

Abstract
Synaptic integrity is now recognized as a central component of Alzheimer's disease. Surprisingly, however, the structural status of glutamatergic synapses in Alzheimer's disease is unclear, despite the fact that glutamate is the major excitatory transmitter of the CNS and has key roles in excitotoxicity and long-term potentiation. The identification of specific markers of glutamatergic neurons now allows an assessment of the structural involvement of the glutamatergic system across progressive stages of the Alzheimer's pathology, an opportunity not afforded by previously used neurochemical approaches. Glutamatergic presynaptic bouton density and dystrophic neurite abundance were quantified in midfrontal gyrus brain tissue from subjects with no cognitive impairment, mild cognitive impairment, or mild- or severe-stage Alzheimer's disease. Our study demonstrates a striking pathology-dependent pattern of glutamatergic synaptic remodeling with disease progression. Subjects with mild cognitive impairment display a paradoxical elevation in glutamatergic presynaptic bouton density, a situation akin to that observed in the cholinergic system, which then depletes and drops with disease progression. This pattern of synaptic remodeling mirrors our previous findings in transgenic animal models and is of major relevance to current transmitter-based therapeutics.
Keywords: Alzheimer's disease, mild cognitive impairment, glutamatergic, presynaptic bouton, dystrophic neurite, MMSE
Introduction
Mounting evidence suggests that synapses, as opposed to plaques or tangles, are key to the clinical presentation of Alzheimer's disease (AD) [for review, see Selkoe (2002) and Tanzi (2005)]. Indeed, synaptic density is the strongest pathological correlate of cognitive ability (Perry et al., 1978; Masliah et al., 1989; DeKosky and Scheff, 1990; Scheff et al., 1990; Terry et al., 1991; DeKosky et al., 1992; Masliah and Terry, 1993) [for review, see Terry (1996, 1997)]. Early AD studies have focused on the cholinergic system because of its tangible pathological involvement. The cholinergic focus clearly benefited the AD field; however, it also led to an underappreciation of the roles of the noncholinergic transmitter systems. In the context of glutamate, the most abundant excitatory neurotransmitter in the cortex and hippocampus, the brain areas implicated in AD, this information is especially relevant. The hippocampal and cortical atrophy visible in AD brains demonstrates the degeneration of brain areas predominated by glutamatergic large pyramidal neurons. The involvement of said neurons is of immense relevance to the pathological and clinical presentation of AD given glutamate's central role in long-term potentiation (LTP) and excitotoxicity (Greenamyre et al., 1988).
Previous investigations largely used neurochemical approaches, as reliable immunohistochemical markers of glutamatergic synapses were unavailable. Because neurochemical data can be influenced by postmortem interval, tissue preparation, and premortem use of CNS-acting drugs, many studies yielded conflicting results. K+-evoked neuronal glutamate release in temporal neocortical AD brain tissue samples has been reported as unchanged (Smith et al., 1983). However, other groups observed a loss of cortical and hippocampal glutamatergic uptake sites, which were interpreted as nerve terminal loss (Palmer et al., 1986; Cross et al., 1987; Hardy et al., 1987; Procter et al., 1987; Cowburn et al., 1988a,b). NMDA receptor levels are also conflicting, with some groups showing a decrease in AD subjects (Greenamyre et al., 1985, 1987) and others showing no change (Geddes et al., 1986; Monaghan et al., 1987; Cowburn et al., 1988b). The observed reductions in glutamate concentration in AD brain tissue (Ellison et al., 1986; Sasaki et al., 1986; Hyman et al., 1987) make no distinction between neuronal and metabolic pools, limiting potential interpretations. Resultantly, the involvement of glutamatergic neurons in AD remains unclear, despite their central role in the very symptoms that characterize the disease. Fortuitously, the recent identification of the vesicular glutamate transporter 1 (VGluT1) (Takamori et al., 2000), a reliable marker of glutamatergic presynaptic boutons, now allows such an investigation.
Dystrophic neurite generation and reduced presynaptic bouton densities detrimentally influence neurotransmission and cognitive function (McKee et al., 1991; Terry et al., 1991). To determine the involvement of glutamatergic neurons in AD and the relevance of the observed changes to clinical presentation, presynaptic bouton density and dystrophic neurite abundance were quantified in the midfrontal gyrus of subjects with no cognitive impairment (NCI), mild cognitive impairment (MCI), or mild- or severe-stage AD (mAD or sAD). Our study reveals a novel and therapeutically relevant pattern of cortical synaptic remodeling in response to progressive stages of the AD pathology.
Materials and Methods
General demographics of Religious Orders Study subject population.
Participants were elderly members of the Catholic clergy (nuns, priests, and brothers) who took part in the Religious Orders Study, a longitudinal clinicopathologic study of aging and AD. Each subject signed an informed consent and an anatomical gift act donating his/her brain to investigators at the time of death. The study was approved by the institutional Review Board of Rush University Medical Center. Table 1 shows the characteristics of the sample population by diagnostic group. The NCI, MCI, and mild and severe AD groups were similar in postmortem interval, education, and interval between last date of Mini Mental State Examination (MMSE) and death. An ANOVA found a significant difference in age between groups (F(3,62) = 4.87; p < 0.005). Post hoc analysis revealed no statistically significant difference between MCI, mAD, and sAD (p > 0.05); however, NCI was significantly lower (p < 0.05) than all three remaining groups. As expected, MMSE scores were significantly different between groups, as detected by an ANOVA (F(3,62) = 196.9; p < 0.0001). One-way ANOVA of glutamatergic presynaptic bouton densities grouped by apolipoprotein E (apoE) genotype revealed no significant difference between groups (F(4,64) = 1.79; p > 0.05), nor did an unpaired Student's t test comparison between glutamatergic presynaptic bouton densities in subjects who were or were not an apoE4 carrier (t(62) = 1.775; p > 0.05).
Table 1.
General demographics of Religious Orders Study subject population
| NCI | MCI | mAD | sAD | Total | ANOVA | ||
|---|---|---|---|---|---|---|---|
| Group size | 12 | 16 | 18 | 20 | 66 | ||
| Age (years) | Mean ± SD | 82.28 ± 6.83 | 88.84 ± 6.22 | 88.35 ± 5.31 | 89.53 ± 4.35 | 87.72 ± 6.05 | F(3,62) = 4.87 |
| Range | 73–93 | 78–101 | 80–100 | 83–99 | 73–101 | p = 0.0046 | |
| PMI (h) | Mean ± SD | 6.92 ± 4.59 | 7.18 ± 5.56 | 5.74 ± 2.94 | 6.98 ± 5.87 | 6.68 ± 4.84 | F(3,62) = 0.31 |
| Range | 3.0–16.25 | 2.5–19.5 | 1.75–13 | 0.75–21.25 | 0.75–21.25 | p = 0.82 | |
| MMSE (0–30) | Mean ± SD | 28.08 ± 1.56 | 26.25 ± 2.49 | 20 ± 3.68 | 3.8 ± 4.12 | 18.08 ± 10.44 | F(3,62) = 196.9 |
| Range | 25–30 | 20–29 | 14–27 | 0–11 | 0–30 | p < 0.0001 | |
| Education (years) | Mean ± SD | 17.75 ± 1.91 | 18.31 ± 3.63 | 18.28 ± 2.27 | 16.06 ± 3.94 | 17.52 ± 3.25 | F(3,62) = 2.14 |
| Range | 15–22 | 9–24 | 16–22 | 8–25 | 8–25 | p = 0.1042 | |
| Gender | Male | 2 (16.67%) | 2 (12.5%) | 3 (16.67%) | 7 (35%) | 14 | χ2(3) = 3.37 |
| Female | 10 (83.33%) | 14 (87.5%) | 15 (83.33%) | 13 (65%) | 52 | p > 0.05 | |
| Interval | Mean ± SD | 217.2 ± 159 | 200.8 ± 114.5 | 203.8 ± 93.5 | 185.9 ± 107.5 | 207.3 ± 116 | F(3,62) = 1.07 |
| Range | 25–595 | 21–379 | 78–345 | 1–354 | 1–595 | p = 0.3686 |
Interval, Interval between last MMSE test date and death.
Clinical evaluation.
Each subject underwent a uniform structured baseline clinical evaluation that included a medical history, neurologic examination, and neuropsychological performance testing. Details of the clinical evaluation have been described previously (Bennett et al., 2002). Based on the results of the evaluation, participants were classified as having dementia and AD according to the joint working group of the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association (McKhann et al., 1984). Blood was collected for DNA, and genotyping was performed for apoE allele status, as described previously (Bennett et al., 2003). Follow-up evaluations were performed annually by examiners blinded to previously collected data. At the time of death, all available clinical data were reviewed, and a summary diagnosis was made of the most likely clinical status at the time of death. The summary diagnosis was made blinded to all postmortem data as described previously (Bennett et al., 2003).
Neuropsychologic performance tests.
At each evaluation, 20 cognitive performance tests were administered. Details of the individual tests have been previously described in detail (Wilson et al., 2002). Briefly, the MMSE was used to describe the cohort and stratify persons with AD into mild (MMSE 11–27) and severe (MMSE <11) dementia. The remaining 19 tests assessed five cognitive domains, including episodic memory (seven tests), semantic memory (three tests), working memory (four tests), perceptual speed (three tests), and visuospatial ability (two tests). The 19 tests were converted to z-scores, based on the mean and SD from all subjects at baseline, and averaged to yield a global measure of cognition [global cognitive score (GCS)] as reported previously (Bennett et al., 2002; Barnes et al., 2003; Wilson et al., 2003).
Neuropathologic evaluation and tissue processing.
All brains were removed, weighed, cut into 1-cm-thick coronal slabs, and immersion fixed in 4% paraformaldehyde for 72 h as described previously (Bennett et al., 2003, 2005). A complete neuropathologic evaluation was performed. Briefly, for these analyses, tissue blocks from two adjacent slabs from the dorsolateral prefrontal cortex (Brodmann area 46/9) were dissected and embedded in paraffin and cut into 6 and 20 μm sections. One 6 μm section was stained with modified Bielschowsky silver stain, and counts of neuritic plaques, diffuse plaques, and neurofibrillary tangles were made as described previously (Bennett et al., 2005). Two 20 μm sections, one each from two adjacent slabs, were immunostained for amyloid-β peptide (MO0872; 1:100) and phosphorylated tau (AT8; 1:800), and amyloid load and the density of paired helical filament (PHF)-tau tangles were determined as described previously (Bennett et al., 2004).
Immunohistochemistry.
All sections were labeled with a random eight-digit numeric code, which was only revealed after acquisition of final data to ensure unbiased processing. Sections were stained 25 slides at a time under identical conditions. Sections were subject to rehydration and deparaffinization, a 30 min incubation in methanol and H2O2 (30%) to eliminate endogenous peroxidase activity, and a 15 min incubation in full-strength (88%) formic acid to expose the amyloid epitope. Extensive washings in either PBS or PBS plus Triton X-100 (PBST) were performed before all incubations (save for blocking). Sections underwent the following treatment: block in 5% normal goat serum (NGS; Vector Laboratories, Burlingame, CA) and 5% normal donkey serum (NDS; Vector Laboratories) PBST solution (45 min), incubation with McSA1 mouse-generated anti-Aβ monoclonal antibodies (Grant et al., 2000) (1:1000; 1 h), overnight incubation with both anti-Aβ McSA1 (1:1000) and guinea pig-generated anti-VGluT1 (1:5000; Millipore, Billerica, MA) primary antibodies, incubation with 5% NDS and biotinylated donkey anti-guinea pig secondary antibodies (1 h; 1:500; Jackson ImmunoResearch, West Grove, PA), incubation in ABC complex kit (1 h; Vector Laboratories), and development with 0.6% 3,3′-diaminobenzidine and 0.03% H2O2. Second staining was achieved via subsequent incubation in 5% NGS and goat anti-mouse IgG secondary antibodies (1 h; 1:80; MP Biomedicals, Solon, OH), 1 h MAP/HRP incubation [monoclonal mouse anti-peroxidase antibody (1:30; Medicorp, Montreal, Quebec, Canada) containing 5 μg/ml horseradish peroxidase (Sigma-Aldrich, St. Louis, MO) type IV], and final development with Vector SG kit. Sections were dehydrated and coverslipped using Permount mounting medium.
Digital imaging and quantification.
Bright-field digital images were captured using a Zeiss (Montreal, Quebec, Canada) Axioplan 2 imaging microscope, equipped with motorized stage and focus and a Zeiss AxioCamHRC digital camera coupled to the AxioVision 4 software program (Zeiss). For studies investigating presynaptic bouton density, digital images from lamina III of the midfrontal gyrus were obtained, because this particular laminar area has been associated with AD-related synaptic changes in previous studies (Paula-Barbosa et al., 1986; DeKosky and Scheff, 1990). Eight images were captured per section, and three sections were included per case, for a total of 24 images per person and 1584 quantified images. For studies involving dystrophic neurites, ∼24 images from laminas I–VI were included per case, for a total of 1056 pictures.
After image acquisition, photos were uploaded into the MCID Elite Image analysis system (Imaging Research, St. Catharine's, Ontario, Canada) and quantified essentially as described previously (Bell et al., 2003, 2006) and as depicted in Figures 1 and 3. Bright-field tagged image files were transformed into MCID digital files (see Figs. 1F, 3E), which improves the computer's ability to accurately isolate specific areas of interest from the background. This process is dependent on the establishment of specific detection parameters such as color [the detection of brown colored pixels (terminals) as opposed to blue (amyloid)], pixel saturation and intensity, form factor (roundness), and size (presynaptic boutons were detected if they were larger than 0.2 μm2 but <1 μm2, whereas dystrophic neurites were detected if they were larger than 1.25 μm2). These parameters were adjusted until the most accurate level of overlap between immunohistochemical staining and computer detection was obtained. As described previously (Bell et al., 2003, 2006; Hu et al., 2003), an area of 8200 μm2 was scanned, areas which met the established parameters (terminals) were detected by the computer, tallied (quantified), and yielded in numeric format as the number of presynaptic boutons per square area or “presynaptic bouton density” (see Fig. 1E–G). For ease of presentation, we have presented our density data as the number of presynaptic boutons per 1000 μm2. The quantification of dystrophic neurite abundance differed from past protocols used by our group (Bell et al., 2003, 2006), because unlike in transgenic animal models, dystrophic neurites in the human AD pathology tend to reside inside the plaque border, as opposed to its periphery. As a result, we used a circular quantification approach, which allowed us to quantify dystrophic neurites both within and outside of the plaque border, as shown in Figure 3F. Because of the computer's difficulty in separating out individual dystrophic neurites, data are presented as “abundance,” which refers to the combined or total area occupied by dystrophic neurites within the quantified circular area. Note that the size of the circle has no bearing on yielded data because the specific detection parameters (color, saturation, intensity, roundness, and size) ensure that only dystrophic neurites are detected. Dystrophic neurite abundance was then correlated to resident plaque size, by tracing the plaque's outer border and scanning the enclosed inner area for size. As stated previously, K.F.S.B. was blind to the source of all material and, in an effort to reduce variability, was the only individual to perform quantification.
Figure 1.

Immunohistochemical staining of the glutamatergic presynaptic bouton sites in human midfrontal gyrus tissue using antibodies directed against the glutamatergic presynaptic bouton site-specific marker vesicular glutamate transporter 1. A–D, Stainings are from subjects with no cognitive impairment (A), mild cognitive impairment (B), mild AD (C), or severe AD (D). Note the elevation in terminal number in B, taken from a subject with mild cognitive impairment, and the decreased presynaptic bouton-immunoreactivities in C and D, taken from subjects with mild and severe AD, respectively. E–G, The quantification protocol used to determine glutamatergic presynaptic bouton density in the midfrontal gyrus. Original digital images as shown in E were transformed into a file type that increases the computer's accuracy of detection (F). This transformed file format is then quantified by the computer using precise inclusion and exclusion criteria (as described in Materials and Methods) to accurately detect the elements of interest. G, The quantified image, in which dark blue coloration indicates those elements that were quantified, and light blue coloration indicates elements that failed to meet the required criteria and were hence omitted. Quantified data are then tallied and yielded in number format. Scale bars: D (for A–D), G (for E–G), 10 μm.
Figure 3.

Double-immunohistochemical staining of glutamatergic dystrophic neurites (brown) and amyloid plaques (blue) in the midfrontal gyrus brain region. Note the predominant localization of dystrophic neurites to the inner areas of amyloid plaques, as opposed to the plaque periphery. Note also the increase in abundance of glutamatergic dystrophic neurites with increasing plaque size (A–C). D–F show the manner in which the relative abundance (total area of occupation) of glutamatergic dystrophic neurites was determined. E, The transformed file, which, as previously described, increases the computer's accuracy of detection. F, The encircled quantified area, in which dark blue coloration shows elements that met the required inclusion and exclusion criteria (size, color, intensity, saturation, and roundness) and were therefore included in the quantification. The light blue coloration shows elements that failed to match the necessary criteria and that were therefore excluded. Note the close level of overlap between computer detection and dystrophic neurite abundance, as well as the omission of smaller terminals or plaque fragments. Scale bar, 10 μm.
Statistics.
All comparisons between the four diagnostic groups (NCI, MCI, mAD, and sAD) were compared statistically via ANOVA, followed by Post hoc Tukey's comparison test to evaluate between subject group differences. Pearson's correlation test was used to examine relationships between the primary variables of interest. Comparisons between two independent variables were performed using independent Student's t tests. All statistical comparisons were performed using the GraphPad (San Diego, CA) Prism software program, and significance was set at p < 0.05.
Results
The use of highly specific antibodies against vesicular glutamate transporter-1 allowed, for the first time, the microscopic identification of glutamatergic presynaptic elements (boutons) and glutamatergic dystrophic neurites in human AD brain material.
Elevated glutamatergic synapses in mild cognitive impairment, depletion with progression to Alzheimer's disease
Immunohistochemical staining of midfrontal gyrus brain tissue from persons with progressive stages of dementia indicated an apparent upregulation of glutamatergic presynaptic boutons during MCI and a depletion in number during mild- and severe-stage AD (Fig. 1A–D). Subsequent computer-assisted quantification (Fig. 1E,F) (described in detail in Materials and Methods) and statistical analysis confirmed a significant between-group difference in glutamatergic presynaptic bouton density across the four cohort groupings (ANOVA, F(3,62) = 25.57; p < 0.0001) (Fig. 2A). More specifically, Tukey's post hoc comparison analysis confirmed our qualitative observations; persons with MCI displayed a significant and paradoxical elevation in glutamatergic presynaptic bouton density (p < 0.05) (Fig. 2A), whereas subjects with mAD and sAD showed a significant depletion in density (p < 0.05 and p < 0.001, respectively) (Fig. 2A).
Figure 2.
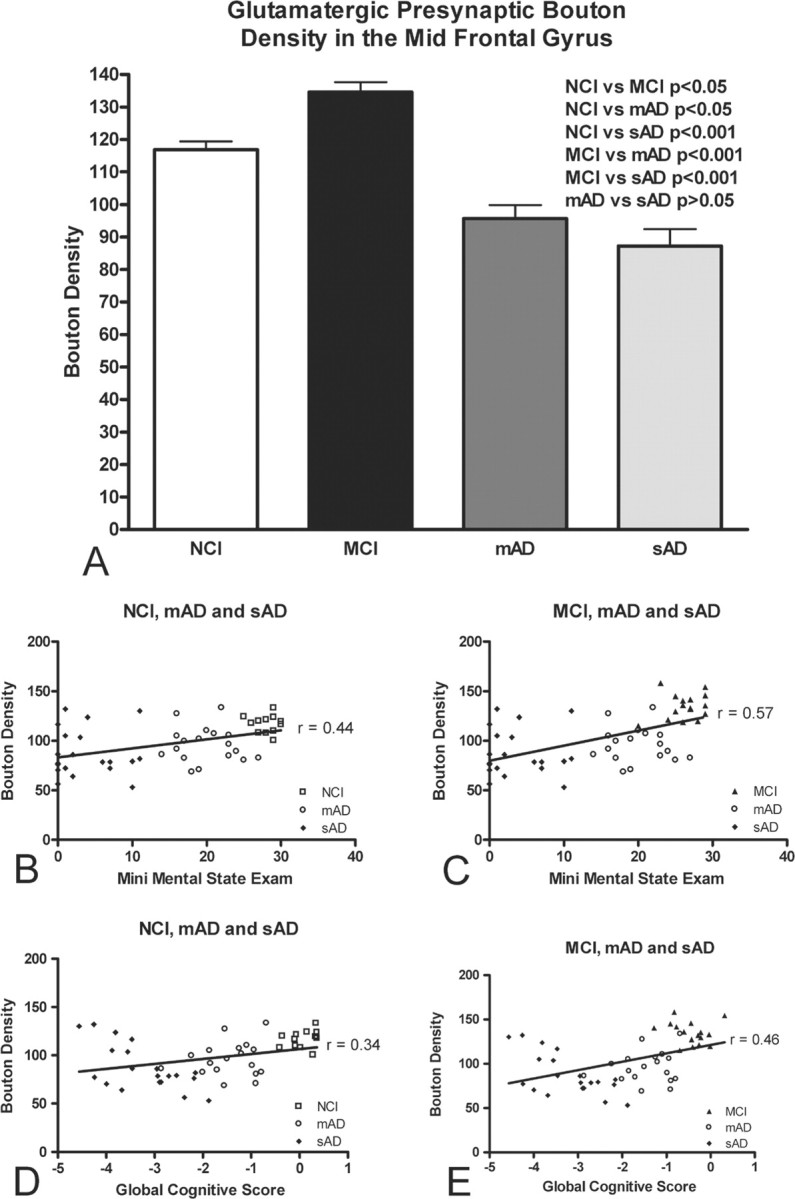
Graphic depiction of the relationship between glutamatergic presynaptic bouton density (number of presynaptic boutons per 1000 μm2 area) and cognitive ability. A, Mean midfrontal gyrus glutamatergic presynaptic bouton densities in subjects grouped by cognitive ability (NCI, MCI, mAD, and sAD). Means were compared by ANOVA followed by post hoc Tukey's test. B–E, Positive associations between increased glutamatergic presynaptic bouton density and cognitive performance for both the Mini Mental State Examination (B, C) and Global Cognitive Test score (D, E). Data were grouped by cognitive ability as NCI, mAD, and sAD (B, D) or MCI, mAD, and sAD (C, E). Note that cognitive performance declines with decreasing glutamatergic presynaptic bouton density across all groupings and test types.
Glutamatergic presynaptic bouton density correlates to cognitive function
To determine whether glutamatergic presynaptic bouton density might influence cognitive ability, glutamatergic presynaptic bouton density was correlated with GCS and MMSE scores obtained proximate to death. The unexpected upregulation in density during MCI prevents the possibility of performing a correlation analysis with all four groupings. Thus, only three of the four diagnostic groupings were evaluated at a time (either NCI, mAD, and sAD or MCI, mAD, and sAD). Given that this approach is somewhat unconventional, all correlations throughout the paper are performed using an identical manner of grouping. Correlation analyses reveal a significant relationship between cognitive ability (MMSE and GCS) and glutamatergic presynaptic bouton density (Fig. 2B–E, Table 2). Increased glutamatergic presynaptic bouton density correlates with improved cognition in all groups, save for persons with MCI, in which the increase in glutamatergic presynaptic bouton density is paradoxically associated with a decreased cognitive ability. Correlation analyses investigating the MCI and NCI groups alone only show a significant correlation when glutamatergic presynaptic bouton density is correlated with GCS (r = −0.4, p < 0.05; for correlation with MMSE, r = −0.1; p > 0.05).
Table 2.
Correlation between pathological and clinical markers of the Religious Orders Study Cohort
| Pathological parameter | MMSE score |
Global cognitive score |
||
|---|---|---|---|---|
| NCI, mAD, sAD | MCI, mAD, sAD | NCI, mAD, sAD | MCI, mAD, sAD | |
| VGluT PB density | r = 0.44*; p < 0.01 | r = 0.57*; p < 0.0001 | r = 0.34; p < 0.05 | r = 0.46*; p < 0.001 |
| Amyloid plaque load | r = −0.42; p < 0.01 | r = −0.42; p < 0.01 | r = −0.43*; p < 0.01 | r = −0.44; p < 0.001 |
| NFT load | r = −0.35; p < 0.05 | r = −0.33; p < 0.05 | r = −0.38; p < 0.01 | r = −0.36; p < 0.01 |
| Neuritic plaque number | r = −0.31; p < 0.05 | r = −0.31; p < 0.05 | r = −0.34; p < 0.05 | r = −0.34; p < 0.05 |
| Diffuse plaque number | r = −0.27; p > 0.05 | r = −0.14; p > 0.05 | r = −0.30; p < 0.05 | r = −0.15; p > 0.05 |
| NFT number | r = −0.40; p < 0.01 | r = −0.42; p < 0.01 | r = −0.41; p < 0.01 | r = −0.43; p < 0.01 |
PB, Presynaptic bouton; NFT, neurofibrillary tangle; r, Pearson correlation factor;
*represents strongest correlation across parameter of interest.
Hallmark pathological indices and cognitive ability
Hallmark pathological lesions of AD such as amyloid plaques and neurofibrillary tangles are widely accepted as contributing factors to resultant cognitive ability. In an effort to determine whether glutamatergic presynaptic bouton density might also contribute to clinical presentation, we compared the strength of our correlations to those obtained from the more traditional indices of the AD pathology (Table 2). As stated earlier, to remain consistent with the above previous analyses, cohort groupings were performed in an identical manner (e.g., NCI, mAD, and sAD or MCI, mAD, and sAD). For both groupings, significant inverse correlations were observed between the number of neuritic plaques, the number of neurofibrillary tangles, the total amyloid load, and the density of PHF-containing tau tangles and both MMSE and GCS cognitive assessment tests (Table 2). Diffuse plaque number failed to correlate with MMSE and was only found to significantly correlate with GCS in the NCI, mAD, and sAD grouping (Table 2). Interestingly, glutamatergic presynaptic bouton density was a stronger correlate for MMSE scores than any of the plaque- or tangle-based parameters. This was also the case for correlations between GCS and glutamatergic presynaptic bouton density in the MCI inclusive grouping. Although our results do not prove a causative link between decreased glutamatergic presynaptic bouton and decreased neurotransmission, the strength of the observed correlation (e.g., greater than that obtained with more traditional pathological markers widely accepted as having profound influence on cognition) would suggest that the novel pattern of cortical remodeling observed here would most likely contribute to AD clinical presentation.
Glutamatergic dystrophic neurite abundance correlates with resident plaque size
Persons diagnosed as NCI rarely displayed plaques and were hence omitted from analyses involving glutamatergic dystrophic neurite abundance. In the included MCI, mAD, and sAD cohorts, glutamatergic dystrophic neurites were most often located within or on the border of amyloid plaques and were only very rarely visible in the plaque-free neuropile (Fig. 3A–C). Qualitative observations suggested that larger plaques were surrounded by more glutamatergic dystrophic neurites than smaller plaques (Fig. 3A–C). To confirm this, we quantified the abundance of glutamatergic dystrophic neurites (demonstrated in Fig. 3D–F) (described in Materials and Methods) and correlated the obtained value with resident plaque size. This analysis revealed a significant association between larger plaque size and increasing glutamatergic dystrophic neurite abundance (r = 0.62, p < 0.0001, for MCI, mAD, and sAD combined) (Fig. 4D). This is particularly intriguing given the recent finding that fibrillar Aβ causes the LIM kinase 1-dependent inactivation of both actin-depolymerizing factor and cofilin, ultimately leading to dystrophic neurite formation (Heredia et al., 2006). Interestingly, the strength of the correlation was found to increase with disease progression, as seen by comparing the strength of the correlation factors when each grouping is investigated separately (MCI, r = 0.36, p < 0.0001; mAD, r = 0.67, p < 0.0001; sAD, r = 0.69, p < 0.0001) (Fig. 4A–C). This finding plausibly stems from the fact that plaques increase in size with age (Tsai et al., 2004) and in number with disease progression, ultimately serving to increase the concentration of extracellular fibrillar Aβ.
Figure 4.

Graphic depiction of the association between glutamatergic dystrophic neurite abundance and amyloid plaque size in the midfrontal gyrus of subjects with MCI (A), mAD (B), sAD (C), or all three subject groups combined (MCI, mAD, and sAD; D). Note the positive association between increasing plaque size and increasing glutamatergic dystrophic neurite abundance in all three groups. Note also that amyloid plaque size appears to increase with disease progression, as does the strength of the Pearson's correlation, thus suggesting that increasing levels of fibrillar Aβ correlate with increased glutamatergic terminal toxicity.
Discussion
This study brings to light a new role for the glutamatergic system in the AD and MCI pathologies. It also presents for the first time a comprehensive description of the structural changes occurring in glutamatergic synapses and glutamatergic dystrophic neurites as the AD pathology progresses from NCI to MCI (now considered prodromic of AD) and finally to mild- and severe-stage AD. Previous studies investigating the status of the glutamatergic system in AD were challenged by a lack of specific markers for glutamatergic neurons and as a result are often contradictory, do not provide data on the abundance of glutamatergic synapses, and do not demonstrate changes that occur in parallel with disease progression (because the majority of investigations involved very late-stage AD subjects). Moreover, the glutamatergic system has not been investigated in the context of MCI. To remedy this missing information, we took advantage of recently developed neuronal markers of glutamatergic terminals, specifically to characterize the status of the glutamatergic terminals across progressive stages of AD using cognitively and pathologically well characterized human brain material from the Religious Orders Study.
Our study demonstrates that the AD pathology includes the generation of glutamatergic dystrophic neurites, an event that is known to delay or reduce synaptic connectivity, ultimately contributing to impaired cognitive capacity. Indeed, the correlation between increasing plaque size and dystrophic neurite abundance becomes stronger with disease progression. Our observation that glutamatergic presynaptic boutons are significantly depleted in subjects with mild- and severe-stage AD compared with controls is in line with predicted outcomes. However, the unexpected and paradoxical observation of an elevation in glutamatergic presynaptic bouton density in persons with MCI is most intriguing. The majority of our correlations indicate that an increase in presynaptic bouton density equates with improved cognitive ability, which would agree with previous findings (Perry et al., 1978; Masliah et al., 1989; DeKosky and Scheff, 1990; Scheff et al., 1990; Terry et al., 1991; DeKosky et al., 1992; Masliah and Terry, 1993) [for review, see Terry (1996, 1997)]. Indeed, glutamatergic presynaptic bouton density was a better pathological correlate of cognitive capacity than the more classically associated plaque- and tangle-based lesions of AD, in almost all of the investigated parameters. This would suggest that the decrease in glutamatergic presynaptic bouton density would likely contribute to the decline in cognitive capacity exhibited by AD patients; however, complimentary experimental approaches in transgenic models would be required to demonstrate this unequivocally. Our finding that the MCI subjects (who by virtue of their grouping designation are mildly cognitively impaired) actually have more midfrontal glutamatergic presynaptic boutons than the NCI group is remarkable.
This elevation in presynaptic terminal number can essentially be interpreted in one of two ways. Either the upregulation indicates a type of compensatory upregulation intended to counter the effects of preexisting synaptotoxicity (potentially Aβ induced), or, alternatively, the upregulated terminals are indicative of an uncoordinated aberrant response not representative of concerted synaptic plasticity. We believe the former possibility to be the case, given our previous observation that transgenic animal models of the amyloid pathology also display an upregulation of glutamatergic, cholinergic, and GABAergic presynaptic boutons during early stages of the amyloid pathology (Bell et al., 2003; Hu et al., 2003). Moreover, like the glutamatergic system, the cholinergic system displays a transient upregulation during the MCI state, as shown by the increase in choline acetyl transferase (ChAT) activity in persons with MCI (DeKosky et al., 2002). In fact, Ikonomovic et al. (2003) identified a pattern of hippocampal ChAT activity across progressive stages of dementia that parallels our present findings: an upregulation in subjects with MCI followed by a successive depletion with progression to mild- and severe-stage AD.
Amyloid has been shown to reduce potassium-evoked acetylcholine release (Kar et al., 1996) and glutamatergic transmission (Kamenetz et al., 2003) and has recently been shown to promote internalization of NMDA receptors in cortical neurons (Snyder et al., 2005). Therefore, a concerted upregulation of presynaptic terminals in areas in which pathology-induced synaptic disconnection is present would seem a plausible compensatory response if the aim is to improve transmission probability. A main caveat of our findings, however, is that the upregulation in presynaptic bouton number does not necessarily equate with an increase in glutamate release, which would ultimately be required if the upregulation was indeed intended to increase transmission probability. In this regard, however, it is interesting to note that the synaptic transmission- and transport-related genes GAP-43 and VGluT1 become upregulated during MCI (Williams et al., 2006), whereas GAP-43 (Callahan et al., 1994), VGluT1 (Kirvell et al., 2006), and synaptophysin (Terry et al., 1991; Callahan et al., 2002) protein levels are decreased in AD. Furthermore, imaging studies have demonstrated a significant upregulation in medial temporal lobe activation in subjects with MCI, but not in subjects with NCI or AD (Dickerson et al., 2005), and subjects with MCI display a higher number of activation changes (indicative of increased brain function) in a larger number of brain structures (including the cortex) than subjects with AD, suggesting that the synapses in these brain areas are indeed more active during MCI than in AD (Goekoop et al., 2006). Finally, the TgCRND8 transgenic mouse model displays increased levels of both EPSP and LTP (Jolas et al., 2002), at an age at which our group detected a significant upregulation of glutamatergic presynaptic boutons in this same model (Bell et al., 2003), thus highlighting the probability that the upregulation in terminal number is indeed functional and likely represents a compensatory response to the Aβ-induced internalization of NMDA receptors (Snyder et al., 2005).
The presented findings are also relevant given the recent approval of NMDA receptor antagonist memantine for the treatment of moderate-to-severe AD, the only noncholinergic-based drug currently approved to treat AD. Moreover, given that LTP is a key factor in memory and learning, the very functions affected by AD (Greenamyre et al., 1988), our data are highly relevant. In addition to the potential benefit of increasing transmission probability, it must also be noted that elevated levels of extracellular glutamate might also increase excitotoxicity [an event with established links to the cognitive impairment and neuronal death visible in AD (Greenamyre et al., 1988)] or activate extrasynaptic prodeath NMDA receptors (for review, see Hardingham and Bading, 2003), potentially contributing to the subsequent depletion in terminal numbers seen with progression to AD. In short, our report illustrates an important and hitherto underappreciated involvement of the cortical glutamatergic terminals in the AD pathology. The findings support the possibility of a compensatory upregulation of cortical terminals during the MCI state and highlight the valuable therapeutic window offered therein to prevent potential neuronal damage occurring upstream of clinical AD diagnosis.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP-37996 (A.C.C.) and National Institute on Aging Grants P30AG10161 and R01AG15819 (D.A.B.). K.F.S.B. was a recipient of a CIHR Doctoral Research Award, and A.C.C. is the holder of the Charles E. Frosst Merck endowed Chair. We thank the many participants of the Religious Orders Study and the staff of the Rush Alzheimer's Disease Center.
References
- Barnes LL, Wilson RS, Schneider JA, Bienias JL, Evans DA, Bennett DA. Gender, cognitive decline, and risk of AD in older persons. Neurology. 2003;60:1777–1781. doi: 10.1212/01.wnl.0000065892.67099.2a. [DOI] [PubMed] [Google Scholar]
- Bell KF, de Kort GJ, Steggerda S, Shigemoto R, Ribeiro-da-Silva A, Cuello AC. Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci Lett. 2003;353:143–147. doi: 10.1016/j.neulet.2003.09.027. [DOI] [PubMed] [Google Scholar]
- Bell KF, Ducatenzeiler A, Ribeiro-da-Silva A, Duff K, Bennett DA, Cuello AC. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol Aging. 2006;27:1644–1657. doi: 10.1016/j.neurobiolaging.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Aggarwal NT, Arnold SE, Cochran EJ, Berry-Kravis E, Bienias JL. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology. 2003;60:246–252. doi: 10.1212/01.wnl.0000042478.08543.f7. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–384. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Bienias JL, Evans DA, Wilson RS. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–841. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- Callahan LM, Selski DJ, Martzen MR, Cheetham JE, Coleman PD. Preliminary evidence: decreased GAP-43 message in tangle-bearing neurons relative to adjacent tangle-free neurons in Alzheimer's disease parahippocampal gyrus. Neurobiol Aging. 1994;15:381–386. doi: 10.1016/0197-4580(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:384–395. doi: 10.1093/jnen/61.5.384. [DOI] [PubMed] [Google Scholar]
- Cowburn R, Hardy J, Roberts P, Briggs R. Presynaptic and postsynaptic glutamatergic function in Alzheimer's disease. Neurosci Lett. 1988a;86:109–113. doi: 10.1016/0304-3940(88)90192-9. [DOI] [PubMed] [Google Scholar]
- Cowburn R, Hardy J, Roberts P, Briggs R. Regional distribution of pre- and postsynaptic glutamatergic function in Alzheimer's disease. Brain Res. 1988b;452:403–407. doi: 10.1016/0006-8993(88)90048-0. [DOI] [PubMed] [Google Scholar]
- Cross AJ, Slater P, Simpson M, Royston C, Deakin JF, Perry RH, Perry EK. Sodium dependent D-[3H]aspartate binding in cerebral cortex in patients with Alzheimer's and Parkinson's diseases. Neurosci Lett. 1987;79:213–217. doi: 10.1016/0304-3940(87)90699-9. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Harbaugh RE, Schmitt FA, Bakay RA, Chui HC, Knopman DS, Reeder TM, Shetter AG, Senter HJ, Markesbery WR. Cortical biopsy in Alzheimer's disease: diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Intraventricular Bethanecol Study Group. Ann Neurol. 1992;32:625–632. doi: 10.1002/ana.410320505. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, Cochran EJ, Kordower JH, Mufson EJ. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison DW, Beal MF, Mazurek MF, Bird ED, Martin JB. A postmortem study of amino acid neurotransmitters in Alzheimer's disease. Ann Neurol. 1986;20:616–621. doi: 10.1002/ana.410200510. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Chang-Chui H, Cooper SM, Lott IT, Cotman CW. Density and distribution of NMDA receptors in the human hippocampus in Alzheimer's disease. Brain Res. 1986;399:156–161. doi: 10.1016/0006-8993(86)90611-6. [DOI] [PubMed] [Google Scholar]
- Goekoop R, Scheltens P, Barkhof F, Rombouts SA. Cholinergic challenge in Alzheimer patients and mild cognitive impairment differentially affects hippocampal activation—a pharmacological fMRI study. Brain. 2006;129:141–157. doi: 10.1093/brain/awh671. [DOI] [PubMed] [Google Scholar]
- Grant SM, Ducatenzeiler A, Szyf M, Cuello AC. Abeta immunoreactive material is present in several intracellular compartments in transfected, neuronally differentiated, P19 cells expressing the human amyloid beta-protein precursor. J Alzheimers Dis. 2000;2:207–222. doi: 10.3233/jad-2000-23-403. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, Young AB, D'Amato CJ, Hicks SP, Shoulson I. Alterations in L-glutamate binding in Alzheimer's and Huntington's diseases. Science. 1985;227:1496–1499. doi: 10.1126/science.2858129. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Penney JB, D'Amato CJ, Young AB. Dementia of the Alzheimer's type: changes in hippocampal L-[3H]glutamate binding. J Neurochem. 1987;48:543–551. doi: 10.1111/j.1471-4159.1987.tb04127.x. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Maragos WF, Albin RL, Penney JB, Young AB. Glutamate transmission and toxicity in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12:421–430. doi: 10.1016/0278-5846(88)90102-9. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cowburn R, Barton A, Reynolds G, Lofdahl E, O'Carroll AM, Wester P, Winblad B. Region-specific loss of glutamate innervation in Alzheimer's disease. Neurosci Lett. 1987;73:77–80. doi: 10.1016/0304-3940(87)90034-6. [DOI] [PubMed] [Google Scholar]
- Heredia L, Helguera P, de Olmos S, Kedikian G, Sola VF, LaFerla F, Staufenbiel m, de Olmos J, Busciglio J, Caceres A, Lorenzo A. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid β-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer's disease. J Neurosci. 2006;26:6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Wong TP, Cote SL, Bell KF, Cuello AC. The impact of Abeta-plaques on cortical cholinergic and non-cholinergic presynaptic boutons in Alzheimer's disease-like transgenic mice. Neuroscience. 2003;121:421–432. doi: 10.1016/s0306-4522(03)00394-4. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR. Alzheimer's disease: glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol. 1987;22:37–40. doi: 10.1002/ana.410220110. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Mufson EJ, Wuu J, Cochran EJ, Bennett DA, DeKosky ST. Cholinergic plasticity in hippocampus of individuals with mild cognitive impairment: correlation with Alzheimer's neuropathology. J Alzheimers Dis. 2003;5:39–48. doi: 10.3233/jad-2003-5106. [DOI] [PubMed] [Google Scholar]
- Jolas T, Zhang XS, Zhang Q, Wong G, Del Vecchio R, Gold L, Priestley T. Long-term potentiation is increased in the CA1 area of the hippocampus of APP(swe/ind) CRND8 mice. Neurobiol Dis. 2002;11:394–409. doi: 10.1006/nbdi.2002.0557. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kar S, Seto D, Gaudreau P, Quirion R. β-Amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal slices. J Neurosci. 1996;16:1034–1040. doi: 10.1523/JNEUROSCI.16-03-01034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirvell SL, Esiri M, Francis PT. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem. 2006;98:939–950. doi: 10.1111/j.1471-4159.2006.03935.x. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry R. The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathol. 1993;3:77–85. doi: 10.1111/j.1750-3639.1993.tb00728.x. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett. 1989;103:234–239. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- McKee AC, Kosik KS, Kowall NW. Neuritic pathology and dementia in Alzheimer's disease. Ann Neurol. 1991;30:156–165. doi: 10.1002/ana.410300206. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Geddes JW, Yao D, Chung C, Cotman CW. [3H]TCP binding sites in Alzheimer's disease. Neurosci Lett. 1987;73:197–200. doi: 10.1016/0304-3940(87)90017-6. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Procter AW, Stratmann GC, Bowen DM. Excitatory amino acid-releasing and cholinergic neurones in Alzheimer's disease. Neurosci Lett. 1986;66:199–204. doi: 10.1016/0304-3940(86)90190-4. [DOI] [PubMed] [Google Scholar]
- Paula-Barbosa MM, Saraiva A, Tavares MA, Borges MM, Verwer RW. Alzheimer's disease: maintenance of neuronal and synaptic densities in frontal cortical layers II and III. Acta Neurol Scand. 1986;74:404–408. doi: 10.1111/j.1600-0404.1986.tb03533.x. [DOI] [PubMed] [Google Scholar]
- Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procter AW, Palmer AM, Bowen DM, Murphy E, Neary D. Glutamatergic denervation in Alzheimer's disease—a cautionary note. J Neurol Neurosurg Psychiatry. 1987;50:825. doi: 10.1136/jnnp.50.6.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Muramoto O, Kanazawa I, Arai H, Kosaka K, Iizuka R. Regional distribution of amino acid transmitters in postmortem brains of presenile and senile dementia of Alzheimer type. Ann Neurol. 1986;19:263–269. doi: 10.1002/ana.410190307. [DOI] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Smith CC, Bowen DM, Sims NR, Neary D, Davison AN. Amino acid release from biopsy samples of temporal neocortex from patients with Alzheimer's disease. Brain Res. 1983;264:138–141. doi: 10.1016/0006-8993(83)91131-9. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R. Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature. 2000;407:189–194. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci. 2005;8:977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- Terry RD. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- Terry RD. The pathology of Alzheimer's disease: numbers count. Ann Neurol. 1997;41:7. doi: 10.1002/ana.410410104. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Williams C, Shai RM, Wu Y, Miller C. Expression of mRNAs regulating local translation in synaptic terminals of MCI and early AD brain. Neurobiol Aging. 2006;2:S505. [Google Scholar]
- Wilson RS, Beckett LA, Barnes LL, Schneider JA, Bach J, Evans DA, Bennett DA. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging. 2002;17:179–193. [PubMed] [Google Scholar]
- Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer's disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]