were washed, and media were replaced with DMEM [10% fetal bovine serum (FBS)]. Inhibition of targeted gene expression was confirmed by Western blot.
Lentiviral constructs and short hairpin RNA production.
TIMP-3 constructs were introduced into plasmid FSW lentiviral vector under the synapsin promoter. To generate the TIMP-3 short hairpin RNA (shRNA), target sequences were designed against rat TIMP-3 and used to synthesize oligonucleotides comprising a forward and reverse sequence separated by a linker sequence (CGAA). Lentiviral vectors expressing shRNA were then constructed using the BLOCK-iT system according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Briefly, oligonucleotides were ligated into the pENTR/U6 vector and then transferred to the pLenti6/BLOCK-iT-DEST vector via LR recombination. Two target sequences (2A, GCAAGATCAAGTCCTGCTACT; 3A, GCTACTACTTGCCTTGCTTTG) proved competent for reduction of TIMP-3 expression compared with cells transduced with lentivirus containing a nonsense control sequence (GCCTAGACGCGATAGTATGGA). To produce the lentivirus, 293FT cells were transiently transfected with the following constructs: 5 μg of VSVg (vesicular stomatitis virus glycoprotein), 15 μg of δ-8.9, and 20 μg of promoter-reporter plasmid. Approximately 4 h after transfection, medium was changed and incubated for an additional 48–60 h. Virus was concentrated and stored at −80°C. Primary neurons were infected with FSW vector, TIMP-3, or TIMP-3 shRNA for 5 d.
Gene array.
Analysis of RNA expression by Affymetrix (Santa Clara, CA) array was performed as described previously (Massaro et al., 2006). Briefly, total RNA was isolated from SY5Y cells using TRIzol (Invitrogen) and purified with RNeasy mini kit (Qiagen, Chatsworth, CA). Superscript Choice System (Invitrogen) was used to convert RNA to double-stranded DNA (dsDNA). dsDNA was purified by phenol/chloroform extraction and used for transcription with the Enzo (New York, NY) Bioarray RNA transcription-labeled kit. Resulting biotin-labeled cRNA was purified using RNeasy, randomly fragmented, and hybridized using an Affymetrix Fluidics Station 400 and scanned using a Hewlett Packard (Palo Alto, CA) G2500/7 Gene Array scanner (Massaro et al., 2006).
Reverse transcription-PCR of TIMP-3.
Total RNA was isolated from COS7, SY5Y, primary neurons, and primary glia using RNeasy mini kit (Qiagen). Reverse transcription (RT)-PCR was performed from 0.5 μg of RNA (Omniscript RT; Qiagen) using an oligo-dT primer. Amplification was performed on a thermal cycler (PCR Express; Hybaid, Ashford, UK) using HotStarTaq DNA polymerase (Qiagen). After generating cDNA, quantitative RT-PCR was then performed with custom primers for TIMP-3 from the Applied Biosystems library (Mm00441825m1 for mouse TIMP-3; Mm99999915g1 for mouse GAPDH; Hs00165949m1 for human TIMP-3; Hs99999905m1 for human GAPDH).
Primary and secondary cell lines and culture conditions.
COS7, SY5Y, Neuro2A, and HEK293 cells were maintained in Opti-MEM (Invitrogen) with 10% FBS. TIMP-3 and control conditioned media (CMs) were prepared as described previously (Chen et al., 2004). Briefly, COS7 cells were transfected with TIMP-3 plasmids or with an empty pSecTag2/HygroB vector. After 24 h, cells were incubated in Opti-MEM serum-free media and conditioned media collected at 48 h after transfection. Conditioned media was concentrated 30-fold using a Centricon centrifugal filter with a 10,000 molecular weight cutoff (Millipore, Billerica, MA), aliquoted, and stored at −80°C.
Primary mouse embryonic cortical neuron cultures were prepared from embryonic day 16 Swiss-Webster mice as described previously (Qiu et al., 2002). Brain cortices were chopped and trypsinized for 10 min at 37°C. After trypsinization, 0.4 μg/ml trypsin inhibitor, 0.025% DNase, and 12 mm MgSO4 were added and mixed until tissue was thoroughly homogenized. Cells were then transferred to Neurobasal medium containing B27 serum supplement, 1 mm glutamine, gentamycin, and AraC. Neurons were seeded on 50 μg/ml poly-d-lysine-coated 12-well tissue culture plates at a density of 2 × 106 cells per well.
Primary hippocampal neurons from embryonic day 18–19 Sprague Dawley rats were cultured at 150 cells/mm2 as described previously (Pak et al., 2001). Neurons were transfected at 10 or 12 d in vitro (DIV) by calcium phosphate precipitation (4–5 μg of DNA per well). Six or eight days after transfection, we analyzed cell surface expression levels of APP and endocytosis of APP in vitro (DIV 16–18) (see below).
Primary astrocyte cultures were prepared from cerebral cortex of 1- to 2-d-old Swiss-Webster mice (LaDu et al., 2000). Cells were maintained in Eagle's minimum essential medium (EMEM; Invitrogen) containing 10% FBS, 10% horse serum (Invitrogen), gentamycin (Invitrogen), Fungizone (250 μg/ml; 1:100; Invitrogen), antibiotics (10,000 U/ml penicillin G, 25 μg/ml amphotericin B, and 10,000 μg/ml streptomycin; 1:100), and epidermal growth factor (EGF) (1:1000; Sigma-Aldrich, St. Louis, MO). For experiments, secondary glial cells were seeded on 50 μg/ml poly-d-lysine-coated 12-well tissue culture plates at a density of 2 × 106 per well. After 24 h, cells were washed twice with PBS to remove serum and incubated in serum-free EMEM containing EGF. Each set of primary cells was treated either with LXR agonist (TO-901317; 5 μm) or vehicle (ethanol) at a final concentration of 0.05%, and cell lysates were collected after 24 h.
Antibodies.
The following antibodies were used: anti-HA (Abcam, Cambridge, MA), anti-c-myc (Abcam), anti-APP (Sigma-Aldrich), anti-sAPPα (Immuno-Biological Laboratories, Takasaki, Japan), and anti-TIMP-3 (Millipore). For analysis of APP, antibody 22C11 recognizing the extracellular domain of APP (Millipore) and 6E10 identifying sAPPα and β-CTF (Signet, Dedham, MA) were used. Dr. Paul Mathews (Nathan S. Kline Institute, Orangeburg, NY) provided antibody c1/6.1, and Dr. Sam Gandy (Thomas Jefferson University, Philadelphia, PA) provided antibody 369, both of which recognize the C terminus of APP. Rabbit polyclonal antibody 5810 against mouse ApoER2 was the gift from Dr. Uwe Beffert (University of Texas, Southwestern, Dallas, TX).
Measurement of ApoER2 and APP proteolytic fragments by Western blot and ELISA.
COS7 cells were transiently transfected with APP770 or ApoER2 (containing a C-terminal HA tag) (Hoe and Rebeck, 2005) using Fugene 6 (Roche Applied Science, Indianapolis, IN) according to the manufacturer's protocol and cultured in DMEM containing 10% FBS for 24 h. Cells were maintained for another 24 h in serum-free media with or without TIMP-3 conditioned media. Proteins from cell extracts or conditioned media were separated using Tris-glycine PAGE. Proteins were transferred onto polyvinylidene difluoride (PVDF) membrane and probed with relevant primary antibodies for 1 h at room temperature. Horseradish peroxidase-conjugated secondary antibody was visualized by ECL detection system (Pierce, Rockford, IL) and exposed to film. Secreted fragments were determined from the media (sAPPα, 6E10 antibody; sApoER2, 5810 antibody). CTF were measured by Western blot from cell lysates (APP CTF, C1/6.1; APP β-CTF, 6E10; ApoER2 CTF, HA antibodies). Aβ40 and Aβ42 levels in the conditioned media were determined by ELISA, using 1A10 (anti-Aβ40) or 1C3 (anti-Aβ42) as capture antibodies and 12B2, which recognizes both mouse and human Aβ, as a detection antibody (Immuno-Biological Laboratories) (Horikoshi et al., 2004).
Biotin-labeled cell surface proteins.
COS7 cells were transiently transfected with APP and vector, APP and TIMP-3, ApoER2 and vector, or ApoER2 and TIMP-3, and cultured for 24 h in DMEM containing 10% FBS. After 24 h, serum-free medium was added with or without TIMP-3 conditioned media. After 24 h, cells were washed, and surface proteins were labeled with Sulfo-NHS-SS-Biotin at 500 μg/ml PBS (Pierce) under gentle shaking at 4°C for 30 min. After quenching, cells were lysed, disrupted by sonication, and clarified by centrifugation (10,000 × g for 2 min). To isolate biotin-labeled proteins, lysate was added to immobilized NeutrAvidin gel and incubated for 1 h at room temperature. Gels were washed and incubated with SDS-PAGE sample buffer including 50 mm dithiothreitol (DTT). Eluants were analyzed by immunoblotting.
Live-cell surface staining.
HEK cells were washed with PBS and incubated with primary antibody diluted in ECS (145 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 5 mm HEPES, 5 mm glucose, 15 mm sucrose, 0.25 mg/L phenol red, and 10 μm d-serine (Sigma-Aldrich), pH 7.4, for 8 min at room temperature. Antibodies used were 6E10 (recognizing the N terminus of APP) or 5810 (recognizing the N terminus of ApoER2). After washing with ECS, one of the fluorescent secondary antibodies Texas Red goat anti-mouse antibody or Alexa 488 goat anti-rabbit antibody (both from Invitrogen) was incubated for 8 min at room temperature. Cells were fixed with 4% paraformaldehyde for 5 min. Stained cells were imaged on an Axioskop FS microscope (Zeiss, Oberkochen, Germany) equipped with a 63×, 0.9 numerical aperture (NA), Achroplan, water-immersion objective or with a Nikon (Tokyo, Japan) E600 microscope equipped with a 60×, 1.0 NA objective.
For hippocampal neurons, surface immunostaining was performed as described previously (Fu et al., 2007). Briefly, live neuronal cultures were incubated with 6E10 (10 μg/ml in conditioned medium) for 10 min to specifically label surface APP and then lightly fixed for 5 min in 4% paraformaldehyde (nonpermeabilizing conditions). After fixation, the surface-remaining antibody labeled APP was measured with Alexa Fluor 555-linked α-mouse secondary antibodies for 1 h.
Quantitation and image analysis.
Images were collected using a Zeiss LSM510 confocal microscope. Confocal z-series image stacks encompassing entire neurons were analyzed using MetaMorph software (Universal Imaging, Downingtown, PA) (Fu et al., 2007).
Pulse-chase analysis of APP.
COS7 cells were transfected with either APP V5 and vector or APP V5 and TIMP-3 myc using Fugene 6 (Roche Applied Science) as per the manufacturer's instructions. Forty-eight hours after transfection, cells were starved for 1 h in serum-free DMEM without cysteine or methionine before being pulsed with DMEM containing Promix [35S] cysteine/methionine (GE Healthcare, Piscataway, NJ) at 200 μCi/well for 3 h. After the pulse, cells were washed once in PBS and incubated with complete DMEM with 10% FBS, with wells harvested in triplicate at 0, 30, 60, 120, and 240 min time points. Cells were washed and lysed in lysis buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris HCl, pH 7.6, plus 0.25% NP-40 and 1× complete protease inhibitors) for 30 min on ice. Lysates were precleared with 30 μl of 50% protein G agarose slurry (Sigma-Aldrich) for 1 h at 4°C and spun down at 5000 × g for 5 min. The resulting supernatants were incubated with 2 μg of anti-V5 monoclonal antibody (Invitrogen) for 2 h at 4°C, with 30 μl of 50% protein G-agarose slurry being added for an additional 2 h. Agarose/antibody complexes were pulled down (5000 × g for 5 min) and washed five times with 400 μl of radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris HCl, 150 mm NaCl, pH 7.4, plus 0.1% SDS, 1% deoxycholate, 1% Triton X-100, phosphatase inhibitors, 1× complete protease inhibitors, and proteasome inhibitors). Bound protein was eluted by boiling for 10 min in 2 × Laemmli sample buffer plus 5% β-mercaptoethanol (Bio-Rad, Hercules, CA) and samples separated using SDS-PAGE on 4–12% gels as described previously. After transfer to PVDF (Millipore), membranes were exposed to a phosphoscreen for between 24 and 48 h and scanned using a Storm 840 phosphoimager (GE Healthcare). Resulting bands were quantified using ImageQuant software (GE Healthcare). Data were analyzed, standard curves were generated, and half-lives were calculated using Prism 4 (GraphPad, San Diego, CA).
Mouse and human brain tissue.
To measure the effects of LXR agonist on APP processing in vivo, male Swiss-Webster mice (Taconic, Rockville, MD) were administered 50 mg/kg TO-901317 by oral gavage in 0.25% methyl cellulose and 1% Tween 80 at a final volume of 5 ml/kg. Twenty-four hours after drug administration, mice were killed, and brains were removed and frozen at −80°C until assay. For a mouse model of AD, we used triple-transgenic mice overexpressing APP, presenilin, and tau, which exhibit pathological changes similar to those found in AD patients (Oddo et al., 2003). Brain tissue from mice of different ages was fixed in paraformaldehyde, sectioned at 30 micrometers, and used for immunohistochemistry. Tissue was blocked with MOM reagent (Vector Laboratories, Burlingame, CA) and probed with a monoclonal antibody against TIMP-3. The presence of the TIMP-3 antibody was detected using biotinylated secondary antibody and HRP-linked avidin, using ABC development with 3,3-diaminobenzidine as chromogen (Vector Laboratories). For immunoblots, frozen brain tissue was extracted in RIPA buffer for analysis of TIMP-3 and ABCA1 (Novus Biologicals, Littleton, CO).
Age-matched human AD and control tissue (between 66 and 94 years of age) was obtained from the Department of Neuropathology, Johns Hopkins University (Baltimore, MD). Parkinson's disease (PD) control tissue was obtained from the National Institute on Aging. Protein was extracted from human frontal cortex in 100 mmol/L KCl, 5 mmol/L MgCl2, 10 mmol/L HEPES, pH 7.0, 0.5% Nonidet P-40, 1 mmol/L DTT, and complete protease inhibitor mixture.
Statistical analyses.
All data were analyzed with GraphPad Prism 4 software using either Student's t test or one-way ANOVA followed by a Newman–Keuls post hoc test. Descriptive statistics were displayed as an expressed mean ± SEM. Differences were deemed significant when p < 0.05.
Results
Regulation of TIMP-3 by an LXR agonist
To identify genes regulated by LXR activation, we treated SY5Y human neuroblastoma cells with an LXR agonist, TO-901317 (5 μm), for 24 h and screened for changes in mRNA expression using Affymetrix gene chips in triplicate. The largest significant increases were observed for genes known to be induced by LXR, including abcg1, abca1, and srebp (Table 1). The largest significant decrease (65%) was observed for the gene encoding the metalloprotease inhibitor TIMP-3, a gene not previously associated with LXR regulation.
Table 1.
Gene expression profile analysis of SY5Y cells treated with TO-901317
| Genes identified | Control (%) | t test p value |
|---|---|---|
| abcg1 | 418 | 0.029 |
| abca1 | 264 | 0.049 |
| srebp1 | 230 | 0.018 |
| TIMP-3 | 35 | 0.041 |
SY5Y cells were treated with vehicle (ethanol) or the LXR agonist TO-901317 (5 μm). Relative mRNA levels in comparison to control were measured in triplicate for 20,000 genes by Affymetrix microarray analyses. Significant changes in abcg1, abca1, srebp-1, and TIMP-3 mRNA levels are shown here expressed as a percentage of control levels, with corresponding p values.
We tested whether the reduction in TIMP-3 message after TO-901317 treatment observed by gene chip analysis was reproducible, using RT-PCR in various cells: primary neurons, SY5Y, and COS7 cells. Cells were treated in triplicate with 5 μm TO-901317 for 16 h, and TIMP-3 mRNA was measured. Compared with cells treated with vehicle alone, TO-901317-treated cells showed significant reductions in TIMP-3 mRNA: by 66% in primary glia, 55% in SY5Y cells, and 42% in COS7 cells (p < 0.05 for all groups) (Fig. 1A). To further test this transcriptional regulation of TIMP-3, we used quantitative real-time PCR of cDNA from primary glial cells and primary neuronal cells treated with 5 μm TO-901317 for 16 h. Using this third measure of TIMP-3 mRNA, we found similar and significant decreases in TIMP-3 message after TO-901317, by 57% in primary neurons and 79% in primary glia (p < 0.05).
Figure 1.

LXR agonist decreases levels of TIMP-3. A, Primary glia, SH-SY5Y cells, or COS7 cells were treated in triplicate with control (ethanol; C) or 5 μm TO-901317 (TO) for 16 h. RT-PCR of isolated RNA shows that TIMP-3 mRNA levels were reduced after TO-901317 treatment in primary glia, SY5Y, and COS7 cells. As a control, GAPDH mRNA levels were tested in COS7 cells and found unchanged by TO-901317. B, Primary neurons and primary glia were treated in triplicate with vehicle (ethanol) or 5 μm TO-901317 for 16 h. Real-time PCR showed that TIMP-3 message was significantly decreased in both glia and neurons by TO-901317 (p < 0.05). B, COS7 cells were transfected with empty vector (lane 1) or TIMP-3 (lane 2). Secreted TIMP-3 was measured in conditioned media (15 μl) with anti-myc antibody. Secreted TIMP-3 was detected as 25 kDa (lane 2). C, COS7 cells were transfected with empty vector (lane 1) or TIMP-3 (lane 2). TIMP-3 immunoreactivity in cell lysate (20 μg) was detected as 50 kDa (endogenous) and 25 kDa (overexpressed) (arrows). D, N2a cells were transfected with control siRNA (−) or TIMP-3 siRNA (+) at the indicated concentrations (10–40 nm). Top, Immunoblotting with anti-TIMP-3 antibody revealed a clear reduction in the TIMP-3 50 kDa band after TIMP-3 siRNA treatment in a dose-responsive manner. Bottom, There were no significant changes in the amount of β-actin after any siRNA treatments. E, Primary glia or primary neurons were treated with control (ethanol; lanes 1, 3) or 5 μm TO-901317 (lanes 2, 4) for 16 h. Immunoblotting of cell lysates (20 μg) with anti-TIMP-3 antibody revealed a reduction in the level of 50 kDa TIMP-3 after TO-901317 treatment. β-Actin levels were not altered by TO-901317.
We transfected COS7 cells with a TIMP-3 construct tagged with a myc epitope and observed the appearance of a 20–25 kDa band in conditioned media, consistent with the expected size of TIMP-3 (Fig. 1B). In Western blots of the cell extracts from COS7 cells, we observed only a 50 kDa band with an anti-TIMP-3 antibody (Fig. 1C, lane 1). Previous studies have shown that TIMP-3 appears in Western blots as a 50 kDa band (Plumb et al., 2005). Transfected COS7 cells also showed an appearance of a 20–25 kDa band in cell lysates, although the level of the 50 kDa band was unchanged (Fig. 1C, lane 2). To explore the immunoreactivity of TIMP-3 in CNS cells, we used Neuro2A and treated them with increasing concentrations of TIMP-3 siRNA. The 50 kDa TIMP-3 band was also present in Neuro2A cell extracts, and it was reduced in a dose-dependent manner by the TIMP-3 siRNA (Fig. 1D); β-actin levels were unaffected by the siRNA. Thus, we conclude that TIMP-3 is made and secreted as a 20–25 kDa protein, but can remain cell associated, perhaps as a stable dimer.
We then asked whether TO-901317 treatment decreased TIMP-3 protein levels. TIMP-3 immunoblotting of primary glia and neuron extracts revealed only the 50 kDa band (Fig. 1E). Treatment with 5 μm TO-901317 significantly decreased TIMP-3 protein expression in primary glia (by 98%; p < 0.01). The decrease in TIMP-3 protein levels in primary neuronal cells after treatment with TO-901317 was, however, less marked (Fig. 1E): expression was reduced by 34%, which did not reach statistical significance. Together, these protein data are consistent with the mRNA data, demonstrating that TIMP-3 expression is negatively regulated by the LXR agonist TO-901317.
TIMP-3 affects APP processing
ADAM-10 and ADAM-17 have been defined as α-secretases acting on APP. Because TIMP-3 inhibits ADAM-10 and ADAM-17 (Amour et al., 1998, 2000; Allinson et al., 2004), we examined the impact of TIMP-3 on APP processing. COS7 cells were transfected with either APP and vector (Fig. 2A, lanes 1, 3) or APP and TIMP-3 (lanes 2, 4). Secreted APP was measured in conditioned media, and full-length APP and APP CTF were measured in cell lysates. Transfection with TIMP-3 decreased sAPPα and APP CTF without affecting levels of total APP (Fig. 2A, top and middle). This apparent decrease in α-secretase cleavage in APP was also accompanied by an increase in β-secretase cleavage in APP, as evidenced by increased APP β-CTF (Fig. 2A, bottom). We also tested whether TIMP-3 conditioned media had a similar affect on APP processing. COS7 cells transfected with APP were treated with media from TIMP-3-overexpressing cells for 16 h. As with overexpression of TIMP-3, exogenous TIMP-3 decreased secreted APP and APP CTF (Fig. 2B).
Figure 2.

TIMP-3 inhibits α-cleavage of APP. A, COS7 cells were transfected with APP and vector (lanes 1, 3) or APP and TIMP-3 (lanes 2, 4). Secreted APP was measured in conditioned media (15 μl) with antibody 6E10. Cell proteins (20 or 50 μg) were measured with C1/6.1 [full-length (FL) APP and α-CTF] or anti-6E10 (β-CTF). TIMP-3 treatment decreased secreted APP and APP α-CTF levels and increased β-CTF levels. B, COS7 cells were transfected with APP and then treated with TIMP-3-containing conditioned medium for 16 h. TIMP-3 treatment decreased secreted APP and APP CTF. C, COS7 cells were transfected with APP (lanes 1–3) and ADAM-10 cDNA (lanes 2, 3). Cells expressing APP and ADAM-10 were treated with TIMP-3 CM (lane 3). ADAM-10 expression increased cleavage fragments of APP (lane 2). Extracellular TIMP-3 blocked the increased APP cleavage by ADAM-10 (lane 3). D, Primary cortical neurons were infected with TIMP-3 and TIMP-3 shRNA for 5 d. Top, TIMP-3 levels in neuronal lysates were measured by Western blot. Bottom histogram, Aβ40 levels in the conditioned media were determined by ELISA. TIMP-3 significantly increased secreted Aβ40 by 96% (p < 0.05), whereas inhibition of endogenous TIMP-3 significantly decreased secreted Aβ40 by 48% compared with control lentivirus vector (p < 0.05).
Because TIMP-3 affected β-processing of APP, we measured another product of APP β-cleavage, Aβ. COS7 cells were cotransfected with APP and vector or APP and TIMP-3, and secreted Aβ40 and Aβ42 were measured in conditioned media by ELISA. Cotransfection with TIMP-3 significantly increased Aβ40 (by 134%; p < 0.01) and Aβ42 (by 91%; p < 0.05). TIMP-3 conditioned media had a similar effect as cotransfection with APP and TIMP-3 in COS7 cells, leading to significantly increased levels of Aβ40 (by 69%; p < 0.05) and Aβ42 (by 98%; p < 0.05). Thus, the presence of TIMP-3 decreased α-cleavage and increased β-cleavage of APP, increasing the production of Aβ isoforms.
We directly tested whether TIMP-3 affected ADAM-10 cleavage of APP. We transfected COS7 cells with APP with and without ADAM-10 cDNA (Fig. 2C, lanes 1, 2). As expected, ADAM-10 increased the production of sAPPα and APP CTF (Fig. 2C, lane 2). The presence of TIMP-3 conditioned media prevented the increased cleavage of APP by ADAM-10, decreasing the levels of sAPP and APP CTF (Fig. 2C, lane 3), directly demonstrating the effect of TIMP-3 on APP cleavage via inhibition of ADAM-10.
To test whether inhibition of endogenous TIMP-3 affected APP processing and Aβ production, we constructed lentiviral vectors with TIMP-3 and TIMP-3 shRNA (construct 3A), and infected primary cortical neurons. This approach successfully altered TIMP-3 levels (Fig. 2D, top blot). We measured Aβ production 5 d after lentiviral infection. We found that overexpression of TIMP-3 significantly increased Aβ production (by 96%; p < 0.05), and inhibition of TIMP-3 significantly decreased Aβ production (by 48%; p < 0.05) (Fig. 2D).
TIMP-3 affects ApoER2 processing
ApoE receptors undergo α-cleavage similar to APP (Hoe and Rebeck, 2005). To determine whether TIMP-3 had a similar affect on ApoE receptor processing, we tested the effects on one apoE receptor, ApoER2. We treated ApoER2-transfected cells with TIMP-3 conditioned medium and measured secreted ApoER2 in the media and ApoER2 CTF in cell lysates. The presence of TIMP-3 (Fig. 3A, lanes 2, 4) decreased secreted ApoER2 and ApoER2 CTF compared with cells treated with control media (Fig. 3A, lanes 1, 3).
Figure 3.

TIMP-3 inhibits α-cleavage of ApoER2. A, COS7 cells were transfected with ApoER2 and treated with control media (lanes 1, 3) or TIMP-3 CM (lanes 2, 4). Secreted ApoER2 was detected in conditioned media with antibody 5810, and ApoER2 CTFs were detected from cell lysates with anti-HA antibody. The presence of TIMP-3 decreased secreted ApoER2 and ApoER2 CTF. B, COS7 cells were transfected with ApoER2 (lanes 1–3) or ADAM-10 cDNA (lanes 2, 3). Cells expressing ApoER2 and ADAM-10 were treated with TIMP-3 conditioned media (lane 3). ADAM-10 expression increased cleavage fragments of ApoER2 (lane 2). The presence of extracellular TIMP-3 blocked the increased ApoER2 cleavage by ADAM-10 (lane 3).
Similar to our experiment with APP, we questioned whether ADAM-10 was a secretase responsible for cleaving ApoER2. Again, cotransfection of cells with ADAM-10 greatly increased the production of soluble ApoER2 and ApoER2 CTF (Fig. 3B, lane 2). The presence of TIMP-3-containing conditioned media prevented the cleavage of ApoER2 by ADAM-10, reducing levels of sApoER2 and ApoER2 CTF (Fig. 3B, lane 3). Thus APP and ApoER2 are both cleaved by ADAM-10, and both processes are inhibited by TIMP-3.
TIMP-3 decreases cell surface expression of APP, ApoER2, and ADAM-10
To assess whether TIMP-3 alters the physical interaction of APP and α-secretase, we examined the effects of TIMP-3 overexpression on cell surface levels of APP. ADAM-10 and ADAM-17 are both responsible for proteolysis of a number of cell surface proteins other than APP, and the majority of α-secretase activity is thought to occur on the cell surface (Reiss et al., 2006). COS7 cells were transfected with APP or ApoER2 and cotransfected with TIMP-3. Cell surface proteins were biotin labeled and isolated from cell lysates with avidin beads. The resulting protein samples were then immunoblotted for APP or ApoER2. Cell surface levels of both APP and ApoER2 were decreased by TIMP-3 (Fig. 4A, top), whereas total cell-associated APP and ApoER2 levels remained similar (Fig. 4A, bottom). Quantification showed that surface protein levels of APP and ApoER2 were significantly decreased to between 20 and 50% of control (p < 0.01).
Figure 4.
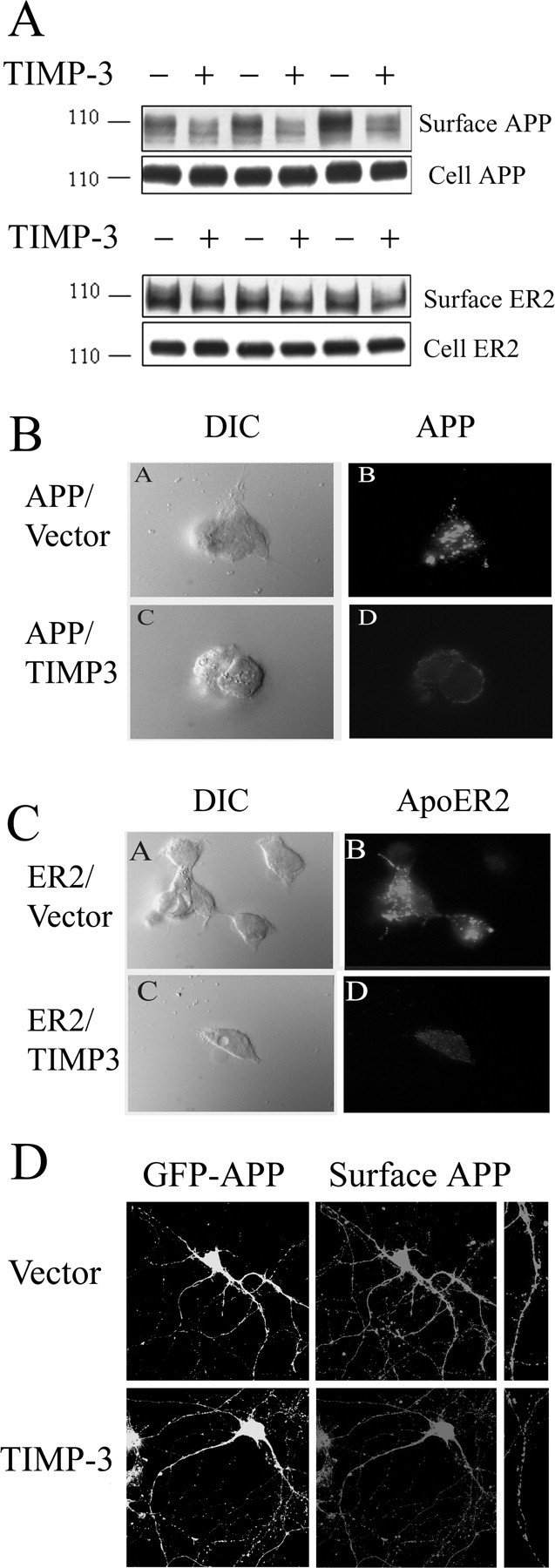
TIMP-3 decreases cell surface levels of APP and ApoER2. A, COS7 cells were transfected with APP (top blots) or ApoER2 (bottom blots) with control vector (−) or with TIMP-3 cDNA (+). Cell surface proteins were biotin labeled, isolated with avidin beads, and immunoblotted with 6E10 (anti-APP) or 5810 (anti-ApoER2) antibody. Top, TIMP-3 decreased surface levels of APP and ApoER2. Bottom, Levels of full-length APP or ApoER2 were unaffected by TIMP-3. B, HEK cells were transfected with APP and vector (BA, BB) or APP and TIMP-3 (BC, BD). Cells were incubated with anti-APP and secondary antibody Alexa Fluor 594 anti-mouse. BA, BC, Cells were imaged with DIC. BB, BD, Confocal laser-scanning microscopy demonstrated that TIMP-3 decreased surface staining levels of APP. C, HEK cells were transfected with ApoER2 and vector (CA, CB) or ApoER2 and TIMP-3 (CC, CD). Cells were incubated with antibody 5810 (anti-ApoER2) and secondary antibody Alexa Fluor 555 anti-rabbit. CB, CD, Confocal laser-scanning microscopy demonstrated that TIMP-3 decreased surface staining levels of ApoER2. D, Cultured hippocampal neurons were transfected at DIV 12 with either GFP-APP and empty vector (top) or GFP-APP and TIMP-3 (bottom). Surface APP was measured with 6E10 at DIV 18 by immunofluorescence of live cells. Left, GFP-APP; right, surface APP (far right, higher magnification showing individual processes).
We confirmed these analyses with another assay of surface protein levels. Live transfected HEK293 cells were incubated with anti-APP or antibody 5810 (anti-ApoER2). Antibodies only label the proteins on the plasma membrane, and not internal APP or ApoER2, as demonstrated by the lack of staining of the endoplasmic reticulum or Golgi. Whole cells were visualized by differential interference contrast (DIC) (Fig. 4B,C, left). TIMP-3 again decreased cell surface levels of APP and ApoER2 compared with control (Fig. 4B,C, right). Thus, at the site of α-secretase cleavage on the cell membrane, TIMP-3 reduces available APP and ApoER2. These data suggest that TIMP-3 reduces the level of substrate available for α-secretase cleavage as well as direct inhibition of α-secretase proteins.
To test the effects in neurons, we cotransfected primary hippocampal neurons with APP and empty vector (top) or APP and TIMP-3 (bottom) (Fig. 4D). Transfections of cell with APP allowed us to measure surface APP by live-cell immunostaining. Surface APP was observed in neuronal processes as well as on the cell body. Consistent with the COS7 cells and HEK cells data, cotransfection with TIMP-3 decreased surface levels of APP (Fig. 4D, bottom). Quantification demonstrated that TIMP-3 overexpression significantly decreased surface levels of APP by 63% (p < 0.05).
To test whether the effect of TIMP-3 on surface APP and ApoER2 was mimicked by changes to α-secretase activity, we analyzed whether surface ADAM-10 levels were also affected. We transfected COS7 cells with ADAM-10 with and without TIMP-3 and measured cell surface levels of ADAM-10 using biotinylation as above. In the presence of TIMP-3, surface levels of ADAM-10 were reduced (Fig. 5, top), with no effect on total cellular levels of ADAM-10 (Fig. 5, bottom). Quantitation of triplicate experiments showed that this reduction was 89% (p < 0.05). Thus, TIMP-3 reduced surface levels of at least one α-secretase and two of its targets.
Figure 5.

TIMP-3 decreases surface levels of ADAM-10. COS7 cells were transfected with ADAM-10 cDNA (HA tagged on its N terminus) and either TIMP-3 or a control vector. Surface proteins were labeled with a water-soluble biotinylating agent and isolated using avidin beads. Surface levels of ADAM-10 were measured from the precipitated proteins using anti-HA antibody. Total ADAM-10 levels were measured in complete cell lysates. Cotransfection with TIMP-3 decreased surface levels of ADAM-10.
TIMP-3 and APP turnover
To investigate whether TIMP-3 played a role in the turnover of the APP holoprotein, we performed pulse-chase labeling experiments. COS7 cells transfected with APP and empty vector or with APP and TIMP-3 were pulsed with [35S], and the half-life of radiolabeled APP within the cell was calculated (Fig. 6A,B). There was no significant difference between the measured half-life of APP in the cells cotransfected with TIMP-3 (58 min) and that for APP in cells transfected with APP and an empty vector (63 min). Thus, although TIMP-3 affects the availability of APP in the α- and β-proteolytic pathways, it does not alter the rate of bulk APP holoprotein turnover.
Figure 6.

TIMP-3 does not affect the turnover rate of the APP holoprotein. A, Cells transfected with APP and vector (left) or APP and TIMP-3 (right) were pulsed with [35S] and APP immunoprecipitated at indicated times and measured. The turnover of APP was followed over a 4 h time course (representative image from 3 replicates). B, Bands were quantified using ImageQuant software, expressed as a percentage of radiolabeled APP at time 0 and the mean value for each time point calculated. These data were plotted, and one-phase exponential decay curves were generated for both conditions. T1/2 for APP/vector was 63 min; T1/2 for APP/TIMP-3 was 58 min.
TO-901317 treatment decreases TIMP-3 protein levels in vivo
To test whether an LXR agonist induces changes in TIMP-3 protein levels in vivo as well as in vitro, we treated mice with TO-901317 (50 mg/kg) for 24 h. To ensure TO-901317 had entered the brain and activated LXR at this time point, we measured protein levels of ABCA1 and found that this cholesterol transporter was increased by 124% (p < 0.05) (Fig. 7A, top). Consistent with our previous findings in vitro (Table 1, Fig. 1), we found that TO-901317 treatment significantly decreased TIMP-3 protein level in vivo (Fig. 7A, middle). β-Actin levels were not altered by TO-901317 (Fig. 7A, bottom).
Figure 7.

TIMP-3 protein levels were significantly decreased after LXR activation in vivo. A, Mice received a single dose of the LXR agonist TO-901317 (TO; 50 mg/kg) or vehicle control (C), and 24 h later, ABCA1 and TIMP-3 protein levels were measured in cortical hemispheres. ABCA1 protein levels were measured to ensure that TO-901317 was activating LXR within 24 h. Top, Treatment increased ABCA1 levels by 124%. Middle, In contrast, TIMP-3 levels (arrow) were dramatically decreased in TO-901317-treated mice. Bottom, β-Actin levels were unchanged. B, Top, Secreted APPα levels were not significantly changed in TO-901317-treated mice after 24 h. Middle, Full-length APP and APP CTF did not change in the brains of animals treated with TO-901317 compared with vehicle.
Previous studies have shown that TO-901317 decreases Aβ levels in vivo (Koldamova et al., 2005). To determine whether this effect might be mediated through an effect on α-secretase, we measured secreted APPα, full-length APP, and APP CTF in brain lysates after TO-901317 treatment. We found that LXR activation did not significantly affect sAPPα levels in vivo (Fig. 7B) (increased by 13%; not significant). We found that full-length APP and APP CTF did not change in TO-901317-treated mice after 24 h (Fig. 7B).
TIMP-3 expression in vivo
TIMP-3 expression has been demonstrated within developing embryonic neural tissues and within astrocytes and neurons of the adult rat brain (Jaworski and Fager, 2000). In situ hybridization data demonstrate a predominantly neuronal expression of TIMP-3 in the brain, including neurons of the cortex, hippocampus, and cerebellum (www.brain-map.org). To determine whether TIMP-3 expression was altered in a mouse model of AD, we examined immunostaining of TIMP-3 in triple-transgenic AD mice overexpressing APP, PS-1, and tau (Oddo et al., 2003). TIMP-3 antibodies diffusely stained the brain tissue and weakly identified neurons throughout the brain, including cortex, hippocampus, and striatum, in agreement with existing in situ hybridization data. At ages before pathology is seen (6 months), only weak neuronal staining was observed in the mouse brain tissue (Fig. 8A). However, at later ages as AD pathology accumulates (Oddo et al., 2003), there was increased neuronal TIMP-3 immunoreactivity (12 months, Fig. 8B; 20 months, Fig. 8C). Higher magnification of TIMP-3 immunoreactivity showed that TIMP-3 was present in perinuclear vesicles in neurons (Fig. 8D). There were also occasional glial cells that were immunopositive for TIMP-3 (data not shown). Western blots for TIMP-3 protein on brain extracts from aged wild-type and triple-transgenic mice showed an increase in both the 50 and 25 kDa TIMP-3-immunoreactive bands in this mouse model of AD at 1 and 2 years of age (Fig. 8E, lanes 2, 3), supporting the immunostaining data.
Figure 8.
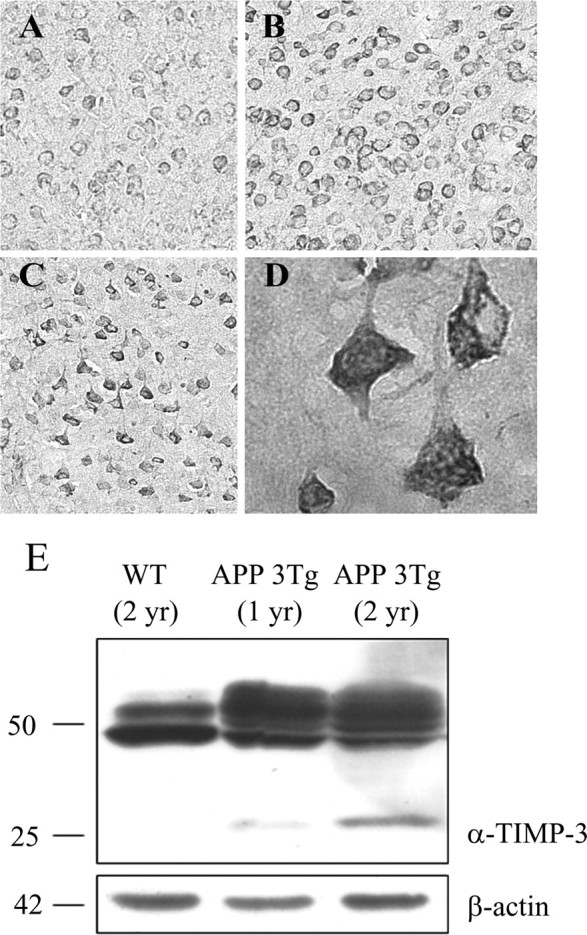
TIMP-3 immunoreactivity is increased in a mouse model of AD. Brain tissue from a mouse model of AD was immunostained for TIMP-3. Neuronal TIMP-3 expression increased with the age of these mice (A, 6 months; B, 12 months; C, 20 months; cortical neurons, original magnification, 20×). TIMP-3 immunostaining was perinuclear and punctate in cortical neurons. D, There was also a strong increase in TIMP-3 immunostaining in hippocampal neurons of the AD mouse model (63×). E, Brain tissue from wild-type (WT) or APP triple-transgenic (APP 3Tg) mice was extracted in RIPA buffer, and proteins were analyzed by Western blot with an anti-TIMP-3 antibody. Top, Both 25 and 50 kDa TIMP-3 species were increased in the AD mouse model compared with the wild-type mouse. Bottom, β-Actin was measured as a loading control.
We also tested whether TIMP-3 was increased in human AD brains. We homogenized cortical tissue from five AD brains and age-matched controls and examined TIMP-3 protein levels by immunoblot. TIMP-3 protein levels were elevated in AD brain compared with controls, with β-actin used as a loading control (Fig. 9A). Quantification of the Western blots showed an 87% increase in AD brain (p < 0.05). To test whether this increase in TIMP-3 was caused by a nonspecific upregulation common to neurodegenerative diseases, we examined brain samples from 13 Parkinson's disease patients and age-matched controls in the same manner. In contrast to the AD samples analyzed, PD brains showed a (nonsignificant) 50% decrease in TIMP-3 protein levels compared with control samples (Fig. 9B). Thus, TIMP-3 was increased in both a mouse model of AD and in human AD brain.
Figure 9.

TIMP-3 immunoreactivity is increased in AD brains. A, Brain tissue (frontal cortex) from AD patients (n = 5) and age-matched controls (C; n = 5) were extracted in lysis buffer, and proteins were analyzed by Western blot with an anti-TIMP-3 antibody. The 50 kDa immunoreactive band was increased in all AD brains tested (4 C–AD pairs shown). β-Actin was measured as a loading control. B, Brain tissue (frontal cortex) from control (C; n = 13) or PD (n = 13) individuals were extracted in lysis buffer, and proteins were analyzed by Western blot with an anti-TIMP-3. The immunoreactive band of 50 kDa was not increased in PD brains (2 of 13 C–PD pairs shown). β-Actin was measured as a loading control.
Discussion
The processing of APP is important to Alzheimer's disease, most profoundly demonstrated by the presence of disease causing mutations at cleavage sites in APP and mutations in the presenilins, components of gamma secretase (Wolfe, 2006). The importance of APP in AD research was also recently underscored in the finding that numerous polymorphisms in the SORL1 gene alter the risk of AD (Rogaeva et al., 2007); SORL1 encodes for a protein important in APP trafficking and is structurally related to lipoprotein receptors (Andersen et al., 2006). An increased knowledge of the enzymes that cleave APP and the means by which those activities are regulated is essential to our understanding of the pathogenesis of AD. The research reported here examined one way that APP processing is organized within the cell: the extracellular matrix protein TIMP-3 enhances endocytosis of APP from the cell surface, away from α-secretase (Reiss et al., 2006) and toward endosomes, in which β-secretase cleavage of APP is thought to be enhanced (Huse et al., 2000). We identified TIMP-3 in a screen of genes in human neuroblastoma cells that were reduced after LXR activation (Table 1, Fig. 1). We also found that TIMP-3 protein levels significantly decreased after LXR activation in vivo (Fig. 7). TIMP-3 had been previously identified as an inhibitor of ADAM-10 and ADAM-17 (Amour et al., 1998, 2000), two α-secretase activities (Allinson et al., 2004). Our cell culture experiments showed that TIMP-3 decreases cleavage of APP to release the extracellular domain and membrane-bound CTFs (Fig. 2), demonstrating that TIMP-3 can alter processing of APP.
Cellular overexpression of TIMP-3 or treatment of cells with TIMP-3 conditioned medium led to decreased α-cleavage of APP and increased β-cleavage (Fig. 2A–C) and an increase in the levels of Aβ40 and Aβ42 in COS7 cells. We also found that overexpression of TIMP-3 increased Aβ, whereas inhibition of endogenous TIMP-3 decreased Aβ in primary cortical neurons (Fig. 2D). Thus, although ADAMs were initially identified as a target for TIMP-3, the mechanism of the effects of TIMP-3 on Aβ is not only through α-secretase inhibition. Instead, TIMP-3 also decreases surface levels of ADAM-10 (Fig. 5) and APP (Fig. 4). We found that TIMP-3 increased endocytosis of APP (data not shown), thus trafficking it through the endosomal pathway, where increased β-secretase cleavage and Aβ production is thought to occur (Huse et al., 2000).
TIMP-3 protein was significantly increased in a mouse model of Alzheimer's disease (Fig. 8A,B) and in human AD brain (Fig. 9A), with the majority of this staining occurring in neurons. Neurons in early stages of AD pathogenesis demonstrate enlargement of endosomes, which may be sites of abnormal APP proteolysis by β-secretase and Aβ accumulation (Huse et al., 2000). The higher levels of TIMP-3 may thus contribute to increased conversion of APP to Aβ, contributing to the pathogenesis of AD. We expected TIMP-3 levels might be decreased in AD brains, because there are data that the endogenous agonist of LXR, 24-hydroxycholesterol, is increased in AD (Lutjohann et al., 2000; Papassotirpoulos et al., 2000). However, levels of 24-hydroxycholesterol are decreased in late-stage AD and in AD cases with the APOE-ε4 genotype (Papassotirpoulos et al., 2000), suggesting that LXR activation may be quite variable in AD. In addition, TIMP-3 expression is undoubtedly influenced by mechanisms unrelated to cholesterol homeostasis. For example, TIMP-3 mRNA and protein are increased in brain neurons after ischemia (Wallace et al., 2002) and in the spinal cord during experimental autoimmune encephalomyelitis (Plumb et al., 2005). It is possible that the endosomal abnormalities in AD noted above also contribute to a decrease in TIMP-3 degradation. Investigation into these mechanisms may help determine why TIMP-3 levels are increased in AD and whether this increase affects APP trafficking and processing in vivo.
TIMP-3 also prevented cleavage and decreased cell surface levels of a lipoprotein receptor, ApoER2 (Fig. 3), potentially affecting cholesterol uptake from apoE-lipoproteins. Cellular cholesterol homeostasis is regulated by several systems. When cholesterol levels are high, cholesterol converted to hydroxycholesterol binds to the nuclear hormone receptor LXR and promotes transcription of genes necessary for cholesterol efflux, such as abca1, abcg1, and apoE. When cellular cholesterol levels are low, SREBP is cleaved, and a cytoplasmic fragment acts as a transcriptional activator to increase cholesterol synthesis and import [3-hydroxy-3-methylglutaryl-coenzyme A reductase and the low-density lipoprotein (LDL) receptor] (Dietschy and Turley, 2004). In addition to these gene transcription-based mechanisms, cellular cholesterol levels also affect trafficking of the LDL receptor to the cell surface, thus altering the amount of cholesterol that can be internalized by the cell (Lagace et al., 2006). Here we report that LXR activation, which can result from excess cellular cholesterol, decreases levels of TIMP-3. We have shown that TIMP-3 decreases the levels of the lipoprotein receptor ApoER2 on the cell surface, which may reduce the amount of cholesterol entering cells via lipoprotein-mediated pathways. Thus regulation of TIMP-3 may be another mechanism by which LXR controls cellular cholesterol homeostasis.
Previous in vivo studies of LXR activation on APP processing have shown consistent reductions in Aβ (Koldamova et al., 2005), but have been mixed on whether this effect is modulated via α-secretase. Koldamova et al. (2005) found that TO-901317 significantly increased sAPPα in a transgenic mouse model of AD, whereas a later study found no differences in a similar mouse model (Riddell et al., 2007). We found that TO-901317 had minimal effects on APP processing, despite considerably reducing TIMP-3 levels. However, given other effects of LXR activation (on apoE levels, on cellular cholesterol, and on inflammation) (Joseph et al., 2003; Burns et al., 2006), dissecting an effect of TIMP-3 regulation on Aβ production is difficult. It will be worthwhile to examine the specific effects of TIMP-3 on APP processing in vivo in future experiments using some of the methods we have developed here for examining TIMP-3 in the absence of altered cholesterol metabolism (i.e., lentiviral systems with TIMP-3 and TIMP-3 shRNA).
The downregulation of TIMP-3 via an LXR mechanism is unusual because most attention has been on genes induced by LXR (Kino and Chrousos, 2004). Interestingly, LXR also mediates downregulation of MMP-9 in macrophages (Castrillo et al., 2003), suggesting that MMP-9 and an inhibitor TIMP-3 may be coregulated. LXR also inhibits expression of inflammatory genes (Blaschke et al., 2006). LXR bound to oxysterol forms a heterodimer with the retinoid X receptor (RXR) nuclear hormone receptor bound to retinoic acid. This heterodimer then binds to a defined LXR response element in the DNA. In addition to LXR, RXR also forms heterodimers with other nuclear hormone receptors (Giguere, 1999), promoting gene transcription of other classes of genes. For example, activation of LXR increases its binding of RXR, decreasing the association of RXR with peroxisome proliferator-activated receptor β, decreasing expression of angiopoietin-like protein 3 (Matsusue et al., 2006). It is also possible that binding of LXR-RXR to the promoter region of the TIMP-3 gene blocks binding of other elements normally promoting gene transcription, as demonstrated for LXR downregulation of myeloperoxidase (Reynolds et al., 2006). Gene regulation via LXR is now being recognized as a complex regulatory system, recruiting activators and repressors in competitive manner (Albers et al., 2006).
TIMP-3 joins several other extracellular matrix molecules that strongly regulate the processing of APP and ApoER2. F-spondin is an extracellular matrix protein important to neuronal and axonal migration during development (Burstyn-Cohen et al., 1999). It was identified as a protein that bound the extracellular domain of APP (Ho and Sudhof, 2004), leading to increased levels of cell surface APP, increased α-cleavage of APP, and decreased β-cleavage of APP (Hoe et al., 2005). Similarly, the extracellular matrix protein Reelin caused increased surface APP and a preference for α-cleavage over β-cleavage (Hoe et al., 2006b). Both of these proteins, F-spondin (Hoe et al., 2005) and Reelin, interact with ApoER2 (Jossin et al., 2004). APP and ApoER2 also share a number of cytoplasmic adaptor proteins (such as Dab1 and FE65), suggesting that their trafficking and proteolysis are regulated together (Hoe et al., 2006a,b). That proteolysis of both of these proteins are affected by TIMP-3 further supports their common regulation.
The data described in this study are the first to demonstrate the impact of TIMP-3 on the α-secretase processing of APP and ApoER2; they are also the first to describe regulation of surface protein cleavage through the LXR cholesterol homeostasis system. TIMP-3 treatment prevents α-cleavage of APP and promotes β-cleavage, with significant increases in Aβ production. The striking increase in TIMP-3 in AD brains (but not in another neurodegenerative disease) raises the possibility that TIMP-3 contributes to the elevated Aβ levels in AD, making its inhibition a potential target for therapeutic approaches to reducing Aβ.
Footnotes
This work was supported by National Institutes of Health Grant AG14473 (G.W.R.), the Intramural Aging Grant at Georgetown University (H.-S.H.), and the Alzheimer's Research Fund in memory of Bill and Marie Drach. We thank Yasuji Matsuoka for assistance with mouse brain tissue and Aβ assays and Bio-Rad (Hercules, CA) for immunoblotting reagents.
References
- Albers M, Blume B, Schlueter T, Wright MB, Kober I, Kremoser C, Deuschle U, Koegl M. A novel principle for partial agonism of liver X receptor ligands. Competitive recruitment of activators and repressors. J Biol Chem. 2006;281:4920–4930. doi: 10.1074/jbc.M510101200. [DOI] [PubMed] [Google Scholar]
- Allinson TM, Parkin ET, Condon TP, Schwager SL, Sturrock ED, Turner AJ, Hooper NM. The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. Eur J Biochem. 2004;271:2539–2547. doi: 10.1111/j.1432-1033.2004.04184.x. [DOI] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ, Murphy G. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, Docherty AJ, Murphy G. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000;473:275–279. doi: 10.1016/s0014-5793(00)01528-3. [DOI] [PubMed] [Google Scholar]
- Andersen OM, Schmidt V, Spoelgen R, Gliemann J, Behlke J, Galatis D, Mckinstry WJ, Parker MW, Masters CL, Hyman BT, Cappai R, Willnow TE. Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry. 2006;45:2618–2628. doi: 10.1021/bi052120v. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Takata Y, Caglayan E, Collins A, Tontonoz P, Hsueh WA, Tangirala RK. A nuclear receptor corepressor-dependent pathway mediates suppression of cytokine-induced C-reactive protein gene expression by liver X receptor. Circ Res. 2006;99:88–99. doi: 10.1161/01.RES.0000252878.34269.06. [DOI] [PubMed] [Google Scholar]
- Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- Burns MP, Vardanian L, Pajoohesh-Ganji A, Wang L, Cooper M, Harris DC, Duff K, Rebeck GW. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem. 2006;98:792–800. doi: 10.1111/j.1471-4159.2006.03925.x. [DOI] [PubMed] [Google Scholar]
- Burstyn-Cohen T, Tzarfaty V, Frumkin A, Feinstein Y, Stoeckli E, Klar A. F-Spondin is required for accurate pathfinding of commissural axons at the floor plate. Neuron. 1999;23:233–246. doi: 10.1016/s0896-6273(00)80776-x. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J Biol Chem. 2003;278:10443–10449. doi: 10.1074/jbc.M213071200. [DOI] [PubMed] [Google Scholar]
- Chen K, Ochalski PG, Tran TS, Sahir N, Schubert M, Pramatarova A, Howell BW. Interaction between Dab1 and CrkII is promoted by Reelin signaling. J Cell Sci. 2004;117:4527–4536. doi: 10.1242/jcs.01320. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Fu Z, Lee SH, Simonetta A, Hansen J, Sheng M, Pak DT. Differential roles of Rap1 and Rap2 small GTPases in neurite retraction and synapse elimination in hippocampal spiny neurons. J Neurochem. 2007;100:118–131. doi: 10.1111/j.1471-4159.2006.04195.x. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- Giguere V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- Hashimoto G, Aoki T, Nakamura H, Tanzawa K, Okada Y. Inhibition of ADAMTS4 (aggrecanase-1) by tissue inhibitors of metalloproteinases (TIMP-1, 2, 3, and 4) FEBS Lett. 2001;494:192–195. doi: 10.1016/s0014-5793(01)02323-7. [DOI] [PubMed] [Google Scholar]
- Ho A, Sudhof TC. Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci USA. 2004;101:2548–2553. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe HS, Rebeck GW. Regulation of ApoE receptor proteolysis by ligand binding. Brain Res Mol Brain Res. 2005;137:31–39. doi: 10.1016/j.molbrainres.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Wessner D, Beffert U, Becker AG, Matsuoka Y, Rebeck GW. F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol Cell Biol. 2005;25:9259–9268. doi: 10.1128/MCB.25.21.9259-9268.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe HS, Magill LA, Guenette S, Fu Z, Vicini S, Rebeck GW. FE65 interaction with the ApoE receptor ApoEr2. J Biol Chem. 2006a;281:24521–24530. doi: 10.1074/jbc.M600728200. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem. 2006b;281:35176–35185. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- Horikoshi Y, Sakaguchi G, Becker AG, Gray AJ, Duff K, Aisen PS, Yamaguchi H, Maeda M, Kinoshita N, Matsuoka Y. Development of Abeta terminal end-specific antibodies and sensitive ELISA for Abeta variant. Biochem Biophys Res Commun. 2004;319:733–737. doi: 10.1016/j.bbrc.2004.05.051. [DOI] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000;275:33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- Jaworski DM, Fager N. Regulation of tissue inhibitor of metalloproteinase-3 (Timp-3) mRNA expression during rat CNS development. J Neurosci Res. 2000;61:396–408. doi: 10.1002/1097-4547(20000815)61:4<396::AID-JNR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Iqnatova N, Hiesberger T, Herz J, Lambert de Rouvroit C, Goffinet AM. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J Neurosci. 2004;24:514–521. doi: 10.1523/JNEUROSCI.3408-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Chrousos GP. Combating atherosclerosis with LXR and PPAR agonists: is rational multitargeted polypharmacy the future of therapeutics in complex diseases? Mol Interv. 2004;4:254–257. doi: 10.1124/mi.4.5.5. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand TO901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Shah JA, Reardon CA, Getz GS, Bu G, Hu J, Guo L, van Eldik LJ. Apolipoprotein E receptors mediate the effects of beta-amyloid on astrocyte cultures. J Biol Chem. 2000;275:33974–33980. doi: 10.1074/jbc.M000602200. [DOI] [PubMed] [Google Scholar]
- Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Andersen NN, Ho YK, Hammer RE, Horton JD. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995–3005. doi: 10.1172/JCI29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loechel F, Fox JW, Murphy G, Albrechtsen R, Wewer UM. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278:511–515. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- Lu TT, Repa JJ, Mangelsdorf DJ. Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J Biol Chem. 2001;276:37735–37738. doi: 10.1074/jbc.R100035200. [DOI] [PubMed] [Google Scholar]
- Lutjohann D, Papassotiropoulos A, Bjorkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, Bergmann KV, Heun R. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41:195–198. [PubMed] [Google Scholar]
- Massaro D, Alexander E, Reiland K, Hoffman EP, Massaro GD, Clerch LB. Rapid onset of gene expression in lung, supportive of formation of alveolar septa, induced by refeeding mice after calorie restriction. Am J Physiol Lung Cell Mol Physiol. 2006;292:L1313–L1326. doi: 10.1152/ajplung.00146.2006. [DOI] [PubMed] [Google Scholar]
- Matsusue K, Miyoshi A, Yamano S, Gonzalez FJ. Ligand-activated PPARbeta efficiently represses the induction of LXR-dependent promoter activity through competition with RXR. Mol Cell Endocrinol. 2006;256:23–33. doi: 10.1016/j.mce.2006.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G, Willenbrock F, Crabbe T. Regulation of matrix metalloproteinase activity. Ann NY Acad Sci. 1994;732:31–41. doi: 10.1111/j.1749-6632.1994.tb24722.x. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Papassotirpoulos A, Lutjohann D, Bagli M, Locatelli S, Jessen F, Rao ML, Maier W, Bjorkhem I, Bergmann KV, Heun R. Plasma 24S-hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer's disease. NeuroReport. 2000;9:1959–1962. doi: 10.1097/00001756-200006260-00030. [DOI] [PubMed] [Google Scholar]
- Plumb J, Cross AK, Surr J, Haddock G, Smith T, Bunning RA, Woodroofe MN. ADAM-17 and TIMP3 protein and mRNA expression in spinal cord white matter of rats with acute experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:1–9. doi: 10.1016/j.jneuroim.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Strickland DK, Hyman BT, Rebeck GW. alpha 2-Macroglobulin exposure reduces calcium responses to N-methyl-d-aspartate via low density lipoprotein receptor-related protein in cultured hippocampal neurons. J Biol Chem. 2002;277:14458–14466. doi: 10.1074/jbc.M112066200. [DOI] [PubMed] [Google Scholar]
- Reiss K, Ludwig A, Saftig P. Breaking up the tie: disintegrin-like metalloproteinases as regulators of cell migration in inflammation and invasion. Pharmacol Ther. 2006;111:985–1006. doi: 10.1016/j.pharmthera.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Reynolds WF, Kumar AP, Piedrafita FJ. The human myeloperoxidase gene is regulated by LXR and PPARalpha ligands. Biochem Biophys Res Commun. 2006;349:846–854. doi: 10.1016/j.bbrc.2006.08.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg 2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Alexander S, Estrada EY, Hines C, Cunningham LA, Rosenberg GA. Tissue inhibitor of metalloproteinase-3 is associated with neuronal death in reperfusion injury. J Cereb Blood Flow Metab. 2002;22:1303–1310. doi: 10.1097/01.WCB.0000040943.89393.c1. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Weber LW, Boll M, Stampfl A. Maintaining cholesterol homeostasis: sterol regulatory element-binding proteins. World J Gastroenterol. 2004;10:3081–3087. doi: 10.3748/wjg.v10.i21.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS. The gamma-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry. 2006;45:7931–7939. doi: 10.1021/bi060799c. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhu F, Liu J, Wang W, Zhang R, Garlisi CG, Liu YH, Wang S, Shah H, Wan Y, Umland SP. Catalytic activity of human ADAM33. J Biol Chem. 2004;279:9818–9830. doi: 10.1074/jbc.M309696200. [DOI] [PubMed] [Google Scholar]